

Abstract
The broad roles of RNA in performing and regulating fundamental life processes are being uncovered at a rapid pace. This has sparked profound questions about how RNA function is enacted through RNA structures that orchestrate processes in the complex cellular environment. These questions are being answered with technologies that use massively parallel sequencing to interrogate RNA structure at unprecedented throughput. In this article, we place the growing number of these technologies on a common conceptual framework, review their technical underpinnings, discuss the meaning and use of their data, and highlight their application to new areas of RNA biology.
Introduction
RNA molecules adopt intricate folds that underlie their central roles in regulating, maintaining and defending the genomes of all organisms1,2. For example, messenger RNA is a key focal point for regulating gene expression, and non-coding RNA (ncRNA) functions range from transcription and translation regulation and promoting messenger stability regulation in prokaryotes, to gene silencing, epigenetic regulation, splicing regulation and molecular scaffolding in eukaryotes3–7. RNAs also catalyze some of the fundamental chemical reactions of life, including peptidyl transfer in the ribosome8.
The functional diversity of RNA stems from its ability to form intricate structures that can change dynamically in response to cellular signals, such as ligands, proteins, temperature and the general cellular chemical environment9,10. For example, the formation of simple secondary structures can directly impact gene expression by occluding regulatory binding sites. More sophisticated tertiary structures precisely position RNA helices, loops, bulges and junctions into architectures that define selective ligand binding pockets11 and enzymatic active sites12. Given the rapid pace at which new functional RNAs are being discovered2, there is great opportunity to build off of this knowledge to uncover the structural principles that underlie the enormous functional diversity of RNA.
Biophysical characterization of RNA structure has yielded an atomic resolution understanding of the chemical and structural basis of RNA functions. However, experimental requirements often make these approaches low throughput. Biochemical approaches13–20 (Figure 1) complement this limitation by sacrificing resolution for increased experimental flexibility and throughput. Efforts to establish biochemical probing as a quantitative measurement of RNA conformation21–31 have yielded significant advances including the ability to interrogate RNA structures in the complex cellular environment32,33. Additionally, the recent coupling of RNA structure probing with high-throughput sequencing (HTS)34–36 has transformed the measurement of RNA structure into the ‘omics’ era by allowing thousands of RNAs, and even whole transcriptomes37–40, to be studied simultaneously.
Figure 1 |. A brief history of biochemical RNA structure probing.

Biochemical probing of RNA structure began even before the diverse and important roles of RNA were fully appreciated162. Early methods used sequence and structure dependent cleavage by nuclease enzymes, which gave the first insights into how tRNA structures decode the genetic code13. Chemical probes were developed later14,15,18, followed by methods that used RT primer extension to read out adduct position16,17. Almost two decades later, biochemical probing data was incorporated into RNA folding algorithms to give more accurate models of RNA structure163. The development of SHAPE chemistry in 200524 launched efforts to make RNA structure probing quantitative. The coupling of enzymatic34,35,41 and chemical36 probing to high throughput sequencing, and the development of probes that penetrate cell membranes32,33 brought RNA structure probing into the ‘omics’ era. This allowed rapid advances in probing entire transcriptomes37–40, high resolution modelling of tertiary structures48, mapping of nascent RNA cotranscriptional folding pathways75, and uncovering the role of RNA structures in mammalian RNA processing164. Rapid technological advances such as new adduct mapping methods44,54 promise to continue to increase the accuracy and power of these methods.
This review seeks to present a unified framework for high throughput RNA structure probing by describing recent developments and applications of these techniques in the context of their common conceptual core. We start with an overview of the experimental elements shared by all techniques and discuss how the resulting probe ‘reactivity’ data is generated. We next discuss several ways in which reactivities can be directly analyzed as well as incorporated within computational RNA structure prediction methods to generate more accurate models of RNA structures. We then discuss recent applications and practical considerations for using these techniques in several exciting new areas of RNA biology. Finally, we end with a roadmap for combining these techniques into an even more powerful suite of methods for uncovering the role of RNA structure in some of the most fundamental aspects of biology.
Probing RNA structures with sequencing
All techniques that use high throughput sequencing to characterize RNA structures share a common conceptual framework to encode RNA structural information into a pool of complementary DNA (cDNA) molecules that are sequenced and analyzed to recover that information (Figure 2) (Table 1)34–55: in vitro or in vivo folding of RNA, structure-dependent RNA modification, recording of modifications by reverse transcription, sequencing and sequencing read alignment to map modification positions, and using these modification positions to calculate a ‘reactivity’ for each nucleotide in the RNA that reflects the underlying RNA structure. The use of HTS and bioinformatics enables simultaneous probing and mapping of modifications in complex mixtures of RNA molecules and whole transcriptomes. In the following sections, we discuss the different experimental choices for each step to enable users to adjust these techniques to ask different biological questions.
Figure 2 |. A common conceptual core for determining RNA structures with high throughput sequencing.

High throughput sequencing allows thousands of RNAs in complex mixtures to be structurally interrogated in a single experiment. In vitro or in vivo RNA mixtures (a) are first probed with chemical reagents that covalently modify the RNAs (red pin), or enzymes that modify the RNAs through cleavage (SI Figure 1), in a structure-dependent fashion (b) (Box 1). Modified RNAs are then converted into DNA (c) through reverse transcription that either stops or causes a mutation at each modification position (Fig. 3). The resulting pool of DNA molecules thus encode the original probe positions, and are formatted and sequenced to map the distribution of probe modifications using sequence alignment algorithms (d) (Fig. 4). The frequencies of mapped positions are used to estimate a reactivity value for each nucleotide of each RNA molecule (Box 2). High reactivities indicate nucleotides that are unstructured, while low reactivities indicate nucleotides that are constrained by RNA structures, tertiary interactions, ligand binding, or protein interactions. Reactivity information is then used to infer RNA structures (a) using a variety of computational methods.
Table 1 |. High Throughput RNA Structure Probing Techniques.
High throughput RNA structure determination methods combine enzymatic or chemical probes of RNA structures with high throughput sequencing to map structural information for up to thousands of RNAs in a single experiment. There has been a recent explosion in these techniques, which share a common conceptual core (Figure 2) to address a range of biological questions from determining RNA ligand interactions, uncovering cellular RNA structures and even cotranscriptional RNA folding pathways. The table below summarizes each of these techniques, the experimental choices and data analysis methods used in each, the level of RNA structure probed with each technique, and demonstrated applications. While many techniques were originally presented as a specific combination of experimental steps, due to their conceptual similarities many techniques are compatible with different probes and experimental strategies. Many techniques are also applicable to a wide array of RNA structural biology questions beyond their demonstrated applications. RNA structure modelling approaches that have used data from these techniques to predict RNA structures are listed in Supplementary Table 1.
| Method | Demonstrated RNA Structure Level Probed | Probe | Modification Detection | Priming Strategy | Library Prep | Demonstrated Applications |
|---|---|---|---|---|---|---|
| Enzymatic Probes | ||||||
| PARS 34,165 and PARTE166 | In vitro Secondary | RNase V1 (dsRNA) and S1 (ssRNA) | RT mapping of fragment ends | Linker defined | RNA Fragmentation, Ligation, Gel size selection | Transcriptome-wide in vitro RNA structure probing in S. cerevisiae34, Impact of SNPs on in vitro RNA structure of human transcriptomes115,165, Effect of temperature on RNA structures166 |
| FragSeq35 | In vitro secondary | P1 nuclease (ssRNA) | RT mapping of fragment ends | Linker defined | Ligation, Gel size selection | Transcriptome-wide in vitro RNA structure probing in M. musculus cell lines |
| dsRNA-seq and ssRNA-seq41,42 | In vitro secondary | RNase ONE (ssRNase) and RNase V1 (dsRNase) | Read coverage of fragments from digested RNAs | Linker defined | RNA fragmentation, Gel size selection, Adapter ligation | Transcriptome-wide in vitro RNA structure probing in Arabidopsis thaliana, Drosophila melanogaster, and Caenorhabditis elegans |
| Small Molecule Probes | ||||||
| SHAPE-Seq36,43,50,75,85 | In vitro/in vivo secondary/tertiary, RNA-ligand/RNA/protein interactions | 1M7, BzCN, DMS | RT-Stop | Defined43,50, Linker defined43,75, Multiple defined85 | Ligation, Bead size selection43 PCR Selection 50,75 | In vitro equilibrium refolding of target RNAs36,43 in vivo probing in E. coli50,96,167, in vitro cotranscriptional RNA folding75,76,79, RNA-guided assembly of mammalian RNA nuclear export assemblies168 |
| DMS-seq37 | In vitro and in vivo secondary | DMS | RT-Stop | Linker defined | RNA fragmentation, gel size selection, adapter ligation, cDNA circularization | Transcriptome wide in vivo and in vitro RNA structure probing in S. cerevisiae and H. sapiens cell lines37, in vivo structures of G-quadruplexes in mammalian cells and E. coli128, structure of operon mRNAs in E. coli127 |
| Mod-seq40 | In vivo secondary | DMS | RT-Stop | Linker defined | RNA fragmentation, custom adapter ligation to detect RT-stops due to fragmentation, bead enrichment, circularization | Transcriptome wide in vivo RNA structure probing in S. cerevisiae, RNA-protein interaction footprinting |
| CIRS-seq46 | In vitro secondary | DMS, CMCT | RT-Stop | Random | Ligation, Gel size selection | Transcriptome wide in vitro RNA structure probing in M. musculus cell lines |
| Structure-Seq38,52,54,84 | In vivo secondary | DMS | RT-Stop38,52,84, RT-Stop and RT-Mutate54 | Random | Ligation38 with hairpin adapter improvements52, Gel size selection or biotin-streptavidin pull down52 | Transcriptome wide in vivo RNA structure probing in A. thaliana38 and Rice52, Connection to mRNA structure and protein structure in A. thaliana169. In vivo probing of RNA structure of Xist in M. musculus54,84 |
| ChemModSeq45,53 | In vivo secondary, RNA-protein interactions | DMS, 1M7, NAI | RT-Stop | Random | Ligation, Gel size selection, Random barcode to remove PCR duplicates | RNA-protein interactions governing ribosome biogenesis in S. cerevisiae |
| MAP-Seq47,80,170 | In vitro secondary | DMS, CMCT, 1M7 | RT-Stop | Defined | Ligation, Bead purification allowing 96-well plate formats, Fluorescent quantification of libraries and ligation efficiency pre-sequencing | In vitro folding of model natural47 and synthetic RNAs170, direct detection of in vitro RNA secondary structures with mutate-and-map read out (M2-Seq)145 |
| SHAPE-MaP44 | In vitro/in vivo secondary/tertiary, RNA-ligand/RNA/protein interactions | 1M7, 1M6, NMIA44, DMS81 | RT-Mutate | Defined, Random | No ligations if using defined priming, PCR or second strand synthesis to add all adapter sequences. Random priming requires adapter ligation. | In vitro folding of targeted RNAs, viral genomes44,86, in vivo RNA-protein interactions59, in vivo lncRNA structures and interactions (Xist)60, rRNA structure171 |
| RING-MaP81 | In vitro tertiary structure, RNA structure subpopulations | DMS | RT-Mutate | Defined | Same as SHAPE-MaP | In vitro folded model RNA systems and RNA-ligand interactions |
| icSHAPE39 | In vivo secondary | NAI-N3 | RT-Stop | Linker Defined | Biotin pull down of modified RNAs, RNA fragmentation, circularization, gel size selection | Transcriptome wide probing of RNA structure in M. musculus, effect of RNA modification on RNA structure |
| MOHCA-Seq48 | In vitro tertiary | Targeted OH Radicals | RT-Stop | Linker Defined | Ligation, Bead purification | Tertiary reconstruction of in vitro folded model RNA systems |
| SHAPES172 | In vitro secondary | NPIA | RT-Stop | Defined, Random | Biotin pull down, Ligation | In vitro probing of RNase P and 16S rRNA |
| DMS-MaP-Seq51 | In vivo secondary structure | DMS | RT-Mutate | Linker defined | RNA fragmentation and gel size selection, linker ligation for RT, circularization, Random barcode to remove PCR duplicates | Global transcriptome analysis of S. cerevisiae RNA structure, Targeted RNA structures in Drosophila ovaries and mammalian cell lines51, analysis of 3’ UTR structure in mammalian genes164 |
| Ligation-Based Methods | ||||||
| RNA Proximity Ligation134 | In vivo RNA-RNA interactions | T4 RNA Ligase 1 | Mapping of ligated junctions | Linker defined | TruSeq RNA-Seq library preparation | Transcriptome wide analysis of S. cerevisiae and H. sapiens cell lines, targeted analysis of select RNAs |
| LIGR-Seq136 | In vivo RNA-RNA interactions | AMT crosslinking and circRNA ligase | Mapping of ligated junctions | Linker defined | RNA fragmentation, Size selection, Adapter ligation | Transcriptome-wide in vivo analysis in H. sapiens |
| PARIS135 | In vivo RNA-RNA interactions | AMT crosslinking and T4 RNA ligase I | Mapping of ligated junctions | Linker defined | RNA digestion and 2D gel purification, Adapter ligation, Size selection | Transcriptome-wide in vivo analysis in H. sapiens and M. musculus |
| SPLASH137 | In vivo RNA-RNA interactions | Biotinylated psoralen (biopsoralen) crosslinking and T4 RNA ligase I | Mapping of ligated junctions | Linker defined | RNA fragmentation, Bead enrichment, Size selection, Adapter ligation | Transcriptome-wide in vivo analysis in H. sapiens, E. coli, and S. cerevisiae |
Abbreviations:
1M7, 1-methyl-7-nitroisatoic anhydride
1M6, 1-methyl-6-nitroisatoic anhydride
AMT, 4’-aminome-thyltrioxaie
BzCN, Benzoyl Cyanide
CIRS, Chemical Inference of RNA Structures
COHCOA, Closure-based *OH correlation analysis
CMCT, 1-cyclohexyl(2-morpholinoethyl)carbodiimide metho-p-toluene sulfonate
DMS, dimethyl sulfate
icSHAPE, in vivo click SHAPE
LIGR, LIGation of interaction RNA
MAP, Multiplexed Accessibility Probing
MaP, Mutational Profiling
MOCHA, Multiplexed *OH Cleavage Analysis
NAI, 2-methylnicotinic acid imidazole
NMIA, N-methyl isatoic anhydride
PARS, Parallel Analysis of RNA Structure
PARIS, Psoralen Analysis of RNA Interactions and Structures
PARTE, Parallel Analysis of RNA Structure with Temperature Elevation
REEFFIT, RNA Ensemble Extraction From Footprinting Insights Technique
RING, RNA Interaction Groups
SHAPE, Selective 2’-Hydroxyl Acylation Analysed by Primer Extension
SHAPES, SHAPE Selection
SPLASH, Sequencing of Psoralen crosslinked Ligated And Selected Hybrids
Different probes for different biological questions
The design of a high-throughput RNA structure probing experiment begins with the biological question being asked. This determines the experimental context, which in turn influences the choice of probe or combination of probes used.
Biochemical RNA structural probes fall into two broad categories: enzymatic and chemical probes. Enzymatic probing uses nuclease enzymes that cleave RNAs in structure- and base-specific patterns of fragments. Chemical probes react with RNA molecules to encode the structure of the RNA as a pattern of covalent adducts that can then be mapped (Figure 2). Since chemical probes are much smaller than comparatively bulky enzymatic probes, they often offer higher resolution RNA structural information56,57. In addition, chemical probes have been tailored to allow probing of RNA structures within cells19,20,32,33,39,50,58–63, which is not the case for enzymatic probes due to the difficulty of transporting bulky nuclease enzymes across cell walls in a controlled fashion. The reactivity of chemical probes can also be manipulated, giving rise to a wide array of probe choices that can be tailored to specific questions (Box 1). We therefore focus on chemical probes in this review, while enzymatic probes are covered in other reviews56,57,64.
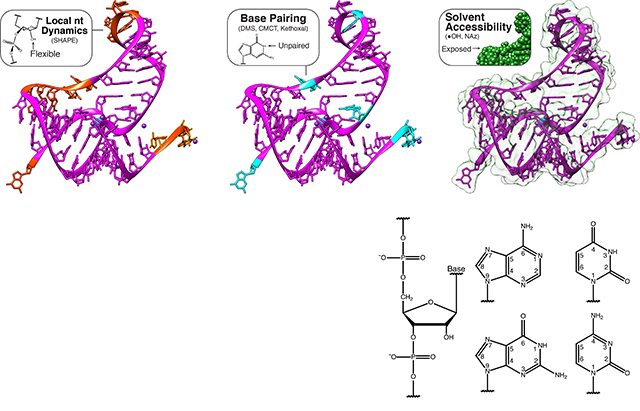
Box 1 |. A palette of chemical probes interrogate many layers of RNA structure.
Chemical probes offer great flexibility in designing experiments that interrogate many facets of RNA structure. Different chemical probes interrogate multiple contexts of RNA structure including base pairing, local nucleotide dynamics and solvent accessibility. In the figure, levels of structural information accessed by chemical probes are mapped onto the structure of the aptamer domain of the T. petrophila fluoride riboswitch (PDB: 4ENC) that is shown alongside chemical structures of the bases (part a). The chemical structures of an array of commonly used probes, along with the structure of the adduct formed (red), the leaving group (blue), and reagent half-life (t1/2) where appropriate, are shown in a table (part b).
When choosing which chemical probe to use to address a particular biological question, care must be taken to properly account for the detailed nuances of each probe’s chemistry to ensure the resulting reactivity data can be properly analyzed, interpreted and compared between different conditions and experiments. Several details are particularly important. First, while probes generally fall into broad categories of chemical reactivity patterns, each probe chemistry is specific and it is generally being established that the most informative experiments use multiple probes to obtain more complete RNA structural information25,48,72,80,151. Second, probes can often react with base specificities other than that commonly reported. For example, DMS can also react with the N7 position of G’s65, and some reports detect DMS modification of the N3 position of U and N1 and N3 positions of G81,82. In addition, SHAPE reagents such as NMIA and 1M7 show minor, but significant, dependencies on base identity159, though these measurements have not been performed for the newer SHAPE probes. Third, the measured reactivity can depend on the timescale of the probe reactions (Box 2). For probes that need to be quenched with specific quenching reactions (see part b) this is particularly important, as the measured reactivity will increase with the longer duration of the probing step before quenching. For probes that naturally quench through the reaction with water, the t1/2,hydrolysis (t1/2 in the figure) is an important parameter that governs the timescale of RNA folding being probed. Specifically, t1/2,hydrolysis can directly govern the measured reactivity of a specific position if the timescale of ribose puckering transitions are much slower than SHAPE adduct formation25 (Box 2). This fact can be exploited to reveal particularly slow conformational changes in RNA72,74 and lead to more accurate models of RNA dynamics151. In addition, t1/2,hydrolysis can be influenced by ionic strength and pH for some SHAPE reagents such as NMIA71, so care must be taken to account for this when designing folding conditions. It is also generally unknown how side reactions with other cellular components may influence the general timescales, and therefore observed reactivities of in vivo probing with SHAPE reagents, which should be a topic of future research. Fourth, the formation of covalent adducts with the RNA could change the underlying RNA structure being probed82,93, especially under conditions where multiple modifications are being sought81, and control experiments or careful data interpretation are needed to take this into account. Finally, some probing reagents are toxic or have toxic side products so care must be taken to perform probing experiments safely.
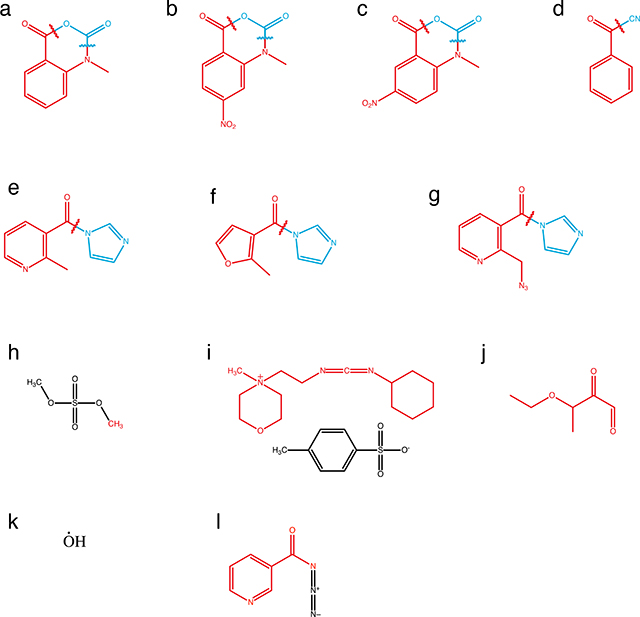
Chemical Structures
| Probe | Chemical Structure (Adduct Red, Leaving Group Blue). Letters refer to Box_l_ProbeStructures. | Primary Modification Sites | Half-Life | Reference | |
|---|---|---|---|---|---|
| N-methyl-isatoic anhydride (NMIA) | a | 2’ OH, all nts | 430 s | Merino et al., 2005 | |
| 1-methyl-7-nitroisatoic anhydride (1M7) | b | 2’ OH, all nts | 14 s | Mortimer and Weeks, 2007 | |
| 1-methyl-6-nitroisatoic anhydride (1M6) | c | 2’ OH, all nts | 31 s | Steen et al., 2012 | |
| SHAPE | Benzoyl cyanide (BzCN) | d | 2’ OH, all nts | 0.25 s | Mortimer and Weeks, 2008 |
| 2-methylnicotinic acid imidazolide (NAI) | e | 2’ OH, all nts | 33 min | Spitale et al., 2013 | |
| 2-methyl-3-furoic acid imidazolide (FAI) | f | 2’ OH, all nts | 73 min | Spitale et al., 2013 | |
| 2-methylnicotinic acid imidazolide azide (NAI-N3) | g | 2’ OH, all nts | 33 min | Spitale et al., 2015 | |
| Dimethyl sulfate (DMS) | h | G N7, A N1, C N3 | N/A | Peattie and Gilbert, 1980; Tijerina et al., 2007 | |
| Base Specific | N-cyclohexyl-N’-(2-morpholinoethyl)carbodiimide metho-p-toluenesulfonate | i | G N1, U N3 | N/A | Gilham, 1962; Ho and Gilham 1967 |
| Kethoxal, other 1,2-dicarbonyl compounds | j | G N1 and N4 | N/A | Litt and Hancock, 1967; Mitchell et al., 2018 | |
| Solvent Accessibility | Hydroxyl Radical | k | backbone | N/A | Latham and Cech, 1989; Wang and Padgett, 1989 |
| Nicotinoyl Azide (Naz) | l | G C8, A C8 | N/A | Feng et al., 2018 |
Chemical probes fall into two main classes (Box 1): base-specific probes and generalist probes. Base-specific probes react with base moieties and are therefore directly sensitive to base-pairing interactions or solvent accessibility. For example, dimethyl sulfate (DMS) reacts preferentially with the Watson-Crick face of adenine (N1 position) and cytosine (N3 position) as well as the N7 position of guanine65, 1-cyclohexyl(2-morpholinoethyl)carbodiimide metho-p-toluene sulfonate (CMCT) reacts with the Watson-Crick face of guanine (N1 position) and uracil (N3 position)56, kethoxal reacts with the N1 and N2 positions of guanine to form a new ring structure56, and glyoxal derivatives react with the amidine moieties on the Watson crick faces of G, A and C62. On the other hand, N,N-(dimethylamino)dimethyl chlorosilane (DMAS-Cl)66 reacts with the N2 position of guanine and nicotinoyl azide (NAz) 63 react with the C8 position of guanine and adenine to measure solvent accessibility (Box 1). While there is a large palette of base-specific probes67 that enable direct interrogation of the structural state of the base, it is necessary to use multiple reagents if structural information for every nucleotide is desired.
Generalist probes react with the RNA backbone to interrogate RNA structure and can therefore be used to monitor every nucleotide simultaneously (Box 1). For example, hydroxyl radicals (*OH) probe solvent accessibility through backbone cleavage18,68. SHAPE probes (Selective 2’-hydroxyl acylation analyzed by primer extension)24 are electrophilic reagents that interrogate local nucleotide dynamics by reacting with the ribose 2’-hydroxyl of each backbone position25,27,28,69. SHAPE probes self-quench by reacting with water and are typically characterized by hydrolysis half-life (t1/2,hydrolysis) which governs the timescale of RNA dynamics probed, the type of environment they can be used in (in vitro vs. in vivo), and the ultimate meaning of the reactivity measured from these probes (Box 1,2). The mechanism of SHAPE probing can make reactivity interpretation challenging. For example, unpaired bases in an RNA hairpin loop can still be constrained by base stacking interactions that make them weakly reactive to SHAPE probes70. However, if interpreted appropriately this can reveal higher order structures and tertiary contacts which can manifest themselves as regions of intermediate to weak reactivities that change when different reagents or divalent cation concentrations are used in the folding conditions 71,72.
The availability of diverse chemical probes enables tailoring of probe choice to specific biological questions. Probes that react with RNA rapidly are well-suited to studying RNA folding dynamics. For example, the renaturation folding pathway of RNase P RNA has been studied with the fast SHAPE reagent benzoyl cyanide (BzCN) which revealed that the forming of a single stacking interaction is the rate limiting step of folding this RNA73,74. BzCN was also used to study how nascent RNAs fold as they exit RNA polymerase cotranscriptionally75,76. More recently, a fast probing technique called light-activated structural examination of RNA (LASER) was developed 63. Here, UV light is used to activate nicotinoyl azide (NAz) into an electrophilic aroyl nitrenium ion, with both excitation and quenching occurring on a ps timescale, which promises to open new doors to accessing even faster timescales of RNA loop and interhelical dynamics 10with chemical probes.
There is also a wide pallet of probes that can be used to study RNA folding in vivo. Probes that react with RNA slowly naturally allow this, since their long reaction time allows them to penetrate cellular and nuclear membranes. Specifically, probes such as DMS, 1-methyl-nitroisatoic anhydride (1M7), 2-methyl-3-furoic acid imidazolide (FAI), 2-methylnicotinic acid imidazolide (NAI), NAI-N3 and glyoxal have been used in a number of studies to ask fascinating questions about the role of the cellular environment on RNA folding and function (see below)19,20,32,33,39,50,58–62. In addition, since NAz can pass through cellular membranes and be subsequently activated with light, LASER joins hydroxyl radical footprinting 77 as a fast in vivo probe of solvent accessibility, tertiary structure and RNA-protein interactions though without the need to use synchrotron radiation. We discuss additional applications of the diverse chemical probes available to biological questions below.
Approaches to detecting probe modifications
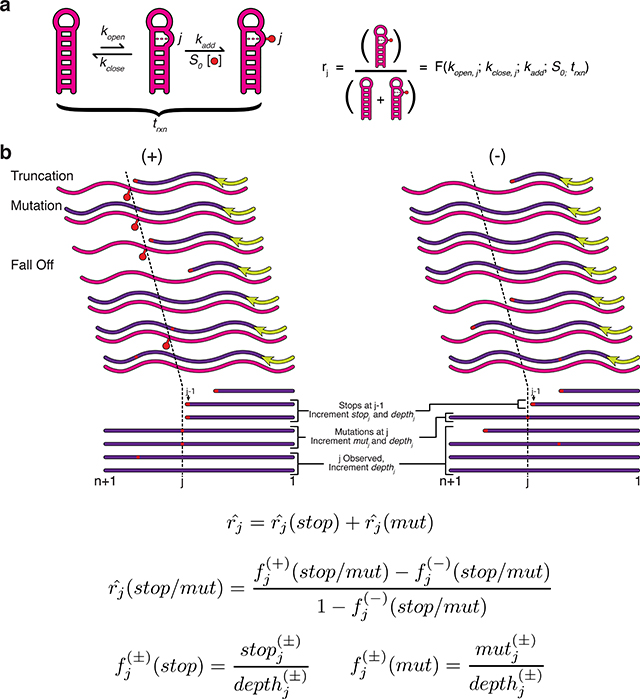
Following probe selection, a method for RNA modification detection must be chosen. Current strategies for chemical adduct detection use reverse transcription (RT) to record modifications in the resultant cDNA sequences as either truncations (RT-stop) or mutations (RT-mutate) (Figure 3c).
Figure 3 |. Reverse transcription strategies for detecting RNA modifications.

Chemical probing experiments detect the presence of RNA modifications using reverse transcription (RT). Three important variables to consider are the RNA processing steps used before RT, the RT priming strategy, and the method used to detect modifications (Table 1). Optional processing steps before RT include fragmenting the RNA, linker ligation for downstream RT priming, or modifying adducts to allow purifications (a). Several RT priming strategies can be used (b). RNA defined priming uses an RT priming site within the RNA, while linker defined primes off of the ligated linker. Multiple RT primers are used for long RNAs, and random primers can be used to tile across long RNAs, or to probe complex mixtures of RNAs and transcriptomes. Two methods are used to detect modification positions (c). In RT-stop methods, RT stops one nucleotide before the RNA modification, leading to a truncated cDNA product. In RT-mutate methods, RTs read through the modification and cause a mutation in the cDNA. Both methods result in premature truncations and background mutations in both the probed (+) and control (−) reactions that should be accounted for in downstream data analysis (Box 2).
RT-stop methods are based on the tendency for chemical adducts to halt reverse transcriptases one nucleotide prior to the modification24,67. Historically, RT-stop has been the detection strategy of choice17 because truncated cDNAs are easily separated by gel electrophoresis. The first sequencing-based strategies also relied on RT-stops to encode RNA modifications36 but used HTS to map cDNA ends thereby enabling multiplexed probing. RT-stop methods are amenable to the least expensive HTS kits because they require very short DNA sequencing reads to map each fragment. However only one modification can be detected per cDNA because RT has a propensity to stop at the first modification encountered. Therefore, when using RT-stop methods, probing conditions should be optimized to balance the desire for single-hit kinetics while still modifying the RNAs sufficiently to allow good data quality78. RT-stop methods also require signal decay correction during data analysis since longer cDNA fragments are naturally less abundant due to the higher chance of encountering a previous stop 79,80 (Box 2).
RT-mutate methods are based on the tendency for reverse transcriptases to misincorporate at chemical adducts under specific reaction conditions44. Modification detection by RT-mutate is specifically enabled by HTS due to the requirement of sequencing to read mutations. In these Mutational Profiling (MaP) methods, the encoding of modifications as mutations rather than truncations during RT is promoted by the use of Mn2+ as the divalent cation in place of Mg2+. Because misincorporation under these conditions does not tend to halt RT, multiple RNA modifications can be detected per molecule44,51. This allows for more advanced data analysis approaches that separate sequencing reads into different groups according to mutation pattern before downstream analysis to uncover the signatures of tertiary interactions, or multiple subpopulations of RNA structures in the probed ensemble81, or even to detect paired bases82 (see “Towards Tertiary Interactions” below). RT-mutate methods have also be used to detect structural changes in an RNA due to single-nucleotide differences by splitting reads according to these differences before structural analysis 83. When using RT-mutate methods, it is important to use DNA sequencing read lengths that cover the entire RNA region of interest to obtain complete information on its structure.
While the properties of the two modification detection methods may appear to favor RT-mutate methods, two recent reports suggest that both may suffer from biases in their ability to detect specific RNA modifications. Sexton et al.54 and Novoa et al.55 performed in cell DMS modification and detected both RT truncations and mutations. They found several important features of the resulting data: (i) RT stops are detected even in conditions that favor mutations; (ii) distinct biases are observed for each encoding strategy, with RT-mutations favoring DMS-modification detection at C and RT-stops favoring detection at A (observable in many previous DMS probing datasets37,38,45,81); and (iii) biases in modification recording are both reverse transcriptase and sequence context dependent. Overall this suggests that RT-stop and RT-mutate methods should be used simultaneously to give the most accurate mapping of DMS modifications54,55 (Box 2). More work is needed to confirm that these conclusions apply broadly to the palette of chemical probes, and to study these effects in the context of other well-defined RNA folding benchmarks.
A key aspect of experimental design for both RT-stop and RT-mutate approaches is the choice of RT priming strategy (Figure 3b). Defined priming uses DNA primers that bind to specific parts of the RNAs under study, and is well suited to studying isolated RNAs, or specific RNAs that are present in a complex mixture36,40,43,44,47,49,50,54,58–60,80. Here, the priming sites can be either internal to the RNA48,50,51,54,59,84 or within a defined region that can be included in the RNA 3’ end and that is present during folding and probing44,47. Ligation of a linker sequence to the RNA after probing enables the addition of a defined priming site after probing40,43,48,49,51,75. For RT-stop methods, each priming site can be used to generate useful information for a ~300–400 nt window beyond which the signal becomes too weak due to signal decay85. For RT-mutate methods, longer regions are possible but are limited by current sequencing read lengths. For RNAs that exceed these length limitations, multiple defined priming sites can be used and reactivity data stitched together within the different windows84,85. Multiple priming can also be performed using random RT primers that can bind at many positions throughout a mixed population of RNAs, which is particularly useful for transcriptome-wide studies38,45,46,52, or studying long RNAs44,60,85,86. The use of stronger binding LNA primers can be advantageous for random priming44. We note that some methods perform RNA fragmentation before or after RNA modification and before reverse transcription to address issues with RT drop off in long fragments37,39,40. If this is done, special care must be taken to account for the presence of RT stops due to the fragmentation process instead of adduct formation, which can confound the calculation of reactivities40. Fragmentation can also be used with RT-mutate approaches to study long RNAs51.
Finally, for both RT-stop and RT-mutate approaches, another key aspect of experimental design is the inclusion of a no-reagent control sample (often referred to as a (−) channel sample). This RNA sample is folded in the same conditions, but treated with a control solvent without modifying reagent prior to RT in the exact same steps as the modified ((+) channel) RNA sample. The inclusion of the (−) channel allows data analysis to remove false positive reactivity signal that could be due to natural RT fall off or mutations in the specific sequence context of the RNAs being studied (see Box 2 below).
Multiplexing with high throughput sequencing
The conversion of modified RNA into cDNA enables sequence analysis by HTS. The use of HTS to directly sequence cDNAs immediately expanded the scope of previous structure probing experiments (Figure 1), as the DNA sequencing information could be used to bioinformatically distinguish reads that arise from thousands of different RNA molecules all probed simultaneously.
While powerful, care must be taken when designing experiments using HTS as many additional experimental steps are required to ‘format’ cDNAs into ‘libraries’ that are compatible with specific sequencing platforms. Specifically, most HTS platforms require that cDNAs contain platform-specific adapter sequences on either end of the molecule that are used by the instrument for a variety of purposes such as localizing DNA molecules to imaging surfaces, facilitating amplification, or serving as sequencing priming sites87. Adapter sequences can be added during reverse transcription as an RT primer tail, through ligations, by PCR, or by a combination of these approaches (Figure 4).
Figure 4 |. Strategies for next generation sequencing library preparation.

Several biochemical steps are needed in order to convert cDNA products from reverse transcription (Fig. 3) into a format compatible with sequencing instruments. After reverse transcription, a series of ligation, circularization and PCR steps are performed in different combinations to append adapter sequences needed for the sequencing process (a), and an optional set of barcodes and indices that can be used for multiplexing and downstream analysis (b). Adapter sequences are often required on both ends of the cDNA. One adapter can be conveniently included as a tail of the RT primer. The second adapter can then be ligated on through a single stranded DNA ligation, or through a circularization if the RT primer tail contained elements of both adapter sequences. Note that ligations/circularizations must be used for RT-stop techniques since each RNA modification position leads to a different cDNA end which must be mapped to recover the position. In RT-mutate methods, the presence of full length products due to RT read through allows PCR to be used to add the necessary adapter sequences. Many techniques contain additional enzymatic processing (c) and purification (d) steps throughout library preparation that get rid of excess oligonucleotides or unwanted side products that can impact downstream steps or the number of informative reads from the library. Each additional step has potential biases that must be considered when estimating reactivities (Box 2).
A major consideration when selecting a library formatting strategy is the biochemical biases that can be introduced at every stage of library preparation, including during the linker ligation used for RT priming88, the RT priming strategy itself78 (Figure 3), the ligation/circularization step used to add additional adapter sequences89 (Figure 4) and PCR amplification43. Bias in DNA adapter ligation is of particular importance in RT-stop methods, since the cDNA 3’ end that the adapter is ligated to is precisely the sequence that needs to be accurately mapped in order to recover the location of RNA modification (Fig. 3c). Common concerns for each of these steps is that the reactions do not proceed to completeness, and have biases towards specific sequences that would allow those contexts to be unequally sampled, which would lead to biased reactivity data. To address this, several studies have undertaken careful optimizations of some of the key steps of library preparation including using quality control analysis to directly assess ligation efficiency47, and the particularly promising approach of using optimized adapter formats and conditions to reduce adapter ligation bias52,89. Lastly, while PCR amplification of sequencing libraries enables the use of low amounts of input material, there is a concern that amplification could lead to bias. The limited studies to directly assess this have concluded this is not the case43, though extensive comparisons for all variations of library preparation were not made. A particularly promising approach for eliminating PCR bias has recently been used by several studies that incorporate a random barcode within the library adapter sequences51,55 (Fig. 4B). This barcode is designed to be theoretically long enough to uniquely tag each cDNA product for bioinformatic filtering so that each molecule is only counted once.
Another important consideration is the removal of unwanted side products. Many library preparation steps use excess primer, linker or adapter oligonucleotides to help drive reactions to completion. This excess can interfere with downstream library preparation steps or result in adapter dimers that use up valuable sequencing reads. Gel extraction protocols have been used to remove these excess oligonucleotides and side products. However, this approach is laborious and inefficient. To remedy this several bead selection approaches have been developed to select against unwanted products by targeting specific sequences of interest through complementary oligonucleotides to the target RNA 47, streptavidin binding to biotinylated positions in the cDNA added during RT52 or streptavidin binding to biotin positions introduced through click chemistry approaches on specially designed SHAPE reagents such as NAI-N339. Finally, PCR selection approaches can be used whereby PCR primers are designed to include a mismatch in unwanted side products that prevents their amplification50.
Experimental challenges
Since all high-throughput RNA structure probing techniques share this conceptual umbrella, they share several common challenges that should be considered to understand the complexities and limitations of the data generated. Perhaps the most significant is that all techniques inherently project the sophisticated three-dimensional nature of RNA structures onto a one-dimensional vector of reactivity information. This means that in order to recover or infer models of RNA structures from this data, additional information needs to be added – either through computational methods or multiple experiments, which are both discussed below. In addition, these techniques are inherently ensemble measurements – they probe a population of RNA molecules and structural states, meaning the resulting data reflects an average over this population. Computational methods90–92 or recently developed experimental strategies81 are needed to extract information about different RNA structures that may exist in the population. Furthermore, the fact that these techniques probe structure by directly chemically modifying RNA has two important implications: covalent adducts can alter the underlying RNA structure being probed 82,93 meaning care must be taken in interpreting data resulting from multiple modifications per molecule, and these techniques are inherently destructive meaning that there is no way to continually probe the time trajectory of RNA folding of the same sample. Finally, while some work has been done to identify what sequencing depth of coverage is required for accuracy44,78,94,95, more work is needed to fully establish best practices, particularly in the case of transcriptome-wide experiments. Nevertheless, the benefits of determining RNA structures with high throughput sequencing outweigh these challenges and the rapid pace of innovation is already starting to improve the technique.
Data analysis: defining and calculating reactivities
The ultimate goal of RNA structure probing experiments is the quantitative measurement of RNA structural information. This measurement is most naturally reported as ‘reactivity’ that describes how reactive each RNA nucleotide is to a given probe. Since probes tend to react with unpaired positions, high reactivities generally correlate with unstructured regions of the RNA, while low reactivities correlate with structured regions. However, the definition of reactivity more deeply links the nuances of the probe chemistry to the pattern of observed DNA sequencing reads (Box 2), and informs the interpretation of reactivities for structural insights and modeling.
‘Reactivity’ links chemistry with RNA structure
Chemical and enzymatic probing are governed by the chemical reaction of the probe with an RNA. Consequently, the quantitative definition of reactivity lies within the chemical kinetics that govern a probing reaction. The inherent fact exploited by RNA probing methods is that the rate of modification is structure-dependent. Nucleases depend on the propensity for specific enzymes to cleave either single or double stranded RNA34,35,56, whereas chemical probes tend to react faster with unstructured nucleotides. For some chemical probes, models of the chemical kinetics have been developed that directly link reactivity to the rate constants that describe the structural context of a nucleotide and the chemical reaction itself23–25 (Box 2). The implication is that if the rate constants of the chemical reaction are known, then reactivities can be interpreted as quantitative kinetic parameters about nucleotide-level RNA structural properties. For example, when defined in this way, reactivity values that are in between the extreme high and low ends of the scale reveal information about nuanced structural contexts such as base stacking70, slow structural fluctuations in specific regions of the molecules25,74, regions of the molecule which undergo melting transitions69,96,97, tertiary interactions69,71,72,98, and others, which highlight the richness of chemical probing reactivity data.
Calculating reactivities from sequencing reads
Chemical kinetics naturally establishes the reactivity of a nucleotide as the fraction of molecules with a modification at that position at the end of the probing reaction (Box 2). Thus, reactivity information is inherently encoded in the distribution of modifications across an RNA molecule – if many modifications are observed at a given nucleotide, the reactivity will be higher at that position and vice versa. In the absence of complicating factors, the reactivity of a nucleotide is the number of modifications observed at that position divided by the number of RNA molecules probed, or the fraction of adduct formed at that nucleotide. However, because current methods for modification detection are indirect, this calculation must be adjusted to account for experimental factors in order to estimate the true reactivity value (Box 2).
Experimental factors can be accounted for by calculating reactivities as a corrected formula for the fraction of adduct formed at each position (Box 2), and represent a statistical estimate of the true reactivity values determined by chemical kinetics. Consequently, the kinetic parameters that describe RNA structural context should be quantitatively extractable from these reactivity estimates. Furthermore, these estimates define a natural and absolute scale for reactivity that could be used to make comparisons across different experiments, laboratories, and even conditions80 if the kinetic parameters and biases of different experimental strategies are taken into account. Many methods propose normalization schemes whereby highly reactive positions are normalized to a value of one. While normalized data can still be used for relative comparisons within a molecule, the normalization breaks the link to the chemical kinetics definition of reactivity. Furthermore, alternative methods for calculating reactivity that do not estimate fraction of adduct formed also break the links to the underlying chemistry of the probing reaction.
Data analysis: interpreting and using reactivities
Structural probing reactivities can be analyzed in different ways. Most directly, reactivity values can be interpreted based on their magnitude to roughly identify structured and unstructured regions of the RNA. Reactivities can also be compared across different RNAs or across different experimental conditions to identify regions that change structure in these contexts. More complete models of RNA structure can be generated using computational algorithms that incorporate reactivity data in their calculations. Below we discuss each of these areas.
Interpreting reactivity information
The direct interpretation of reactivity values can be a powerful starting point when analyzing high throughput structure probing data. Highly structured or unstructured regions can frequently be observed by visual analysis of the reactivity patterns across the molecule. For simple RNA structures, characteristic reactivity patterns often emerge. For example, strong hairpins often result in a ‘low-high-low’ pattern where low reactivity regions correspond to the stem and the high reactivity pattern indicates the loop (Figure 5). However, complex RNA structures often yield reactivity patterns that are difficult to directly interpret and may necessitate computational modeling for a deeper understanding (see below). Nevertheless, these complex patterns can often be understood through the lens of additional structural information. For example, comparing reactivity patterns to known crystal structures can reveal how base stacking or non-canonical base pairing are manifest as reactivity signatures27,28,36,44,48,70,72. More recently, comparison of reactivity data to detailed molecular dynamics simulations revealed that reactivities correlated with features of RNA structural dynamics such as how frequent the ribose backbone samples the C2’-endo configuration, and suggested mechanistic roles for SHAPE reagent stacking with the backbone and specific nucleotide bases to facilitate reagent binding prior to reaction 93. It is important to note that the interpretation of intermediate reactivities will depend on the reactivity scale and any normalization schemes used (Box 2). As community efforts to share RNA structural mapping datasets grow99–101 this creates an exciting opportunity for a deeper analysis of reactivity patterns to aid in their direct interpretation.
Figure 5 |. Applications of high-throughput structure probing.

The development of high-throughput RNA structure probing methods has enabled new and diverse applications to previously inaccessible biological questions. The development of new chemical probes and analysis frameworks has enabled the characterization of in vivo RNA structure as it pertains to RNA protein interactions59, RNA functional states, and post-transcriptional interactions39,50 (a). The extension of in vivo chemical probing experiments to transcriptome wide studies has identified functional roles for RNA structure in mRNA cleavage and polyadenylation164 and the structural organization of operon mRNAs127, and has found that G-Quadruplexes are globally unfolded in eukaryotic cells128 (b). The combination of high-throughput RNA structure probing and multiplexed in vitro transcription allows measurement of nascent RNA folding pathways75 and immunoprecipitation of Pol II has been used to isolate cellular nascent RNAs for structure probing146(c). MOHCA-Seq 48 and RING-MaP81 leverage the information richness of high-throughput RNA structure probing to infer higher order RNA structure (d). Structural subpopulations can be identified through spectral clustering of RING-MaP data81 and by multiple structure prediction using SLEQ computational analysis methods 120 (e).
Comparative reactivity analysis
A strength of chemical probing experiments is their flexibility, which permits the controlled manipulation of probing conditions in vitro such as temperature, salt concentration, and the presence of interacting ligands and proteins32,49,69,72,96,102, and even allows probing in cellular conditions32,33,37–40,50,51,58–61,96. A direct comparison between reactivities in different conditions is therefore a powerful way to study how these conditions affect RNA structure. Comparative reactivity analysis typically involves subtracting values between specific conditions to find RNA regions that are higher or lower reactivity (less structured or more structured) in particular conditions. For example, several studies have applied in vitro probing to study how ligand binding changes riboswitch structure48,49,72,102, and to find differences between kinetically folded and equilibrium refolded RNAs75,76. Comparative methods have been recently developed that make use of sliding window averages and statistical tests to give more confidence in the discovery of protein-RNA interactions59. Comparative methods have also been coupled to high-throughput targeted mutagenesis to dissect RNA structural ensembles and the mechanism of ligand-mediated structural switching in a regulatory RNA 103. A particular exciting area is merging comparative reactivity analysis with multiple sequence alignment, which has been used to identify structural RNA regulatory elements within viral genomes86. While powerful, care should be taken in these analyses to separate the effects of changes in reactivity due to structural changes of the RNA, or to ligand binding and other cellular interactions.
Using reactivity information to model RNA structures
The accuracy of computational RNA secondary and tertiary structure prediction can be improved by incorporating reactivity data104. Reactivities can be used within RNA secondary structure prediction methods as restraints to return structures that are more consistent with the data, by either using reactivities to modify the underlying model parameters in the algorithm104–109, using data to select structures from a set of generated possibilities 92,110, or a combination of both90,91,111. For example, SHAPE data incorporation into thermodynamic RNA folding models have be done through addition of defined pseudo-free energy terms 104, or iteratively perturbing the energy model to generate structures that better match the SHAPE data 106. Specifically, pseudo-free energy terms that use reactivities (r) when calculating free energies of nucleotides involved in base stacking have typically taken the form:
where is the pseudo-free energy term at nucleotide i, m and b are constant parameters, and r(i) is the reactivity at nucleotide i 104. With m positive and b negative, this term effectively penalizes nucleotides within structures that have high reactivity, and thus favors nucleotides with low reactivities to be in structured positions. As an alternative to specifying a functional form for a free energy derived from reactivities, iterative approaches add small perturbations to each free energy term in order to minimize the difference between estimated experimental base pairing probabilities derived from reactivities and predicted base pairing probabilities using the pseudo-free energy terms 106.
Once reactivity information is incorporated into the energy model, there are a variety of computational methods to predict the experimentally-restrained structural state. Minimum free energy (MFE) approaches return the most thermodynamically favored conformation, which is theoretically the structure most occupied by an RNA in equilibrium112. On the other hand, maximum expected accuracy (MEA) methods use a probabilistic model to select the most probable structure across the ensemble 105,113,114. Multiple structures consistent with the experimental data can be predicted with these two types of methods by reporting the next best MFE or MEA structure(s)109. Computational approaches that sample multiple RNA structures from the equilibrium thermodynamic probability distribution can also be used to model an ensemble of structures as a way to extract population level structural information from reactivity data, and can be used to look at how structural populations shift when conditions are changed or SNPs are introduced83,90,91,96,115. There are also methods that can use reactivity information to restrain tertiary structure models48,116–120. For more details, there are excellent reviews of RNA structure prediction algorithms that incorporate experimental structure probing data 109,121.
Many of the methods that incorporate experimental probing data have been shown to improve predictive accuracy 49,122. To date, most accuracy assessments are done by comparing the base pairing patterns of structural predictions to solved RNA crystal structures since the latter reveal both secondary and tertiary structure base pairing partners 43,123. In addition, experimentally restrained structural predictions have been made to structures derived from comparative sequence analysis to determine the accuracy of these methods on highly conserved RNAs 122. In each case, accuracy is quantified through measures of sensitivity (true positives divided by the sum of true positives and false negatives) and positive predictive value (PPV, true positives divided by the sum of true positives and false positives), with current techniques improving each measure broadly from around 60% for predictions without experimental data to above 85% when experimental data is included 105. For all methods, the accuracy improvement of each algorithm varies based on the RNA, and to date there is no algorithm that can use experimental data to predict the structure of every RNA with perfect accuracy. This is due to several factors. First there are inherent limitations on the computational structural models that may limit accuracy. These include the fact that free energy parameters are optimized to predict structures in specific in vitro folding conditions (temperature, salt concentration, pH) that may not match well to the experimental probing conditions, and the fact that certain non-canonical base pairing interactions are not included in most models. Second, predictive accuracy is likely influenced by experimental data quality, though the relationship between data quality and prediction accuracy has been mostly assessed using simulated data. Finally, accuracy assessments have only been done on a limited set of benchmark RNAs that are not representative of all biologically relevant RNAs across all organisms 100,101,124,125 which could reveal important classes of structures where method improvement is needed. The continual improvement of these data-informed RNA structure modeling methods, and an extension into modeling RNA folding dynamics 93, is an exciting area of research and will likely evolve with the improvements in experimental techniques.
Enabling new RNA structural biology
The advent of techniques to probe RNA structures with HTS has given rise to fundamentally new ways to interrogate the role of RNA structures in a wide range of biological processes from transcription to macromolecular assembly. Below we highlight several areas of RNA structural biology that are being revolutionized and in some cases pioneered by these techniques.
Examining how the cellular environment affects structure
The ability to perform RNA structure probing within different experimental conditions has allowed some of the first insights into how the complex environment in the cell impacts RNA folding (Figure 5A). These studies make use of the fact that some older chemical probes32,37,38 and some newly developed probes33,39,62 can traverse cellular membranes. By comparing in vivo and in vitro measurements, several studies have found general impacts of the cellular environment on RNA structure, with some concluding that RNAs in vivo are more32, or less37 structured than in vitro. Other studies have found that RNA structure in cells depends on the context39, for example in regulatory regions where low reactivities were observed upstream of splice sites for unspliced RNAs38. Other studies have started to uncover clues as to how specific factors such as protein and ligand binding, or RNA modifications alter RNA structures in the cell. These include the observation of protein binding to sites in regulatory and catalytic RNAs such as snRNPs, SRP RNA, rRNAs and RNase MRP45,53,58,59,126, hints that m6A modification affects RNA structures39, views on the role of structure on differential translation rates within mRNA operons127, and intriguing suggestions that G quadruplexes are globally unfolded in eukaryotic cells128. When performing cellular probing, care should be taken when deciding which probe to use61, the priming strategy to target specific RNAs or interrogate structures transcriptome-wide, and the depth of coverage needed to accurately estimate reactivities78,94,129.
RNA ‘structuromics’: probing RNA structure transcriptome-wide
All RNAs have an inherent structure and thus studying RNAs transcriptome-wide could provide a systems level view of RNA structure (Figure 5B). The first transcriptome-wide structure probing experiments used in vitro enzymatic methods34,35,41,42 and the subsequent application of chemical probing37–39,130 enabled these measurements to be made within the cell. These methods have been applied across diverse systems and provide insight into the global regulation of RNAs through structure. Comparison of in vivo and in vitro measurements has started to address questions such as how RNA binding proteins affect RNA structure and the structural accessibility of coding sequences at transcriptome scale37–39,131. Developments in in silico computational screens for conserved RNA structures have also been used to enhance the discovery of functional structured RNAs expressed across the genome and have been shown to agree with in vitro structure probing data 132,133. Alternative to direct structural probing, sequencing-based proximity ligation methods have also been developed to more directly determine RNA-RNA interactions134–137 and those mediated by RNA binding proteins138–141. Depth and coverage are a large consideration for transcriptome-wide studies since not only do reads need to span the transcriptome, there should also be a high coverage of reads to accurately estimate reactivities for each RNA. New computational methods that incorporate the estimation of transcript isoform abundance along with reactivities are an important advance for the analysis of experimental transcriptome-wide studies142. In addition, the ability to use SHAPE-MaP to detect structural differences between different RNA alleles 83 may become important for understanding how genome sequence variation impacts cellular functions through changes in RNA folding.
Towards Tertiary Interactions
RNA chemical probing has emerged as a tool that can provide insights into complex RNA folds using simple biochemical measurements that are compatible with diverse experimental contexts. Interestingly, even though chemical probing is lower resolution than biophysical methods, recent work has leveraged the information richness of HTS to infer higher order structure. In particular, the insight that RT-mutate methods can extract the locations of chemical adducts at multiple positions within a single RNA has enabled the implementation of a probing experiment to identify RNA interaction groups by mutational profiling (RING-MaP)81. RING-MaP heavily modifies a target RNA using DMS, which results in a pattern of mutations in each cDNA. Nucleotide positions that are closer to each other in space can cause modification patterns to correlate with each other, for example in a breathing motion where multiple positions may become reactive. The identification of such RNA interaction groups thus facilitates tertiary structure modeling (Figure 5D). Alternatively, if cDNAs are first clustered into groups before reactivity calculation, structural information about RNA subpopulations may be extracted from the data81. Another approach to uncovering tertiary interactions jointly applies Multiplexed OH (radical) Cleavage Analysis with paired-end sequencing (MOHCA-seq) to infer tertiary contacts by measuring solvent accessibility of positions48. MOHCA-Seq uses 2’-NH2 modified RNAs coupled to isothiocyanobenzyl-Fe(III)•EDTA treatment such that hydroxyl radicals generated by fenton chemistry cleave positions that are near the modified base. By using HTS to map both the modification position and the cleavage position, through-space connections can be mapped. The multidimensional chemical mapping (MCM) pipeline combines MOHCA-Seq with the mutate-and-map approach98,143–145 to first infer a secondary structure model through systematic mutagenesis and chemical probing, which then creates a rich dataset that can be used with three-dimensional modeling up to 1 nm resolution (Figure 5D). For RING-MaP, it is important for the RNA to be sufficiently modified and sequenced at sufficient depth to extract the most higher order structural information. Similarly, the size of the RNA target is an important consideration when applying the MCM pipeline as its multidimensional nature makes the experimental cost scale quadratically with RNA length48.
Uncovering the fleeting structures of nascent RNAs
The folding of RNA molecules into functional structures begins during transcription, however technical barriers have limited the measurement of nascent RNA intermediates at nucleotide resolution. Because the coupling of chemical probing to HTS enables the interrogation of RNA mixtures in solution, it is ideal for mapping intermediate nascent RNA folds (Figure 5C). We recently developed methods to simultaneously probe every intermediate transcript of a target RNA in an in vitro transcription reaction75,76. The basis of cotranscriptional RNA structure probing is the stable distribution of transcription elongation complexes across every position of a DNA template prior to rapid chemical modification. The resulting data can be used to infer the folds that a nascent RNA could pass through during transcription and its relation to its function, and exciting new computational techniques are able to use this data to estimate the proportion of RNAs that undergo a particular folding trajectory within the population 120 (Figure 5E). New methods that couple these techniques to RNA polymerase pull downs are also giving promising early views on nascent RNA structure in vivo146 (Figure 5C). While studies of nascent RNA structure using high-throughput chemical probing are in their infancy, early results emphasize the importance of metastable intermediate structures, particularly in the context of riboswitch-mediated transcription regulation. The compatibility of chemical probing with the cellular environment makes in vivo nascent RNA structure probing an intriguing prospect. However, stably halting cellular transcription for the duration of chemical modification while minimally perturbing nascent RNA structure poses a significant challenge. Given the dynamic nature of cotranscriptional folding it is also important to consider the timescale of the probing reaction when designing experiments and interpreting data.
Future perspective and conclusions
Massively parallel DNA sequencing technologies have transformed efforts to understand the molecular nature of life by enabling information rich biochemical measurements at unprecedented scale147. This is especially true for RNA biology, where high throughput RNA structure determination technologies are quickly becoming an engine for discovering the RNA structures and interactions that mediate fundamental cellular functions. When applied in well-designed and innovative experiments, these tools have generated a renewed appreciation for the importance of gaining a complete understanding of the physicochemical basis of RNA function, which must include knowledge of cellular RNA folding.
This next chapter of RNA biology though is not without challenges. As the application of high-throughput structure probing increases, a major challenge is to improve experimental accuracy. To do this, high quality benchmarks to systematically evaluate these techniques are needed. Current benchmarks43,104,122,148,149 use a panel of natural RNAs with sophisticated structures, and evaluate techniques by whether reactivities can be incorporated into computational structural models to accurately predict those structures. While important, this is an indirect assessment of experimental accuracy that is confounded by the computational modeling approaches. In contrast, a more direct method that evaluates accuracy at the level of the reactivity data itself is needed. For example, developing a panel of simpler RNAs to directly test the ability of techniques to uncover the chemical kinetic parameters of the probing reaction (Box 2), and verify these using complementary approaches, would be a simpler and more direct measure of accuracy. Once established, this benchmark would provide a route to solving the challenge of reducing bias and maximizing the accuracy of measured reactivities. While there has been progress to this end, no study to date has directly compared all of the protocol choices on the same pool of modified and unmodified RNA. There is also the challenge of the depth of sequencing needed to accurately recover reactivity information78,94,129, which varies by approach. Finally, in vivo studies must address how the timescale of chemical probing can impact measurements made in the dynamic cellular environment and in the presence of cellular factors.
Future innovation in high-throughput RNA structure probing will combine advances in probe chemistry, adduct detection, and multi-level computational models to address new frontiers in RNA biology. Techniques for direct RNA sequencing150 that could provide a direct measurement of adduct position could greatly simplify the complexity, and cost needed to uncover reactivity information. In addition, the new emphasis on using multiple probes to extract maximal information44,48,72,151 with advanced modeling48 creates an exciting prospect for in vivo probing to uncover higher resolution cellular RNA structure models. It is also foreseeable that the creation of troves of probing data99–101 will create a rich resource for machine learning approaches to infer these higher resolution RNA structure models.
There is also great potential for combining high throughput RNA structure probing with complementary approaches to enhance and expand our view of the RNA structure/function relationship. For example, merging these methods with single molecule biophysical approaches could link the structural signatures uncovered by chemical probing to RNA folding dynamics152–154. In vivo probing techniques could also be merged with optical methods for nucleic acid detection155,156 that could create enormously rich views of not only what, but where RNA structures are in the cell.
The adaptation of high throughput RNA structure probing methods to new biological and application contexts also has great potential to address longstanding biological questions and facilitate medical advances. For example, we anticipate the application to studying RNA folding dynamics will create a new frontier in RNA biology to uncover the importance of nascent RNA structure. There is also vast potential for these techniques to uncover therapeutic RNA targets and help engineer treatments as evidenced by a new wave of emerging biotechnology companies that are treating RNAs as druggable targets, or using engineered antisense oligonucleotides as therapeutic drugs themselves157,158. An efficient future pipeline for developing RNA centric therapies will likely involve a combination of high-throughput characterization and screening approaches to identify targets and uncover antisense oligonucleotide design principles.
The questions about how RNA structure mediates biological function are some of the oldest of molecular biology. While our ability to ask and answer these questions has been transformed by high throughput RNA structure determination technologies, it is likely that many more waves of innovation are yet to come. Given the history of discovery in RNA biology1,2,6, we are likely on the cusp of another expansion in our understanding of the roles that RNA molecules play in life.
Supplementary Material
Box 2 |. Estimating chemical kinetic ‘reactivities’ from statistical analysis of sequencing reads.
The concept of ‘reactivity’ quantitatively links the chemical probing reaction to the underlying RNA structures. Reactivity is therefore defined from the coupled kinetics of RNA structural fluctuations10 and probe chemistry (Box 1), but must in practice be statistically estimated from the observed sequencing reads. This creates two views of reactivities that should be linked to allow the maximum amount of RNA structural information to be obtained.
Chemical kinetics view
Under a coarse-grained model25, different bases fluctuate between reactive (open), and unreactive (closed) conformations with rates kopen and kclose, respectively (part a). The open state can react with the probe (red) with a rate constant kadd, and the reaction proceeds for a time trxn (set by either t1/2,hydrolysis or by the quenching time (Box 1)). The reactivity of site j (denoted rj) is then defined as the fraction of RNA molecules in the population that is modified at site j, and can be written as a function of all the rates, trxn and the concentration of reagent used, S0. Under this model, the reactivity is linked to RNA structure through the dependence on kopen and kclose25. For example, when S0kadd is slower than kopen and kclose, rj can be calculated to be25
though this form can change and be exploited to gain deeper structural information25,72,74. More work is needed to incorporate features of the nuanced mechanisms of probe reactions into this model27,28,93. This definition of reactivity has been established through modelling SHAPE probes24,25,28. However, since any RNA nucleotide can dynamically fluctuate10, similar considerations could apply to all probes.
Read statistics view
Reactivities must ultimately be estimated from counts of the sequencing reads observed in a chemical probing experiment 79. This is most rigorously done by using statistical methods to estimate the percentage (or fraction, f) of adducts observed at any given position to give an accurate estimate of reactivity at that given position j (rj^) that should correspond to the chemical kinetic reactivity (rj) (part b) 160. While the most simple estimate of reactivity would be to divide the number of RT-stops or RT-mutations at a specific position by the total number of observed reads, more accurate statistical estimates must account and correct for the many steps of sequencing library construction used (Figure 4) that can give rise to multiple sources of bias and noise that must be accounted for:
Bias in adduct detection: An adduct can either cause RT to stop (stopj) or introduce a mutation (mutj) at a specific position160, and the propensity towards one or the other changes based on RT enzyme and conditions54,55. It was recently suggested that a more accurate estimate of total reactivity should use both signatures to count adducts 54,55, which can be rigorously incorporated as a sum of the reactivities due to stops (r^j(stop)) plus that due to mutations (r^j(mut)) 160.
RT drop off bias: RTs prematurely terminate due to encountering an adduct, a sequence/structure context, or other processes. This causes fewer long cDNAs, which requires a correction for single 79,161 or multiple85 RT priming positions, and should also be used for random priming. Specifically, to estimate the fraction of adduct observed at each position, fj, adduct counts stopj and mutj are divided by the observed read depth at that position, depthj, rather than the total reads observed 160.
Experimental noise due to background RT stops and mutations: This noise can be corrected for by performing a control reaction (−) where no adduct is used, and subtracting percent adduct estimates from the reaction with adduct (+).
Context biases in library construction: These could be reduced through experimental optimization or techniques to measure and correct for specific biases47.
Reactivities calculated from sequencing data are estimates to the true chemical kinetics reactivity, and the specific interpretation of what constitutes the values of ‘high’, ‘low’, and intermediate reactivities is nuanced. By estimating reactivities that correspond to the fraction of adduct observed 160, reactivity values should most closely align with the kinetics of the chemical probing reaction, which should allow a deeper understanding of data from high throughput RNA structure probing experiments.
Acknowledgements
We would like to thank Aaron Coraor, Adam Silverman and Rob Batey for informative discussions about the chemical kinetic view of reactivities. We also thank Chaitan Khosla for inspiring the connections between the chemical and statistical perspectives of reactivities. We also thank Molly Evans for helpful comments on the manuscript. This work was supported by an Arnold and Mabel Beckman Foundation Postdoctoral Fellowship [to EJS], the Tri-Institutional Training Program in Computational Biology and Medicine (via NIH training grant T32GM083937) [to AMY], a New Innovator Award through the National Institute of General Medical Sciences of the National Institutes of Health [grant number 1DP2GM110838 to JBL], and Searle Funds at the Chicago Community Trust [to JBL]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Glossary
- Chemical Probe
A small molecule that chemically reacts with RNA molecules in a structure-dependent fashion. Reactions produce adducts which can be detected to give a measure of an RNA’s structure
- cDNA
The complementary single stranded DNA molecule that is produced when RNA is converted to a DNA molecule by a reverse transcriptase enzyme. This conversion allows RNAs to be indirectly sequenced by HTS
- High Throughput Sequencing (HTS)
A suite of technologies that can be used to sequence millions to billions of DNA molecules simultaneously. Many experiments can be performed at once since bioinformatics can be used to distinguish signals between experiments
- Positive Predictive Value (PPV)
A measure of accuracy of RNA structure prediction equal to the number of true positive pairs predicted divided by the sum of true positive and false positive pairs predicted. PPV is often used in combination with sensitivity to assess predictive accuracy of RNA structure models. PPV is equivalent to one minus the false discovery rate
- Reactivity
A measure of a chemical probing reaction that contains RNA structural information. Typically high reactivities indicate unstructured regions while low reactivities indicate structured regions
- Reverse Transcription (RT)
The process by which RNA is enzymatically converted into complementary DNA. RT proceeds in the RNA in the 3’->5’ direction
- RT-Stop
An event where reverse transcriptase stops when encountering a chemical probe adduct on an RNA. This produces a truncated cDNA that can be used to map the adduct position
- RT-Mutate
An event where reverse transcriptase produces a mutation when encountering a chemical probe adduct on an RNA. This mutation can be used to map the adduct position
- Secondary Structure
The pattern of base pairing interactions in an RNA that create helices, loops, bulges, junctions and single stranded regions. In addition to Watson-Crick base pairs, RNAs can pair in many non-canonical patterns
- Sensitivity
A measure of accuracy of RNA strucutre prediction equal to the number of true positive pairs predicted divided by the sum of true positive and false negative pairs predicted. Sensitivity is often used in combination with positive predictive value (PPV) to assess predictive accuracy of RNA structure models
- Shape
Selective 2’-hydroxyl acylation analyzed by primer extension. A class of chemical probes that modifies the RNA backbone. SHAPE probes can be used to interrogate RNA structure at single nucleotide resolution
- Tertiary Structure
The three dimensional orientation of secondary structure elements and nucleotides that give rise to sophisticated three dimensional structures. Tertiary structures can be stabilized by non-covalent interactions and divalent cations
Footnotes
Competing interests
The authors declare no competing financial interests.
References
- 1.Sharp PA The centrality of RNA. Cell 136, 577–580, doi: 10.1016/j.cell.2009.02.007 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Cech TR & Steitz JA The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94, doi: 10.1016/j.cell.2014.03.008 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Strobel EJ, Watters KE, Loughrey D & Lucks JB RNA systems biology: uniting functional discoveries and structural tools to understand global roles of RNAs. Curr Opin Biotechnol 39, 182–191, doi: 10.1016/j.copbio.2016.03.019 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gottesman S & Storz G Bacterial small RNA regulators: versatile roles and rapidly evolving variations. Cold Spring Harb Perspect Biol 3, doi: 10.1101/cshperspect.a003798 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Luco RF & Misteli T More than a splicing code: integrating the role of RNA, chromatin and non-coding RNA in alternative splicing regulation. Curr Opin Genet Dev 21, 366–372, doi: 10.1016/j.gde.2011.03.004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rinn JL & Chang HY Genome regulation by long noncoding RNAs. Annu Rev Biochem 81, 145–166, doi: 10.1146/annurev-biochem-051410-092902 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chappell J et al. The centrality of RNA for engineering gene expression. Biotechnol J 8, 1379–1395, doi: 10.1002/biot.201300018 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cech TR Structural biology. The ribosome is a ribozyme. Science 289, 878–879 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Al-Hashimi HM & Walter NG RNA dynamics: it is about time. Curr Opin Struct Biol 18, 321–329, doi: 10.1016/j.sbi.2008.04.004 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mustoe AM, Brooks CL & Al-Hashimi HM Hierarchy of RNA functional dynamics. Annu Rev Biochem 83, 441–466, doi: 10.1146/annurev-biochem-060713-035524 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hermann T & Patel DJ Adaptive recognition by nucleic acid aptamers. Science 287, 820–825 (2000). [DOI] [PubMed] [Google Scholar]
- 12.Fedor MJ & Williamson JR The catalytic diversity of RNAs. Nat Rev Mol Cell Biol 6, 399–412, doi: 10.1038/nrm1647 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Holley RW et al. Structure of a Ribonucleic Acid. Science 147, 1462–1465 (1965). [DOI] [PubMed] [Google Scholar]
- 14.Noller HF & Chaires JB Functional modification of 16S ribosomal RNA by kethoxal. Proc Natl Acad Sci U S A 69, 3115–3118 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peattie DA & Gilbert W Chemical probes for higher-order structure in RNA. Proc Natl Acad Sci U S A 77, 4679–4682 (1980). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qu HL, Michot B & Bachellerie JP Improved methods for structure probing in large RNAs: a rapid ‘heterologous’ sequencing approach is coupled to the direct mapping of nuclease accessible sites. Application to the 5’ terminal domain of eukaryotic 28S rRNA. Nucleic Acids Res 11, 5903–5920 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moazed D, Stern S & Noller HF Rapid chemical probing of conformation in 16 S ribosomal RNA and 30 S ribosomal subunits using primer extension. J Mol Biol 187, 399–416 (1986). [DOI] [PubMed] [Google Scholar]
- 18.Latham JA & Cech TR Defining the inside and outside of a catalytic RNA molecule. Science 245, 276–282 (1989). [DOI] [PubMed] [Google Scholar]
- 19.Moazed D, Robertson JM & Noller HF Interaction of elongation factors EF-G and EF-Tu with a conserved loop in 23S RNA. Nature 334, 362–364, doi: 10.1038/334362a0 (1988). [DOI] [PubMed] [Google Scholar]
- 20.Climie SC & Friesen JD In vivo and in vitro structural analysis of the rplJ mRNA leader of Escherichia coli. Protection by bound L10-L7/L12. J Biol Chem 263, 15166–15175 (1988). [PubMed] [Google Scholar]
- 21.Lavery R & Pullman A A new theoretical index of biochemical reactivity combining steric and electrostatic factors. An application to yeast tRNAPhe. Biophys Chem 19, 171–181 (1984). [DOI] [PubMed] [Google Scholar]
- 22.Soukup GA & Breaker RR Relationship between internucleotide linkage geometry and the stability of RNA. RNA 5, 1308–1325 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chamberlin SI & Weeks KM Mapping local nucleotide flexibility by selective acylation of 2 ‘-amine substituted RNA. Journal of the American Chemical Society 122, 216–224, doi:DOI 10.1021/ja9914137 (2000). [DOI] [Google Scholar]
- 24.Merino EJ, Wilkinson KA, Coughlan JL & Weeks KM RNA structure analysis at single nucleotide resolution by selective 2’-hydroxyl acylation and primer extension (SHAPE). J Am Chem Soc 127, 4223–4231, doi: 10.1021/ja043822v (2005). [DOI] [PubMed] [Google Scholar]
- 25.Gherghe CM, Mortimer SA, Krahn JM, Thompson NL & Weeks KM Slow conformational dynamics at C2’-endo nucleotides in RNA. J Am Chem Soc 130, 8884–8885, doi: 10.1021/ja802691e (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gherghe CM, Shajani Z, Wilkinson KA, Varani G & Weeks KM Strong correlation between SHAPE chemistry and the generalized NMR order parameter (S2) in RNA. J Am Chem Soc 130, 12244–12245, doi: 10.1021/ja804541s (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vicens Q, Gooding AR, Laederach A & Cech TR Local RNA structural changes induced by crystallization are revealed by SHAPE. RNA 13, 536–548, doi: 10.1261/rna.400207 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGinnis JL, Dunkle JA, Cate JH & Weeks KM The mechanisms of RNA SHAPE chemistry. J Am Chem Soc 134, 6617–6624, doi: 10.1021/ja2104075 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das R, Laederach A, Pearlman SM, Herschlag D & Altman RB SAFA: semi-automated footprinting analysis software for high-throughput quantification of nucleic acid footprinting experiments. Rna 11, 344–354, doi: 10.1261/rna.7214405 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitra S, Shcherbakova IV, Altman RB, Brenowitz M & Laederach A High-throughput single-nucleotide structural mapping by capillary automated footprinting analysis. Nucleic Acids Res 36, e63, doi: 10.1093/nar/gkn267 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkinson KA et al. High-throughput SHAPE analysis reveals structures in HIV-1 genomic RNA strongly conserved across distinct biological states. PLoS Biol 6, e96, doi: 10.1371/journal.pbio.0060096 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tyrrell J, McGinnis JL, Weeks KM & Pielak GJ The cellular environment stabilizes adenine riboswitch RNA structure. Biochemistry 52, 8777–8785, doi: 10.1021/bi401207q (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spitale RC et al. RNA SHAPE analysis in living cells. Nat Chem Biol 9, 18–20, doi: 10.1038/nchembio.1131 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kertesz M et al. Genome-wide measurement of RNA secondary structure in yeast. Nature 467, 103–107, doi: 10.1038/nature09322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Underwood JG et al. FragSeq: transcriptome-wide RNA structure probing using high-throughput sequencing. Nat Methods 7, 995–1001, doi: 10.1038/nmeth.1529 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lucks JB et al. Multiplexed RNA structure characterization with selective 2’-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Proc Natl Acad Sci U S A 108, 11063–11068, doi: 10.1073/pnas.1106501108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rouskin S, Zubradt M, Washietl S, Kellis M & Weissman JS Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505, 701–705, doi: 10.1038/nature12894 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding Y et al. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505, 696–700, doi: 10.1038/nature12756 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Spitale RC et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 519, 486–490, doi: 10.1038/nature14263 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Talkish J, May G, Lin Y, Woolford JL Jr. & McManus CJ Mod-seq: high-throughput sequencing for chemical probing of RNA structure. RNA 20, 713–720, doi: 10.1261/rna.042218.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng Q et al. Genome-wide double-stranded RNA sequencing reveals the functional significance of base-paired RNAs in Arabidopsis. PLoS Genet 6, e1001141, doi: 10.1371/journal.pgen.1001141 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li F et al. Global analysis of RNA secondary structure in two metazoans. Cell Rep 1, 69–82, doi: 10.1016/j.celrep.2011.10.002 (2012). [DOI] [PubMed] [Google Scholar]
- 43.Loughrey D, Watters KE, Settle AH & Lucks JB SHAPE-Seq 2.0: systematic optimization and extension of high-throughput chemical probing of RNA secondary structure with next generation sequencing. Nucleic Acids Res 42, doi: 10.1093/nar/gku909 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Siegfried NA, Busan S, Rice GM, Nelson JA & Weeks KM RNA motif discovery by SHAPE and mutational profiling (SHAPE-MaP). Nat Methods 11, 959–965, doi: 10.1038/nmeth.3029 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hector RD et al. Snapshots of pre-rRNA structural flexibility reveal eukaryotic 40S assembly dynamics at nucleotide resolution. Nucleic Acids Res 42, 12138–12154, doi: 10.1093/nar/gku815 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Incarnato D, Neri F, Anselmi F & Oliviero S Genome-wide profiling of mouse RNA secondary structures reveals key features of the mammalian transcriptome. Genome Biol 15, 491, doi: 10.1186/s13059-014-0491-2 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seetin MG, Kladwang W, Bida JP & Das R Massively parallel RNA chemical mapping with a reduced bias MAP-seq protocol. Methods Mol Biol 1086, 95–117, doi: 10.1007/978-1-62703-667-2_6 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Cheng CY et al. Consistent global structures of complex RNA states through multidimensional chemical mapping. Elife 4, e07600, doi: 10.7554/eLife.07600 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Watters KE, Yu AM, Strobel EJ, Settle AH & Lucks JB Characterizing RNA structures in vitro and in vivo with selective 2’-hydroxyl acylation analyzed by primer extension sequencing (SHAPE-Seq). Methods 103, 34–48, doi: 10.1016/j.ymeth.2016.04.002 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Watters KE, Abbott TR & Lucks JB Simultaneous characterization of cellular RNA structure and function with in-cell SHAPE-Seq. Nucleic Acids Res 44, e12, doi: 10.1093/nar/gkv879 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zubradt M et al. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat Methods 14, 75–82, doi: 10.1038/nmeth.4057 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ritchey LE et al. Structure-seq2: sensitive and accurate genome-wide profiling of RNA structure in vivo. Nucleic Acids Res 45, e135, doi: 10.1093/nar/gkx533 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Burlacu E et al. High-throughput RNA structure probing reveals critical folding events during early 60S ribosome assembly in yeast. Nat Commun 8, 714, doi: 10.1038/s41467-017-00761-8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sexton AN, Wang PY, Rutenberg-Schoenberg M & Simon MD Interpreting Reverse Transcriptase Termination and Mutation Events for Greater Insight into the Chemical Probing of RNA. Biochemistry 56, 4713–4721, doi: 10.1021/acs.biochem.7b00323 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Novoa EM, Boeudoin J-D, Giraldez AJ, Mattick JS & Kellis M Best practices for genome-wide RNA structure analysis: combination of mutational profiles and drop-off information. BioRxiv, 1–24, doi: 10.1101/176883 (2017). [DOI] [Google Scholar]
- 56.Ehresmann C et al. Probing the structure of RNAs in solution. Nucleic Acids Res 15, 9109–9128 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knapp G Enzymatic approaches to probing of RNA secondary and tertiary structure. Methods Enzymol 180, 192–212 (1989). [DOI] [PubMed] [Google Scholar]
- 58.Kwok CK, Ding Y, Tang Y, Assmann SM & Bevilacqua PC Determination of in vivo RNA structure in low-abundance transcripts. Nat Commun 4, 2971, doi: 10.1038/ncomms3971 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Smola MJ, Calabrese JM & Weeks KM Detection of RNA-Protein Interactions in Living Cells with SHAPE. Biochemistry 54, 6867–6875, doi: 10.1021/acs.biochem.5b00977 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smola MJ et al. SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc Natl Acad Sci U S A 113, 10322–10327, doi: 10.1073/pnas.1600008113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee B et al. Comparison of SHAPE reagents for mapping RNA structures inside living cells. RNA 23, 169–174, doi: 10.1261/rna.058784.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mitchell D 3rd et al. Glyoxals as In Vivo RNA Structural Probes of Guanine Base Pairing. RNA 24, 114–124, doi: 10.1261/rna.064014.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng C et al. Light-activated chemical probing of nucleobase solvent accessibility inside cells. Nat Chem Biol 14, 276–283, doi: 10.1038/nchembio.2548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wan Y, Kertesz M, Spitale RC, Segal E & Chang HY Understanding the transcriptome through RNA structure. Nat Rev Genet 12, 641–655, doi: 10.1038/nrg3049 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tijerina P, Mohr S & Russell R DMS footprinting of structured RNAs and RNA-protein complexes. Nat Protoc 2, 2608–2623, doi: 10.1038/nprot.2007.380 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mortimer SA, Johnson JS & Weeks KM Quantitative analysis of RNA solvent accessibility by N-silylation of guanosine. Biochemistry 48, 2109–2114, doi: 10.1021/bi801939g (2009). [DOI] [PubMed] [Google Scholar]
- 67.Brunel C & Romby P Probing RNA structure and RNA-ligand complexes with chemical probes. Methods Enzymol 318, 3–21 (2000). [DOI] [PubMed] [Google Scholar]
- 68.McGinnis JL, Duncan CD & Weeks KM High-throughput SHAPE and hydroxyl radical analysis of RNA structure and ribonucleoprotein assembly. Methods Enzymol 468, 67–89, doi: 10.1016/S0076-6879(09)68004-6 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilkinson KA, Merino EJ & Weeks KM RNA SHAPE chemistry reveals nonhierarchical interactions dominate equilibrium structural transitions in tRNA(Asp) transcripts. J Am Chem Soc 127, 4659–4667, doi: 10.1021/ja0436749 (2005). [DOI] [PubMed] [Google Scholar]
- 70.Bindewald E et al. Correlating SHAPE signatures with three-dimensional RNA structures. RNA 17, 1688–1696, doi: 10.1261/rna.2640111 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mortimer SA & Weeks KM A fast-acting reagent for accurate analysis of RNA secondary and tertiary structure by SHAPE chemistry. J Am Chem Soc 129, 4144–4145, doi: 10.1021/ja0704028 (2007). [DOI] [PubMed] [Google Scholar]
- 72.Steen KA, Rice GM & Weeks KM Fingerprinting noncanonical and tertiary RNA structures by differential SHAPE reactivity. J Am Chem Soc 134, 13160–13163, doi: 10.1021/ja304027m (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mortimer SA & Weeks KM Time-resolved RNA SHAPE chemistry. J Am Chem Soc 130, 16178–16180, doi: 10.1021/ja8061216 (2008). [DOI] [PubMed] [Google Scholar]
- 74.Mortimer SA & Weeks KM C2’-endo nucleotides as molecular timers suggested by the folding of an RNA domain. Proc Natl Acad Sci U S A 106, 15622–15627, doi: 10.1073/pnas.0901319106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Watters KE, Strobel EJ, Yu AM, Lis JT & Lucks JB Cotranscriptional folding of a riboswitch at nucleotide resolution. Nat Struct Mol Biol 23, 1124–1131, doi: 10.1038/nsmb.3316 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Strobel EJ, Watters KE, Nedialkov Y, Artsimovitch I & Lucks JB Distributed biotin-streptavidin transcription roadblocks for mapping cotranscriptional RNA folding. Nucleic Acids Res 45, e109, doi: 10.1093/nar/gkx233 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Adilakshmi T, Lease RA & Woodson SA Hydroxyl radical footprinting in vivo: mapping macromolecular structures with synchrotron radiation. Nucleic Acids Res 34, e64, doi: 10.1093/nar/gkl291 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aviran S & Pachter L Rational experiment design for sequencing-based RNA structure mapping. RNA 20, 1864–1877, doi: 10.1261/rna.043844.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Aviran S, Lucks J & Pachter L RNA structure characterization from chemical mapping experiments. Proc. 49th Annual Allerton Conf. on Communication, Control, and Computing 1743–1750 (2011). [Google Scholar]
- 80.Kladwang W et al. Standardization of RNA chemical mapping experiments. Biochemistry 53, 3063–3065, doi: 10.1021/bi5003426 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Homan PJ et al. Single-molecule correlated chemical probing of RNA. Proc Natl Acad Sci U S A 111, 13858–13863, doi: 10.1073/pnas.1407306111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Krokhotin A, Mustoe AM, Weeks KM & Dokholyan NV Direct identification of base-paired RNA nucleotides by correlated chemical probing. RNA 23, 6–13, doi: 10.1261/rna.058586.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lackey L, Coria A, Woods C, McArthur E & Laederach A Allele-specific SHAPE-MaP assessment of the effects of somatic variation and protein binding on mRNA structure. Rna, doi: 10.1261/rna.064469.117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fang R, Moss WN, Rutenberg-Schoenberg M & Simon MD Probing Xist RNA Structure in Cells Using Targeted Structure-Seq. PLoS Genet 11, e1005668, doi: 10.1371/journal.pgen.1005668 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Watters KE et al. Probing of RNA structures in a positive sense RNA virus reveals selection pressure for structural elements. Nucleic Acids Res (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mauger DM et al. Functionally conserved architecture of hepatitis C virus RNA genomes. Proc Natl Acad Sci U S A 112, 3692–3697, doi: 10.1073/pnas.1416266112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Metzker ML Sequencing technologies - the next generation. Nat Rev Genet 11, 31–46, doi: 10.1038/nrg2626 (2010). [DOI] [PubMed] [Google Scholar]
- 88.Hafner M et al. RNA-ligase-dependent biases in miRNA representation in deep-sequenced small RNA cDNA libraries. RNA 17, 1697–1712, doi: 10.1261/rna.2799511 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kwok CK, Ding Y, Sherlock ME, Assmann SM & Bevilacqua PC A hybridization-based approach for quantitative and low-bias single-stranded DNA ligation. Anal Biochem 435, 181–186, doi: 10.1016/j.ab.2013.01.008 (2013). [DOI] [PubMed] [Google Scholar]
- 90.Kutchko KM et al. Multiple conformations are a conserved and regulatory feature of the RB1 5’ UTR. RNA 21, 1274–1285, doi: 10.1261/rna.049221.114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Woods CT et al. Comparative Visualization of the RNA Suboptimal Conformational Ensemble In Vivo. Biophys J 113, 290–301, doi: 10.1016/j.bpj.2017.05.031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Spasic A, Assmann SM, Bevilacqua PC & Mathews DH Modeling RNA secondary structure folding ensembles using SHAPE mapping data. Nucleic Acids Res, doi: 10.1093/nar/gkx1057 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mlynsky V & Bussi G Molecular Dynamics Simulations Reveal an Interplay between SHAPE Reagent Binding and RNA Flexibility. The journal of physical chemistry letters 9, 313–318, doi: 10.1021/acs.jpclett.7b02921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Choudhary K et al. Metrics for rapid quality control in RNA structure probing experiments. Bioinformatics 32, 3575–3583, doi: 10.1093/bioinformatics/btw501 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Busan S & Weeks KM Accurate detection of chemical modifications in RNA by mutational profiling (MaP) with ShapeMapper 2. RNA, doi: 10.1261/rna.061945.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Meyer S, Carlson PD & Lucks J Characterizing the structure-function relationship of a naturally ocurring RNA thermometer. Biochemistry, doi: 10.1021/acs.biochem.7b01170 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Choi EK, Ulanowicz KA, Nguyen YAH, Frandsen JK & Mitton-Fry RM SHAPE analysis of the htrA RNA thermometer from Salmonella enterica. RNA 23, 1569–1581, doi: 10.1261/rna.062299.117 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kladwang W, VanLang CC, Cordero P & Das R A two-dimensional mutate-and-map strategy for non-coding RNA structure. Nat Chem 3, 954–962, doi: 10.1038/nchem.1176 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rocca-Serra P et al. Sharing and archiving nucleic acid structure mapping data. RNA 17, 1204–1212, doi: 10.1261/rna.2753211 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cordero P, Lucks JB & Das R An RNA Mapping DataBase for curating RNA structure mapping experiments. Bioinformatics 28, 3006–3008, doi: 10.1093/bioinformatics/bts554 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yesselman JD et al. Updates to the RNA mapping database (RMDB), version 2. Nucleic Acids Res 46, D375–D379 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Steen KA, Malhotra A & Weeks KM Selective 2’-hydroxyl acylation analyzed by protection from exoribonuclease. J Am Chem Soc 132, 9940–9943, doi: 10.1021/ja103781u (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Tian S, Kladwang W & Das R Allosteric mechanism of the V. vulnificus adenine riboswitch resolved by four-dimensional chemical mapping. Elife 7, doi: 10.7554/eLife.29602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Deigan KE, Li TW, Mathews DH & Weeks KM Accurate SHAPE-directed RNA structure determination. Proc Natl Acad Sci U S A 106, 97–102, doi: 10.1073/pnas.0806929106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu Y et al. Improved prediction of RNA secondary structure by integrating the free energy model with restraints derived from experimental probing data. Nucleic Acids Res 43, 7247–7259, doi: 10.1093/nar/gkv706 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Washietl S, Hofacker IL, Stadler PF & Kellis M RNA folding with soft constraints: reconciliation of probing data and thermodynamic secondary structure prediction. Nucleic Acids Res 40, 4261–4272, doi: 10.1093/nar/gks009 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lorenz R, Hofacker IL & Stadler PF RNA folding with hard and soft constraints. Algorithms Mol Biol 11, 8, doi: 10.1186/s13015-016-0070-z (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lorenz R, Luntzer D, Hofacker IL, Stadler PF & Wolfinger MT SHAPE directed RNA folding. Bioinformatics 32, 145–147, doi: 10.1093/bioinformatics/btv523 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lorenz R, Wolfinger MT, Tanzer A & Hofacker IL Predicting RNA secondary structures from sequence and probing data. Methods 103, 86–98, doi: 10.1016/j.ymeth.2016.04.004 (2016). [DOI] [PubMed] [Google Scholar]
- 110.Ouyang Z, Snyder MP & Chang HY SeqFold: genome-scale reconstruction of RNA secondary structure integrating high-throughput sequencing data. Genome Res 23, 377–387, doi: 10.1101/gr.138545.112 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tan Z, Sharma G & Mathews DH Modeling RNA Secondary Structure with Sequence Comparison and Experimental Mapping Data. Biophys J 113, 330–338, doi: 10.1016/j.bpj.2017.06.039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Seetin MG & Mathews DH RNA structure prediction: an overview of methods. Methods Mol Biol 905, 99–122, doi: 10.1007/978-1-61779-949-5_8 (2012). [DOI] [PubMed] [Google Scholar]
- 113.Sahoo S, Switnicki MP & Pedersen JS ProbFold: a probabilistic method for integration of probing data in RNA secondary structure prediction. Bioinformatics 32, 2626–2635, doi: 10.1093/bioinformatics/btw175 (2016). [DOI] [PubMed] [Google Scholar]
- 114.Zarringhalam K, Meyer MM, Dotu I, Chuang JH & Clote P Integrating chemical footprinting data into RNA secondary structure prediction. PLoS One 7, e45160, doi: 10.1371/journal.pone.0045160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Corley M, Solem A, Qu K, Chang HY & Laederach A Detecting riboSNitches with RNA folding algorithms: a genome-wide benchmark. Nucleic Acids Res 43, 1859–1868, doi: 10.1093/nar/gkv010 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Parisien M & Major F The MC-Fold and MC-Sym pipeline infers RNA structure from sequence data. Nature 452, 51–55, doi: 10.1038/nature06684 (2008). [DOI] [PubMed] [Google Scholar]
- 117.Gherghe CM, Leonard CW, Ding F, Dokholyan NV & Weeks KM Native-like RNA tertiary structures using a sequence-encoded cleavage agent and refinement by discrete molecular dynamics. J Am Chem Soc 131, 2541–2546, doi: 10.1021/ja805460e (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ding F, Lavender CA, Weeks KM & Dokholyan NV Three-dimensional RNA structure refinement by hydroxyl radical probing. Nat Methods 9, 603–608, doi: 10.1038/nmeth.1976 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Magnus M et al. Computational modeling of RNA 3D structures, with the aid of experimental restraints. RNA Biol 11, 522–536, doi: 10.4161/rna.28826 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Li H & Aviran S Statistical modeling of RNA structure profiling experiments enables parsimonious reconstruction of structure landscapes. Nat Commun 9, 606, doi: 10.1038/s41467-018-02923-8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Sloma MF & Mathews DH Improving RNA secondary structure prediction with structure mapping data. Methods Enzymol 553, 91–114, doi: 10.1016/bs.mie.2014.10.053 (2015). [DOI] [PubMed] [Google Scholar]
- 122.Hajdin CE et al. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc Natl Acad Sci U S A 110, 5498–5503, doi: 10.1073/pnas.1219988110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sloma MF & Mathews DH Base pair probability estimates improve the prediction accuracy of RNA non-canonical base pairs. PLoS Comput Biol 13, e1005827, doi: 10.1371/journal.pcbi.1005827 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rose PW et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res 45, D271–d281, doi: 10.1093/nar/gkw1000 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kalvari I et al. Rfam 13.0: shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res 46, D335–d342, doi: 10.1093/nar/gkx1038 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.McGinnis JL et al. In-cell SHAPE reveals that free 30S ribosome subunits are in the inactive state. Proc Natl Acad Sci U S A 112, 2425–2430, doi: 10.1073/pnas.1411514112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Burkhardt DH et al. Operon mRNAs are organized into ORF-centric structures that predict translation efficiency. Elife 6, doi: 10.7554/eLife.22037 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Guo JU & Bartel DP RNA G-quadruplexes are globally unfolded in eukaryotic cells and depleted in bacteria. Science 353, doi: 10.1126/science.aaf5371 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Choudhary K, Ruan L, Deng F, Shih N & Aviran S SEQualyzer: interactive tool for quality control and exploratory analysis of high-throughput RNA structural profiling data. Bioinformatics 33, 441–443, doi: 10.1093/bioinformatics/btw627 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Mustoe AM et al. Pervasive Regulatory Functions of mRNA Structure Revealed by High-Resolution SHAPE Probing. Cell 173, 181–195.e118, doi: 10.1016/j.cell.2018.02.034 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Silverman IM et al. RNase-mediated protein footprint sequencing reveals protein-binding sites throughout the human transcriptome. Genome Biol 15, R3, doi: 10.1186/gb-2014-15-1-r3 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Seemann SE et al. The identification and functional annotation of RNA structures conserved in vertebrates. Genome Res 27, 1371–1383, doi: 10.1101/gr.208652.116 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kawaguchi R & Kiryu H Parallel computation of genome-scale RNA secondary structure to detect structural constraints on human genome. BMC Bioinformatics 17, 203, doi: 10.1186/s12859-016-1067-9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ramani V, Qiu R & Shendure J High-throughput determination of RNA structure by proximity ligation. Nat Biotechnol 33, 980–984, doi: 10.1038/nbt.3289 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lu Z et al. RNA Duplex Map in Living Cells Reveals Higher-Order Transcriptome Structure. Cell 165, 1267–1279, doi: 10.1016/j.cell.2016.04.028 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sharma E, Sterne-Weiler T, O’Hanlon D & Blencowe BJ Global Mapping of Human RNA-RNA Interactions. Mol Cell 62, 618–626, doi: 10.1016/j.molcel.2016.04.030 (2016). [DOI] [PubMed] [Google Scholar]
- 137.Aw JG et al. In Vivo Mapping of Eukaryotic RNA Interactomes Reveals Principles of Higher-Order Organization and Regulation. Mol Cell 62, 603–617, doi: 10.1016/j.molcel.2016.04.028 (2016). [DOI] [PubMed] [Google Scholar]
- 138.Nguyen TC et al. Mapping RNA-RNA interactome and RNA structure in vivo by MARIO. Nat Commun 7, 12023, doi: 10.1038/ncomms12023 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sugimoto Y et al. hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 519, 491–494, doi: 10.1038/nature14280 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Melamed S et al. Global Mapping of Small RNA-Target Interactions in Bacteria. Mol Cell 63, 884–897, doi: 10.1016/j.molcel.2016.07.026 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kudla G, Granneman S, Hahn D, Beggs JD & Tollervey D Cross-linking, ligation, and sequencing of hybrids reveals RNA-RNA interactions in yeast. Proc Natl Acad Sci U S A 108, 10010–10015, doi: 10.1073/pnas.1017386108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Li B, Tambe A, Aviran S & Pachter L PROBer Provides a General Toolkit for Analyzing Sequencing-Based Toeprinting Assays. Cell Syst 4, 568–574 e567, doi: 10.1016/j.cels.2017.04.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kladwang W, Cordero P & Das R A mutate-and-map strategy accurately infers the base pairs of a 35-nucleotide model RNA. RNA 17, 522–534, doi: 10.1261/rna.2516311 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Tian S, Cordero P, Kladwang W & Das R High-throughput mutate-map-rescue evaluates SHAPE-directed RNA structure and uncovers excited states. RNA 20, 1815–1826, doi: 10.1261/rna.044321.114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cheng CY, Kladwang W, Yesselman JD & Das R RNA structure inference through chemical mapping after accidental or intentional mutations. Proc Natl Acad Sci U S A 114, 9876–9881, doi: 10.1073/pnas.1619897114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Saldi T, Fong N & Bentley DL Transcription elongation rate affects nascent histone pre-mRNA folding and 3’ end processing. Genes & development 32, 297–308, doi: 10.1101/gad.310896.117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Wold B & Myers RM Sequence census methods for functional genomics. Nat Methods 5, 19–21, doi: 10.1038/nmeth1157 (2008). [DOI] [PubMed] [Google Scholar]
- 148.Kladwang W, VanLang CC, Cordero P & Das R Understanding the errors of SHAPE-directed RNA structure modeling. Biochemistry 50, 8049–8056, doi: 10.1021/bi200524n (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Leonard CW et al. Principles for understanding the accuracy of SHAPE-directed RNA structure modeling. Biochemistry 52, 588–595, doi: 10.1021/bi300755u (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ayub M, Hardwick SW, Luisi BF & Bayley H Nanopore-based identification of individual nucleotides for direct RNA sequencing. Nano Lett 13, 6144–6150, doi: 10.1021/nl403469r (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Rice GM, Leonard CW & Weeks KM RNA secondary structure modeling at consistent high accuracy using differential SHAPE. RNA 20, 846–854, doi: 10.1261/rna.043323.113 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Frieda KL & Block SM Direct observation of cotranscriptional folding in an adenine riboswitch. Science 338, 397–400, doi: 10.1126/science.1225722 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rinaldi AJ, Lund PE, Blanco MR & Walter NG The Shine-Dalgarno sequence of riboswitch-regulated single mRNAs shows ligand-dependent accessibility bursts. Nat Commun 7, 8976, doi: 10.1038/ncomms9976 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Xie Z, Srividya N, Sosnick TR, Pan T & Scherer NF Single-molecule studies highlight conformational heterogeneity in the early folding steps of a large ribozyme. Proc Natl Acad Sci U S A 101, 534–539, doi: 10.1073/pnas.2636333100 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cabili MN et al. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol 16, 20, doi: 10.1186/s13059-015-0586-4 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Shah S et al. Single-molecule RNA detection at depth by hybridization chain reaction and tissue hydrogel embedding and clearing. Development 143, 2862–2867, doi: 10.1242/dev.138560 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Nomakuchi TT, Rigo F, Aznarez I & Krainer AR Antisense oligonucleotide-directed inhibition of nonsense-mediated mRNA decay. Nat Biotechnol 34, 164–166, doi: 10.1038/nbt.3427 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Finkel RS et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: a phase 2, open-label, dose-escalation study. Lancet 388, 3017–3026, doi: 10.1016/S0140-6736(16)31408-8 (2016). [DOI] [PubMed] [Google Scholar]
- 159.Wilkinson KA et al. Influence of nucleotide identity on ribose 2’-hydroxyl reactivity in RNA. RNA 15, 1314–1321, doi: 10.1261/rna.1536209 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yu AM, Evans ME, & Lucks JB Estimating RNA structure chemical probing reactivities from reverse transcriptase stops and mutations. BioRxiv, doi: 10.1038/XXX (2018). [DOI] [Google Scholar]
- 161.Aviran S et al. Modeling and automation of sequencing-based characterization of RNA structure. Proc Natl Acad Sci U S A 108, 11069–11074, doi: 10.1073/pnas.1106541108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Gilbert W The RNA World. Nature 319, 618 (1986). [Google Scholar]
- 163.Mathews DH et al. Incorporating chemical modification constraints into a dynamic programming algorithm for prediction of RNA secondary structure. Proc Natl Acad Sci U S A 101, 7287–7292, doi: 10.1073/pnas.0401799101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wu X & Bartel DP Widespread Influence of 3’-End Structures on Mammalian mRNA Processing and Stability. Cell 169, 905–917 e911, doi: 10.1016/j.cell.2017.04.036 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wan Y et al. Landscape and variation of RNA secondary structure across the human transcriptome. Nature 505, 706–709, doi: 10.1038/nature12946 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Wan Y et al. Genome-wide measurement of RNA folding energies. Mol Cell 48, 169–181, doi: 10.1016/j.molcel.2012.08.008 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Takahashi MK et al. Using in-cell SHAPE-Seq and simulations to probe structure-function design principles of RNA transcriptional regulators. RNA 22, 920–933, doi: 10.1261/rna.054916.115 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Bai Y, Tambe A, Zhou K & Doudna JA RNA-guided assembly of Rev-RRE nuclear export complexes. Elife 3, e03656, doi: 10.7554/eLife.03656 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Tang Y, Assmann SM & Bevilacqua PC Protein Structure Is Related to RNA Structural Reactivity In Vivo. J Mol Biol 428, 758–766, doi: 10.1016/j.jmb.2015.11.012 (2016). [DOI] [PubMed] [Google Scholar]
- 170.Cordero P & Das R Rich RNA Structure Landscapes Revealed by Mutate-and-Map Analysis. PLoS Comput Biol 11, e1004473, doi: 10.1371/journal.pcbi.1004473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lavender CA et al. Model-Free RNA Sequence and Structure Alignment Informed by SHAPE Probing Reveals a Conserved Alternate Secondary Structure for 16S rRNA. PLoS Comput Biol 11, e1004126, doi: 10.1371/journal.pcbi.1004126 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Poulsen LD, Kielpinski LJ, Salama SR, Krogh A & Vinther J SHAPE Selection (SHAPES) enrich for RNA structure signal in SHAPE sequencing-based probing data. RNA 21, 1042–1052, doi: 10.1261/rna.047068.114 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.