

Abstract
Background & Aims:
Many genetic and environmental factors, including family history, dietary fat, and inflammation, increase risk for colon cancer development. Peroxisome proliferator-activated receptor alpha (PPARα) is a nuclear receptor that regulates systemic lipid homeostasis. We explored the role of intestinal PPARα in colon carcinogenesis.
Methods:
Colon cancer was induced in mice with intestine-specific disruption of Ppara (PparaΔIE), Pparafl/fl (control), and mice with disruption of Ppara that express human PPARA (human PPARA transgenic mice), by administration of azoxymethane with or without dextran sulfate sodium (DSS). Colons were collected from mice and analyzed by immunoblots, quantitative PCR, and histopathology. Liquid chromatography coupled with mass spectrometry-based metabolomic analyses were performed on urine and colons. We used molecular biology and biochemical approaches to study mechanisms in mouse colons, primary intestinal epithelial cells, and colon cancer cell lines. Gene expression data and clinical features of patients with colorectal tumors were obtained from Oncomine, and human colorectal tumor specimens and adjacent normal tissues were collected and analyzed by immunohistochemistry.
Results:
Levels of Ppara mRNA were reduced in colon tumors from mice. PparaΔIE mice developed more and larger colon tumors than control mice following administration of azoxymethane, with or without DSS. Metabolomic analyses revealed increases in methylation-related metabolites in urine and colons from PparaΔIE mice, compared with control mice, following administration of azoxymethane, with or without DSS. Levels of DNA methyltransferase 1 (DNMT1) and protein arginine methyltransferase 6 (PRMT6) were increased in colon tumors from PparaΔIE mice, compared with colon tumors from control mice. Depletion of PPARα reduced the expression of retinoblastoma protein (RB1), resulting in increased expression of DNMT1 and PRMT6. DNMT1 and PRMT6 decreased expression of the tumor suppressor genes Cdknla (P21) and Cdknlb (p27) via DNA methylation and histone H3R2 dimethylation-mediated repression of transcription, respectively. Fenofibrate protected human PPARA transgenic mice from azoxymethane and DSS-induced colon cancer. Human colon adenocarcinoma specimens had lower levels of PPARA and RB1 and higher levels of DNMT1 and PRMT6 than normal colon tissues.
Conclusions:
Loss of PPARα from the intestine promotes colon carcinogenesis by increasing DNMT1-mediated methylation of P21 and PRMT6-mediated methylation of p27 in mice. Human colorectal tumors have lower levels of PPARA mRNA and protein than non-tumor tissues. Agents that activate PPARα might be developed for chemoprevention or treatment of colon cancer.
Keywords: mouse model, transcriptional regulation, epigenetic
Lay Summary
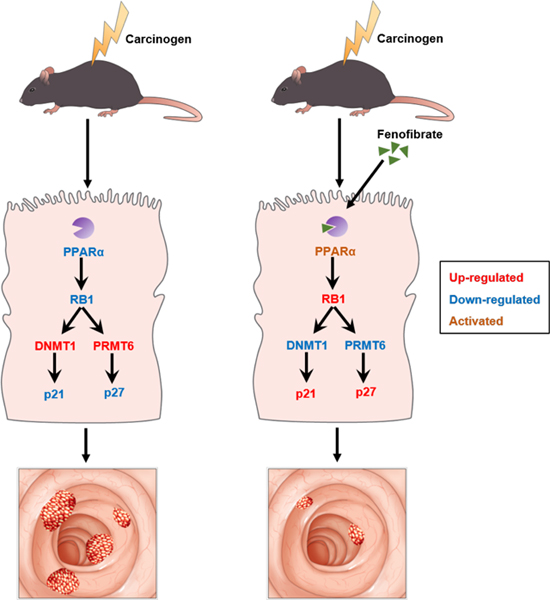
Intestine-specific PPARα deficiency promotes colon carcinogenesis through modulation of DNMT1-mediated methylation of P21 and PRMT6-mediated methylation of P27.
Graphical abstract
Introduction
Colorectal cancer (CRC) is the third most common cancer and is the third leading cause of cancer death in the United States according to 2018 American Cancer Society statistics.1 Colorectal cancer is associated with diverse etiological components including both genetic and environmental factors. The most commonly found genetic mutations include APC (adenomatous polyposis coli), KRAS, and TP53.2 Environmental factors, including inflammation, obesity, diabetes, and diet also play important roles in colon cancer.3, 4 Currently the major treatment option for CRC is a combination of colectomy with chemotherapy employing agents such as 5-fluorouracil (5-FU). Unfortunately, the 5-y survival rate for metastatic colon cancer is only about 13.3%.5 Therefore, it is necessary to further understand the cancer biology driving colon carcinogenesis in order to identify novel drug targets for both the prevention and treatment of this disease. Although significant efforts have been undertaken to understand the pathogenesis of colon cancer, the molecular mechanisms are not fully understood.
Peroxisome proliferator-activated receptor α (PPARα) is a nuclear receptor that serves as a xenobiotic and lipid sensor to regulate energy combustion, lipid homeostasis, and inflammation.6 PPARα is a ligand-dependent transcription factor and forms a heterodimer with the obligate nuclear receptor, retinoid X receptor (RXR).7 The PPARα-RXR heterodimer binds to peroxisome proliferator response elements (PPREs) located in the promotor region of target genes. Fatty acids are endogenous ligands for PPARα and the synthetic fibrate class of agonists, which includes fenofibrate and gemfibrozil, have been used for several decades as hypolipidemic therapeutic agents.8 In addition to their well-known lipid-lowing effect, pharmacological agents that target PPARα may have therapeutic applications in cancer and inflammatory diseases. Conflicting results have emerged regarding the role of PPARα in colon carcinogenesis. Several reports have shown the potential beneficial chemopreventive effect of PPARa ligands on colon carcinogenesis,9–13 while two other studies suggested a cancer-promoting role for PPARα ligands.14, 15 These results illustrate that the precise influence of PPARα on colon carcinogenesis remains elusive.
PPARα is expressed in several tissues including liver, kidney, adipose, and intestine. A comprehensive analysis of PPARα’s role in inflammatory diseases and tumorigenesis can only be achieved by considering the tissue-specific context of PPARα expression. In the current study, villin-cre Pparafl/fl (PparaΔIE) mice were generated and the role of intestinal PPARα in colon carcinogenesis was first studied.
Methods
Animals and Chemicals
Villin-cre mice were obtained from Deborah L. Gumucio, University of Michigan.16 Pparafl/fl mice, on the C57BL/6N genetic background, were generated as previously described.17 Pparafl/fl mice were crossed with Villin-cre mice to produce intestine-specific Ppara null mice (PparaΔIE). Human PPARA transgenic mice, on the Sv129 background, with the complete human PPARA transgene and the mouse Ppara-null allele, were described previously.18 Mice were maintained under a standard 12-h light/12-h dark cycle with water and chow provided ad libitum. All animal experiments were carried out in accordance with Institute of Laboratory Animal Resources guidelines and protocols approved by the National Cancer Institute Animal Care and Use Committee. AOM was purchased from Syncon (Anaheim, CA). 5-Aza-2’-deoxycytidine (5-Aza) and PD0332991 were purchased from Sigma-Aldrich (St. Louis, MO) and DSS (molecular mass 36,000–50,000 Da) was from MP Biochemicals (Solon, OH). EPZ020411 (EPZ) was purchase from Cayman Chemical (Ann Arbor, MI). EGF, R-Spondin 1, and Noggin were purchase from PeproTech (Rocky Hill, NJ).
Animal Studies
For the AOM model, 8- to 10-week-old Pparafl/fl and PparaΔIE mice were intraperitoneally injected with sterile saline with or without AOM (10 mg/kg.bw) once a week for 6 weeks. Urine samples were collected at month 1, 3, and 5 after the last AOM injection by placing mice in metabolic cages for 24 hours. Mice were killed by CO2 asphyxiation 5 months after the last AOM injection then serum samples and colons were collected. For the AOM and DSS model, 8- to 10-week-old Pparafl/fl and PparaΔIE mice were divided into a control group and an AOM/DSS group. Mice in the AOM/DSS group were intraperitoneally injected with 10 mg/kg.bw AOM followed by 1.5% DSS in drinking water for 1 week and then given regular drinking water, while mice in the control group received a saline injection and administered regular drinking water. At 12 weeks after AOM administration, all mice were killed and serum samples and colons were collected. To determine the effect of fenofibrate on colon carcinogenesis, human PPARA transgenic mice were intraperitoneally injected with 5 mg/kg.bw AOM followed by 1.5% DSS in drinking water for 1 week. Mice were fed either standard NIH31 diet or a modified NIH31 diet containing 0.5% fenofibrate (Bioserv, Frenchtown, NJ) ad libitum throughout the study. At 17 weeks after AOM administration, all mice were killed and serum samples and colons were collected.
Analysis of Human Patient Datasets from Oncomine
Gene expression data and clinical features of patients were downloaded from Oncomine (https://www.oncomine.com). 90 normal colons and 284 colon adenocarcinomas were included in TCGA Colorectal 2 dataset and PPARA gene copy number was analyzed by reporter 22–044971739. 28 normal colons and 50 colon adenocarcinomas were included in Ki colon dataset. The expression of PPARA and ACOX1 were analyzed by reporter AI380344 and H65660, respectively.
Human Cohort
Colorectal mucosal biopsies were taken from five groups of patients: (1) hyperplastic polyp, n=5; (2) low grade adenoma, n=19; (3) high grade adenoma, n=17; (4) adenocarcinoma in adenoma, n=10; (5) adenocarcinoma, n=16. Normal mucosae were taken 5 cm away from the adenocarcinoma foci in part of the patients diagnosed with colon adenocarcinoma. Genders and ages are summarized in Supplementary Figure 5. This study was approved by the Conjoint Health Research Ethics Board of the Shanghai Tenth People’s Hospital of Tongji University, China, and written informed consent was given to all individuals before participation in this study. All individuals fulfilled the following inclusion criteria: (i) no familial adenomatous polyposis; (ii) no inflammatory bowel disease; (iii) no thyroid dysfunction; (iv) no Lynch syndrome; (v) no pregnancy; and (vi) do not have a condition that is unsuitable for biopsy as judged by the clinician. The biopsies were used for immunohistochemical staining.
Statistics
Statistical analysis was performed using Prism version 7.0 (GraphPad Software, San Diego, CA). Experimental values are presented as mean ± S.E.M. Statistical significance between two groups was determined using two-tailed Student’s t-test. One-way ANOVA followed by Tukey’s post hoc correction was applied for multi-group comparisons. The statistical significance of gene expression in matched tumors and adjacent non-tumor tissues were calculated using two-tailed paired t-test. A P-value less than 0.05 was considered statistically significant.
Results
PPARα Expression in Human and Mouse Colon Tumors
To determine the expression of PPARα in human CRC, two online datasets including gene expression data and clinical features of patients were downloaded from Oncomine (https://www.oncomine.com). Analysis of the online TCGA Colorectal 2 dataset2 revealed that PPARA gene copy number was lower in human colon adenocarcinomas (n=284) compared with normal colon tissues (n=90), and tended to decrease with disease stage progression (Figure 1A and B). Another online Ki colon dataset19 showed that the expressions of PPARA mRNA, and its target gene ACOX1 mRNA were suppressed in colon adenocarcinomas (n=50) compared with normal colon tissues (n=28) (Figure 1 C and D). To measure the protein levels of PPARα in human CRC, colorectal-tumor specimens collected from patients at different stages of CRC were used for IHC staining with an anti-PPARα antibody. Compared with normal mucosae (n=11), PPARα tended to decrease without significance at low grade adenomas (n=19), while significantly down-regulated at high grade adenomas (n=17), adenocarcinomas in adenomas (n=10), and adenocarcinomas (n=14). No significant change was observed in hyperplastic polyps (n=5) (Figure 1 E and F). Consistent with the human data, the mRNA and protein levels of PPARα and ACOX1 were significantly lower in the AOM-induced mouse colon tumors as compared to adjacent non-tumor tissues (Figure 1G and H).
Figure 1. The expression of PPARα was decreased in human and mouse colon tumors.

(A) Analysis of PPARA gene expression from the online TCGA Colorectal 2 dataset and (B) differential PPARA gene expression based on disease stages. Analysis of PPARA (C) and ACOX1 (D) gene expression from the online Ki colon dataset. (E) Representative IHC staining of PPARα on human normal mucosa and adenocarcinoma. Scale bars: upper, 300 μm; bottom, 200 μm. (F) The IHC staining intensity of PPARα on human colon specimens. (G) Ppara and Acoxl mRNA levels and (H) PPARα and ACOX1 protein levels in mouse colon tumors (T) and adjacent non-tumor tissues (NT). N = 5/group, for mRNA analysis. * P< 0.05, ** P< 0.01, *** P< 0.001.
Intestinal PPARα Deficiency and Colon Carcinogenesis
To investigate the role of intestinal PPARα in colon carcinogenesis, villin-cre Pparafl/fl (PparaΔIE) mice were generated by crossing villin-cre mice with Pparafl/fl mice. A decrease in Ppara mRNA level was found in the small intestine and colon of PparaΔIE mice compared to Pparafl/fl mice, but not in extra-intestinal tissues (Supplementary Figure 1A). Western blot analysis of colon extracts revealed no significant PPARα protein levels in PparaΔIE mice (Supplementary Figure 1B). The colonic epithelia of PparaΔIE mice remained morphologically normal and showed similar proliferation as Pparafl/fl mice (Supplementary Figure 1C and D).
AOM administration resulted in colon cancer, while mice on the C57BL/6J genetic background were relatively resistant to AOM-induced colon carcinogenesis.20 In agreement with this result, the current study showed that a standard AOM administration regime in Pparafl/fl mice only resulted in 36.8% tumor incidence compared to a rate of 57.9% in PparaΔIE mice (Supplementary Figure 1). Total tumor multiplicity in PparaΔIE mice administered with AOM was 1.47 compared with 0.58 in Pparafl/fl mice administered with AOM (Figure 2D). Quantitatively, 15.8% Pparafl/fl and 52.6% PparaΔIE mice administered with AOM had > 3mm diameter tumors, while 31.6% Pparafl/fl and 42.1% PparaΔIE mice administered with AOM had < 3mm diameter tumors (Figure 2E and F). Among the mice bearing colon tumors, the prevalence of low-grade adenomas, high-grade adenomas and adenocarcinomas in AOM-administered Pparafl/fl mice were 57.1%, 28.6%, and 14.3%, respectively, while the corresponding ratios in AOM-administered PparaΔIE mice were 45.4%, 27.3% and 27.3% (Figure 2B). Colon lengths were similar in AOM-administered Pparafl/fl and PparaΔIE mice (Figure 2C). These results suggested that intestinal PPARα deficiency increased AOM-induced colon tumorigenesis and tumor growth.
Figure 2. Intestine-specific knockout of PPARα increased AOM and AOM and DSS induced colon carcinogenesis.

For AOM model, (A) Representative gross pictures (upper) and H&E staining (bottom) of colon sections. Scale bars: upper, 1.5 mm; bottom, 100 μm. (B) Prevalence of low-grade adenomas, high-grade adenomas and adenocarcinomas. (C) Colon length. (D) Tumor number. (E) Number of tumors with diameters > 3 mm. (F) Number of tumors with diameters < 3mm. For AOM and DSS model, (G) Colon length. (H) Tumor number. (I) Prevalence of low-grade adenomas, high-grade adenomas and adenocarcinomas. (J) Representative gross pictures (upper) and H&E staining (bottom) of colon sections. Scale bars: upper, 1.5 mm; bottom, 100 μm. * P< 0.05.
To investigate whether these changes were model dependent, another experimental colitis-mediated mouse colon cancer model generated by administration of AOM followed by DSS was used. AOM combined with DSS resulted in 100% incidence of colon tumors in both Pparafl/fl and PparaΔIE mice (Supplementary Figure 2). None of the mice in the Pparafl/fl AOM/DSS group had anorectal prolapse, while 33.3% mice in the PparaΔIE AOM/DSS group developed anorectal prolapse (Supplementary Figure 2). PparaΔIE mice administered AOM and DSS also showed a significantly higher tumor multiplicity and shorter colon length compared to the AOM and DSS-administered Pparafl/fl mice (Figure 2G and H). In this model, 9.1%, 45.5%, and 45.5% of Pparafl/fl mice were diagnosed as low-grade adenomas, high-grade adenomas and adenocarcinomas, respectively, while 58.3% of PparaΔIE mice were diagnosed as adenocarcinomas and the other 41.7% of PparaΔIE mice were diagnosed as high-grade adenomas (Figure 2I). To evaluate cell proliferation and apoptosis in the colon tumors, BrdU and TUNEL staining were performed, respectively. Incorporation of BrdU in the colon tumors was increased in AOM and DSS-administered PparaΔIE mice with an average labeling index of 15.4% compared to an average labeling index of 8.6% in AOM and DSS-administered Pparafl/fl mice (Supplementary Figure 2B). No significant change in apoptosis between AOM and DSS-administered Pparafl/fl and PparaΔIE mice was observed (Supplementary Figure 2C). Collectively, these results suggested that intestinal PPARα deficiency increased both AOM-induced and AOM and DSS-induced colon carcinogenesis.
Metabolic Profile Analysis of Mouse Urine and Colon
The metabolic profile of urine samples collected at 5 months after the last injection from saline-administered or AOM-administered Pparafl/fl mice and PparaΔIE mice was analyzed by UPLC/QTOFMS. Unsupervised PCA of the creatinine-normalized urinary metabolomics data acquired with HILIC showed some separation between Pparafl/fl AOM and PparaΔIE AOM groups (Supplementary Figure 3A), indicating that these two groups have different metabolic profiles. Supervised OPLS-DA was then performed to maximize differences in urinary metabolite profiles from AOM-administered Pparafl/fl and AOM-administered PparaΔIE mice. The variable importance in projection (VIP) score analysis of OPLS-DA revealed that several ions, which were then identified by fragmentation pattern and retention time comparison with authentic standards (Supplementary Figure 4) as methylation-related metabolites, including methylcytosine, dimethylarginine, methylnicotinamide, carnitine and betaine, contributed to the separation with VIP scores > 1.5 (Supplementary Figure 3). S-adenosylmethionine (SAM), which is the only direct active methyl donor and affects all the methylation reactions in vivo,21 was also identified, although the VIP score was low. Urinary excretion of methylcytosine, dimethylarginine, methylnicotinamide, carnitine, betaine, and SAM were increased in AOM-administered PparaΔIE mice compared with AOM-administered Pparafl/fl mice (Supplementary Figure 3B).
Time course changes of methylation-related metabolites were then quantified in urine samples collected at 1, 3 and 5 months after the last AOM injection (Supplementary Figure 3C). The levels of carnitine and SAM were significantly higher in AOM-administered PparaΔIE mice compared with saline-administered PparaΔIE mice from 1 month, while the rest showed significant changes between these two groups from 3 months or 5 months, suggesting a higher level of these methylation-related metabolites in colon tumor-prone mice. When comparing between AOM-administered Pparafl/fl and AOM-administered PparaΔIE mice, carnitine was higher in AOM-administered PparaΔIE mice at 1 month, while asymmetric dimethylarginine (ADMA), 1-methylnicotinamide, betaine, and SAM were higher at 3 months. At 5 months, all the metabolites, except symmetric dimethylarginine (SDMA), showed higher levels in AOM-administered PparaΔIE mice. In serum, AOM-administered PparaΔIE mice showed higher levels of ADMA, but not SDMA, when compared with similarly administered Pparafl/fl mice (Supplementary Figure 5).
Next, colon metabolomics was analyzed. PCA yielded a good separation between AOM-administered or AOM and DSS-administered Pparafl/fl and PparaΔIE mice (Figure 3 A and C). In both models, the abundance of methylcytosine, dimethylarginine, methylnicotinamide, and SAM were elevated in the colons of carcinogen-administered PparaΔIE mice compared with similarly administered Pparafl/fl mice, which was consistent with corresponding urine samples. Carnitine was increased in the colons of AOM-administered PparaΔIE mice compared with similarly-administered Pparafl/fl mice, while no significant changes were found between AOM and DSS-administered Pparafl/fl and similarly administered PparaΔIE mice. Betaine was elevated in carcinogen-administered mice while remaining comparable between the different genotypes (Figure 3B and D). These observations indicate that changes in urinary metabolic signatures reflect extensive reprogramming of metabolic pathways in tumor tissue.
Figure 3. Several methylation-related metabolites were increased in the colon of tumor-bearing PparaΔIE mice.

(A) Score scatter plot for principal components analysis of data obtained from HILIC analysis of colon samples from AOM mouse model. N=6 mice/group. (B) Relative levels of indicated metabolites in the colon samples described in (A). (C) Score scatter plot for principal components analysis of data obtained from HILIC analysis of colon samples from AOM and DSS mouse model. N=3–6 mice/group. (D) The relative levels of indicated metabolites in the colon samples described in (C). * P< 0.05, ** P< 0.01, *** P< 0.001.
Analysis of Gene Expression Patterns and Protein Levels of Methyltransferases
In colon tumors, DNA methyltransferase Dnmt1, RNA methyltransferases Dnmt2, Nsun2, and Nsun4, protein arginine methyltransferases Prmt1, Prmt5, and Prmt6 mRNAs were significantly increased (Figure 4A). Only Dnmt1 and Prmt6 mRNAs were significantly increased in the colons of PparaΔIE mice as compared to Pparafl/fl mice (Figure 4B). The protein levels of DNMT1 and PRMT6 were also up-regulated in colon tumors (Figure 4C) and PparaΔIE mice (Figure 4D) when compared with adjacent non-tumor and Pparafl/fl mice, respectively. DNMT1 catalyzes the transfer of a methyl group from the universal methyl donor SAM to the 5-position of the cytosine residue in DNA, which is essential for mammalian development and reported to be associated with human tumorigenesis.22 PRMT6 is a type I protein arginine methyltransferase that carries out asymmetric dimethylation and is implicated in tumor progression and metastasis.23 The mRNA levels of Ppara and its target gene Acox1 were decreased in colon tumors, while Dnmt1 and Prmt6 mRNAs were increased in the tumor tissue of Pparafl/fl mice and further potentiated in PparaΔIE mice (Figure 4E). The mRNA (Figure 4F) and protein (Figure 4G) levels of DNMT1 and PRMT6 were significantly higher in primary intestinal epithelial cells isolated from PparaΔIE mice than those from Pparafl/fl mice. DNMT1 and PRMT6 were decreased after forced expression of PPARα in primary intestinal epithelial cells (Figure 4H and I). These observations collectively support the findings of increased expression of DNMT1 and PRMT6 by intestinal PPARα disruption and may explain the elevated 5-methylcytosine and ADMA levels in the colon and urine of PparaΔIE mice.
Figure 4. Intestine-specific knockout of PPARα increased the expression of DNMT1 and PRMT6.

(A) The mRNA levels of methyltransferases in mouse colon tumors (T) and adjacent non-tumor tissues (NT). N = 5/group. (B) The mRNA levels of methyltransferases in the colon samples from Pparafl/fl and PparaΔIE mice. N=5/group. (C) Western blot analysis of indicated proteins in mouse colon tumors (T) and adjacent non-tumor tissues (NT). (D) Western blot analysis of indicated proteins in colon samples from Pparafl/fl and PparaΔIE mice. (E) The mRNA levels of Ppara, Acox1, Dnmt1, and Prmt6 in colon tumors (T) and adjacent non-tumor tissues (NT) from AOM-administered Pparafl/fl and PparaΔIE mice (n=5/group). (F) The mRNA (n=3) and (G) protein levels of DNMT1 and PRMT6 in primary intestinal epithelial cells isolated from Pparafl/fl and PparaΔIE mice. (H) Primary intestinal epithelial cells isolated from wild-type mice were cultured and infected with control adenovirus (Ad-GFP), adenovirus expressing PPARα (Ad-PPARα), or not infected (No-Ad). The mRNA (n=2 for No-Ad group, and n=4 for Ad-GFP group and Ad-PPARα group) and (I) protein levels of indicated genes were analyzed. * P < 0.05, ** P < 0.01, *** P < 0.001.
DNMT1 and PRMT6 Decrease Expression of Tumor Suppressor Genes
Levels of two tumor suppressor gene mRNAs Cdkn1a, encoding p21, and Cdkn1b, encoding p27, were decreased while oncogenic Ctnnbl1, encoding β-catenin, was increased, in both colon tumors and PparaΔIE mice, compared with adjacent non-tumor and Pparafl/fl mice, respectively (Figure 5A and B). Similar changes were found with p21 and p27 proteins (Figure 4C and D). Moreover, in primary intestinal epithelial cells, the mRNA and protein levels of p21 and p27 were increased after adenoviral overexpression of PPARα (Figure 4H and I). It was suggested that DNA methylation plays a major role in enhancing transcriptional silencing, especially of tumor suppressor genes.22 PRMT6 is the primary enzyme responsible for H3R2 asymmetric dimethylation in mammalian cells, which usually leads to transcriptional repression because of its ability to counteract the activator function of H3K4me3.24 These reports prompted examination of whether p21 and p27 are targets of DNMT1 and PRMT6 in the intestine. Treatment of HCT116 cells with the DNMT1 inhibitor 5-aza-2’-deoxycytidine (5-Aza) decreased DNMT1 and increased p21 protein levels, while forced overexpression of DNMT1 in HCT116 cells increased DNMT1 and decreased p21 mRNA and protein levels, suggesting that p21 was a target of DNMT1 (Figure 5C and D). Exposure of HCT116 cells to the PRMT6 inhibitor EPZ020411 (EPZ) decreased PRMT6 activity (shown by decreased levels of H3R2me2a) and increased p27 protein levels, while forced overexpression of PRMT6 in HCT116 cells increased PRMT6 and decreased p27 mRNA and protein levels, indicating that p27 was a target of PRMT6 (Figure 5E and F). Moreover, in HCT116 cells transfected with Dnmt1 siRNA, p21 mRNA and protein were increased (Supplementary Figure 6A and B), while in HCT116 cells transfected with Prmt6 siRNA, p27 mRNA and protein were increased (Supplementary Figure 6C and D), further supporting that p21 and p27 were targets of DNMT1 and PRMT6, respectively.
Figure 5. DNMT1 and PRMT6 suppressed the expression of p21 and p27 by DNA methylation and histone arginine methylation, respectively.

(A) The mRNA levels of tumorigenesis related genes in mouse colon tumors (T) and adjacent non-tumor tissues (NT). N = 5/group. (B) The mRNA levels of tumorigenesis related genes in the colon samples from Pparafl/fl and PparaΔIE mice. N=5/group. (C) The mRNA (n=4/group) and (D) protein levels of indicated genes in HCT116 cells transfected with empty vector pcDNA3 or plasmid expressing DNMT1 (pDNMT1), or exposed to DMSO (Ctrl) or 5-Aza. (E) The mRNA (n=4/group) and (F) protein levels of indicated genes in HCT116 cells transfected with empty vector pCMV or plasmid expressing PRMT6 (pPRMT6), or administered with DMSO (Ctrl) or EPZ020411 (EPZ). (G) Genomic DNA was isolated from HCT116 cells described in (D) and subjected to MeDIP analysis (n=3/group). (H) Genomic DNA was isolated from colon tissues of mice described in Figure 3A and subjected to MeDIP analysis (n=3/group). Values were calculated as percentages of input DNA and expressed as relative enrichment compared to control group, which was equated to 1. (I) Histone ChIP assay was performed using chromatins prepared from HCT116 cells described in (F) (n=3/group). (J) Histone ChIP assay was performed using chromatins prepared from colon tissues of mice described in Figure 3A (n=3/group). Values were expressed as relative enrichment compared to histone H3. * P < 0.05, ** P < 0.01, *** P < 0.001.
To further test whether DNMT1 directly methylates p21, genomic DNA isolated from the HCT116 cells described in Figure 5D or colon tissues of mice described in Figure 3A was immunoprecipitated with an anti-m5C antibody or IgG. The presence of p21 promoter DNA in the IP materials was analyzed by real-time qPCR. 5-Aza treatment decreased, while forced expression of DNMT1 increased the enrichment of m5C in the promoter of p21 in HCT116 cells, supporting that p21 was a methylation target of DNMT1 (Figure 5 G). Enrichment of m5C in the p21 promotor was increased in the colon tissues of AOM-administered Pparafl/fl mice compared with saline-administered Pparafl/fl mice, and further increased in AOM-administered PparaΔIE mice (Figure 5H), providing in vivo support for the methylation machinery. Similar to the expression pattern of PRMT6, H3R2me2a was up-regulated in colon tumors when compared with adjacent non-tumor tissue (Figure 4C). To test whether PRMT6 methylates the H3R2 residue around the p27 promoter region, histone chromatin-immunoprecipitation (ChIP) assays were performed using chromatin prepared from the HCT116 cells described in Figure 5F or colon tissues from mice described in Figure 3A with an anti-H3R2me2a antibody, anti-histone H3 antibody, or IgG. The relative enrichment of H3R2me2a compared to histone H3 on the p27 promoter in the IP material was analyzed by real-time qPCR. EPZ treatment decreased, while forced expression of PRMT6 increased the enrichment of H3R2me2a on the promoter of p27 in HCT116 cells, supporting that p27 was a methylation target of PRMT6 (Figure 5I). Enrichment of H3R2me2a on p27 promotor was also increased in the colon of AOM-administered Pparafl/fl mice compared with saline-administered Pparafl/fl mice, and further increased in AOM-administered PparaΔIE mice (Figure 5J), supporting that this methylation machinery functions in vivo.
To determine if intestinal PPARα regulates the expression of p21 and p27 through DNMT1 and PRMT6, primary intestinal epithelial cells isolated from Pparafl/fl or PparaΔIE mice were cultured and treated with 5-Aza or EPZ. 5-Aza, and EPZ reversed the reduction of p21 (Supplementary Figure 7A) and p27 (Supplementary Figure 7B), respectively, induced by PPARα disruption. These data demonstrated that PPARα deficiency in the intestine decreased p21 and p27 expression at least partially through DNMT1-mediated DNA methylation and PRMT6-mediated H3R2 methylation, respectively.
Intestinal PPARα Regulates DNMT1 and PRMT6 Expression via the RB1/E2F Pathway
DNMT1 was reported to be transcriptionally repressed by retinoblastoma protein’s (RB1) interaction with the E2F transcription factor.25 Many potential E2F binding sites exist in the Prmt6 promoter as predicted by Genomatix software (data not shown). RB1 is a well-known tumor suppressor gene and cyclin D:Cdk4/6 complex inactivates RB1 by phosphorylation, to release E2F transcription factors.26 Treatment of primary intestinal epithelial cells with the Cdk4/6 inhibitor PD0332991, which reduced the phosphorylation of RB1 and enhanced RB1/E2F binding, resulted in a decrease of DNMT1 and PRMT6 and an increase of p21 and p27 mRNA and protein levels (Figure 6A and B). On the contrary, treatment of HCT116 cells with Rb1 siRNA resulted in increased DNMT1 and PRMT6 and decreased p21 and p27 mRNA and protein levels (Supplementary Figure 8). These data suggested that DNMT1 and PRMT6 could be regulated by the RB1/E2F pathway. The RB1 mRNA and protein were down-regulated in colon tumors (Supplementary Figure 9A and B) and PparaΔIE mice (Supplementary Figure 9C and D), compared with adjacent non-tumor and Pparafl/fl mice, respectively. The RB1 mRNA and protein were also lower in primary intestinal epithelial cells isolated from PparaΔIE mice than those from Pparafl/fl mice (Supplementary Figure 9E and F). When overexpressing PPARα in primary intestinal epithelial cells, the mRNA and protein levels of RB1 were increased (Supplementary Figure 9G and H), indicating that the Rb1 gene might be a target of PPARα in the intestine. There are four putative PPREs in the promoter of mouse Rb1 as predicted by Genomatix software. Two Rb1 promoter fragments covering the four potential PPREs were designed and cloned into pGL4.10 luciferase reporter vector (Figure 6C). Treatment of HCT116 cells, with the PPARα agonist WY-14643 after co-transfection with the reporter vector bearing Rb fragment B significantly induced the luciferase activity, an effect diminished by adding the PPARα antagonist GW-6471 (Figure 6D). ChIP assays revealed that activation of PPARα by Wy-14643 administration increased enrichment of PPARα on the promoters of Rb1 as well as the ‘classic’ PPARα target genes Acoxl, Acotl, and Pdk4 in both MC38 (Figure 6E–G) and HCT116 (Supplementary Figure 10) cells, an effect diminished by adding the PPARα antagonist GW-6471, demonstrating that Rb1 is a PPARα target gene.
Figure 6. Intestinal PPARα regulated the expression of DNMT1 and PRMT6 via RB1/E2F pathway.

(A) The mRNA (n=4/group) and (B) protein levels of indicated genes in primary intestinal epithelial cells exposed to DMSO (Ctrl) or PD0332991. (C) Schematic diagram of the mouse Rb1 promotor illustrating the potential PPREs in the regulatory region and the fragments used for luciferase reporter assay. The upstream regions were numbered in relation to the transcription initiation site, which was designated +1. (D) Luciferase reporter assay of mouse Rb1 promoter activity (n=3/group). ChIP assays on chromatins isolated from MC38 cells for Rb1 (E), Acoxl (F), and Acotl (G). N=3/group. * P< 0.05, ** P< 0.01, *** P< 0.001.
Fenofibrate Activates PPARα and Suppresses Colon Carcinogenesis
Sustained activation of PPARα leads to the development of liver tumors in rats and mice, while humans are resistant to the induction of peroxisome proliferation and the development of liver cancer by fibrate drugs.27 To assess the effect of PPARα activation on colon carcinogenesis and avoid any confounding effects caused by liver cancer, human PPARA transgenic mice were administered with AOM followed by DSS to induce colitis-related colon cancer while maintained on a chow or fenofibrate diet. Fenofibrate-fed mice exhibited higher liver weights (Supplementary Figure 11B), but no liver tumors, consistent with previous reports that WY-14643 and fenofibrate administration led to hepatomegaly in human PPARA transgenic mice.28 Although AOM and DSS treatment resulted in 100% incidence of colon tumors in both chow and fenofibrate-fed mice, fenofibrate-fed mice displayed markedly lower tumor multiplicity (Figure 7B) and longer colon length (Figure 7A). Gross and histological analysis also showed less tumors in fenofibrate-fed mice (Figure 7D). Fenofibrate administration decreased the prevalence of colon adenocarcinomas in human PPARA transgenic mice (26.7%, 4/15) compared to chow-fed mice (60%, 9/15) (Figure 7C). None of the fenofibrate-fed mice had anorectal prolapse, while 6 out of 15 mice in the chow diet group had anorectal prolapse (Supplementary Figure 4). Fenofibrate administration decreased the incorporation of BrdU with average labeling indices of 5.43% and 12.1% in the colon of fenofibrate-and chow diet-fed mice (Figure 7E), respectively, while no significant changes in the percentage of apoptotic cells was observed (Supplementary Figure 11C). Furthermore, PPARα activation increased RB1, p21, and p27, and decreasedDNMT1 and PRMT6 mRNAs (Figure 7F) and proteins (Figure 7G) in human PPARA transgenic mouse colons. Thus, PPARα activation by fenofibrate suppressed AOM and DSS-induced colon carcinogenesis.
Figure 7. Fenofibrate decreased AOM and DSS-induced colon carcinogenesis in human PPARA transgenic mice.

(A) Colon length. (B) Tumor number. (C) Prevalence of low-grade adenomas, high-grade adenomas and adenocarcinomas. (D) Representative gross pictures (upper) and H&E staining (bottom) of colon sections. Scale bars: upper, 1.5 mm; bottom, 100 μm. (E) Left, Representative BrdU staining of colon sections. Scale bars: 50 μm. Right, BrdU labeling index. (F) The mRNA (n=8) and (G) protein levels of indicated genes in the colon samples of AOM and DSS-administered human PPARA transgenic mice fed on chow diet or fenofibrate diet. * P< 0.05, ** P< 0.01, *** P< 0.001.
PPARα in Human Colon Carcinogenesis
To determine whether the PPARα regulatory pathway explored in mice also functions during human colon carcinogenesis, colorectal-tumor specimens from patients at different stages of CRC were IHC stained with anti-RB1, DNMT1, PRMT6, p21, p27, and Ki67 antibodies. The percentage of Ki67 positive cells increased in all four stages of colon tumors and the hyperplastic polyps when compared with normal mucosae, indicating enhanced proliferation in those samples (Supplementary Figure 12A). RB1 expression decreased, while DNMT1 and PRMT6 expression increased in tumor samples from low-grade adenomas to adenocarcinomas compared with normal mucosae (Supplementary Figure 12B–D). p21 and p27 were expressed at high levels in normal mucosae and were decreased in tumor samples from low-grade adenomas to adenocarcinomas (Supplementary Figure 12E and F). DNMT1 was increased in hyperplastic polyps compared with normal mucosae, while RB1, PRMT6, p21, and p27 remained unchanged. Collectively, these data suggested that the PPARα-DNMT1/PRMT6-p21/p27 regulatory pathway may also function at an early stage during human colorectal carcinogenesis.
Discussion
Intestinal disruption of PPARα increases AOM or AOM and DSS-induced colon carcinogenesis with larger tumor size as well as higher tumor multiplicity and malignancy. DNMT1 and PRMT6 are up-regulated in the colon of PparaΔIE mice via the RB1/E2F pathway, which may account for the increase in methylation-related metabolites including 5-methylcytosine and ADMA in the urine and colon tissues from carcinogen-administered PparaΔIE mice. p21 and p27 are in vivo methylation targets of DNMT1 and PRMT6, respectively. Increased expression of DNMT1 and PRMT6 in PparaΔIE mice leads to decreased levels of p21 and p27, thus promoting cell proliferation and colon carcinogenesis. Our data demonstrate a critical role for intestinal PPARα in colon cancer development and progression. This regulatory pathway may also occur in human colorectal carcinogenesis as suggested by IHC staining of human colorectal specimens.
PPARA mRNA levels were reported to decrease by about 60% in patients with tubular adenomas.9 Another study showed that PPARA expression, assessed at both the mRNA and protein level, were significantly lower in 26 human colorectal tumors compared with matched non-malignant tissues.29 On the contrary, PPARA mRNA expression in colon tumors and adjacent normal mucosa from 17 colon cancer patients was increased between 2- and 32-fold in 41% of all patients, while decreased by 50% in one patient. No obvious changes were seen in the other 52% patients.30 A larger sample size is needed to confirm the change of PPARA in colon cancer patients. To this end, two datasets from Oncomine with relatively large sample size were analyzed. Both TCGA colorectal 2 and Ki colon datasets showed decreased PPARA expression in human colon tumors, suggesting a tumor-suppressive role of PPARα in colon cancer.
There are several reports suggesting PPARα ligands as potential chemopreventive agents in colon carcinogenesis. Bezafibrate, a PPARα ligand, is able to suppress AOM and AOM and DSS-induced aberrant crypt foci formation in rat colon,31 decrease intestinal polyp formation in Apc-deficient mice,32 and inhibit AOM and DSS-induced colon carcinogenesis in mice.12 In addition, this PPARα agonist was reported to prevent colon cancer in patients with coronary artery disease.13 The PPARα activator methylclofenapate was also shown to reduce polyp formation in Apcmin/+ mice.29 MCC-555, a dual ligand for PPARα and PPARγ, was demonstrated to suppress AOM-induced colorectal tumorigenesis.11 However, the exact mechanism by which PPARα activation suppressed colon carcinogenesis was not clear. Consistent with these findings, the current study showed that disruption of intestinal PPARα promoted carcinogen-induced colon tumorigenesis. Moreover, administration of the PPARα agonist fenofibrate protected human PPARA transgenic mice from AOM and DSS-induced colon tumorigenesis. Cell proliferation was suppressed in the colon of fenofibrate-administered human PPARA transgenic mice, likely due to the increased expression of p21 and p27 in the colon after fenofibrate administration as p21 and p27 are negative regulators of the cell cycle. Mice nullizygous for p21 or p27 had increased colon tumors in Apc-deficient mice or in mice fed a Western diet.33–35
Cyclooxygenase-2 (COX-2) expression was up-regulated in human colorectal cancer tissues.36 Activation of PPARα by WY-14643 or LY-171883 diminished COX-2 transcription via inhibition of activator protein-1 in the colon cancer cell line SW620.10 However, two other studies showed that WY-14643 administration increased COX-2 transcription in colon cancer cell lines HT29 or HCT116, suggesting a cancer promoting role for PPARα ligands.14, 15 In the current study, Cox2 mRNA levels were not changed between carcinogen-administered PparaΔIE mice and similarly administered Pparafl/fl mice (data not shown).
Aberrant methylation is associated with the development of several cancers.37 Elevation of some methylation-related metabolites including SDMA, ADMA, betaine, and SAM were found in the urine and colon of colon cancer mouse models and colon cancer patients.38 Consistent with this, in the current study, increased metabolites involved in methylation machinery were found along with overexpression of genes encoding methyltransferases in the colons from carcinogen-administered mice compared with controls. In addition, carcinogen-administered PparaΔIE mice showed higher urinary and colonic levels of methylcytosine, dimethylarginine, and SAM compared with similarly-administered Pparafl/fl mice along with higher expression of DNA methyltransferase DNMT1 and protein arginine methyltransferase PRMT6.
There are several mechanisms by which mammalian cells regulate DNMT1 levels, including both transcriptional and post-transcriptional regulation.39 However, to date, little is known about the regulation of PRMT6. The current study found several potential E2F binding sites in the promoter region of Prmt6. Inhibition of the RB1/E2F pathway by the selective Cdk4/6 inhibitor PD0332991 down-regulated DNMT1 and PRMT6 expression in primary intestinal epithelial cells, suggesting that PRMT6 is a novel downstream target. Other possible regulatory pathways need further investigation.
It is well established that genetic mutations drive colon carcinogenesis.2 Therefore, exome-sequencing was performed to check the genetic mutations in tumors derived from Pparafl/fl and PparaΔIE mice. Generally, there were more mutated genes in AOM and DSS-administered PparaΔIE mice when compared with similarly administered Pparafl/fl mice (see “Supplementary Discussion”). These mutations may also contribute to the enhanced colorectal tumorigenesis in PparaΔIE mice. Whether these mutations contribute to or work in concert with the current mechanism outlined in this study (i.e., increased expression of methyltransferases DNMT1 and PRMT6 and decreased expression of tumor suppressor genes p21 and p27) is unknown. Further data mining and experiments will be performed to investigate this question.
Colorectal cancer is a disease with heterogeneous outcomes and drug responses. An international consortium that included a panel of expert research groups agreed on four consensus molecular subtypes (CMS1–4) of CRC with distinctive biological features and clinical outcomes.40 Stratification of CRC into subtypes is critical to the development of more personalized and precise therapies.40, 41 Further studies are needed to investigate the CMS subtype/s of human CRC where PPARα dysregulation may act as a contributing factor.
In conclusion, the present work identifies a pathway by which intestinal PPARα deficiency contributes to CRC progression. Activation of PPARα by fenofibrate administration protects human PPARA transgenic mice from chemical-induced colon cancer, providing a potential target for colon cancer prevention and treatment.
Supplementary Material
What You Need to Know.
Background and Context:
Peroxisome proliferator-activated receptor α (PPARα) is a nuclear receptor that serves as a xenobiotic and lipid sensor to regulate lipid homeostasis and inflammation. Agents that affect PPARα activity might be used in treatment or prevention of colon cancer.
New Findings:
Intestinal depletion of PPARα promotes colon carcinogenesis via up-regulation of DNMT1 and PRMT6 in mice. Activation of PPARα by fenofibrate protects mice that express the human PPARA gene from colon carcinogenesis.
Limitations:
We studied only 1 pathway regulated by PPARα, in mice with carcinogen-induced colon cancer and with limited human data. Further studies are needed to explore additional mechanisms of PPARα and its ability to prevent colon carcinogenesis in humans.
Impact:
We identified preventative effects of intestinal PPARα in colon carcinogenesis and potential strategies for colon cancer prevention and treatment.
Acknowledgments
Funding
This study was supported by the National Cancer Institute Intramural Research Program and grants from the National Natural Science Foundation of China (Grant No. 81572061), and the Outstanding academic leaders plan of Shanghai (Grant No. 2018BR07).
Abbreviations:
- 5-Aza
5-aza-2’-deoxycytidine
- ADMA
asymmetric dimethylarginine
- AOM
azoxymethane
- APC
adenomatous polyposis coli
- BrdU
bromodeoxyuridine
- ChIP
chromatin-immunoprecipitation
- CMS
consensus molecular subtypes
- COX-2
cyclooxygenase-2
- CRC
colorectal cancer
- DNMT1
DNA methyltransferase 1
- DSS
dextran sulfate sodium
- 5-FU
5-fluorouracil
- H&E
hematoxylin and eosin
- HILIC
hydrophilic interaction liquid chromatography
- IHC
immunohistochemistry
- MeDIP
methyl-DNA immunoprecipitation
- OPLS-DA
orthogonal projection to latent structures multivariate data analysis
- PCA
principal component analysis
- PPARα
peroxisome proliferator-activated receptor α
- PPRE
peroxisome proliferator responsive element
- PRMT6
protein arginine methyltransferase 6
- RB1
retinoblastoma protein
- RXR
retinoid X receptor
- SAM
s-adenosylmethionine
- SDMA
symmetric dimethylarginine
- UPLC/QTOFMS
ultra-high performance liquid chromatography/Xevo G2 quadrupole-time-of-flight mass spectrometry
Footnotes
Supplementary Materials
Supplementary methods, supplementary discussion, 14 supplementary figures, and 8 supplementary tables are provided.
Conflicts of interest
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012;487:330–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Onitilo AA, Stankowski RV, Berg RL, et al. Type 2 diabetes mellitus, glycemic control, and cancer risk. Eur J Cancer Prev 2014;23:134–40. [DOI] [PubMed] [Google Scholar]
- 4.Yehuda-Shnaidman E, Schwartz B. Mechanisms linking obesity, inflammation and altered metabolism to colon carcinogenesis. Obes Rev 2012;13:1083–95. [DOI] [PubMed] [Google Scholar]
- 5.Siegel RL, Miller KD, Fedewa SA, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin 2017;67:177–193. [DOI] [PubMed] [Google Scholar]
- 6.Pyper SR, Viswakarma N, Yu S, et al. PPARalpha: energy combustion, hypolipidemia, inflammation and cancer. Nucl Recept Signal 2010;8:e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell 1995;83:841–50. [DOI] [PubMed] [Google Scholar]
- 8.Bays H, Stein EA. Pharmacotherapy for dyslipidaemia--current therapies and future agents. Expert Opin Pharmacother 2003;4:1901–38. [DOI] [PubMed] [Google Scholar]
- 9.Matthiessen MW, Pedersen G, Albrektsen T, et al. Peroxisome proliferator-activated receptor expression and activation in normal human colonic epithelial cells and tubular adenomas. Scand J Gastroenterol 2005;40:198–205. [DOI] [PubMed] [Google Scholar]
- 10.Grau R, Punzon C, Fresno M, et al. Peroxisome-proliferator-activated receptor alpha agonists inhibit cyclo-oxygenase 2 and vascular endothelial growth factor transcriptional activation in human colorectal carcinoma cells via inhibition of activator protein-1. Biochem J 2006;395:81–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imchen T, Manasse J, Min KW, et al. Characterization of PPAR dual ligand MCC-555 in AOM-induced colorectal tumorigenesis. Exp Toxicol Pathol 2013;65:919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohno H, Suzuki R, Sugie S, et al. Suppression of colitis-related mouse colon carcinogenesis by a COX-2 inhibitor and PPAR ligands. BMC Cancer 2005;5:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tenenbaum A, Boyko V, Fisman EZ, et al. Does the lipid-lowering peroxisome proliferator-activated receptors ligand bezafibrate prevent colon cancer in patients with coronary artery disease? Cardiovasc Diabetol 2008;7:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikawa H, Kameda H, Kamitani H, et al. Effect of PPAR activators on cytokine-stimulated cyclooxygenase-2 expression in human colorectal carcinoma cells. Exp Cell Res 2001;267:73–80. [DOI] [PubMed] [Google Scholar]
- 15.Oshio H, Abe T, Onogawa T, et al. Peroxisome proliferator-activated receptor alpha activates cyclooxygenase-2 gene transcription through bile acid transport in human colorectal cancer cell lines. J Gastroenterol 2008;43:538–49. [DOI] [PubMed] [Google Scholar]
- 16.Madison BB, Dunbar L, Qiao XT, et al. Cis elements of the villin gene control expression in restricted domains of the vertical (crypt) and horizontal (duodenum, cecum) axes of the intestine. J Biol Chem 2002;277:33275–83. [DOI] [PubMed] [Google Scholar]
- 17.Brocker CN, Yue J, Kim D, et al. Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am J Physiol Gastrointest Liver Physiol 2017;312:G283–G299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang Q, Nagano T, Shah Y, et al. The PPAR alpha-humanized mouse: a model to investigate species differences in liver toxicity mediated by PPAR alpha. Toxicol Sci 2008;101:132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ki DH, Jeung HC, Park CH, et al. Whole genome analysis for liver metastasis gene signatures in colorectal cancer. Int J Cancer 2007;121:2005–12. [DOI] [PubMed] [Google Scholar]
- 20.Nambiar PR, Girnun G, Lillo NA, et al. Preliminary analysis of azoxymethane induced colon tumors in inbred mice commonly used as transgenic/knockout progenitors. Int J Oncol 2003;22:145–50. [PubMed] [Google Scholar]
- 21.Chiang PK, Gordon RK, Tal J, et al. S-Adenosylmethionine and methylation. FASEB J 1996;10:471–80. [PubMed] [Google Scholar]
- 22.Daniel FI, Cherubini K, Yurgel LS, et al. The role of epigenetic transcription repression and DNA methyltransferases in cancer. Cancer 2011;117:677–87. [DOI] [PubMed] [Google Scholar]
- 23.Yoshimatsu M, Toyokawa G, Hayami S, et al. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer 2011;128:562–73. [DOI] [PubMed] [Google Scholar]
- 24.Guccione E, Bassi C, Casadio F, et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature 2007;449:933–7. [DOI] [PubMed] [Google Scholar]
- 25.McCabe MT, Davis JN, Day ML. Regulation of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res 2005;65:3624–32. [DOI] [PubMed] [Google Scholar]
- 26.Narasimha AM, Kaulich M, Shapiro GS, et al. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. Elife 2014;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ashby J, Brady A, Elcombe CR, et al. Mechanistically-based human hazard assessment of peroxisome proliferator-induced hepatocarcinogenesis. Hum Exp Toxicol 1994;13 Suppl 2:S1–117. [DOI] [PubMed] [Google Scholar]
- 28.Cheung C, Akiyama TE, Ward JM, et al. Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res 2004;64:3849–54. [DOI] [PubMed] [Google Scholar]
- 29.Jackson L, Wahli W, Michalik L, et al. Potential role for peroxisome proliferator activated receptor (PPAR) in preventing colon cancer. Gut 2003;52:1317–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feilchenfeldt J, Brundler MA, Soravia C, et al. Peroxisome proliferator-activated receptors (PPARs) and associated transcription factors in colon cancer: reduced expression of PPARgamma-coactivator 1 (PGC-1). Cancer Lett 2004;203:25–33. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka T, Kohno H, Yoshitani S, et al. Ligands for peroxisome proliferator-activated receptors alpha and gamma inhibit chemically induced colitis and formation of aberrant crypt foci in rats. Cancer Res 2001;61:2424–8. [PubMed] [Google Scholar]
- 32.Niho N, Takahashi M, Kitamura T, et al. Concomitant suppression of hyperlipidemia and intestinal polyp formation in Apc-deficient mice by peroxisome proliferator-activated receptor ligands. Cancer Res 2003;63:6090–5. [PubMed] [Google Scholar]
- 33.Yang W, Bancroft L, Nicholas C, et al. Targeted inactivation of p27kip1 is sufficient for large and small intestinal tumorigenesis in the mouse, which can be augmented by a Western-style high-risk diet. Cancer Res 2003;63:4990–6. [PubMed] [Google Scholar]
- 34.Philipp-Staheli J, Kim KH, Payne SR, et al. Pathway-specific tumor suppression. Reduction of p27 accelerates gastrointestinal tumorigenesis in Apc mutant mice, but not in Smad3 mutant mice. Cancer Cell 2002;1:355–68. [DOI] [PubMed] [Google Scholar]
- 35.Yang WC, Mathew J, Velcich A, et al. Targeted inactivation of the p21(WAF1/cip1) gene enhances Apc-initiated tumor formation and the tumor-promoting activity of a Western-style high-risk diet by altering cell maturation in the intestinal mucosal. Cancer Res 2001;61:565–9. [PubMed] [Google Scholar]
- 36.Eberhart CE, Coffey RJ, Radhika A, et al. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology 1994;107:1183–8. [DOI] [PubMed] [Google Scholar]
- 37.Feinberg AP, Koldobskiy MA, Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet 2016;17:284–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manna SK, Tanaka N, Krausz KW, et al. Biomarkers of coordinate metabolic reprogramming in colorectal tumors in mice and humans. Gastroenterology 2014;146:1313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kinney SR, Pradhan S. Regulation of expression and activity of DNA (cytosine-5) methyltransferases in mammalian cells. Prog Mol Biol Transl Sci 2011;101:311–33. [DOI] [PubMed] [Google Scholar]
- 40.Guinney J, Dienstmann R, Wang X, et al. The consensus molecular subtypes of colorectal cancer. Nat Med 2015;21:1350–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dienstmann R, Vermeulen L, Guinney J, et al. Consensus molecular subtypes and the evolution of precision medicine in colorectal cancer. Nat Rev Cancer 2017;17:79–92. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.