

Abstract
Background
Perineal trauma, due to spontaneous tears, surgical incision (episiotomy), or in association with operative vaginal birth, is common after vaginal birth, and is often associated with postpartum perineal pain. Birth over an intact perineum may also lead to perineal pain. There are adverse health consequences associated with perineal pain for the women and their babies in the short‐ and long‐term, and the pain may interfere with newborn care and the establishment of breastfeeding. Aspirin has been used in the management of postpartum perineal pain, and its effectiveness and safety should be assessed. This is an update of the review, last published in 2017.
Objectives
To determine the effects of a single dose of aspirin (acetylsalicylic acid), including at different doses, in the relief of acute postpartum perineal pain.
Search methods
For this update, we searched the Cochrane Pregnancy and Childbirth's Trials Register (4 October 2019), ClinicalTrials.gov, the WHO International Clinical Trials Registry Platform (ICTRP) (4 October 2019) and screened reference lists of retrieved studies.
Selection criteria
Randomised controlled trials (RCTs), assessing single dose aspirin compared with placebo, no treatment, a different dose of aspirin, or single dose paracetamol or acetaminophen, for women with perineal pain in the early postpartum period. We planned to include cluster‐RCTs, but none were identified. We excluded quasi‐RCTs and cross‐over studies.
Data collection and analysis
Two review authors independently assessed study eligibility, extracted data and assessed the risk of bias of the included RCTs. Data were checked for accuracy. The certainty of the evidence for the main comparison (aspirin versus placebo) was assessed using the GRADE approach.
Main results
We included 17 RCTs, 16 of which randomised 1132 women to aspirin or placebo; one RCT did not report numbers of women. Two RCTs (of 16) did not contribute data to meta‐analyses. All women had perineal pain post‐episiotomy, and were not breastfeeding. Studies were published between 1967 and 1997, and the risk of bias was often unclear, due to poor reporting.
We included four comparisons: aspirin versus placebo (15 RCTs); 300 mg versus 600 mg aspirin (1 RCT); 600 mg versus 1200 mg aspirin (2 RCTs); and 300 mg versus 1200 mg aspirin (1 RCT).
Aspirin versus placebo
Aspirin may result in more women reporting adequate pain relief four to eight hours after administration compared with placebo (risk ratio (RR) 2.03, 95% confidence interval (CI) 1.69 to 2.42; 13 RCTs, 1001 women; low‐certainty evidence). It is uncertain whether aspirin compared with placebo has an effect on the need for additional pain relief (RR 0.25, 95% CI 0.17 to 0.37; 10 RCTs, 744 women; very low‐certainty evidence), or maternal adverse effects (RR 1.08, 95% CI 0.57 to 2.06; 14 RCTs, 1067 women; very low‐certainty evidence), four to eight hours after administration. Analyses based on dose did not reveal any clear subgroup differences.
300 mg versus 600 mg aspirin
It is uncertain whether over four hours after administration, 300 mg compared with 600 mg aspirin has an effect on adequate pain relief (RR 0.82, 95% CI 0.36 to 1.86; 1 RCT, 81 women) or the need for additional pain relief (RR 0.68, 95% CI 0.12 to 3.88; 1 RCT, 81 women). There were no maternal adverse effects in either aspirin group.
600 mg versus 1200 mg aspirin
It is uncertain whether over four to eight hours after administration, 600 mg compared with 1200 mg aspirin has an effect on adequate pain relief (RR 0.85, 95% CI 0.52 to 1.39; 2 RCTs, 121 women), the need for additional pain relief (RR 1.32, 95% CI 0.30 to 5.68; 2 RCTs, 121 women), or maternal adverse effects (RR 3.00, 95% CI 0.13 to 69.52; 2 RCTs, 121 women).
300 mg versus 1200 mg aspirin
It is uncertain whether over four hours after administration, 300 mg compared with 1200 mg aspirin has an effect on adequate pain relief (RR 0.62, 95% CI 0.29 to 1.32; 1 RCT, 80 women) or need for additional pain relief (RR 2.00, 95% CI 0.19 to 21.18; 1 RCT, 80 women). There were no maternal adverse effects in either aspirin group.
None of the included RCTs reported on neonatal adverse effects.
No RCTs reported on secondary review outcomes of: prolonged hospitalisation due to perineal pain; re‐hospitalisation due to perineal pain; fully breastfeeding at discharge; mixed feeding at discharge; fully breastfeeding at six weeks; mixed feeding at six weeks; perineal pain at six weeks; maternal views; or maternal postpartum depression.
Authors' conclusions
Single dose aspirin may increase adequate pain relief in women with perineal pain post‐episiotomy compared with placebo. It is uncertain whether aspirin has an effect on the need for additional analgesia, or on maternal adverse effects, compared with placebo. We downgraded the certainty of the evidence because of study limitations (risk of bias), imprecision, and publication bias.
Aspirin may be considered for use in non‐breastfeeding women with post‐episiotomy perineal pain. Included RCTs excluded breastfeeding women, so there was no evidence to assess the effects of aspirin on neonatal adverse effects or breastfeeding.
Future RCTs should be designed to ensure low risk of bias, and address gaps in the evidence, such as the secondary outcomes established for this review. Current research has focused on women with post‐episiotomy pain; future RCTs could be extended to include women with perineal pain associated with spontaneous tears or operative birth.
Plain language summary
Aspirin (single dose) for relief of perineal pain after childbirth
What is the issue?
Can aspirin be given to women who experience perineal pain following childbirth to relieve the pain, without causing side effects for either the women or their babies?
Why is this important?
Many women experience pain in the perineum (the area between the vagina and anus) following childbirth. The perineum may be bruised or torn during childbirth, or have a cut made to help the baby to be born (an episiotomy). After childbirth, perineal pain can interfere with women's ability to care for their newborns and establish breastfeeding. If perineal pain is not relieved effectively, longer‐term problems for women may include painful sexual intercourse, pelvic floor problems resulting in incontinence, prolapse, or chronic perineal pain. Aspirin may be given to women who have perineal pain after childbirth, but its effectiveness and safety had not been assessed in a systematic review. This is an update of a review last published in 2017. This is part of a series of reviews looking at drugs to help relieve perineal pain in first few weeks after childbirth.
What evidence did we find?
We searched for evidence in October 2019, and included 17 randomised controlled studies, involving 1132 women, published between 1967 and 1997. All women had perineal pain following an episiotomy (usually within 48 hours after birth), and were not breastfeeding. The women received either aspirin (doses ranging from 300 mg to 1200 mg) or fake pills (placebo), by mouth. The methodological quality of the studies was often unclear. Two studies did not contribute any data for analyses.
Aspirin compared with placebo may increase adequate pain relief for mothers four to eight hours after administration (low‐certainty evidence). It is uncertain whether aspirin compared with placebo has an effect on the need for additional pain relief, or on adverse effects for mothers, in the four to eight hours after administration (both very low‐certainty evidence).
The effects of administering 300 mg versus 600 mg aspirin (1 study), 600 mg versus 1200 mg aspirin (2 studies), or 300 mg versus 1200 mg aspirin (1 study) are uncertain for adequate pain relief, the need for additional pain relief, or adverse effects for the mother.
No studies reported on adverse effects of aspirin for the baby, or other outcomes we planned to assess: prolonged hospital stay, or readmission to hospital due to perineal pain; perineal pain six weeks after childbirth, women's views, or postpartum depression.
What does this mean?
A single dose of aspirin may help with perineal pain following episiotomy for women who are not breastfeeding, when measured four to eight hours after administration.
We found no information to assess the effects of aspirin for women who are breastfeeding.
Summary of findings
Summary of findings 1. Aspirin compared with placebo for perineal pain in the early postpartum period.
| Aspirin compared with placebo for perineal pain in the early postpartum period | ||||||
|
Patient or population: women with perineal pain in the early postpartum period Settings: hospitals in USA, Venezuela, Belgium, Canada, India Intervention: aspirin (single dose) Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Assumed risk for placebo | Corresponding risk for aspirin | |||||
|
Adequate pain relief as reported by the woman (4 to 8 hours) |
Study population | RR 2.03 (1.69 to 2.42) | 1001 (13 RCTs) | ⊕⊕⊝⊝ lowa | ||
| 253 per 1000 | 513 per 1000 (427 to 612) | |||||
|
Need for additional pain relief (4 to 8 hours) |
Study population | RR 0.25 (0.17 to 0.37) | 744 (10 RCTs) | ⊕⊝⊝⊝ very lowa,b | ||
| 267 per 1000 | 67 per 1000 (45 to 99) | |||||
|
Maternal adverse effects (4 to 8 hours) |
Study population | RR 1.08 (0.57 to 2.06) | 1067 (14 RCTs) | ⊕⊝⊝⊝ very lowa,c | ||
| 27 per 1000 | 29 per 1000 (15 to 55) | |||||
| Neonatal adverse effects | (0 RCTs) | Not reported by any of the included RCTs | ||||
| Perineal pain at six weeks postpartum | (0 RCTs) | Not reported by any of the included RCTs | ||||
| *The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RCT: randomised controlled trial; RR: risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: Further research is likely to have an important impact on our confidence in the estimate of effect, and may change the estimate. Low certainty: Further research is very likely to have an important impact on our confidence in the estimate of effect, and is likely to change the estimate. Very low certainty: We are very uncertain about the estimate. | ||||||
aWe downgraded 2 levels for very serious limitations in study design: most of the trials contributing data were at unclear risk of selection bias bWe downgraded 1 level for serious limitations in publication bias: visual inspection of funnel plot indicates likely publication bias cWe downgraded 1 level for serious limitations in imprecision: there were few events and wide 95% CI around the pooled estimate that includes no effect
Background
Description of the condition
Perineal trauma may result from naturally occurring tears, surgical incisions, such as episiotomy (cutting of the perineum to enlarge the vaginal opening during the second stage of labour), or in association with operative vaginal births (vacuum or forceps assisted births); and is frequently associated with acute perineal pain in the immediate postpartum period (Chou 2009). Birth over an intact perineum is also often associated with acute postpartum perineal pain. Perineal trauma is common, for example, in high‐income countries, such as Australia, only approximately one quarter (24%) of mothers have an intact perineum after vaginal birth, with over half of mothers having either a first or second degree laceration or vaginal graze (53%), and a smaller proportion having third or fourth degree lacerations (3%) or other types of lacerations (8%). Of the approximately one in five mothers (23%) having an episiotomy, approximately 42% have a laceration of some degree (AIHW 2019).
Short‐term morbidities for the mother arising from perineal trauma may include bleeding, infection, haematoma, and acute postpartum perineal pain, which may also interfere with newborn care and the establishment of breastfeeding (Chou 2009; East 2012a). In the longer‐term, women are at an increased risk of dyspareunia (painful sexual intercourse), pelvic floor problems, and chronic perineal pain (Chou 2009; East 2012a).
Various practices can impact on the extent of perineal trauma sustained during birth, and so can influence the degree of perineal pain experienced by the woman in the immediate postpartum period. Cochrane Reviews have shown antenatal digital perineal massage (Beckmann 2013), and the use of warm compresses on the perineum during the second stage of labour (Aasheim 2017), to be effective in preventing perineal trauma and associated pain (WHO 2018).
A variety of practices and agents have also been assessed for the relief of perineal pain in the immediate postpartum period. Cochrane Reviews have reported finding limited evidence to support routine use of local cooling (such as with ice packs or cold gel packs) of the perineum (East 2012b), or the application of topical local anaesthetics to the perineum for postpartum perineal pain relief (Hedayati 2005). Another Cochrane Review found some support for the use of paracetamol to reduce postpartum perineal pain, and decrease the need for additional pain relief. However, the overall quality of included studies was assessed as unclear, and adverse effects were not assessed (Chou 2013). Other practices and agents that have been systematically reviewed and shown to have varied effectiveness in relieving postpartum perineal pain include: methods and materials for suturing perineal tears or episiotomies, therapeutic ultrasound, and rectal analgesia (East 2012a; Hay‐Smith 1998; Hedayati 2003; Kettle 2012). For example, in regard to perineal suturing after childbirth, a Cochrane Review showed that continuous suturing techniques for perineal closure, compared with interrupted methods, are associated with less short‐term pain; if continuous suturing is used for all layers (vagina, perineal muscles and skin), the reduction of pain has been reported to be even greater (Kettle 2012).
Description of the intervention
The history of aspirin began thousands of years ago, with early uses of extracts from plants and herbs containing salicylates (Vane 2003). In the 1870s, it was demonstrated that salicin and salicylic acid from white willow bark could reduce fever, pain, and inflammation in people with rheumatic fever (Maclagan 1879). The success of salicylic acid prompted the German pharmaceutical manufacturer, Bayer, to search for a derivative that was equally, or more effective. Felix Hoffman, a young chemist at Bayer, motivated by his father's inability to take salicylic acid for his arthritis due to its adverse effects (particularly vomiting), found a way to acetylate the hydroxyl group on the benzene ring of salicylic acid to form acetylated salicylic acid (Vane 2003).
In the first decades of the 1900s, acetylsalicylic acid, or 'aspirin' was considered the supreme analgesic (pain reliever); for three quarters of the 20th century, its use was solely as an analgesic and antipyretic (fever reducing) agent (Vane 2003). In the 1970s and 1980s, as part of his Nobel Prize‐winning work, Sir John Vane demonstrated that aspirin could inhibit the formation of prostaglandins, associated with pain, fever, and inflammation, providing a physiological rationale for the effectiveness of one of the world's most widely used medication. As part of this work, Vane also discovered prostacyclin, an important prostaglandin that plays a vital role in the process of blood coagulation. The potential for aspirin to prevent a range of serious, life‐threatening conditions, including heart attacks and stroke, was recognised following this discovery (Smith 2014).
Aspirin is now considered to be one of the most effective and versatile medications in the world. It is commonly recommended to be taken in the lowest effective dose to avoid adverse effects secondary to higher doses. For example, low‐dose aspirin (75 mg to 150 mg daily) has been shown to provide substantial benefit for preventing serious cardiovascular events (heart attacks, stroke, and vascular death) in people with pre‐existing cardiovascular disease, or with a history of events (secondary prevention; ATT Collaboration 2002). In primary prevention, for people without a history of events or previous disease, the value of low‐ and high‐dose aspirin (75 mg to 500 mg daily) remains uncertain (ATT Collaboration 2009). There is increasing evidence that aspirin may reduce the risk of some cancers, and certain pregnancy complications. Long‐term low‐dose aspirin (at least 75 mg daily) has been shown to reduce colorectal cancer incidence and mortality (Rothwell 2010). Low‐dose aspirin is reported to have small‐to‐moderate benefits in preventing pre‐eclampsia and its consequences (Duley 2019): 75 mg daily is recommended for pregnant women at high risk of developing the condition (WHO 2011).
How the intervention might work
Perineal pain is transmitted primarily through the pudendal nerve, a somatic sensory and motor nerve that innervates the external genitalia, as well as the bladder and rectum sphincters (Cunningham 2005). Although a detailed description of the mechanism of action and pharmacology of aspirin is beyond the scope of this review, we have outlined the basic concepts.
The mechanisms by which aspirin exerts its analgesic, anti‐inflammatory, and antipyretic effects were discovered in the 1970s. Aspirin inhibits the activity of the cyclo‐oxygenase (COX) enzymes (irreversible inhibition of COX‐1 and modification of COX‐2), which play important roles in inflammation and nociceptive processes (the encoding and processing in the nervous system of noxious stimuli), such as through the formation of prostaglandins and thromboxanes (Vane 2003).
Through inhibiting these key enzymes, it has also been demonstrated that aspirin can prevent the production of physiologically important prostaglandins and thromboxanes, including those that protect the stomach mucosa from damage by hydrochloric acid, and those that aggregate platelets when required (Vane 2003). It is through these mechanisms that aspirin has been shown to cause adverse effects, such as gastrointestinal irritation and occult (hidden) blood loss (Derry 2012). The availability of alternative agents with improved tolerability has reduced the use of aspirin for pain relief over recent years, however, in many parts of the world, where alternatives are not available, or are more expensive, aspirin is still the most commonly used analgesic for many different pain conditions (Derry 2012; Vane 2003).
A Cochrane Review that included 67 trials (involving 5743 adults), which were assessed to be "overwhelmingly of adequate or good methodological quality", confirmed single‐dose aspirin (300 mg to 1200 mg) to be an effective analgesic for acute, postoperative, moderate to severe intensity pain (Derry 2012). Higher doses (900 mg to 1000 mg) were shown to be more effective, however, these doses were associated with increased adverse effects, including gastric irritation, nausea, vomiting, drowsiness, and dizziness. The pain relief achieved with aspirin was very similar to paracetamol given at the same dose (Derry 2012). Derry 2012 excluded trials in which pain was due to trauma, such as is often the case for women with acute perineal pain in the immediate postpartum period. It is considered plausible that aspirin may also be effective in relieving acute perineal pain in the early postpartum period after birth.
Why it is important to do this review
Perineal trauma is common after vaginal birth, and frequently associated with acute postpartum perineal pain; birth over an intact perineum is also often associated with perineal pain. Perineal pain may be associated with adverse health consequences for the mother and her baby in the short and long term, such as dyspareunia, pelvic floor problems, and chronic perineal pain, and may also interfere with newborn care, including the establishment of breastfeeding (Chou 2009; East 2012a).
There is currently a dearth of evidence on effective interventions to reduce acute perineal pain in the immediate postpartum period. Previous Cochrane Reviews have assessed practices and agents, including therapeutic ultrasound (Hay‐Smith 1998), rectal analgesia (Hedayati 2003), local cooling (East 2012b), topical anaesthetics (Hedayati 2005), paracetamol (Chou 2013), and most recently, non‐steroidal anti‐inflammatory agents (Wuytack 2016), for the relief of perineal pain in the postpartum period. These reviews have reported mixed results. Therefore, it is important to establish if aspirin may be effective in relieving perineal pain and improving health outcomes for mothers and their babies. Because it is known that salicylate and salicylate metabolites, including aspirin, are excreted in breast milk, there is potential for effects on babies who are breast fed (NIH 2015). Therefore, adverse effects or harms for both mothers and their babies must be assessed.
We assessed the clinical effectiveness and adverse effects of aspirin given to relieve perineal pain in the early postpartum period. This review is one of a series of reviews of drugs for perineal pain in the early postpartum period, all based on the same generic protocol (Chou 2009). This protocol is published in the Cochrane Library, and describes the methods that shaped the production of all the reviews on drugs for perineal pain. It is available for consultation for prospective reviews undertaken on future drugs that may be introduced for this population and indication. This is an update of the review last published in 2017 (Molakatalla 2017).
Objectives
To determine the effects of a single dose of aspirin (acetylsalicylic acid), including at different doses, in the relief of acute postpartum perineal pain.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials, and had planned to include cluster‐randomised controlled trials. We excluded quasi‐randomised controlled trials and cross‐over trials. We planned to include studies published as abstracts only, as well as studies published in full‐text form.
Types of participants
All women with acute perineal pain in the early postpartum period; defined as the first four weeks after giving birth, or as defined by the authors of the studies.
Types of interventions
Single administration of aspirin, used to treat perineal pain due to spontaneous lacerations, episiotomy, or birth over an intact perineum, in the early postpartum period. We included studies in which aspirin was compared with a placebo or no treatment, and where different doses of aspirin (e.g. 75 mg, 300 mg, etc), administered as a single dose, were compared. We also planned to include studies where aspirin was compared with a single dose of paracetamol or acetaminophen for perineal pain in the early postpartum period.
Types of outcome measures
Primary outcomes
Adequate pain relief, as reported by the woman*
Need for additional pain relief in the first 48 hours for perineal pain
Maternal adverse effects, composite of any of the following: nausea, vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness, gastric discomfort, psychological impact
Neonatal adverse effects, composite of any of the following: vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness
* Determined by more than 50% relief of pain, stated by the woman or calculated using a formula; see Data collection and analysis for details.
Secondary outcomes
Prolonged hospitalisation due to perineal pain
Rehospitalisation due to perineal pain
Fully breastfeeding at discharge
Mixed feeding at discharge
Fully breastfeeding at six weeks
Mixed feeding at six weeks
Perineal pain at six weeks
Maternal views (using a validated questionnaire)
Maternal postpartum depression
Search methods for identification of studies
The methods section of this review is based on a standard template used by Cochrane Pregnancy and Childbirth.
Electronic searches
For this update, we searched the Cochrane Pregnancy and Childbirth’s Trials Register by contacting their Information Specialist (4 October 2019).
The Register is a database containing over 25,000 reports of controlled trials in the field of pregnancy and childbirth. It represents over 30 years of searching. For full current search methods used to populate Pregnancy and Childbirth’s Trials Register, including the detailed search strategies for CENTRAL, MEDLINE, Embase, and CINAHL; the list of handsearched journals and conference proceedings, and the list of journals reviewed via the current awareness service, please follow this link.
Briefly, Cochrane Pregnancy and Childbirth’s Trials Register is maintained by their Information Specialist, and contains trials identified from:
monthly searches of the Cochrane Central Register of Controlled Trials (CENTRAL);
weekly searches of MEDLINE Ovid;
weekly searches of Embase Ovid;
monthly searches of CINAHL EBSCO;
handsearches of 30 journals and the proceedings of major conferences;
weekly current awareness alerts for a further 44 journals plus monthly BioMed Central email alerts.
Two people screen the search results, and review the full text of all relevant trial reports, identified through the searching activities described above. Based on the intervention described, they assign each trial report a number that corresponds to a specific Pregnancy and Childbirth review topic (or topics), and then add it to the Register. The Information Specialist searches the Register for each review using this topic number, rather than keywords. This results in a more specific search set that has been fully accounted for in the relevant review sections (Included studies; Excluded studies; Studies awaiting classification).
We also searched ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform (ICTRP; 4 October 2019) for unpublished, planned, and ongoing trial reports, using the terms given in Appendix 1.
Searching other resources
We searched for further studies in the reference lists of the studies identified.
We did not apply language or date restrictions.
Data collection and analysis
For methods used in the previous version of this review, seeMolakatalla 2017.
For this update, we used the following methods to assess the reports that were identified as a result of the updated search.
The following methods section is based on both the generic protocol (Chou 2009) and a standard template used by Cochrane Pregnancy and Childbirth.
Assessment of pain
The number of women achieving adequate pain relief was defined as one of the following.
The number of women reporting 'good' or 'excellent' pain relief, when asked about their level of pain relief four to six hours after receiving their allocated treatment (the data were extracted as dichotomous data).
The number of women who reported 50% pain relief, or greater.
The number of women who achieved 50% pain relief, or greater, as calculated by using derived pain relief scores (TOTPAR (total pain relief), or SPID (summed pain intensity differences)) over four to six hours.
It is common to use categorical or visual analogue scales for pain intensity, and to calculate the results for each participant, over periods of four or six hours, as SPID or TOTPAR (Moore 1996). From these categorical scales, it was possible to convert results into dichotomous data (the proportion of participants achieving at least 50%, or greater, max TOTPAR) using standard formulae (Moore 1996; Moore 1997b). Converting data in this way enabled us to use these data in a meta‐analysis (Moore 1997a; Moore 1997b). We used the following equations to estimate the proportions of women who achieved at least 50% of maximum TOTPAR.
1. Proportion with greater than 50% maxTOTPAR = (1.33 x mean % maxTOTPAR – 11.5)
With % maxTOTPAR = mean TOTPAR x 100/(maximum score x number of hours)
2. Proportion with greater than 50% maxTOTPAR = (1.36 x mean % maxSPID – 2.3)
With % maxSPID = mean SPID x 100/(maximum score x number of hours)
We then calculated the number of participants achieving at least 50% maxTOTPAR, by multiplying the proportions of participants with at least 50% maxTOTPAR by the total number of participants in the treatment groups. We then used the number of participants with at least 50% maxTOTPAR to calculate the relative benefit and number needed to treat to benefit.
Where studies used more than one method of calculating adequate pain relief, preferences for analyses and reporting purposes, in order of decreasing preference, were: i) the proportion with at least 50% maxTOTPAR calculated using SPID; ii) the proportion with at least 50% maxTOTPAR calculated using TOTPAR; and iii) the number of participants reporting 'good' or 'excellent' pain relief/number of participants reporting at least 50% pain relief. We also assessed the number of participants who re‐medicated in four to eight hours, as well as the median time to re‐medication, if data were available.
Selection of studies
Two review authors independently assessed for inclusion, all potential studies we identified as a result of the search strategy. We resolved any disagreement through discussion, or if required, we consulted a third review author.
We created a study flow diagram to illustrate the number of records identified, included, and excluded (Figure 1).
1.
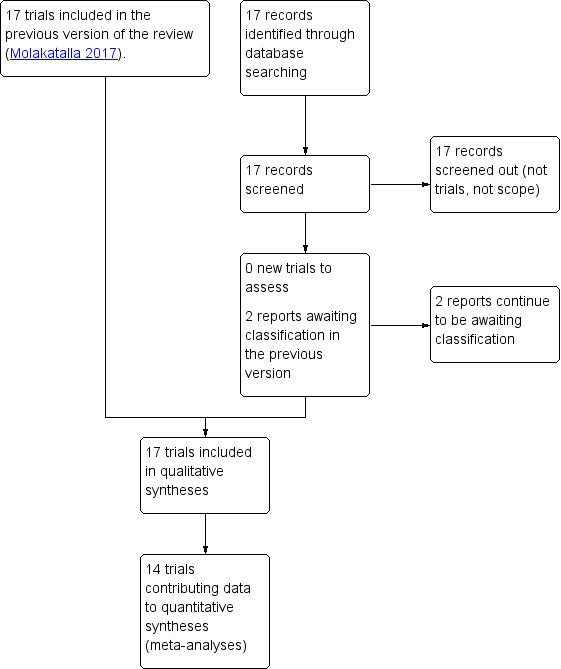
Study flow diagram
Data extraction and management
We designed a form to extract data. At least two review authors extracted data using the agreed form for eligible studies. We resolved discrepancies through discussion, or if required, consultation with another member of the review author team. We entered data into Review Manager 5 software and checked for accuracy (Review Manager 2014). When information regarding any steps was unclear, we attempted to contact authors of the original reports to provide further details.
Assessment of risk of bias in included studies
Two review authors independently assessed risk of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We resolved any disagreement by discussion, or by involving a third assessor.
(1) Random sequence generation (checking for possible selection bias)
For each included study, we described the method used to generate the allocation sequence in sufficient detail to allow an assessment of whether it should produce comparable groups.
We assessed the method as:
low risk of bias (any truly random process, e.g. random number table; computer random number generator);
high risk of bias (any non‐random process, e.g. odd or even date of birth; hospital, or clinic record number);
unclear risk of bias.
(2) Allocation concealment (checking for possible selection bias)
For each included study, we described the method used to conceal allocation to interventions prior to assignment, and assessed whether intervention allocation could have been foreseen in advance of, or during recruitment, or changed after assignment.
We assessed the methods as:
low risk of bias (e.g. telephone or central randomisation; consecutively numbered, sealed, opaque envelopes);
high risk of bias (open random allocation; unsealed or non‐opaque envelopes; alternation; date of birth);
unclear risk of bias.
(3.1) Blinding of participants and personnel (checking for possible performance bias)
For each included study, we described the methods used, if any, to blind study participants and personnel from knowledge of which intervention a participant received. We considered that studies were at low risk of bias if they were blinded, or if we judged that the lack of blinding would be unlikely to affect results.
We assessed the methods as:
low, high, or unclear risk of bias for participants;
low, high, or unclear risk of bias for personnel.
(3.2) Blinding of outcome assessment (checking for possible detection bias)
For each included study, we described the methods used, if any, to blind outcome assessors from knowledge of which intervention a participant received.
We assessed methods used to blind outcome assessment as:
low, high, or unclear risk of bias.
(4) Incomplete outcome data (checking for possible attrition bias due to the amount, nature and handling of incomplete outcome data)
For each included study, and for each outcome or class of outcomes, we described the completeness of data, including attrition and exclusions from the analysis. We stated whether attrition and exclusions were reported, and the numbers included in the analysis at each stage (compared with the total randomised participants), reasons for attrition or exclusion, where reported, and whether missing data were balanced across groups or were related to outcomes. Where sufficient information was reported, or could be supplied by the trial authors, we planned to re‐include missing data in the analyses.
We assessed methods as:
low risk of bias (e.g. no missing outcome data; missing outcome data balanced across groups);
high risk of bias (e.g. numbers or reasons for missing data imbalanced across groups; ‘as treated’ analysis done with substantial departure of intervention received, from that assigned at randomisation);
unclear risk of bias.
(5) Selective reporting (checking for reporting bias)
For each included study, we described how we investigated the possibility of selective outcome reporting bias and what we found.
We assessed the methods as:
low risk of bias (where it was clear that all of the study’s prespecified outcomes, and all expected outcomes of interest to the review were reported);
high risk of bias (where not all the study’s prespecified outcomes were reported; one or more reported primary outcomes were not prespecified; outcomes of interest were reported incompletely and so could not be used; study failed to include results of a key outcome that would have been expected to have been reported);
unclear risk of bias.
(6) Other bias (checking for bias due to problems not covered by points (1) to (5)
For each included study, we described any important concerns we had about other possible sources of bias, including: was the trial stopped early due to some data‐dependent process? Was there extreme baseline imbalance? Did someone claim the study was fraudulent?
We assessed whether each study was free of other problems that could put it at risk of bias:
low risk of other bias;
high risk of other bias;
unclear whether there was risk of other bias.
(7) Overall risk of bias
We made explicit judgements about whether studies were at high risk of bias, according to the criteria given in the Handbook (Higgins 2011). With reference to points (1) to (6), we assessed the likely magnitude and direction of the bias, and whether we considered them likely to impact the findings. We planned to assess the impact of the level of bias through undertaking sensitivity analyses ‐ seeSensitivity analysis.
Measures of treatment effect
Dichotomous data
We presented results as summary risk ratio with 95% confidence intervals for dichotomous data.
Continuous data
We planned to use the mean difference if outcomes were measured in the same way between trials. We planned to use the standardised mean difference to combine trials that measured the same outcome, but used different methods.
Unit of analysis issues
Cluster‐randomised trials
We planned to include cluster‐randomised trials in the analyses along with individually‐randomised trials. If we include cluster‐randomised trials in future updates, we will adjust their sample sizes using the methods described in the Handbook, using an estimate of the intracluster correlation co‐efficient (ICC) derived from the trial (if possible), from a similar trial, or from a study of a similar population (Higgins 2011). If we use ICCs from other sources, we will report this, and conduct sensitivity analyses to investigate the effect of variation in the ICC. If we identify both cluster‐randomised trials and individually‐randomised trials, we plan to synthesise the relevant information. We will consider it reasonable to combine the results from both if there is little heterogeneity between the study designs, and the interaction between the effect of the intervention and the choice of randomisation unit is considered to be unlikely.
We will also acknowledge heterogeneity in the randomisation unit, and perform a sensitivity analysis to investigate the effects of the randomisation unit.
Cross‐over trials
We considered cross‐over trials to be inappropriate for this research question, and excluded them.
Multi‐armed trials
We included all the relevant intervention groups (aspirin) and control groups (placebo) from multi‐arm trials. We excluded other arms that were not relevant to this review.
Dealing with missing data
We noted levels of attrition for the included studies. We planned to explore the impact of including studies with high levels of missing data in the overall assessment of treatment effect, by conducting sensitivity analyses.
We carried out analyses, as far as possible, on an intention‐to‐treat basis for all outcomes. That is, we attempted to include all participants randomised to each group in the analyses, and all participants were analysed in the group to which they were allocated, regardless of whether or not they received the allocated intervention. The denominator for each outcome, in each trial, was the number randomised minus any participants whose outcomes were known to be missing.
Assessment of heterogeneity
We assessed statistical heterogeneity in meta‐analyses using the Tau², I², and Chi² statistics. We regarded heterogeneity as substantial, if an I² was greater than 30%, and either a T² was greater than zero, or there was a low P value (P < 0.10) in the Chi² test for heterogeneity.
Assessment of reporting biases
Because there were 10 or more studies included in the meta‐analyses for 'Adequate pain relief as reported by the woman', 'Need for additional pain relief', and 'Maternal adverse effects', we investigated reporting biases (such as publication bias) using funnel plots. We assessed funnel plot asymmetry visually.
Data synthesis
We carried out statistical analysis using Review Manager 5 software (Review Manager 2014). We used fixed‐effect methods for combining data because we assumed that studies were estimating the same underlying treatment effect.
In future updates of this review, if there is clinical heterogeneity sufficient to expect that the underlying treatment effects differed between trials, or if we detect substantial statistical heterogeneity, we will use the random‐effects method to produce an overall summary. We will treat the random‐effects summary as the average of the range of possible treatment effects, and discuss the clinical implications of treatment effects differing among trials. If the average treatment effect is not clinically meaningful, we will not combine trials. If we use the random‐effects model in future updates, we will present the results as the average treatment effect with 95% confidence intervals, and the estimates of Tau² and I².
Subgroup analysis and investigation of heterogeneity
If we had identified substantial heterogeneity, we planned to investigate possible sources, using subgroup analyses and sensitivity analyses. We planned to consider whether an overall summary was meaningful, and if it was, use the random‐effects model to produce the effect.
We planned to carry out the following subgroup analyses:
primiparous versus multiparous women;
women with perineal trauma versus women who gave birth over intact perineum; and
dose of aspirin (i.e. low‐dose versus high‐dose).
However, due to the absence of relevant data in the included trials, we were able to conduct analyses based on dose only.
Subgroup analyses were restricted to the review's primary outcomes with reported data.
We assessed subgroup differences by interaction tests available in Review Manager 2014. We reported the results of subgroup analyses quoting the Chi² statistic and P value, and the interaction test I² value.
Sensitivity analysis
We planned to conduct sensitivity analyses to explore the effects of risk of bias on the outcomes. We planned to explore the effects of trial quality assessed by allocation concealment and random sequence generation (considering selection bias), by omitting studies rated at high or unclear risk of bias for these components. However, because we assessed all included trials at unclear bias for at least one of these two components, we did not conduct a sensitivity analysis.
We also planned to investigate the effects of the randomisation unit (individual versus cluster) on the outcomes, and the impact of including studies with high levels of missing data. We planned to explore the effects of fixed‐effect or random‐effects models for outcomes with statistical heterogeneity, and the effects of any assumptions made, such as the value of the ICC used for cluster‐randomised trials. However, because we did not include any cluster‐randomised trials, trials with high levels of missing data, or identify outcomes with substantial statistical heterogeneity, we did not conduct sensitivity analyses.
We planned to use only primary outcomes in sensitivity analyses.
Summary of findings and assessment of the certainty of the evidence
We assessed the certainty of the evidence using the GRADE approach, as outlined in the GRADE Handbook, in order to assess the certainty of the body of evidence relating to the following outcomes, for the main comparison: aspirin versus placebo (GRADE Handbook).
Adequate pain relief as reported by the woman
Need for additional pain relief in the first 48 hours for perineal pain
Maternal adverse effects, composite of any of the following: nausea, vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness, gastric discomfort, psychological impact
Neonatal adverse effects, composite of any of the following: vomiting, sedation, constipation, diarrhoea, drowsiness, sleepiness
Perineal pain at six weeks postpartum
However, we could only assess the certainty of the evidence for the first three outcomes, as we had no data from the included trials for outcomes 4 and 5.
We used GRADEpro GDT to import data from Review Manager 5, in order to create ’Summary of findings’ tables (GRADEpro GDT; Review Manager 2014). We produced a summary of the intervention effect and a measure of certainty for each of the above outcomes, using the GRADE approach. The GRADE approach uses five considerations (study limitations, consistency of effect, imprecision, indirectness, and publication bias) to assess the certainty of the body of evidence for each outcome. The evidence can be downgraded from 'high certainty' by one level for serious (or by two levels for very serious) limitations, depending on assessments for risk of bias, indirectness of evidence, serious inconsistency, imprecision of effect estimates, or potential publication bias.
Results
Description of studies
Results of the search
There were no new studies identified in the 2019 updated search (see Figure 1).
Two studies (two records) await classification: in both studies, the method of allocation was not clearly reported (Bhounsule 1990; Sunshine 1989).
Included studies
Settings and dates
We included 17 studies (22 reports) in this review. All were reported to be randomised controlled trials.
Most (11 trials) were conducted in the USA (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; Friedrich 1983; Jain 1978a; Jain 1978b; Jain 1985; London 1983a; London 1983b; Okun 1982), three in Venezuela (Olson 1997; Sunshine 1983a; Sunshine 1983b), and one each in Belgium (Devroey 1978), Canada (Trop 1983) and India (Mukherjee 1980).
Only two of the included trials reported their dates; Bloomfield 1967 was conducted between December 1965 and April 1966, and London 1983b was conducted between July and December 1980. Of the remaining 15 trials, six were published in the 1970s (Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; Devroey 1978; Jain 1978a; Jain 1978b); eight in the 1980s (Friedrich 1983; Jain 1985; London 1983a; Mukherjee 1980; Okun 1982; Sunshine 1983a; Sunshine 1983b; Trop 1983); and one in the 1990s (Olson 1997).
Participants and sample sizes
In total, there were 1132 women in the aspirin and placebo arms of 16 of the 17 included trials, with 617 women randomised to receive aspirin, and 515 to a placebo. Three trials only reported the numbers analysed (not randomised; (Devroey 1978; London 1983a; London 1983b)), and one trial dot report the number of women at all (Trop 1983). The sample sizes of the trials (including only the relevant arms) ranged from 26 (Bloomfield 1970b), to 178 (Mukherjee 1980). We reported the number of women in arms of the trials not included in our analyses in the 'Characteristics of included studies' tables.
All included trials included women with perineal pain in the early postpartum period, post‐episiotomy. One trial recruited and randomised women on the first postoperative morning (Mukherjee 1980), two within 24 hours of birth (Bloomfield 1967; Bloomfield 1970b), one from 16 to 48 hours following induction of anaesthesia (Friedrich 1983), six within 48 hours of birth (Bloomfield 1970a; Bloomfield 1974; Devroey 1978; Jain 1978a; London 1983a; Okun 1982); and seven trials did not specify a time period following birth (Jain 1978a; Jain 1985; London 1983b; Olson 1997; Sunshine 1983a; Sunshine 1983b; Trop 1983). We did not identify any trials that assessed perineal pain associated with naturally occurring tears, or birth over an intact perineum. The intensity of women's pain following episiotomy varied among the included trials; eight trials included women with moderate or severe pain (Bloomfield 1967; Devroey 1978; Friedrich 1983; Jain 1978a; London 1983a; London 1983b; Mukherjee 1980; Sunshine 1983b); three included women with moderate to very severe pain (Bloomfield 1970a; Bloomfield 1974; Okun 1982); one included women with mild to severe pain (Bloomfield 1970b); one included women with at least moderate pain (Jain 1985); and three included women with severe pain (Jain 1978b; Olson 1997; Sunshine 1983a). One trial did not specify pain intensity (Trop 1983). Most trials clearly specified that breastfeeding was an exclusion criterion, and excluded women with known sensitivity or allergy to aspirin, and women who had recently received analgesia.
Interventions and comparisons
Only one trial had two trial arms, comparing aspirin and placebo (Bloomfield 1970b). Another trial compared only aspirin and placebo, but had four trial arms (London 1983b). The remaining 15 trials had between three and five trial arms, and in addition to aspirin, assessed a number of other agents for perineal pain in the early postpartum period. These agents included chlorphenesin 400 mg, 800 mg, and combination aspirin 300 mg and chlorphenesin 400 mg (Bloomfield 1967); flufenisal 300 mg and 600 mg (Bloomfield 1970a); ibuprofen 300 mg and 900 mg (Bloomfield 1974); diflunisal 125 mg, 250 mg, and 500 mg (Devroey 1978), etodolac 25 mg and 100 mg (Friedrich 1983), piroxicam 20 mg and 40 mg (Jain 1978a); combination aspirin 800 mg and caffeine 64 mg (Jain 1978b); indoprofen 50 mg and 100 mg (Jain 1985); fluproquazone 100 mg and 200 mg (London 1983a); dipyrone 500 mg (Mukherjee 1980); fendosal 100 mg, 200 mg, and 400 mg (Okun 1982); potassium 25 mg, 50 mg, and 100 mg (Olson 1997); zomepirac and ibuprofen (Sunshine 1983a); flurbiprofen 25 mg, 50 mg, and 100 mg (Sunshine 1983b); and tiaprofenic acid 200 mg and 400 mg (Trop 1983). For the purposes of the review, we analysed only the aspirin and placebo arms from the included trials.
Fifteen trials included comparisons of aspirin and placebo only; the single, oral doses of aspirin in these were 500 mg (Mukherjee 1980), 600 mg (Bloomfield 1967; Devroey 1978; Jain 1985; Sunshine 1983a; Sunshine 1983b), 648 mg (Jain 1978a), 650 mg (Friedrich 1983; Jain 1978b; London 1983a; Okun 1982; Olson 1997), 900 mg (Bloomfield 1974), and 1200 mg (Bloomfield 1970b). Three trials included two or more aspirin arms (in addition to a placebo arm); Bloomfield 1970a and Trop 1983 compared 600 mg and 1200 mg aspirin, and London 1983b compared 300 mg, 600 mg, and 1200 mg aspirin. The number of aspirin and placebo tablets (and dose of the tablets) taken varied across the trials.
Outcomes
We were a able to extract and meta‐analyse some measure of 'Adequate pain relief as reported by the woman' four to eight hours after drug administration from 13 trials. Data from four trials were not presented in a way that enabled us to include them in the meta‐analysis (Jain 1978a; Jain 1978b; Okun 1982; Trop 1983).
Three trials in the meta‐analysis provided data on adequate pain relief four hours after taking the medication (London 1983b; Olson 1997; Sunshine 1983a); two trials reported this outcome after five hours (Bloomfield 1970b; Jain 1985); seven trials after six hours (Bloomfield 1967; Bloomfield 1974; Devroey 1978; Friedrich 1983; London 1983a; Mukherjee 1980; Sunshine 1983b); and one trial after eight hours (Bloomfield 1970a). SPID scores were used to calculate the number of women with adequate pain relief for the meta‐analysis in 11 trials; (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; Devroey 1978; Friedrich 1983; Jain 1985; London 1983b; Olson 1997; Sunshine 1983a; Sunshine 1983b), one used total pain relief (TOTPAR) scores (Mukherjee 1980), and one used the number of women reporting pain relief to be good or excellent (London 1983a).
Five trials provided both summed pain intensity differences (SPID) and TOTPAR scores (Friedrich 1983; Jain 1985; Olson 1997; Sunshine 1983a; Sunshine 1983b); two trials provided both SPID scores, and the number of women reporting pain relief to be good or excellent (Friedrich 1983; Jain 1985); and five reported SPID scores and the number of women with at least 50% pain relief (or similar; (Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; Devroey 1978; Mukherjee 1980)). In these cases, we used SPID data to calculate the number of women with adequate pain relief, and include in the meta‐analysis. In some cases, the number of women with adequate pain relief according to these different measures did not match, and the reasons for discrepancies were not entirely clear, particularly in numbers of women with adequate pain relief, calculated using the SPID versus TOTPAR scores.
Data on the need for additional analgesia, which could be included in a meta‐analysis, were available from 10 trials (Bloomfield 1970a; Bloomfield 1974; Devroey 1978; Jain 1978a; Jain 1985; London 1983a; London 1983b; Olson 1997; Sunshine 1983a; Sunshine 1983b); 14 trials reported data on any maternal adverse effects suitable for meta‐analysis (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1974; Devroey 1978; Friedrich 1983; Jain 1978a; Jain 1978b; Jain 1985; London 1983a; London 1983b; Mukherjee 1980; Olson 1997; Sunshine 1983a; Sunshine 1983b).
Two trials did not provide any data that could be meta‐analysed (Okun 1982; Trop 1983).
None of the 17 included trials reported on any of the prespecified secondary outcomes.
Funding sources
Two of the trials reported support or funding solely from the National Institutes of Health (Bloomfield 1967), and the United Stated Public Health Service and National Heart Institute (Bloomfield 1970b). Ten of the trials reported at least partial support from pharmaceutical companies or commercial medical research organisations: Merck Sharp and Dohme Research Laboratories (Bloomfield 1970a; Devroey 1978); the Upjohn Company (Bloomfield 1974; Sunshine 1983b); American Home Products, Ives Laboratories and Wyeth Laboratories (Jain 1978b); Adria Laboratories (Jain 1985); Sandoz Inc. (London 1983a); the Ciba‐Geigy Corporation (Olson 1997); Boots Pharmaceuticals (Sunshine 1983b); and Roussel Canada Inc. (Trop 1983). Five of the trials did not report any sources of support or funding (Friedrich 1983; Jain 1978a; London 1983b; Mukherjee 1980; Okun 1982).
Declarations of interests
None of the 17 included trials provided specific declarations of interest for the manuscript authors. We noted that three of the trials had author(s) with affiliations to pharmaceutical companies or commercial medical research organisations: Merck Sharp and Dohme Research Laboratories (Devroey 1978); Analgesic Development Ltd. (Olson 1997); and Roussel Canada Inc. (Trop 1983).
Excluded studies
We excluded 10 studies (11 records) for the following reasons: five trials included mixed populations of women with postpartum pain (such as uterine cramping), and did not report results separately for women with perineal pain (Bruni 1965; Gruber 1979; Moggian 1972; Sunshine 1983c; Sunshine 1985); one was not randomised (Santiago 1959); three assessed combination agents (not aspirin alone; (Gindhart 1971; Prockop 1960; Rubin 1984)); and one assessed twice daily aspirin rather than single dose aspirin (Van der Pas 1984).
Risk of bias in included studies
2.

Risk of bias graph: review authors' judgements about each 'Risk of bias' item presented as percentages across all included studies
3.

Risk of bias summary: review authors' judgements about each 'Risk of bias" item for each included study
Allocation
We judged that only two trials applied adequate sequence generation methods; both used computer‐generated random sequences (Olson 1997; Sunshine 1983b). The remaining 15 trials did not report the methods used for random sequence generation, and simply reported that the women were randomised.
We judged all included trials at unclear risk of selection bias; none of them reported methods of allocation concealment.
Blinding
Of the 17 trials, we judged 14 at low risk of both performance and detection bias; women and study personnel (who were also the outcome assessors) were blinded by using identical placebos. We judged three trials at unclear risk of performance and detection bias, because although the trials were reported to be double‐blind, they provided no information on the nature of the placebos used, for us to determine if blinding was successfully achieved (Friedrich 1983; Jain 1978a; Jain 1978b).
Incomplete outcome data
We judged only three trials at low risk of attrition bias, with no losses, or exclusions (Bloomfield 1970b; Jain 1985; Mukherjee 1980).
We assessed five trials as unclear risk of attrition bias, largely due to unclear reporting regarding any losses and exclusions, reasons for missing data, or both (Friedrich 1983; Jain 1978a; Jain 1978b; London 1983a; Trop 1983).
We assessed nine trials at high risk of attrition bias (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1974; Devroey 1978; London 1983b; Okun 1982; Olson 1997; Sunshine 1983a; Sunshine 1983b). In eight, the trial authors imputed data (i.e. for women requesting additional analgesia, trial authors either used women's pre‐treatment pain intensity or relief scores for all subsequent hours, or used the last observation carried forward method for subsequent hours), which may have introduced bias; in one trial, women who requested additional analgesia were excluded from the analyses, which may have similarly introduced bias (Bloomfield 1967).
Selective reporting
We assessed 12 trials as unclear risk of reporting bias, since we had no access to trial protocols or registrations to confidently assess the risk of selective reporting (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; Friedrich 1983; Jain 1985; London 1983a; London 1983b; Mukherjee 1980; Okun 1982; Olson 1997; Sunshine 1983b).
We assessed five trials at high risk of reporting bias (Devroey 1978; Jain 1978a; Jain 1978b; Sunshine 1983a; Trop 1983). In all five, some of outcome data and results were reported incompletely in the text, which meant, we were unable to extract these data for a meta‐analysis. For example: "The three drugs were much the same for mean onset, duration, and time to peak values. The hypothesis that there is no difference among treatments was rejected at the 0.05 level or better for all variables" (Sunshine 1983a).
The 17 trials reported very few outcome data.
Other potential sources of bias
We assessed nine trials at low risk of other potential sources of bias (Bloomfield 1967; Bloomfield 1970a; Jain 1978a; Jain 1985; Mukherjee 1980; Okun 1982; Olson 1997; Sunshine 1983a; Sunshine 1983b). We assessed eight trials as unclear risk of other bias (Bloomfield 1970b; Bloomfield 1974; Devroey 1978; Friedrich 1983; Jain 1978b; London 1983a; London 1983b; Trop 1983). These trials did not report baseline characteristics in a way that enabled us to assess comparability among groups (with no baseline characteristics reported, or lack of detail reported); one trial reported that most baseline characteristics were similar between groups "However, body weight was not similar in all treatment groups" (Bloomfield 1970a).
Effects of interventions
See: Table 1
Comparison 1. Aspirin versus placebo for perineal pain
Fifteen of the 17 included trials contributed data to meta‐analyses in this comparison (Bloomfield 1967; Bloomfield 1970a; Bloomfield 1970b; Bloomfield 1974; Devroey 1978; Friedrich 1983; Jain 1978a; Jain 1978b; Jain 1985; London 1983a; London 1983b; Mukherjee 1980; Olson 1997; Sunshine 1983a; Sunshine 1983b). Two trials did not provide any data that could be meta‐analysed (Okun 1982; Trop 1983).
Primary outcomes
Adequate pain relief, as reported by the woman
Over four to eight hours after drug administration, aspirin compared with placebo may increase adequate pain relief (risk ratio (RR) 2.03, 95% confidence intervals (CI) 1.69 to 2.42; 13 trials, 1001 women; low‐certainty evidence; Analysis 1.1). Visual inspection of the funnel plot for this outcome suggested no clear evidence of reporting bias (Figure 4).
1.1. Analysis.

Comparison 1: Aspirin versus placebo for perineal pain, Outcome 1: Adequate pain relief as reported by the woman
4.

Funnel plot of comparison: 1 Aspirin versus placebo for perineal pain, outcome: 1.1 Adequate pain relief as reported by the women
Data from the trials not included in the meta‐analysis
Jain 1978a: "By all measurements of drug effect... aspirin 648 mg [was] significantly (P < 0.01) superior to placebo in [its] overall analgesic effect and also at second, third and fourth hours after dosing".
Jain 1978b: "In comparing 650 mg aspirin with placebo, we detected no significant difference at 1, 2, or 3 hr, but at the fourth hour we noted trends toward significant in favour of aspirin (P < 0.10) by each Kruskal‐Wallis analysis for pain analogue, pain intensity, and pain relief scores. The corresponding analysis of covariance at hour 4 showed differences in favour of aspirin for both pain analogue and pain intensity scores (P < 0.02)".
Okun 1982: "In patients with either uterine cramp or episiotomy pain, aspirin... provided greater pain relief (lower mean pain intensity scores) than did placebo from the 2nd through the 8th study hour".
Trop 1983: "When compared to placebo both patient's self‐rating scale and nurse's impression scale have shown a significant reduction in pain following treatment... with ASA".
Need for additional pain relief in the first 48 hours, for perineal pain
It is uncertain whether aspirin compared with placebo has an effect on the need for additional analgesia over four to eight hours after drug administration (RR 0.25, 95% CI 0.17 to 0.37; 10 trials, 744 women; very low‐certainty evidence; Analysis 1.2). Visual inspection of the funnel plot for this outcome indicated possible evidence of reporting bias, which could be due to some smaller trials producing exaggerated intervention effect estimates (Figure 5).
1.2. Analysis.

Comparison 1: Aspirin versus placebo for perineal pain, Outcome 2: Need for additional pain relief
5.

Funnel plot of comparison: 1 Aspirin versus placebo for perineal pain, outcome: 1.2 Need for additional pain relief
Data from the trials not included in the meta‐analysis
Bloomfield 1967: not reported; although one woman in the aspirin group was reported to have been "withdrawn owing to distressing pain unrelieved by the study drugs" compared with no women in the placebo group.
Okun 1982: "The proportion of patients requiring additional analgesic was significantly different... Approximately 71% of patients in the placebo group needed additional analgesic as compared with... 48% in the aspirin group" (these data related to women with uterine cramp or episiotomy pain).
Trop 1983 "None of the patients on… ASA required any additional analgesic during the 4‐hour observation period, but four patients in the placebo group required supplementary medication for pain" (the denominators for each group were not reported).
Maternal adverse effects
It is uncertain whether aspirin compared with placebo has an effect on overall maternal adverse effects over four to eight hours after administration (RR 1.08, 95% CI 0.57 to 2.06; 14 trials, 1067 women; very low‐certainty evidence; Analysis 1.3). Visual inspection of the funnel plot for this outcome suggested no clear evidence of reporting bias (Figure 6).
1.3. Analysis.

Comparison 1: Aspirin versus placebo for perineal pain, Outcome 3: Maternal adverse effects
6.

Funnel plot of comparison: 1 Aspirin versus placebo for perineal pain, outcome: 1.3 Maternal adverse effects
Data from the trials not included in the meta‐analysis
Okun 1982: "The incidence of side effects was not significantly different among the treatment groups... Three patients each in the placebo group... and 5 patients in the aspirin group reported side effects" (these data related to women with uterine cramp or episiotomy pain).
Trop 1983: one woman receiving 1200 mg aspirin (dizziness), no women receiving 600 mg aspirin and two women receiving placebo (nausea) experienced side effects (the denominators for each group were not reported).
Neonatal adverse effects
None of the included trials reported on the primary outcome: neonatal adverse effects.
Subgroup analyses based on dose
We integrated subgroup analyses based on dose, into the main analyses, comparing trials using 300 mg, 500 to 650 mg, 900 mg, and 1200 mg aspirin. We observed no clear subgroup differences based on dose of aspirin for 'Adequate pain relief as reported by the woman (test for subgroup differences: Chi² = 0.75, P = 0.86, I² = 0%; Analysis 1.1); 'Need for additional pain relief' (test for subgroup differences: Chi² = 0.63, P = 0.89, I² = 0%; Analysis 1.2); or 'Maternal adverse effects' (test for subgroup differences: Chi² = 3.76, df = 2 (P = 0.15), I² = 46.8%; Analysis 1.3).
Secondary outcomes
None of the included trials reported on any of the secondary review outcomes: prolonged hospitalisation due to perineal pain; re‐hospitalisation due to perineal pain; fully breastfeeding at discharge; mixed feeding at discharge; fully breastfeeding at six weeks; mixed feeding at six weeks; perineal pain at six weeks; maternal views (using a validated questionnaire); maternal postpartum depression.
Comparison 2. 300 mg aspirin versus 600 mg aspirin for perineal pain
London 1983b contributed data to this comparison.
Primary outcomes
Adequate pain relief as reported by the woman
It is uncertain whether, over four hours after administration, 300 mg has an effect on adequate pain relief, compared with 600 mg aspirin (RR 0.82, 95% CI 0.36 to 1.86; 1 trial, 81 women; Analysis 2.1).
2.1. Analysis.

Comparison 2: 300 mg aspirin versus 600 mg aspirin for perineal pain, Outcome 1: Adequate pain relief as reported by the woman
Need for additional pain relief in the first 48 hours for perineal pain
It is uncertain whether, over four hours after administration, 300 mg has an effect on the need for additional pain relief, compared with 600 mg aspirin (RR 0.68, 95% CI 0.12 to 3.88; 1 trial, 81 women; Analysis 2.2).
2.2. Analysis.

Comparison 2: 300 mg aspirin versus 600 mg aspirin for perineal pain, Outcome 2: Need for additional pain relief
Maternal adverse effects
There were no adverse effects reported among women who received 300 mg or 600 mg aspirin (Analysis 2.3).
2.3. Analysis.

Comparison 2: 300 mg aspirin versus 600 mg aspirin for perineal pain, Outcome 3: Maternal adverse effects
Neonatal adverse effects
London 1983b did not report on the primary outcome: neonatal adverse effects.
Secondary outcomes
London 1983b did not report on any of the secondary review outcomes.
Comparison 3. 600 mg aspirin versus 1200 mg aspirin for perineal pain
Bloomfield 1970a and London 1983b contributed data to this comparison.
Primary outcomes
Adequate pain relief as reported by the woman
It is uncertain whether, over four to eight hours after administration, 600 mg has an effect on adequate pain relief, compared with 1200 mg aspirin (RR 0.85, 95% CI 0.52 to 1.39; 2 trials, 121 women; Analysis 3.1).
3.1. Analysis.

Comparison 3: 600 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 1: Adequate pain relief as reported by the woman
Need for additional pain relief in the first 48 hours for perineal pain
It is uncertain whether, over four to eight hours after administration, 600 mg has an effect on the need for additional pain relief, compared with 1200 mg aspirin (RR 1.32, 95% CI 0.30 to 5.68; 2 trials, 121 women; Analysis 3.2).
3.2. Analysis.

Comparison 3: 600 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 2: Need for additional pain relief
Maternal adverse effects
It is uncertain whether, over four to eight hours after administration, 600 mg has an effect on maternal adverse effects compared with 1200 mg aspirin (RR 3.00, 95% CI 0.13 to 69.52; 2 trials, 121 women; Analysis 3.3).
3.3. Analysis.

Comparison 3: 600 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 3: Maternal adverse effects
Neonatal adverse effects
Bloomfield 1970a and London 1983b did not report on the primary outcome: neonatal adverse effects.
Secondary outcomes
Bloomfield 1970a and London 1983b did not report on any of the secondary review outcomes.
Comparison 4. 300 mg aspirin versus 1200 mg aspirin for perineal pain
London 1983b contributed data to this comparison.
Primary outcomes
Adequate pain relief as reported by the woman
It is uncertain whether, over four hours after administration, 300 mg has an effect on adequate pain relief, compared with 1200 mg aspirin (RR 0.62, 95% CI 0.29 to 1.32; 1 trial, 80 women; Analysis 4.1).
4.1. Analysis.

Comparison 4: 300 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 1: Adequate pain relief as reported by the woman
Need for additional pain relief in the first 48 hours for perineal pain
It is uncertain whether, over four hours after administration, 300 mg has an effect on the need for additional pain relief, compared with 1200 mg aspirin (RR 2.00, 95% CI 0.19 to 21.18; 1 trial, 80 women; Analysis 4.2).
4.2. Analysis.

Comparison 4: 300 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 2: Need for additional pain relief
Maternal adverse effects
There were no adverse effects among women who received 300 mg or 1200 mg aspirin (Analysis 4.3).
4.3. Analysis.

Comparison 4: 300 mg aspirin versus 1200 mg aspirin for perineal pain, Outcome 3: Maternal adverse effects
Neonatal adverse effects
London 1983b did not report on the primary outcome: neonatal adverse effects.
Secondary outcomes
London 1983b did not report on any of the secondary review outcomes.
Discussion
Summary of main results
We included 17 trials; of these, 16 randomised 1132 women to single dose aspirin or placebo for perineal pain in the early postpartum period. Fifteen trials contributed data to four comparisons (aspirin versus placebo; 300 mg versus 600 mg aspirin; 600 mg versus 1200 mg aspirin; 300 mg versus 1200 mg aspirin).
Low‐certainty evidence from 13 trials (1001 women) suggested that women receiving aspirin (doses ranging from 300 mg to 1200 mg) may have an increase in adequate pain relief at four to eight hours after administration compared with placebo (a 105% relative increase, from 25% in the placebo group to 47% in the aspirin group). We were unable to include data from four trials in our meta‐analysis for this outcome (Jain 1978a; Jain 1978b; Okun 1982; Trop 1983). Individual results indicated a benefit from aspirin when compared with placebo. The effects of different doses of aspirin on adequate pain relief, as reported by the women were uncertain (in comparisons of 300 mg and 600 mg (1 trial, 81 women), 600 mg and 1200 mg (2 trials, 121 women), and 300 mg and 1200 mg (1 trial, 80 women)).
Very low‐certainty evidence from 10 trials (744 women) suggested that the effect of aspirin (doses ranging from 300 mg to 1200 mg) compared with placebo was uncertain for reducing the need for additional analgesia for perineal pain over four to eight hours after administration. Individual effects of different doses of aspirin (300 mg and 600 mg (1 trial, 81 women), 600 mg and 1200 mg (2 trials, 121 women), and 300 mg and 1200 mg (1 trial, 80 women)) were also uncertain on the need for additional pain relief.
Very low‐certainty evidence from 14 trials (1067 women) also suggested that the effect of aspirin (doses ranging from 300 mg to 1200 mg) compared with placebo was uncertain for maternal adverse effects over four to eight hours after administration. Individual effects of different doses of aspirin (300 mg and 1200 mg aspirin (2 trials, 121 women)) on maternal adverse effects was also uncertain.
None of the included trials reported on the review primary outcome ‐ neonatal adverse effects, nor any of the secondary review outcomes: prolonged hospitalisation due to perineal pain; re‐hospitalisation due to perineal pain; fully breastfeeding at discharge; mixed feeding at discharge; fully breastfeeding at six weeks; mixed feeding at six weeks; perineal pain at six weeks; maternal views (using a validated questionnaire); and maternal postpartum depression.
Overall completeness and applicability of evidence
The included trials enrolled women with perineal pain in the early postpartum period, post‐episiotomy. Accordingly, results may not be applicable to other women with perineal pain, such as those with pain following naturally occurring tears, or birth over an intact perineum. All included trials compared aspirin with placebo; we were unable to assess the comparative effects of aspirin versus paracetamol, as proposed in the protocol for this review.
Most trials recruited women from the USA (11 trials); three trials were conducted in Venezuela, and one each in Belgium, Canada, and India. Sixteen trials were published before the 1990s (one in the 1960s, six in the 1970s, and nine in the 1980s). Results may not be applicable to all settings or countries worldwide, nor to current clinical practice.
Although there were more than 1000 women and their babies in the included trials, individually, sample sizes were small, ranging from 26 to 178 women. Most trials reported on the review primary outcomes adequate pain relief as reported by the woman (N = 13), need for additional analgesia (N = 10), and maternal adverse effects (N = 14). The included trials only examined three (of 13) prespecified outcomes; there were no data reported for the primary outcome ⎯ neonatal adverse effects ⎯ or for any of the secondary review outcomes.
Breastfeeding was clearly stated as an exclusion criterion in most trials, and as a result, no data were available to determine any neonatal adverse effects or effects on breastfeeding. Guidance for the management of perineal pain, including in breastfeeding women, recommends that if oral analgesia is required, then paracetamol or acetaminophen should be used first, unless contraindicated; if paracetamol is not effective, an oral or rectal non‐steroidal anti‐inflammatory (NSAID) agent, such as ibuprofen, should be considered in the absence of contraindications (NICE 2015; NIH 2015; Reece‐Stremtan 2017). Although some guidance indicates that low‐dose aspirin may be considered as an antiplatelet drug for use in breastfeeding women (Bell 2011), it is generally recommended it be used cautiously, or avoided during breastfeeding, because salicylate and salicylate metabolites are excreted in breast milk. Therefore, there is potential for adverse effects in infants. Longer‐term, high‐dose administration of maternal aspirin has been associated with a report of infant metabolic acidosis, and aspirin administration to infants with viral infections has been associated with Reye's syndrome (NIH 2015).
It is recognised that breastfeeding is an unequalled way to provide the ideal food for infants. International guidance, including from the World Health Organization, recommends (where possible) initiation of breastfeeding within the first hour after birth, and exclusive breastfeeding for the first six months of life, for optimal growth, development, and health, followed by age‐appropriate complementary feeding alongside breastfeeding, for two years or more (WHO 2001; WHO 2003). Therefore, the evidence in this review is not directly applicable to current globally recommended best practice.
Quality of the evidence
Many aspects relating to risk of bias were unclear for several of the included trials (Figure 2; Figure 3). Except for one included trial, all studies were published before the 1990s. We found a lack of methodological detail provided in published reports. Attempts to contact trial authors to obtain additional information were unsuccessful. Of the 17 included trials, we assessed 15 as unclear risk of selection bias, because study reports did not provide detailed methods for sequence generation. We judged all trials as unclear risk of selection bias; because study reports did not provide detailed methods for concealment of allocation. We assessed 14 trials at low risk of performance and detection bias; we judged the risk as unclear for three trials. We judged most trials as unclear or high risk of attrition bias (a number imputing data); and all as unclear or high risk of reporting bias, with many of them reporting very limited outcome data; none had available trial registration or protocols.
We assessed the certainty of the evidence using the GRADE approach, as outlined in the GRADE Handbook, for prespecified outcomes analysed in the main comparison (aspirin versus placebo; (GRADE Handbook). We assessed that the certainty of the evidence was low (adequate pain relief as reported by the women), or very low (need for additional pain relief; maternal adverse effects). Our judgements were based on design limitations in the included trials (all outcomes), possible publication bias (need for additional pain relief), and imprecision (maternal adverse effects). See Table 1.
Potential biases in the review process
We took steps to minimise the introduction of bias during the review process. At least two review authors independently assessed trials for inclusion, performed data extraction, and assessed risk of bias for each of the included trials.
The Information Specialist of Cochrane Pregnancy and Childbirth conducted a detailed, systematic search process, without language or publication status restrictions, to reduce the risk for potential publication bias. We also searched trial registries for unpublished, planned, or ongoing trials. It is possible that additional trials assessing aspirin for perineal pain in the early postpartum period have been published but not identified; and that further trials have been conducted but are not yet published; or both. Should any such studies be identified in the future, we will assess these for inclusion in future updates of this review.
We investigated reporting biases (such as publication bias) using funnel plots for our primary outcomes. Although we found no clear evidence of reporting bias for 'adequate pain relief as reported by the women', and 'maternal adverse effects', the funnel plot for 'need for additional pain relief' demonstrated some asymmetry. This could indicate possible reporting bias, with the smaller published trials reporting exaggerated intervention effect estimates, and the possibility of additional small trials (including those reporting smaller effect estimates) remaining unpublished.
Agreements and disagreements with other studies or reviews
Previous Cochrane Reviews have assessed therapeutic ultrasound (Hay‐Smith 1998); rectal analgesia (Hedayati 2003); local cooling (East 2012b); and topical anaesthetics (Hedayati 2005); for the relief of perineal pain in the postpartum period, revealing mixed results. More recently, following publication of a generic protocol for a series of reviews of drugs for perineal pain in the early postpartum period (Chou 2009), two reviews have assessed paracetamol (Chou 2013), and NSAIDs (Wuytack 2016). Both Chou 2013 (including 10 trials involving 2307 women) and Wuytack 2016 (including 28 trials involving 4181 women) showed benefits for paracetamol and NSAIDs compared with placebo, as an increase in adequate pain relief, and reduced need for additional pain relief for women with perineal pain in the early postpartum period. However, like our review, Chou 2013 and Wuytack 2016 found that the risk of bias was unclear for many of the included studies (most of which were also conducted from the 1960s to the 1990s); adverse effects were often not assessed for women, and were not assessed for infants. Breastfeeding women, and thus breastfeeding outcomes, were not included.
Another Cochrane Review (including 37 trials involving 5743 adults) assessed single dose aspirin (doses ranging from 300 mg to 1200 mg) for acute postoperative pain in adults (Derry 2012). Like our review, Derry 2012 found that compared with placebo, aspirin increased the number who experienced adequate pain relief, and reduced the need for rescue medication. Although Derry 2012 reported that benefits were seen for 600 mg to 650 mg aspirin, 900 mg to 1000 mg aspirin, and 1200 mg aspirin, it was reported that lower doses of aspirin (500 mg) were not significantly different from placebo; however, no formal subgroup interaction tests were performed or reported. Derry 2012 reported no difference in adverse effects when 600 mg to 650 mg aspirin was compared with placebo, but reported an increase in adverse effects with 900 mg to 1000 mg aspirin compared with placebo. No formal subgroup interaction test was performed.
No other reviews were identified that assessed single dose aspirin for perineal pain in the early postpartum period.
Authors' conclusions
Implications for practice.
Single dose aspirin (at doses ranging from 300 mg to 1200 mg) may increase adequate pain relief in women with perineal pain post‐episiotomy in the early postpartum period, compared with placebo. It is uncertain whether aspirin has an effect on the need for additional pain relief, or maternal adverse effects, compared with placebo. We assessed the evidence as low‐ to very low‐certainty; downgrading for study limitations (risk of bias), imprecision, or publication bias, or both.
Current evidence is uncertain regarding the effects of different doses of aspirin on pain relief and maternal adverse effects. All trials to date have excluded women who were breastfeeding. Therefore, there was no evidence to formally assess the effects of single dose aspirin on neonatal adverse effects or breastfeeding outcomes. There was no evidence to assess any of the secondary review outcomes.
With international guidance recommending mothers initiate breastfeeding within one hour of birth, and exclusively breast feed for the first six months, the evidence from this review does not apply to current recommended best practice. Aspirin may be considered for use in non‐breastfeeding women with post‐episiotomy perineal pain.
Implications for research.
Due to current guidance suggesting other analgesics be considered first, particularly for breastfeeding women, and possible ethical concerns regarding withholding pain relief, it is considered unlikely that future trials will be conducted to determine the effects of aspirin compared with placebo. It is also considered unlikely that trials will be conducted in breastfeeding women. If conducted, is most likely that future trials would compare single dose aspirin with other pain relievers. Such trials should be designed to ensure robust methodological quality, and address gaps in the evidence, such as maternal views, postpartum depression, and prolonged hospitalisation or re‐hospitalisation. Because research to date has focused on women post‐episiotomy, future trials could be extended to include women with perineal trauma associated with naturally occurring tears, or birth over an intact perineum.
What's new
| Date | Event | Description |
|---|---|---|
| 4 October 2019 | New search has been performed | Search updated and no new trials identified. Minor updates to reporting throughout review. |
| 4 October 2019 | New citation required but conclusions have not changed | No changes to conclusions. |
History
Protocol first published: Issue 3, 2016 Review first published: Issue 2, 2017
Acknowledgements
As part of the pre‐publication editorial process, three peers (an editor and two referees who are external to the editorial team), a member of the Pregnancy and Childbirth Group's international panel of consumers and the Group's Statistical Adviser (2017) commented on this review.
This project was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to Cochrane Pregnancy and Childbirth. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Evidence Synthesis Programme, the NIHR, National Health Service (NHS), or the Department of Health and Social Care.
We acknowledge the support from the Cochrane Pregnancy and Childbirth editorial team in Liverpool. We thank Sujana Molakatalla for her contribution as an author to previous versions of this review.
Appendices
Appendix 1. Search terms for ICTRP and ClinicalTrials.gov
ICTRP
(searched with all synonyms)
aspirin AND postpartum
aspirin AND perineum
aspirin AND episiotomy
ClinicalTrials.gov
Advanced search
Interventional Studies | Episiotomy Wound | aspirin
perineum | Interventional Studies | aspirin
postpartum | Interventional Studies | aspirin
Data and analyses
Comparison 1. Aspirin versus placebo for perineal pain.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Adequate pain relief as reported by the woman | 13 | 1001 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.03 [1.69, 2.42] |
| 1.1.1 300 mg aspirin | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.60 [0.36, 18.88] |
| 1.1.2 500 mg to 650 mg aspirin | 11 | 800 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.98 [1.64, 2.39] |
| 1.1.3 900 mg aspirin | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.83 [0.84, 3.99] |
| 1.1.4 1200 mg aspirin | 3 | 108 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.75 [1.25, 6.06] |
| 1.2 Need for additional pain relief | 10 | 744 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.25 [0.17, 0.37] |
| 1.2.1 300 mg aspirin | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.16 [0.03, 0.79] |
| 1.2.2 500 mg to 650 mg aspirin | 9 | 569 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.27 [0.17, 0.41] |
| 1.2.3 900 mg aspirin | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.14 [0.01, 2.60] |
| 1.2.4 1200 mg aspirin | 2 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.20 [0.06, 0.70] |
| 1.3 Maternal adverse effects | 14 | 1067 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.08 [0.57, 2.06] |
| 1.3.1 300 mg aspirin | 1 | 53 | Risk Ratio (M‐H, Fixed, 95% CI) | Not estimable |
| 1.3.2 500 mg to 650 mg aspirin | 13 | 892 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.13 [0.51, 2.53] |
| 1.3.3 900 mg aspirin | 1 | 40 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.50 [0.55, 11.41] |
| 1.3.4 1200 mg aspirin | 2 | 82 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.10 [0.01, 1.80] |
Comparison 2. 300 mg aspirin versus 600 mg aspirin for perineal pain.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Adequate pain relief as reported by the woman | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.2 Need for additional pain relief | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.3 Maternal adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 3. 600 mg aspirin versus 1200 mg aspirin for perineal pain.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Adequate pain relief as reported by the woman | 2 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.85 [0.52, 1.39] |
| 3.2 Need for additional pain relief | 2 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.32 [0.30, 5.68] |
| 3.3 Maternal adverse effects | 2 | 121 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.00 [0.13, 69.52] |
Comparison 4. 300 mg aspirin versus 1200 mg aspirin for perineal pain.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Adequate pain relief as reported by the woman | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.2 Need for additional pain relief | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.3 Maternal adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bloomfield 1967.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Cincinnati General Hospital, Ohio, USA Trial dates: December 1965 to April 1966 Inclusion criteria: women with a painful ('moderate' or 'severe') mediolateral episiotomy, within 24 hours following an uncomplicated labour and birth Exclusion criteria: breastfeeding; aged < 18 years; known aspirin sensitivity; 'mild' pain at interview within 24 hours of birth |
|
| Interventions |
Aspirin (N = 17 randomised) 600 mg aspirin; women received a single oral dose in a black capsule Placebo (N = 18 randomised) Women received a single oral dose in a black capsule All women: women did not receive other analgesics during the 6 hours of study, or during the 6 hours before entering the study |
|
| Outcomes |
Adequate pain relief as reported by the woman: pain intensity evaluated by 1 research nurse hourly for 6 hours; women were asked "How much do your stiches hurt you?", and answers were transposed into an ordinal scale from 0 to 3 (0 = no pain; 1 = slight pain; 2 = moderate pain; 3 = severe pain). The difference between a woman's pre‐treatment pain intensity score and each hourly post‐treatment score gave an hourly pain relief score; a total 6‐hour pain relief score was calculated for each woman by adding these scores. Mean Pain Relief scores (equivalent to SPID scores) were used to calculate 'Adequate pain relief as reported by the woman' (taken over 6 hours) Maternal adverse effects: women were asked on the day following treatment whether they noticed any other effects of the treatment; if they answered 'yes' they were asked 'What were they'; no leading questions were asked |
|
| Notes |
Funding: the study was supported in part by USPHS grants HE 05622 and HE 07392 from the National Institutes of Health Declarations of interest: not reported (short ‘About the authors’ section describing affiliations) Additional trial arms: this was a 5‐arm trial, also assessing chlorphenesin 400 mg (N = 18), 800 mg (N = 17), and a combination of aspirin 300 mg and chlorphenesin 400 mg (N = 18); we included only the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "were randomly assigned... according to a predetermined schedule" |
| Allocation concealment (selection bias) | Unclear risk | Not detailed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "double‐blind"; and "All patients received a single dose of coded mediation by mouth in identical black capsules". Assumed that blinding of women and personnel was successful with the use of an identical placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | 1 research nurse evaluated pain intensity and side effects by interviewing women. Assumed that blinding of the research nurse and women was successful with the use of an identical placebo |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 88 women had moderate or severe episiotomy pain; 84 completed the 6 hours of study, and "form the basis of this report"… "Four of the 88 patients entering the trial were withdrawn owing to distressing pain unrelieved by the study drugs" (1/17 from the aspirin group; 0/18 from the placebo group) |
| Selective reporting (reporting bias) | Unclear risk | Very few outcomes reported (pain relief and side effects only); no access to trial registration or protocol to further assess selective reporting |
| Other bias | Low risk | Baseline characteristics were comparable between groups; no other obvious risk of bias identified |
Bloomfield 1970a.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Cincinnati General Hospital, University of Cincinnati College of Medicine, Ohio, USA (assumed from author affiliation) Trial dates: not reported (presented and published in 1970) Inclusion criteria: healthy, consenting, ward patients with moderate to very severe episiotomy pain (mediolateral or midline) within 48 hours of an otherwise uncomplicated birth Exclusion criteria: mild pain; under the age of 18; history of aspirin allergy; breastfeeding; given analgesics within the previous 6 hours |
|
| Interventions |
Aspirin Group 1* (N = 20 randomised) 1200 mg aspirin; women received a single oral dose in capsules Aspirin Group 2* (N = 20 randomised) 600 mg aspirin; women received a single oral dose in a capsule Placebo (N = 19 randomised) Lactose placebo; women received a single oral dose in a capsule All women: lactose capsules were included with medication where necessary to provide a total of 4 capsules per dose. All drugs were administered before breakfast with a full glass of water, and women were instructed to lie on their right side for 2 hours after administration. Stilbestrol and ferrous sulphate were given routinely in the postpartum period, but all other drugs except for the study drugs were avoided, and except for "cleansing: all perineal care was suspended for the 8‐hour study period" |
|
| Outcomes |
Adequate pain relief as reported by the woman: the same trained nurse observer interviewed women hourly for 8 hours; they were asked "How much do the stiches hurt you"; answers were transposed to an ordinal score on a scale of 0 to 4 (0 = no pain; 1 = mild pain; 2 = moderate pain; 3 = severe pain; 4 = very severe pain):
Need for additional pain relief in the first 48 hours for perineal pain: requirement for additional known analgesic medication (codeine or propoxyphene) for inadequate response to study drugs Maternal adverse effects: side effects were evaluated at the last interview by the question, "Did you notice any other effects from today's medicine?" If the answer was "yes", the woman was asked, "What are they?" No other leading questions were asked |
|
| Notes |
Funding: "Supported in part by United Stated Public Health Service Grant HE 05622, and by Merck Sharp & Dohme Research Laboratories. Supplies of flufenisal and other coded medications were provided by Dr. A. W. Vogel, Merck Sharp & Dohme Research Laboratories" Declarations of interest: not reported Additional trial arms: this was a 5‐arm trial, also assessing flufenisal 300 mg (N = 20) and flufenisal 600 mg (N = 21); we included only the relevant arms in this review. Note: we combined the 2 aspirin groups for the main analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Within each of the 3 strata of pain intensity, patients were randomly assigned under double‐blind conditions to one of the 5 treatment groups according to a predetermined balanced allocation schedule" |
| Allocation concealment (selection bias) | Unclear risk | Not detailed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "All test medications were prepackaged in individual patient‐coded vials containing a single oral dose in identical capsules. Lactose capsules were included with medication where necessary to uniformly provide a total of 4 capsules per dose" and "double‐blind conditions" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Subjective evaluation of pain relief and side effects; the same trained nurse observer interviewed women; considered reasonable to assume nurse and women were blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Imputation of data likely to have influenced results. Quote: "Pain relief data collected before additional analgesic was given to each of these 14 patients was included in the analysis without qualification, but interviews were discontinued. By convention each patient’s pain intensity score for each of the residual hours was adjusted to the value of her pretreatment score, and these adjusted scores were used for calculations of pain relief which were then analysed together with the earlier recorded data. Although such an adjustment was arbitrary and tended to underestimate in these 14 patients the analgesic response to the study treatments, bias in the opposite direction, i.e. tending to exaggerate analgesic response to treatments, would have occurred if all or part of the hourly data for these 14 patients would have been excluded from the analysis or no adjustment made" |
| Selective reporting (reporting bias) | Unclear risk | Limited number of outcomes reported; no access to trial registration or protocol to further assess selective reporting |
| Other bias | Unclear risk | Most baseline characteristics were similar among groups Quote "However, body weight was not similar in all treatment groups" |
Bloomfield 1970b.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Cincinnati General Hospital, University of Cincinnati College of Medicine, Ohio, USA (assumed from author affiliation) Trial dates: not reported (presented in part in 1968 and published in 1970) Inclusion criteria: healthy, consenting women with mild to severe episiotomy pain within 24 hours of an otherwise uncomplicated birth Exclusion criteria: allergy to aspirin; receipt of medication during the 6 hours before treatment |
|
| Interventions |
Aspirin (N = 13 randomised) 1200 mg aspirin given in coded single oral dose of 4 capsules Placebo (N = 13 randomised) Identical lactose placebo given in coded single oral dose of 4 capsules All women: no additional medications were received in the 5‐hour period of pain evaluation. Medications were given after a non‐fatty breakfast, and all other foods except water were withheld until after the pain evaluations were completed |
|
| Outcomes |
Adequate pain relief as reported by the woman: the same trained nurse observer interviewed women hourly for 5 hours; they were asked "How much do the stiches hurt you"; answers were transposed to an ordinal score on a scale of 0 to 3 (0 = no pain; 1 = mild pain; 2 = moderate pain; 3 = severe pain):
|
|
| Notes |
Funding: "This investigation was supported in part by USPHS training grant HE‐05622 and by the Special Research Fellowship HE‐34688 of the National Heart Institute" Declarations of interest: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Within each of the three strata of pain intensity… patients were randomly assigned to one of the two treatment groups" |
| Allocation concealment (selection bias) | Unclear risk | Not detailed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "identical lactose placebo, given in a coded single oral dose of four capsules under double‐blind conditions" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Subjective evaluation of episiotomy pain; the same trained nurse observer interviewed women; reasonable to assume women and the nurse were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses or exclusions for pain relief |
| Selective reporting (reporting bias) | Unclear risk | Pain relief was the only outcome reported; no access to trial registration or protocol to further assess selective reporting |
| Other bias | Unclear risk | Very limited methodological details provided; no details of baseline characteristics |
Bloomfield 1974.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: University of Cincinnati Medical Center, Ohio, USA (assumed from author affiliation) Trial dates: not reported (published in 1974) Inclusion criteria: healthy postpartum women with moderate to very severe episiotomy pain (mediolateral or midline) within 48 hours of an otherwise uncomplicated birth Exclusion criteria: mild pain; unmarried, aged < 18 years; history of aspirin allergy; given analgesics, sedatives, or other psychotropic drugs within the previous 6 hours; breastfeeding; known drug dependence |
|
| Interventions |
Aspirin (N = 20 randomised) 900 mg aspirin; single oral dose of 3 tablets of 300 mg each Placebo (N = 20 randomised) Lactose placebo; single oral dose of 3 tablets All women: stilbestrol and ferrous sulphate were given routinely during the postpartum period, however all other drugs were avoided unless necessary, and except for "cleansing", all perineal care was suspended for the 6‐hour study period; women were confined to bed for the first 2 hours and were intermittently out of bed during the last 4 hours. Tablets were administered on demand, with a full glass of water, at approximately the same time of the day throughout the study (2 hours before breakfast) and women were instructed to lie on their right sides for 2 hours afterwards |
|
| Outcomes |
Adequate pain relief as reported by the woman: the same trained nurse observer interviewed women hourly for 6 hours; women estimated the severity of 'stitch' pain on a scale of 0 to 4 (0 = no pain; 1 = mild pain; 2 = medium pain; 3 = severe pain; 4 = very severe pain):
Need for additional pain relief in the first 48 hours for perineal pain: request for additional analgesic medication (codeine or propoxyphene) before the end of the 6‐hour study period Maternal adverse effects: side effects were elicited spontaneously at final interview "with a minimum use of leading questions and without invoking a checklist of possible side effects" |
|
| Notes |
Funding: "Supported in part by United States Public Health Service Grant No. HL‐05622 and by the Upjohn Company. Supplies of ibuprofen and other coded medications were provided by Carter D. Brooks, M.D., The Upjohn Company" Declarations of interests: not reported Additional trial arms: this was a 4‐arm trial also assessing ibuprofen 300 mg (N = 20) and 900 mg (N = 20); we only included the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "On entering the study patients were randomly allocated to one of the 4 groups according to a predetermined schedule. The randomization provided for stratification of patients on the basis of initial intensity of pain" |
| Allocation concealment (selection bias) | Unclear risk | Not detailed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "double‐blind conditions"... "All were in the form of film‐coated tablets identical in appearance and taste, and were prepackaged in code‐numbered individual dose vials" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Changes in pain intensity and side effects associated with the treatments were evaluated subjectively in uniformly conducted interviews"; reasonable to assume blinding of women and interviewer |
| Incomplete outcome data (attrition bias) All outcomes | High risk | No losses or exclusions reported; assumed only 80 women randomised, and all included in analyses; 4 women requested additional analgesic (3 in the placebo group); their pain relief data before additional analgesics were given were included in the analysis, but interviews were then discontinued, and for the remaining hours, each woman’s pain intensity score was adjusted to the value of her pre‐treatment score. Imputation of data likely to have influenced results |
| Selective reporting (reporting bias) | Unclear risk | Very limited outcome data; no access to trial registration or protocol to further assess selective reporting |
| Other bias | Low risk | Baseline characteristics comparable for relevant groups ("Two exceptions were a preponderance of unmarried patients in the ibuprofen 300 mg group compared with the 3 other groups, and body weight, which in the group of patients receiving ibuprofen 900 mg was distinctly higher than in patients in the other 3 groups. These were chance occurrences with an uncertain influence on the results"); no other obvious risk of bias identified |
Devroey 1978.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Department of Obstetrics and Gynaecology, St Christiana Clinic, Dendermonde, Belgium Trial dates: not reported (published in 1977 and 1978) Inclusion criteria: primiparae who had undergone mediolateral episiotomy (3 cm to 5 cm) during the course of an otherwise uncomplicated birth within the previous 48 hours, with moderate to severe pain Exclusion criteria: a more extensive episiotomy (because of forceps birth or other procedures); multigravida women; known allergy to aspirin; breastfeeding; other analgesic therapy within the previous 6 hours; mild pain |
|
| Interventions |
Aspirin (N randomised was unclear; N = 32 analysed) 600 mg; single oral dose in 2 identical capsules Placebo (N randomised was unclear; N = 31 analysed) Placebo; single oral dose in 2 identical capsules |
|
| Outcomes |
Adequate pain relief as reported by the woman: the same trained nurse observer questioned women hourly for 6 hours; women estimated the severity of pain on a scale of 0 to 3 (0 = no pain; 1 = mild pain; 2 = moderate pain; 3 = severe pain):
Need for additional pain relief in the first 48 hours for perineal pain: request for additional analgesic medication 4 hours after administration of study drugs Maternal adverse effects: close observation was made for any "adverse reactions" |
|
| Notes |
Funding: "The statistical assistance of T. COOK, B. RODDA, and C. DAURIO of the Merck Sharp & Dohme Research Laboratories is gratefully acknowledged" Declarations of interests: not reported; though author affiliations include "Merck Sharp & Dohme Research Laboratories" Additional arms: this 5‐arm trial also assessed diflunisal 125 mg (N = 33 analysed), 250 mg (N = 30 analysed), and 500 mg (N = 30 analysed); we only included the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were allocated at random" |
| Allocation concealment (selection bias) | Unclear risk | Not detailed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "double‐blind"… "All test medications were prepackaged in individual patient‐coded vials containing a single oral dose in two identically appearing capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Efficacy parameters and side‐effects were recorded by the investigator or by the same trained nurse observer, who questioned the patient at hourly intervals" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 5/161 women admitted to the trial were excluded from the analysis: 2 as their initial pain was not considered severe enough to meet protocol; 2 due to incomplete data; 1 due to lack of cooperation (unclear from which groups; leaving 156 in total; 32 in the aspirin group, and 31 in the placebo group). 3 women in the placebo group were withdrawn at 4 hours because of severe pain, and 1 woman in the aspirin group at 3 hours for reasons unrelated to pain or the drug; women who dropped out of the study were included in the analysis; they were assigned a pain score of 4, worse than the scores of all women who remained in the study |
| Selective reporting (reporting bias) | High risk | Very limited outcome data reported; no access to trial registration or protocol to further assess selective reporting. Quote: "As pain relief was still very marked in the 500 mg diflunisal group at 6 hours, it was decided to extend the period of observation to 8 hours in 42 patients, who were approximately evenly distributed between the three groups" |
| Other bias | Unclear risk | Few baseline characteristics reported (initial pain score rating; age); limited methodological data reported |
Friedrich 1983.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Department of Obstetrics and Gynaecology, Washington University, St Louis, Missouri, USA (assumed from author affiliation) Trial dates: not reported (published in 1983) Inclusion criteria: women, suffering moderate or severe pain following episiotomy Exclusion criteria: current or recent history of gastrointestinal bleeding; peptic ulcer; other GI disorders; alcohol or drug abuse; disorders of the nervous system, kidney, heart, or blood; known allergies to aspirin or aspirin‐like analgesics; conditions likely to interfere with absorption, distribution, metabolism, or excretion of drugs; other pain requiring narcotic analgesics; acute dermatitis or other skin lesions; past or present malignancies; taking corticosteroids or other NSAIDs, anticoagulants or other drugs that may interfere with study medication; experiencing pain due to other causes; breastfeeding |
|
| Interventions |
Aspirin (N = 39 randomised) 650 mg aspirin Placebo (N = 40 randomised) All women: women received the study medication at the onset or recurrence of moderate or severe pain, at least 16 hours, but not more than 48 hours following induction of anaesthesia |
|
| Outcomes |
Adequate pain relief as reported by the woman: pain intensity and relief at 0.5 hours, then hourly for 8 hours was measured
Maternal adverse effects: patient complaints were reported |
|
| Notes |
Funding: not reported Declarations of interests: not reported Additional arms: this 4‐arm trial also assessed etodolac 25 mg (N = 40) and 100 mg (N = 40); we only included the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "were randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | Not detailed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as "double‐blind" but no description of whether the study medications were identical in appearance, taste, etc |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not detailed |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No losses or exclusions reported, but %/no of women ‘remaining in study’ at 4, 6, and 8 hours reported |
| Selective reporting (reporting bias) | Unclear risk | No access to trial registration or protocol to further assess selective reporting |
| Other bias | Unclear risk | Results report patients were "well matched" for a variety of baseline characteristics, but no table of these characteristics presented |
Jain 1978a.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Department of Medicine, Tulane University School of Medicine, New Orleans, Louisiana, USA (assumed from author affiliation) Trial dates: not reported (published in 1978) Inclusion criteria: women with moderate to severe episiotomy pain within 48 hours of a normal vaginal birth Exclusion criteria: nursing; systemic diseases; allergic to aspirin |
|
| Interventions |
Aspirin (N = 30 randomised) 648 mg aspirin; single oral dose Placebo (N = 30 randomised) Placebo; single oral dose All women: the time between the test drug and previous analgesic, tranquillisers, or sedatives was at least 5 hours |
|
| Outcomes |
Adequate pain relief as reported by the woman: a trained nurse observer rated pain intensity and relief hourly for 4 hours
The results were not reported in a way to enable us to calculate 'Adequate pain relief as reported by the woman.' Figure 2 in manuscript provides patient self‐rating of pain (continuous, analogue scale) Need for additional pain relief in the first 48 hours for perineal pain: need for extra analgesia during the 4‐hour study period Maternal adverse effects: volunteered or observed side effects |
|
| Notes |
Funding: not reported Declarations of interests: not reported Additional arms: this 4‐arm trial also assessed piroxicam 20 mg (N = 31) and 40 mg (N = 29); we only included the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "By random assignment"; no other details described |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind"; no further details provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No details provided |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Did not report on losses to follow‐up or exclusions |
| Selective reporting (reporting bias) | High risk | No access to trial protocol; however results reported incompletely in text |
| Other bias | Low risk | Baseline characteristics were reported to be comparable across groups; no other obvious sources of bias identified |
Jain 1978b.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Department of Medicine, Tulane University School of Medicine, USA (assumed from author affiliation) Trial dates: not reported (published in 1978) Inclusion criteria: women with severe episiotomy pain or severe uterine cramping pain, following an uncomplicated vaginal birth Exclusion criteria: mild or moderate pain, or baseline pain < 60% on a pain analogue; dependent on analgesics or tranquillisers; hypersensitive to salicylates or caffeine; gastrointestinal, hepatic, or renal history, or a history of psychiatric illness; emotionally unstable or overtly anxious |
|
| Interventions |
Aspirin (N = 16 randomised) 650 mg aspirin; single oral dose Placebo (N = 16 randomised) Placebo; single oral dose All women: duration between previous analgesic and test medication was at least 6 hours |
|
| Outcomes |
Adequate pain relief as reported by the woman: a trained nurse observer rated pain intensity and relief hourly for 4 hours
The results were not reported in such a way to calculate 'Adequate pain relief as reported by the woman' Maternal adverse effects: women questioned about adverse effects at the last interview |
|
| Notes |
Funding: "We wish to thank Mr. Garrett Swenson of American Home Products for the double‐blind supplies of test drug, and Dr. Ilbok lee (Ives Laboratories), Dr. Bruce Schneider (Wyeth Laboratories), and Dr. Syliva Wassertheil‐Smoller (Albert Einstein College of Medicine) for their assistance in the statistical analysis of data" Declarations of interest: not reported Additional arms: manuscript reports results of 2 randomised controlled trials; we have excluded the first, as it combined women with uterine and episiotomy pain, and did not report any results separately for the subset of women with episiotomy pain. This 3‐arm trial also assessed 800 mg aspirin and 64 mg caffeine (N = 15); we have only included the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were divided at random"; no further details provided |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "double blind"; no further details provided |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not detailed |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Complete data was not reported and published |
| Selective reporting (reporting bias) | High risk | No access to trial protocol; limited data presented, results reported incompletely in text |
| Other bias | Unclear risk | No baseline characteristics reported |
Jain 1985.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Tulane University School of Medicine, New Orleans, Louisiana, USA (assumed from affiliation) Trial dates: not reported (published in 1985) Inclusion criteria: postpartum women who had undergone episiotomy and requested analgesic medication for pain of at least moderate intensity, aged ≥ 18 years Exclusion criteria: receipt of analgesics or tranquillisers within 4 hours of stud entry; planned to breast feed; history of convulsive disorders, known peptic ulcer, renal, hepatic or haematological disease; known allergic reactions to salicylates or other NSAIDs |
|
| Interventions |
Aspirin (N = 30 randomised) 600 mg aspirin; single dose of 2 matching capsules Placebo (N = 30 randomised) Placebo; single dose of 2 matching capsules All women: the test drug was given in a single dose in the form of 2 matching capsules. |
|
| Outcomes |
Adequate pain relief as reported by the woman: a trained nurse observed recorded pain intensity and relief at 0.5 hours and hourly to 5 hours
Need for additional pain relief in the first 48 hours for perineal pain: need for supplemental analgesia in 5‐hour study period Maternal adverse effects: adverse effects reported by women or observed by the nurse were recorded |
|
| Notes |
Funding sources: "Supported in part by a grant from Adria Laboratories, inc., Columbus, Ohio" Declarations of interests: not reported Additional arms: this 4‐arm trial also assessed indoprofen 50 (N = 30) and 100 mg (N = 30); we included only the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned"; no further details provided |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "double blind" "the test drug was given in a single dose in the form of two matching capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Not specifically stated; reasonable to assume women and the nurse were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Any patients who experienced inadequate pain relief were permitted to remedicate with an alternate analgesic. In such cases pain evaluations were discontinued and for the balance of the study, patients were assigned a pain intensity score equal to that at the time of remedication and pain relief scores of zero" … There were however, no women who required re‐medication |
| Selective reporting (reporting bias) | Unclear risk | No access to trial registration or protocol to further assess selective reporting |
| Other bias | Low risk | Baseline characteristics comparable between groups; no other obvious sources of bias identified |
London 1983a.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Sinai Hospital of Baltimore, MD, USA (assumed from affiliation) Trial dates: not reported (published in 1982 and 1983) Inclusion criteria: women with no systemic medical illness, experiencing moderate to severe episiotomy pain within 48 hours following an otherwise uncomplicated vaginal birth Exclusion criteria: "those used by Hermann et al" |
|
| Interventions |
Aspirin (N randomised was unclear; N = 40 analysed) 650 mg aspirin; single dose in identical capsules Placebo (N randomised was unclear; N = 40 analysed) Placebo; single dose in identical capsules |
|
| Outcomes |
Adequate pain relief as reported by the woman: one investigator assessed pain intensity and relief hourly for 6 hours;
Need for additional pain relief in the first 48 hours for perineal pain: frequency of re‐medication Maternal adverse effects: women were observed hourly for adverse reactions |
|
| Notes |
Funding: "We would like to acknowledge the support of Sandoz, Inc. in this study" Declarations of interests: not reported Additional arms: this 4‐arm trial also assessed fluproquazone 100 mg (N = 41 analysed) and 200 mg (N = 39 analysed); we included only the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "double‐blind"… "All medication was supplied in identical capsules… packaged in individually sealed envelopes" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Considered reasonable to assume blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | In the whole trial, there were 166 who entered, and 160 provided "valid data for analyses"; other losses/exclusions not clearly reported |
| Selective reporting (reporting bias) | Unclear risk | No access to trial protocol to further assess; scales used to assess pain intensity and relief (needed to use SPID and TOTPAR scores to calculate adequate pain relief) were not reported |
| Other bias | Unclear risk | Baseline characteristics not reported |
London 1983b.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Sinai Hospital, Baltimore, MD, USA (assumed from author affiliation) Trial dates: July to December 1980 Inclusion criteria: postpartum women with moderate to severe episiotomy pain Exclusion criteria: allergy to salicylates; asthma; history of chronic use of analgesics, alcohol, tranquillisers, or other drugs; blood dyscrasia; gastrointestinal disorders; hepatic or renal disease, or both; psychiatric illness |
|
| Interventions |
Aspirin group 1(N randomised not reported; N = 40 analysed) 5 grains (300 mg) aspirin; 4 tablet single dose Aspirin group 2 (N randomised not reported; N = 41 analysed) 10 grains (600 mg) aspirin; 4 tablet single dose Aspirin group 3 (N randomised not reported; N = 40 analysed) 20 grains (1200 mg) aspirin; 4 tablet single dose Placebo (N randomised not reported; N = 39 analysed) Placebo; 4 tablet single dose All women: if women had been medicated previously for pain, the experimental protocol was not initiated for 4 hours |
|
| Outcomes |
Adequate pain relief as reported by the woman: trained research nurse investigator questioned women at 0.5 hours and hourly for 4 hours regarding pain intensity, which was recorded on a 4‐point scale (0 = none; 1 = slight pain; 2 = moderate pain; 3 = severe pain). Pain intensity scores were provided in Table 1 from 0 to 4 hours, and were used to calculate SPID scores and 'Adequate pain relief as reported by the woman' (taken over 4 hours) Need for additional pain relief in the first 48 hours for perineal pain: women re‐medicated for episiotomy pain within 4‐hour study period Maternal adverse effects: side effects observed or reported |
|
| Notes |
Funding: not reported Declarations of interests: not reported Note: we combined the 3 aspirin groups for the main analysis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomly assigned" |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "prospective blind study"… "Each single four‐tablet dose was individually packaged and identified only by study and patient number, and all tablets appeared identical" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | As above; considered reasonable to assume blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | "Because of protocol ineffectiveness, participation in the study was terminated if the patient requested additional analgesic medication, topical analgesics, or Sitz baths. In such cases pain intensity was measured at all intervals up to the time of termination"… 6/121 women in the aspirin groups and 10/39 in the placebo group required re‐medication. No clear reporting of other losses or exclusions |
| Selective reporting (reporting bias) | Unclear risk | No access to trial protocol to further assess risk of selective reporting |
| Other bias | Unclear risk | Baseline characteristics not reported |
Mukherjee 1980.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: LNJP Hospital, New Delhi, India (assumed from author affiliation) Trial dates: not reported (published in 1980) Inclusion criteria: women from an otherwise healthy population whose chief complaint was moderate to severe pain following episiotomy on the first postoperative morning Exclusion criteria: known hypersensitivity to dipyrone and aspirin; receipt of any analgesics 8 hours before entry to the study |
|
| Interventions |
Aspirin (N = 90 randomised) 500 mg aspirin; single oral dose in identical tablet form Placebo(N = 88 randomised) Placebo; single oral dose in identical tablet form All women: nothing was permitted to be taken orally for the first hour after treatment administration |
|
| Outcomes |
Adequate pain relief as reported by the woman:
Maternal adverse effects: adverse drug reactions |
|
| Notes |
Funding: not reported Declarations of interests: not reported Additional arms: this 3‐arm trial also assessed dipyrone 500 mg (N = 89); we only included the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "allocated at random" |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "approved double‐blind approach, which was strictly adhered to" "identical tablet form" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Reasonable to assume blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses or exclusions |
| Selective reporting (reporting bias) | Unclear risk | No access to trial protocol to further assess risk selective reporting |
| Other bias | Low risk | Figures reported for baseline characteristics (such as pain severity, age, weight, and height at baseline), and reported "all three groups were also comparable…" |
Okun 1982.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Cedars‐Sinai Medical Center, Los Angeles, California, USA (assumed from author affiliation) Trial dates: not reported (published in 1982) Inclusion criteria: hospitalised women with moderate, severe, or very severe pain due to uterine cramps or episiotomy within 48 hours of delivery (94 women in total with episiotomy pain) Exclusion criteria: breastfeeding; receipt of any analgesic, sedative, or psychotropic medication within 6 hours before administration of the study drug |
|
| Interventions |
Aspirin(N = 20 randomised) 650 mg aspirin; single oral dose of 2 x 325 mg identical looking capsules Placebo (N = 18 randomised) Placebo; single oral dose of 2 identical looking capsules |
|
| Outcomes |
Adequate pain relief as reported by the woman: 1 nurse observer recorded pain intensity hourly for 8 hours; intensity was rated from 1 to 5 (1 = no pain; 2 = mild pain; 3 = moderate pain; 4 = severe pain; 5 = very severe pain); pain intensity differences were calculated, as were SPID scores at 4, 6, and 8 hours; the time of maximum pain relief; duration of pain relief; the proportion of women with at least 50% pain relief 1 and 2 hours after treatment Need for additional pain relief in the first 48 hours for perineal pain: proportion requiring additional analgesics Adverse effects: adverse effects mentioned by women were recorded No data included in the meta‐analyses, as results were not reported separately for women with post‐episiotomy pain |
|
| Notes |
Funding: not reported Declarations of interests: not reported Additional arms: this 5‐arm trial also assessed fendosal 100 mg (N = 19), 200 mg (N = 19), and 400 mg (N = 18); we only included the relevant arms in this review Note: trial also included women with postpartum uterine cramps pain; not included in review (157/250); we were unable to include any data in the meta‐analyses, as results were not reported separately for women with post‐episiotomy pain |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Assignment to treatment was randomised" |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "double‐blind" and "identical looking capsules" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | As above; 1 nurse observer recorded the pain intensity scores prior to and hourly after administration of medication, reasonable to assume that outcome assessment was blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | A total of 250 eligible women were "admitted to the study"… "The PI scores from patients taking another analgesic were handled as treatment failures by substituting the initial PI score for all hours after the analgesic was taken" |
| Selective reporting (reporting bias) | Unclear risk | Results (such as SPID scores) were reported incompletely in text |
| Other bias | Low risk | Baseline characteristics (such as initial intensity and type of pain; age; and weight) were comparable between groups |
Olson 1997.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Hospital Maternidad Concepcion Palacios, Caracas, Venezuela Trial dates: not reported (published in 1997) Inclusion criteria: women of legal age (aged ≥ 18 years), who were able to communicate meaningfully with the nurse‐observer, who were hospitalised and had severe post‐episiotomy pain after an uncomplicated birth and could tolerate oral medications Exclusion criteria: planning to breast feed within 24 hours after administration of the study medications; serious complicating illness or abnormal postpartum bleeding, with active peptic ulcer disease or other gastrointestinal disease associated with blood loss; receipt of any other investigational drug within the 1 month prior; history of drug or alcohol abuse; known allergic sensitivities to aspirin, diclofenac, or other NSAIDs |
|
| Interventions |
Aspirin (N = 50 randomised) 650 mg aspirin; single oral dose of 2 x 325 mg capsules; and 3 placebo tablets Placebo (N = 52 randomised) Placebo; single oral dose of 2 placebo capsule and 3 placebo tablets All women: each woman received a single unit dose consisting of 2 capsules and 3 tablets, with at least 8 ounces of water; women were asked to sit up or lie on their right side for 2 hours after administration. No medications (analgesics, sedatives, hypnotics, tranquillisers) were permitted concomitantly or during the 4 hours prior to taking the medication |
|
| Outcomes |
Adequate pain relief as reported by the woman: the same nurse observer interviewed the women at 0.5 hours, and hourly for 8 hours
Need for additional pain relief in the first 48 hours for perineal pain: re‐medication within 8‐hour study period Maternal adverse effects: adverse effects were recorded if they were observed or volunteered |
|
| Notes |
Funding: "This work was supported in part by a grant from the Ciba‐Geigy Corporation, Summit, NJ" Declarations of interests: not reported; though first and second authors affiliated with "Analgesic Development Ltd." Additional arms: this 5‐arm trial also assessed diclofenac potassium 25 mg (N = 52), 50 mg (N = 50), and 100 mg (N = 51); we only included the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "randomly assigned… computer program generated a random permutation such that two patients received each treatment" |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "double‐blind" and "Each patient received a single‐unit dose consisting of 3 tablets and 2 capsules… all unit doses were identical in appearance and packaging" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | 1 nurse observer involved in outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | If a woman wished to withdraw before the first hour because of inadequate relief, a non‐study analgesic was administered, and she was discontinued from the study (none were re‐medicated in this first hour; therefore, no discontinuations); if a woman required additional analgesic after the first hour, she was included, and relief scores of 0, and intensity scores equal to the pain at time of re‐medication were assumed for the duration (5/50 in the aspirin group; 19/52 in the placebo group) |
| Selective reporting (reporting bias) | Unclear risk | No access to trial protocol to further assess risk of selective reporting |
| Other bias | Low risk | Baseline characteristics reported (age, weight, height, parity, days post‐delivery) were comparable between groups; no other obvious sources of bias identified |
Sunshine 1983a.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Hospital Maternidad Concepcion Palacios, Caracas, Venezuela Trial dates: not reported (published in 1983) Inclusion criteria: women with severe post‐episiotomy pain after an uncomplicated birth; aged ≥18 years; who could tolerate oral medications Exclusion criteria: known allergic sensitivities to study medication; abnormal postpartum bleeding, or complicating illnesses; breastfeeding; history of drug dependence; receipt of other investigational drugs prior to enrolment |
|
| Interventions |
Aspirin (N = 30 randomised) 600 mg aspirin; single oral dose of 1 aspirin capsule and 1 placebo tablet Placebo(N = 30 randomised) Placebo; single oral dose of 1 capsule and 1 tablet All women: as a single dose; women were given the study medication by the nurse observer when their pain was severe; no medications that might alter the response to the study analgesics were permitted concomitantly, or during the 4 hours before the test medication was taken |
|
| Outcomes |
Adequate pain relief as reported by the woman: the same nurse observer interviewed at the time of medication, 0.5 hours, and hourly for 4 hours
Need for additional pain relief in the first 48 hours for perineal pain: re‐medication within 4 hours Maternal adverse effects: adverse reactions were noted if observed or volunteered |
|
| Notes |
Funding: "A grant‐in‐aid and test medication from Upjohn Company made this research possible" Declarations of interests: not reported Additional arms: this 4‐arm trial also assessed zomepirac 100 mg (N = 30) and ibuprofen 400 mg (N = 30); we only included the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "In a randomised study" |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "in a double‐blind fashion. Because the study medications were not identical in appearance, a double‐dummy technique was used"; each woman received one tablet and one capsule as appropriate |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The same nurse observer interviewed the patients |
| Incomplete outcome data (attrition bias) All outcomes | High risk | If before the first hour a woman reported inadequate pain relief, a conventional analgesic was given and she was removed from the study; if a woman requested ‘rescue’ medication after the first hour, she was given it and was included in the evaluation; responses at the time of re‐medication were assumed for the duration of the study; all 120 women who participated in the study were included in the analysis; 5 women who received placebo required rescue medication during the study; no women receiving aspirin required re‐medication |
| Selective reporting (reporting bias) | High risk | Some incomplete reporting "The three drugs were much the same for mean onset, duration, and time to peak values. The hypothesis that there is no difference among treatments was rejected at the 0.05 level or better for all variables" patients rating of overall improvement and of study medication mentioned in methods and not reported |
| Other bias | Low risk | Baseline characteristics presented were comparable between groups; no other obvious sources of bias identified |
Sunshine 1983b.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: Hospital Maternidad Concepcion Palacios, Caracas, Venezuela Trial dates: not reported (published in 1983) Inclusion criteria: women with moderate or severe post‐episiotomy pain after an uncomplicated delivery, who could tolerate oral medication, aged ≥ 18 years Exclusion criteria: breastfeeding; any complicating illness or abnormal postpartum bleeding; receipt of any other investigational drug within 1 month prior to enrolment; history of drug dependence or known allergic sensitivities to prolonic acid derivatives or aspirin |
|
| Interventions |
Aspirin (N = 29 randomised) 600 mg aspirin; single oral dose of 5 identical tablets Placebo (N = 31 randomised) Placebo; single oral dose of 5 identical tablets All women: no medications that might confound the interpretation of the efficacy or adverse effect liability (or both) of the study analgesics were permitted concomitantly or during the 4 hours before taking the study medication |
|
| Outcomes |
Adequate pain relief as reported by the woman: the same nurse observer interviewed the women at the time medication was administered and hourly for 6 hours:
Need for additional pain relief in the first 48 hours for perineal pain: re‐medication within 6‐hour study period Maternal adverse effects: adverse reactions were noted if they were observed or volunteered |
|
| Notes |
Funding: "Supported by a grant from Boots Pharmaceuticals, Inc" Declarations of interests: not reported Additional arms: this 5‐arm trial also assessed flurbiprofen 25 mg (N = 32), 50 mg (N = 29), 100 mg (N = 31); we only included the relevant arms in this review |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "In each successive block of ten patients a computer program generated a random permutation such that, two patient received each treatment" |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind… Each patient was given one dose of five tablets that were identical in appearance and packaging" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Same nurse interviewer interviewed patients at administration and hourly afterwards; reasonable to assume blinding of outcome assessment |
| Incomplete outcome data (attrition bias) All outcomes | High risk | If women reported inadequate pain relief before the first hour, a conventional analgesic agent was given and they were removed from the study; if women requested rescue medication after the first hour, they were given the conventional analgesic and included in the analyses – baseline pain intensity and zero pain relief were assumed for the duration of scheduled observations; 168 women were enrolled in the study; 16 were "dropped from the analysis because they received concomitant oxytoxic medication that the sponsor felt might confound the interpretation of the efficacy of the study drug" 1/32 in the placebo group; 4/33 in the aspirin group; no women re‐medicated in the first hour; 14 re‐medicated in placebo group; 1 re‐medicated in the aspirin group |
| Selective reporting (reporting bias) | Unclear risk | No access to trial protocol to further assess selective reporting |
| Other bias | Low risk | Baseline characteristics reported were balanced between groups; no other obvious risk of bias identified |
Trop 1983.
| Study characteristics | ||
| Methods | RCT | |
| Participants |
Setting: an obstetric and gynecology unit; Montreal, Quebec, Canada (assumed from author affiliation) Trial dates: not reported (published in 1983) Inclusion criteria: women who had an episiotomy Exclusion criteria: receipt of tranquillisers, sedatives, hypnotics, or other analgesics during the 4 hours preceding the study; breastfeeding |
|
| Interventions |
Aspirin (N = not reported) 1200 mg aspirin; single dose of 4 x 300 mg tablets Aspirin (N = not reported) 600 mg aspirin; single dose of 2 x 300 mg tablets and 2 placebo tablets Placebo (N = not reported) Placebo; single dose of 4 placebo tablets All women: the medication was administered 10.5 to 14.4 hours after episiotomy, upon request by the woman, or when pain was judged moderate to severe by the nurse |
|
| Outcomes |
Adequate pain relief as reported by the woman: severity of pain was judged using a 30‐cm visual analogue scale (no pain, slight, moderate, severe, unbearable); the women registered the intensity of pain by putting a stroke on the place on the scale before drug administration and every hour for 4 hours (women were not allowed to see the result of their previous assessment); the research nurse independently recorded her own evaluation of the analgesic effect of the medication (scale of 0 to 4: no pain to worse than before) Need for additional pain relief in the first 48 hours for perineal pain: additional analgesic during 4‐hour period Maternal adverse effects: side effects reported by the women were noted on case report forms |
|
| Notes |
Funding: "This study was supported by a grant from Roussel Canada Inc., Montreal, Quebec, Canada" Declarations of interests: not reported; though last author affiliated to "Roussel Canada Inc." Additional arms: this 5‐arm trial also assessed tiaprofenic acid 200 mg (N = not reported) and 400 mg (N = not reported); we only included relevant arms in this review Note: no data could be included in the meta‐analyses as numbers for each group were not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "randomised" |
| Allocation concealment (selection bias) | Unclear risk | No details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quotes: "double‐blind"… "The drugs were prepared as tablets… all were of identical appearance" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quotes: "double‐blind"… "The drugs were prepared as tablets… all were of identical appearance" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not clearly reported (nor were the numbers of women in each group) |
| Selective reporting (reporting bias) | High risk | Results incompletely reported within text (including nurse’s evaluation of analgesic effect); numbers in each group also not reported |
| Other bias | Unclear risk | Baseline characteristics incompletely reported in text "There were no significant differences among the 5 groups with respect to age, height, weight or vital signs" |
GI: gastro‐intestinal NSAIDs: non‐steroidal anti‐inflammatory drugs RCT: randomised controlled trial SPID: summed pain intensity differences TOTPAR: total pain relief
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bruni 1965 | RCT. Included women with 'postpartum pain', not exclusively women with perineal pain; results not reported separately for women with perineal pain |
| Gindhart 1971 | RCT. Did not assess aspirin for perineal pain, but rather assessed 2 agents: 1 (Darvon), containing propoxyphene with aspirin, phenacetin, and caffeine; and 1 (Fiorinal) combined aspirin, phenacetin, and caffeine with a mild sedative, butalbital |
| Gruber 1979 | RCT. Included women with 'postpartum pain', not exclusively women with perineal pain; results not reported separately for women with perineal pain |
| Moggian 1972 | RCT. Included women with 'postpartum pain', not exclusively women with perineal pain; results not reported separately for women with perineal pain |
| Prockop 1960 | RCT. Did not assess aspirin for perineal pain, but rather assessed an aspirin compound (acetophenetidin acetylsalicylic acid and caffeine); codeine and an aspirin compound; dextropropoxyphene; dextropropoxyphene and an aspirin compound; and a starch placebo |
| Rubin 1984 | RCT. Did not assess aspirin for perineal pain, but rather assessed an aspirin and caffeine combination; an acetaminophen and aspirin combination; acetaminophen alone; and placebo |
| Santiago 1959 | Not RCT: "In consecutive cases of vaginal delivery with episiotomy, orders in alternating patients for analgesic 1 or analgesic 2... were written". This study assessed Darvon Compound (dextro propoxyphene and acetylsalicylic acid compound); and a preparation containing acetylsalicylic acid, acetophenetidin, caffeine and codeine phosphate. Aspirin not assessed |
| Sunshine 1983c | RCT. Included women with 'postpartum pain', not exclusively women with perineal pain; results not reported separately for women with perineal pain |
| Sunshine 1985 | RCT. Included women with 'postpartum pain', not exclusively women with perineal pain; results not reported separately for women with perineal pain |
| Van der Pas 1984 | Did not assess single dose aspirin, but rather assessed twice daily acetylsalicylic acid and naproxen; it was not clear whether this was a randomised controlled trial |
RCT: randomised controlled trial
Characteristics of studies awaiting classification [ordered by study ID]
Bhounsule 1990.
| Methods | Unclear; reported to be "a concurrent comparison under double‐blind conditions" |
| Participants | 100 women with post‐episiotomy pain |
| Interventions | Single oral doses of 400 mg ibuprofen, 500 mg analgin, 500 mg paracetamol, 600 mg aspirin, and placebo |
| Outcomes |
Adequate pain relief as reported by the woman: one investigator assessed pain intensity and relief hourly for 6 hours; women were interviewed before dosing, and 7 times thereafter; at each observation women reported:
Maternal adverse effects: women were asked about 'dizziness, headache, nausea, burning pain in abdomen, sleepiness, or sweating, or any other effect she would like to record' |
| Notes | Awaiting classification, pending further details regarding allocation |
Sunshine 1989.
| Methods | Unclear; reported to be "A double‐blind single dose study" |
| Participants | 300 women with severe post‐episiotomy pain; abstract reports on 199 women who received the below interventions |
| Interventions | Single doses of 200 mg ibuprofen, 400 mg ibuprofen, 500 mg aspirin, and placebo |
| Outcomes |
Adequate pain relief as reported by the woman: reports that "Analgesia was measured on the basis of standard derived variables… SPID, TOTAL, and some hourly measures" Maternal adverse effects: reports "serious adverse effects" |
| Notes | Abstract only; awaiting classification, pending further details regarding allocation |
SPID: summed pain intensity differences TOTPAR: total pain relief
Differences between protocol and review
There are some differences between our published protocol (Molakatalla 2016), and this full review.
Methods, data collection, and analysis − assessment of pain − we clarified that our measure was 50% or greater pain relief (our protocol stated 50%). We also clarified equations used for measures of pain in the review.
Contributions of authors
For this update, Emily Shepherd and Rosalie Grivell re‐assessed the studies awaiting classification. Emily Shepherd drafted the update, with input from Rosalie Grivell.
For the previous version of this review, Sujana Molakatalla and Emily Shepherd assessed studies for inclusion and exclusion; carried out data extraction, and assessed the risk of bias of the included trials. Emily Shepherd entered data into RevMan 5 and performed the analyses. Sujana Molakatalla drafted the review with input from both Emily Shepherd (editorial) and Rosalie Grivell (editorial and clinical).
Sources of support
Internal sources
South Australian Health and Medical Research Institute (SAHMRI), Women and Kids, Australia
Department of Obstetrics and Gynaecology, Flinders University and Flinders Medical Centre, Australia
External sources
No sources of support supplied
Declarations of interest
Emily Shepherd: none known
Rosalie M Grivell: none known
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Bloomfield 1967 {published data only}
- Bloomfield SS, Gaffney TE, Howett M. Comparative analgesic efficacy of chlorphenesin carbamate and acetylsalicylic acid after episiotomy. Anesthesia and Analgesia 1967;46(5):515-20. [PubMed] [Google Scholar]
Bloomfield 1970a {published data only}
- Bloomfield SS, Barden TP, Hille R. Clinical evaluation of flufenisal, a long-acting analgesic. Clinical Pharmacology and Therapeutics 1970;11(5):747-54. [DOI] [PubMed] [Google Scholar]
- Bloomfield SS, Barden TP, Mitchell J. Aspirin and codeine in two postpartum pain models. Clinical Pharmacology and Therapeutics 1976;20(4):499-503. [DOI] [PubMed] [Google Scholar]
Bloomfield 1970b {published data only}
- Bloomfield SS, Hurwitz HN. Tourniquet and episiotomy pain as test models for aspirin-like analgesics. Journal of Clinical Pharmacology 1970;10(6):361-9. [PubMed] [Google Scholar]
Bloomfield 1974 {published data only}
- Bloomfield SS, Barden TP, Mitchell J. Comparative efficacy of ibuprofen and aspirin in episiotomy pain. Clinical Pharmacology and Therapeutics 1974;15(6):565-70. [DOI] [PubMed] [Google Scholar]
Devroey 1978 {published data only}
- Devroey P, Steelman SL, Caudron J. A double blind, placebo controlled, single dose study comparing three dose levels of diflunisal with aspirin and placebo in patients with pain due to episiotomy. Acta Therapeutica 1977;3(3):205-16. [Google Scholar]
- Devroey P. A double-blind comparison of diflunisal and aspirin in the treatment of post-operative pain after episiotomy. Current Medical Research and Opinion 1978;5(7):544-7. [DOI] [PubMed] [Google Scholar]
- Devroey P. The treatment of postoperative pain with a single dose of diflunisal. Clinical Therapeutics 1977;1(Suppl A):30-3. [Google Scholar]
Friedrich 1983 {published data only}
- Friedrich E. A comparison of etodolac (Ultradol) with aspirin and placebo in patients with episiotomy pain. Current Therapeutic Research, Clinical and Experimental 1983;33(1):100-7. [Google Scholar]
Jain 1978a {published data only}
- Jain AK, McMahon FG, Ryan JR, Raphan H, Richard W. Piroxicam, a novel analgesic in postpartum pain. European Journal of Rheumatology and Inflammation 1978;1(3):356-9. [Google Scholar]
Jain 1978b {published data only}
- Jain AK, McMahon FG, Ryan JR, Unger D, Richard W. A comparison of aspirin-caffeine versus aspirin: results of two double-blind placebo controlled studies in postpartum pain. Clinical Pharmacology and Therapeutics 1978;23(1):116. [DOI] [PubMed] [Google Scholar]
- Jain AK, McMahon FG, Ryan JR, Unger D, Richard W. Aspirin and aspirin-caffeine in postpartum pain relief. Clinical Pharmacology and Therapeutics 1978;24(1):69-75. [DOI] [PubMed] [Google Scholar]
Jain 1985 {published data only}
- Jain AK, McMahon FG, Ryan JR, Smith GB. Analgesic efficacy of indoprofen in postpartum episiotomy pain. Current Therapeutic Research, Clinical and Experimental 1985;38(5):677-81. [Google Scholar]
London 1983a {published data only}
- London R, Sundaram GS, Feldman S, Goldstein P. Evaluation of analgesic efficacy and safety of fluproquazone compared to aspirin in the treatment of episiotomy pain. Federation Proceedings 1982;41(4):No. 5573. [Google Scholar]
- London R, Sundaram GS, Feldman S, Goldstein PJ. Episiotomy pain: efficacy and safety of fluproquazone compared to aspirin and placebo. International Journal of Gynaecology and Obstetrics 1983;21(3):251-5. [DOI] [PubMed] [Google Scholar]
London 1983b {published data only}
- London RS, Sundaram GS, Feldman S, Goldstein PJ. Aspirin in the treatment of episiotomy pain. Southern Medical Journal 1983;76(7):844-5. [DOI] [PubMed] [Google Scholar]
Mukherjee 1980 {published data only}
- Mukherjee S, Sood S. A controlled evaluation of orally administered aspirin dipyrone and placebo in patients with post-operative pain. Current Medical Research and Opinion 1980;6(9):619-23. [DOI] [PubMed] [Google Scholar]
Okun 1982 {published data only}
- Okun R. Evaluation of the analgesic effect of fendosal in patients with postpartum uterine cramp or episiotomy pain. Current Therapeutic Research, Clinical and Experimental 1982;32(1):65-73. [Google Scholar]
Olson 1997 {published data only}
- Olson NZ, Sunshine A, Zighelboim I, DeCastro A. Onset and duration of analgesia of diclofenac potassium in the treatment of postepisiotomy pain. American Journal of Therapeutics 1997;4(7-8):239-46. [DOI] [PubMed] [Google Scholar]
Sunshine 1983a {published data only}
- Sunshine A, Olson NZ, Laska EM, Zighelboim I, De Castro A, De Sarrazin C. Analgesic effect of graded doses of flurbiprofen in post-episiotomy pain. Pharmacotherapy 1983;3(3):177-81. [DOI] [PubMed] [Google Scholar]
Sunshine 1983b {published data only}
- Sunshine A, Olson NZ, Laska EM, Zighelboim I, De Castro A, De Sarrazin C. Ibuprofen, zomepirac, aspirin, and placebo in the relief of postepisiotomy pain. Clinical Pharmacology and Therapeutics 1983;34(2):254-8. [DOI] [PubMed] [Google Scholar]
Trop 1983 {published data only}
- Trop D, Nucci C, Elie R, Gareau J. Double-blind comparative evaluation of tiaprofenic acid (Surgam) versus acetylsalicylic acid (ASA) in relieving pain following episiotomy. Current Therapeutic Research, Clinical and Experimental 1983;34(2I):274-9. [Google Scholar]
References to studies excluded from this review
Bruni 1965 {published data only}
- Bruni JR, Holt RE. Controlled double-blind evaluation of three analgesic medications for postpartum discomfort. Obstetrics and Gynecology 1965;25:76-81. [PubMed] [Google Scholar]
Gindhart 1971 {published data only}
- Gindhart JD. A rationale for studying analgesia. A double blind study in postpartum patients. Current Therapeutic Research, Clinical and Experimental 1971;13(4):240-50. [PubMed] [Google Scholar]
Gruber 1979 {published data only}
- Gruber CM, Bauer RO, Bettigole JB, Lash AF, McDonald JS. A multicenter study for analgesia involving fenoprofen, propoxyphene (alone or in combination) with placebo and aspirin controls in postpartum pain. Journal of Medicine 1979;10(1-2):65-98. [PubMed] [Google Scholar]
Moggian 1972 {published data only}
- Moggian G, Cervellati I, Tamburini E. Critical study of the post-partum pain by means of a clinical pharmacology experiment. Bollettino Chimico Farmaceutico 1972;111(8):497-9. [PubMed] [Google Scholar]
Prockop 1960 {published data only}
- Prockop LD, Eckenhoff JE, McElroy RC. Evaluation of dextropropoxyphene, codeine and acetylsalicylic compound. Obstetrics and Gynecology 1960;16:113-8. [PubMed] [Google Scholar]
Rubin 1984 {published data only}
- Rubin A, Winter LJ. A double-blind randomized study of an aspirin/caffeine combination versus acetaminophen/aspirin combination versus acetaminophen versus placebo in patients with moderate to severe post-partum pain. Journal of International Medical Research 1984;12(6):338-45. [DOI] [PubMed] [Google Scholar]
Santiago 1959 {published data only}
- Santiago FS, Danforth DN. Non-narcotic analgesia to simplify postpartum care. Obstetrics and Gynecology 1959;14(1):22-3. [PubMed] [Google Scholar]
Sunshine 1983c {published data only}
- Sunshine A, Olson JZ, Siegel C, Laska EM. Oral analgesic study of ketoprofen, aspirin and placebo in post-partum pain. Clinical Pharmacology and Therapeutics 1983;33:154. [Google Scholar]
- Sunshine A, Zighelboim I, Laska E, Siegel C, Olson NZ, De Castro A. A double-blind, parallel comparison of ketoprofen, aspirin, and placebo in patients with postpartum pain. Journal of Clinical Pharmacology 1986;26(8):706-11. [DOI] [PubMed] [Google Scholar]
Sunshine 1985 {published data only}
- Sunshine A, Zighelboim I, Olson NZ, De Sarrazin C, Laska E. A comparative oral analgesic study of indoprofen, aspirin, and placebo in postpartum pain. Journal of Clinical Pharmacology 1985;25(5):374-80. [DOI] [PubMed] [Google Scholar]
Van der Pas 1984 {published data only}
- Van der Pas HFM, Vertommen F. A double-blind study of naproxen-suppositories for post episitomial pain compared with asa, oxyfenbutazon and placebo. Ars Medici Revue Internationale de Therapie Pratique 1984;39(5):79-81. [Google Scholar]
References to studies awaiting assessment
Bhounsule 1990 {published data only}
- Bhounsule SA, Nevreker PR, Agshikar NV, Pal MN, Dhume VG. A comparison of four analgesics in post-episiotomy pain. Indian Journal of Physiology and Pharmacology 1990;34(1):34-8. [PubMed] [Google Scholar]
Sunshine 1989 {published data only}
- Sunshine A, Zighelboim I, Hoburg A, Byrd W, Olson NZ, Laska E, et al. Ibuprofen, aspirin and placebo in postpartum pain. International Journal of Clinical Pharmacology and Therapeutics 1989;45(2):174. [Google Scholar]
Additional references
Aasheim 2017
- Aasheim V, Nilsen ABV, Reinar LM, Lukasse M, . Perineal techniques during the second stage of labour for reducing perineal trauma. Cochrane Database of Systematic Reviews 2017, Issue 6. Art. No: CD006672. [DOI: 10.1002/14651858.CD006672.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
AIHW 2019
- Australian Institute of Health and Welfare (AIHW). Australia’s mothers and babies 2017—in brief. Perinatal statistics series no. 35. Cat. no. PER 100. Canberra: AIHW 2019.
ATT Collaboration 2002
- Antithrombotic Trialists' Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002;324(7329):71-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
ATT Collaboration 2009
- Antithrombotic Trialists' (ATT) Collaboration. Aspirin in the primary and secondary prevention of vascular disease: collaborative meta-analysis of individual participant data from randomised trials. Lancet 2009;373(9678):1849-60. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Beckmann 2013
- Beckmann MM, Stock OM. Antenatal perineal massage for reducing perineal trauma. Cochrane Database of Systematic Reviews 2013, Issue 4. Art. No: CD005123. [DOI: 10.1002/14651858.CD005123.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bell 2011
- Bell AD, Roussin A, Cartier R, Chan WS, Douketis JD, Gupta A, et al. The use of antiplatelet therapy in the outpatient setting: Canadian Cardiovascular Society Guidelines Executive Summary. Canadian Journal of Cardiology 2011;27(2):208-21. [DOI] [PubMed] [Google Scholar]
Chou 2009
- Chou D, Abalos E, Gyte Gillian ML, Gülmezoglu AM. Drugs for perineal pain in the early postpartum period: generic protocol. Cochrane Database of Systematic Reviews 2009, Issue 3. Art. No: CD007734. [DOI: 10.1002/14651858.CD007734.pub2] [DOI] [Google Scholar]
Chou 2013
- Chou D, Abalos E, Gyte GM, Gulmezoglu AM. Paracetamol/acetaminophen (single administration) for perineal pain in the early postpartum period. Cochrane Database of Systematic Reviews 2013, Issue 1. Art. No: CD008407. [DOI: 10.1002/14651858.CD008407.pub2] [DOI] [PubMed] [Google Scholar]
Cooper 1991
- Cooper SA. Commentary: single-dose analgesic studies: the upside and downside of assay sensitivity. In: Max M, Portenov R, Laska E, editors(s). Advances in Pain Research and Therapy. Vol. 18. New York: Raven Press, 1991:117-24. [Google Scholar]
Cunningham 2005
- Cunningham G. Maternal anatomy. In: Cunningham G, Leveno K, Bloom S, Hauth J, Gilstrap L, Wenstrom K, editors(s). Williams Obstetrics. 22nd edition. New York: McGraw-Hill, 2005:21. [Google Scholar]
Derry 2012
- Derry S, Moore RA. Single dose oral aspirin for acute postoperative pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 4. Art. No: CD002067. [DOI: 10.1002/14651858.CD002067.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Duley 2019
- Duley L, Meher S, Hunter KE, Seidler AL, Askie LM. Antiplatelet agents for preventing pre-eclampsia and its complications. Cochrane Database of Systematic Reviews 2019, Issue 10. Art. No: CD004659. [DOI: 10.1002/14651858.CD004659.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
East 2012a
- East CE, Sherburn M, Nagle C, Said J, Forster D. Perineal pain following childbirth: prevalence, effects on postnatal recovery and analgesia usage. Midwifery 2012;28(1):93-7. [DOI] [PubMed] [Google Scholar]
East 2012b
- East CE, Begg L, Henshall NE, Marchant PR, Wallace K. Local cooling for relieving pain from perineal trauma sustained during childbirth. Cochrane Database of Systematic Reviews 2012, Issue 5. Art. No: CD006304. [DOI: 10.1002/14651858.CD006304.pub3] [DOI] [PubMed] [Google Scholar]
GRADE Handbook
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s). Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach (updated October 2013). GRADE Working Group, 2013. Available from gdt.guidelinedevelopment.org/app/handbook/handbook.html.
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Hamilton (ON): McMaster University (developed by Evidence Prime), accessed. Available at gradepro.org.
Hay‐Smith 1998
- Hay-Smith J. Therapeutic ultrasound for postpartum perineal pain and dyspareunia. Cochrane Database of Systematic Reviews 1998, Issue 3. Art. No: CD000495. [DOI: 10.1002/14651858.CD000495] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hedayati 2003
- Hedayati H, Parsons J, Crowther CA. Rectal analgesia for pain from perineal trauma following childbirth. Cochrane Database of Systematic Reviews 2003, Issue 3. Art. No: CD003931. [DOI: 10.1002/14651858.CD003931] [DOI] [PubMed] [Google Scholar]
Hedayati 2005
- Hedayati H, Parsons J, Crowther CA. Topically applied anaesthetics for treating perineal pain after childbirth. Cochrane Database of Systematic Reviews 2005, Issue 2. Art. No: CD004223. [DOI: 10.1002/14651858.CD004223.pub2] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Kettle 2012
- Kettle C, Dowswell T, Ismail KMK. Continuous and interrupted suturing techniques for repair of episiotomy or second-degree tears. Cochrane Database of Systematic Reviews 2012, Issue 11. Art. No: CD000947. [DOI: 10.1002/14651858.CD000947.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Maclagan 1879
- Maclagan TJ. The treatment of acute rheumatism by salicin and salicylic acid. Lancet 1879;113(2912):875-7. [Google Scholar]
Moore 1996
- Moore A, McQuay H, Gavaghan D. Deriving dichotomous outcome measures from continuous data in randomised controlled trials of analgesics. Pain 1996;66(2-3):229-37. [DOI] [PubMed] [Google Scholar]
Moore 1997a
- Moore A, Moore O, McQuary H, Gavaghan D. Deriving dichotomous outcome measures from continuous data in randomised controlled trials of analgesia: use of pain intensity and visual analogue scales. Pain 1997;69(3):311-5. [DOI] [PubMed] [Google Scholar]
Moore 1997b
- Moore A, McQuay H, Gavaghan D. Deriving dichotomous outcome measures from continuous data in randomised controlled trials of analgesia: verification from independent data. Pain 1997;69(1-2):127-30. [DOI] [PubMed] [Google Scholar]
NICE 2015
- National Institute for Health and Care Excellence (NICE). Postnatal care up to 8 weeks after birth, NICE Clinical Guideline CG37. nice.org.uk/guidance/cg37 (accessed 20 August 2016).
NIH 2015
- US National Library of Medicine, National Institutes of Health, Health and Human Services. LacMed, a ToxNet database. www.toxnet.nlm.nih.gov (accessed 20 June 2016).
Reece‐Stremtan 2017
- Reece-Stremtan S, Campos M, Kokajko L, Academy of Breastfeeding Medicine. ABM clinical protocol #15: analgesia and anesthesia for the breastfeeding mother, Revised 2017. Breastfeeding Medicine 2017;12(9):500-6. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rothwell 2010
- Rothwell PM, Wilson M, Elwin CE, Norrving B, Algra A, Warlow CP, et al. Long-term effect of aspirin on colorectal cancer incidence and mortality: 20-year follow-up of five randomised trials. Lancet 2010;376(9754):1741-50. [DOI] [PubMed] [Google Scholar]
Smith 2014
- Smith T, Elwood P, Keating C, Rothwell P, Detering E, Freedman A, et al. The Aspirin Foundation Scientific Conference: the history, the present state and the future of aspirin prophylaxis. Ecancermedicalscience 2014;8:388. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vane 2003
- Vane JR, Botting RM. The mechanism of action of aspirin. Thrombosis Research 2003;110(5-6):255-8. [DOI] [PubMed] [Google Scholar]
WHO 2001
- World Health Organization. The optimal duration of exclusive breastfeeding. WHO report of an expert consultation. www.who.int/nutrition/publications/infantfeeding/WHO_NHD_01.09/en/ (accessed 17 December 2019).
WHO 2003
- World Health Organization, UNICEF. Global Strategy for Infant and Young Child Feeding. www.who.int/nutrition/publications/infantfeeding/9241562218/en/ (accessed 17 December 2019).
WHO 2011
- World Health Organization. WHO recommendations for prevention and treatment of pre-eclampsia and eclampsia. www.who.int/reproductivehealth/publications/maternal_perinatal_health/9789241548335/en/ (accessed 17 December 2019). [PubMed]
WHO 2018
- WHO Reproductive Health Library. WHO recommendation on techniques for preventing perineal trauma in second stage of labour. https://extranet.who.int/rhl (accessed 17 December 2019).
Wuytack 2016
- Wuytack F, Smith V, Cleary BJ. Oral non-steroidal anti-inflammatory drugs (single dose) for perineal pain in the early postpartum period. Cochrane Database of Systematic Reviews 2016, Issue 7. Art. No: CD011352. [DOI: 10.1002/14651858.CD011352.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Molakatalla 2016
- Molakatalla S, Shepherd E, Grivell RM. Aspirin (single dose) for perineal pain in early postpartum period. Cochrane Database of Systematic Reviews 2016, Issue 3. Art. No: CD012129. [DOI: 10.1002/14651858.CD012129] [DOI] [PMC free article] [PubMed] [Google Scholar]
Molakatalla 2017
- Molakatalla S, Shepherd E, Grivell RM. Aspirin (single dose) for perineal pain in the early postpartum period. Cochrane Database of Systematic Reviews 2017, Issue 2. Art. No: CD012129. [DOI: 10.1002/14651858.CD012129.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]