

Abstract
目的
探讨早发型腓骨肌萎缩症(CMT)的临床特征及遗传变异分析。
方法
以临床诊断为早发型CMT的患儿为研究对象,收集相关临床资料,进行肌电图及CMT相关基因检测并分析。
结果
早发型CMT病例共13例,男9例(69%),女4例(31%),平均就诊年龄4.0±2.1岁,其中12例(92%)患儿起病年龄 < 2岁。9例(69%)诊断为CMT1型(其中Dejerine-Sottas综合征6人),1例(8%)为中间型,3例(23%)为CMT2型。13例患儿的基因检测结果显示6例(46%)患儿存在外周髓鞘蛋白22(PMP22)基因重复突变、3例(23%)髓鞘蛋白零(MPZ)基因插入突变及点突变、3例(23%)线粒体融合蛋白2(MFN2)基因点突变、1例(8%)人轻肽神经丝蛋白(NEFL)基因点突变,其中11例(85%)为已知致病突变,2例(15%)为新变异。MPZ基因新变异c.394C > G(p.P132A)评级为"可能致病的"及MFN2基因新变异c.326A > G(p.K109R)评级为"致病的"。
结论
早发型CMT以PMP22基因重复突变及MPZ基因突变为主,临床分型以CMT1型为主,其中Dejerine-Sottas综合征占有相当比例。
Keywords: 腓骨肌萎缩症, 外周髓鞘蛋白22基因, 髓鞘蛋白零基因, 线粒体融合蛋白2基因, 人轻肽神经丝蛋白基因, 高通量测序, 儿童
Abstract
Objective
To study the clinical characteristics and genetic variation of early-onset Charcot-Marie-Tooth disease (CMT).
Methods
Children with a clinical diagnosis of early-onset CMT were selected for the study. Relevant clinical data were collected, and electromyogram and CMT-related gene detection were performed and analyzed.
Results
A total of 13 cases of early-onset CMT were enrolled, including 9 males (69%) and 4 females (31%). The mean age at consultation was 4.0±2.1 years. Among them, 12 children (92%) had an age of onset less than 2 years, 9 children (69%) were diagnosed with CMT type 1 (including 6 cases of Dejerine-Sottas syndrome), 1 child (8%) with intermediate form of CMT, and 3 children (23%) with CMT type 2. The genetic test results of these 13 children showed 6 cases (46%) of PMP22 duplication mutation, 3 cases (23%) of MPZ gene insertion mutation and point mutation, 3 cases (23%) of MFN2 gene point mutation, and 1 case (8%) of NEFL gene point mutation. Eleven cases (85%) carried known pathogenic mutations and 2 cases (15%) had novel mutations. The new variant c.394C > G (p.P132A) of the MPZ gene was rated as "possibly pathogenic" and the new variant c.326A > G (p.K109R) of the MFN2 gene was rated as "pathogenic".
Conclusions
Early-onset CMT is mainly caused by PMP22 gene duplication mutation and MPZ gene mutations. The clinical phenotype is mainly CMT type 1, among which Dejerine-Sottas syndrome accounts for a considerable proportion.
Keywords: Charcot-Marie-Tooth disease, PMP22 gene, MPZ gene, MFN2 gene, NEFL gene, High-throughput sequencing, Child
腓骨肌萎缩症(Charcot-Marie-Tooth disease, CMT),又称遗传性运动感觉神经病(hereditary motor and sensory neuropathy, HMSN),是一组具有高度临床和遗传异质性的周围神经单基因遗传病,发病率约为1/2 500[1]。CMT的遗传模式有常染色体显性遗传、常染色体隐性遗传及X-连锁遗传。目前发现80余个基因的1 000余个突变均可导致CMT的发生[2-3],其中外周髓鞘蛋白22(peripheral myelin protein 22, PMP22)、间隙连接蛋白β1(gap junction protein beta 1, GJB1)、髓鞘蛋白零(myelin protein zero, MPZ)、线粒体融合蛋白2(mitofusin 2, MFN2)四个基因的突变占总突变的近95%[4]。随着多重连接探针扩增技术(multiplex ligation-dependent probe amplification, MLPA)及二代基因测序技术,如基因panel、全外显子组测序技术等的发展及应用,对于CMT的认识不再局限于传统的临床分型。如何确定突变基因,特别是新变异的致病性,确定基因分型,成为了当今分子诊断的难点所在。而美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)于2015年重新修订的序列变异解读标准和指南[5],基于人群数据、计算数据、功能数据、共分离数据等,对基因的变异进行分级,为解读工作提供了指向。本文通过对13例早发型CMT患儿的临床特征、家族史及基因检测结果进行分析,最终确定患儿的基因分型。
1. 资料与方法
1.1. 研究对象
以2014年4月至2018年3月在浙江大学医学院附属儿童医院临床诊断为早发型CMT的13例患儿为研究对象,其中男9例(69%),女4例(31%),其中12例(92%)患儿起病年龄 < 2岁,平均就诊年龄4.0±2.1岁。
对于早发型CMT采用诊断标准[6-7]:(1)发病年龄 < 10岁;(2)慢性起病,缓慢进展的肢体远端尤其是双下肢远端的肌无力和肌萎缩,典型者成鹤腿畸形;(3)肌电图显示周围神经性受损;(4)神经活检证实周围神经损害;(5)家族史阳性;(6)弓形足或脊柱侧凸;(7)排除以下疾病:慢性炎性脱髓鞘性多发性神经根神经病、糖尿病性周围神经病、酒精中毒性周围神经病、周围神经淀粉样变性、进行性脊髓性肌萎缩症、脊髓亚急性联合变性等。
患儿须具备上述第(1)、(2)、(3)、(7)四条及第(4)、(5)、(6)中任何一条。若家族中已有确诊者,再具备第(1)、(2)条即可诊断。
1.2. CMT临床分型
基于神经电生理和病理特点,CMT的临床分型可初步分为(1)脱髓鞘型(CMT1型):正中神经运动传导速度(motor nerve conduction velocity, MNCV) < 38 m/s,神经活检示显著的节段性脱髓鞘,施万细胞增生,呈“洋葱头”样改变;其中正中神经MNCV < 12 m/s者亦称Dejerine-Sottas综合征(Dejerine-Sottas syndrome, DSS);(2)轴索型(CMT2型):正中神经MNCV > 38 m/s,神经活检示慢性轴索变性和再生,有髓纤维减少,神经再生簇形成;(3)中间型:正中神经MNCV介于25~45 m/s,神经病理兼具脱髓鞘和轴索变性特点。
1.3. 资料采集
收集患儿相关临床资料:性别、就诊年龄、起病年龄、起病症状、家族史及神经系统体征。
1.4. 周围神经病相关基因高通量测序
抽取患儿外周静脉血3 mL,放置EDTA抗凝管中,送至北京金准基因科技有限责任公司,采用周围神经病相关106个基因panel检测。发现基因点突变时,根据选取位点的位置用Primer 5设计、合成引物,进行PCR扩增,对高通量测序结果中的变异位点进行Sanger测序验证;发现基因重复突变时,对捕获区域进行覆盖度计算及均一化处理,检测出可能发生拷贝数变异的区域,经MLPA检测基因(主要针对PMP22基因),确认重复范围;对亲属进行Sanger测序突变位点验证,根据家系共分离结果进一步判断其致病性。
应用人类基因突变数据库(HGMD, http://www.hgmd.cf.ac.uk)确认已报道的致病基因位点。对于新变异位点,应用ESP数据库(http://evs.gs.washington.edu/EVS)、千人数据库(http://browser.1000genomes.org)、ExAC数据库(http://exac.broadinstitute.org)进行正常对照人群变异比对,应用ACMG指南标准对新变异进行评级。
2. 结果
2.1. 临床表现
5例(38%)患儿存在运动发育落后,独走年龄 > 18月,伴行走不稳、平衡能力差、易摔倒、步态异常。13例(100%)患儿均存在远端肢体无力,4例(31%)伴远端肢体萎缩,6例(46%)伴上肢受累,1例(8%)伴脊柱侧凸。见表 1。
1.
13例早发型CMT患儿基本信息及临床表现
| 病例 | 性别 | 就诊年龄(岁) | 起病年龄(岁) | 起病症状 | 临床表现 | |||||
| 足畸形 | 远端肢体无力 | 远端肢体萎缩 | 上肢受累 | 感觉症状 | 脊柱侧凸 | |||||
| 注:+示阳性症状;-示阴性症状。 | ||||||||||
| 1 | 女 | 2.3 | < 2 | 20月独走,下肢无力 | 鹤腿 | + | - | - | 未评 | - |
| 2 | 男 | 3.3 | < 2 | 24月独走,下肢无力 | 弓形足 | + | + | + | 未评 | + |
| 3 | 女 | 1.8 | < 2 | 16月独走,步态异常 | 弓形足 | + | - | - | 未评 | - |
| 4 | 男 | 6 | < 2 | 15月独走,步态异常,走路易摔跤 | 弓形足、鹤腿 | + | + | + | + | - |
| 5 | 男 | 5.4 | < 2 | 13月独走,走路易摔跤 | 弓形足、鹤腿 | + | - | - | - | - |
| 6 | 男 | 4 | < 2 | 20月独走,步态异常,走路易摔跤 | 弓形足 | + | - | + | 未评 | - |
| 7 | 男 | 5 | < 2 | 12月独走,步态异常 | 马蹄内翻足 | + | + | - | + | - |
| 8 | 女 | 9 | 8 | 13月独走,8岁出现下肢无力,步态异常 | 弓形足、鹤腿 | + | + | - | + | - |
| 9 | 男 | 4.7 | < 2 | 15月独走,下肢无力 | 弓形足、鹤腿 | + | - | + | 未评 | - |
| 10 | 男 | 1.3 | < 2 | 16月尚不能独走 | 鹤腿 | + | - | + | 未评 | - |
| 11 | 男 | 3.2 | < 2 | 18月独走,步态异常 | 鹤腿 | + | - | - | 未评 | - |
| 12 | 男 | 2.5 | < 2 | 20月独走,上楼梯步态异常 | 鹤腿 | + | - | + | 未评 | - |
| 13 | 女 | 4.2 | < 2 | 12月独走,15月出现步态异常,走路易摔跤 | 弓形足 | + | - | - | 未评 | - |
2.2. 肌电图结果
肌电图显示正中神经MNCV < 25 m/s者9人,介于25~45 m/s者1人,> 45 m/s者3人,可分为CMT1型9人(69%),中间型1人(8%),CMT2型3人(23%)。9例CMT1型患儿中,有6例正中神经MNCV < 12 m/s,即DSS;有5例(病例1、2、4、8、13)正中神经复合肌肉动作电位(compound muscle action potential, cMAP)均降低,提示合并轴索损害;且正中神经感觉神经传导速度(sensory nerve conduction velocity, SNCV)及感觉神经动作电位(sensory nerve action potential, SNAP)均未能引出。见表 2。
2.
13例早发型CMT患儿的肌电图结果
| 病例 | 正中神经 | 临床分型 | |||
| MNCV (m/s) | cMAP (mV) | SNCV (m/s) | SNAP (μV) | ||
| 注:[NP]未引出。 | |||||
| 1 | 4.9 | 0.68 | NP | NP | CMT1/DSS |
| 2 | 8.8 | 0.38 | NP | NP | CMT1/DSS |
| 3 | 9.4 | 1.5 | NP | NP | CMT1/DSS |
| 4 | 9.2 | 0.2 | NP | NP | CMT1/DSS |
| 5 | 51.3 | 2.9 | 51.3 | 9.0 | CMT2 |
| 6 | 25.5 | 2.6 | 30.1 | 1.63 | 中间型CMT |
| 7 | 51.4 | 3.6 | 41.1 | 4.5 | CMT2 |
| 8 | 23.3 | 1.3 | NP | NP | CMT1 |
| 9 | 11.4 | 3.0 | NP | NP | CMT1/DSS |
| 10 | 6.1 | 2.8 | NP | NP | CMT1/DSS |
| 11 | 52.3 | 6.1 | NP | NP | CMT2 |
| 12 | 18.6 | 3.0 | NP | NP | CMT1 |
| 13 | 15.3 | 1.05 | NP | NP | CMT1 |
2.3. 基因诊断
13例患儿的基因检测结果显示11例(85%)为已知致病突变,2例(15%)为新变异,其中5例(38%)存在家族史。突变基因包含PMP22重复突变(6例,46%)、MPZ基因插入突变及点突变(3例,23%)、MFN2基因点突变(3例,23%)及人轻肽神经丝蛋白(NEFL)基因点突变(1例,8%)。2例新变异分别为MPZ基因c.394C > G(p.P132A)及MFN2基因c.326A > G(p.K109R)。见表 3。
3.
13例早发型CMT患儿突变基因、核苷酸及氨基酸改变信息
| 病例 | 临床分型 | 突变基因 | 核苷酸改变 | 氨基酸改变 | 父母有无变异 | 是否为新变异 |
| 1 | CMT1/DSS | MPZ | c.394C > G | p.P132A | 无 | 是 |
| 2 | CMT1/DSS | MPZ | c.90C > G | p.I30M | 无 | 否 |
| 3 | CMT1/DSS | PMP22 | PMP22基因整体重复 | - | 父 | 否 |
| 4 | CMT1/DSS | PMP22 | PMP22基因整体重复 | - | 无 | 否 |
| 5 | CMT2 | MFN2 | c.326A > G | p.K109R | 无 | 是 |
| 6 | 中间型CMT | NEFL | c.293A > G | p.N98S | 无 | 否 |
| 7 | CMT2 | MFN2 | c.280C > T | p.R94W | 无 | 否 |
| 8 | CMT1 | PMP22 | PMP22基因整体重复 | - | 无 | 否 |
| 9 | CMT1/DSS | PMP22 | PMP22基因整体重复 | - | 父 | 否 |
| 10 | CMT1/DSS | MPZ | c.293C > T | p.R98C | 无 | 否 |
| 11 | CMT2 | MFN2 | c.1090C > T | p.R264W | 母 | 否 |
| 12 | CMT1 | PMP22 | PMP22基因整体重复 | - | 母 | 否 |
| 13 | CMT1 | PMP22 | PMP22基因整体重复 | - | 母 | 否 |
2.4. 新变异分析
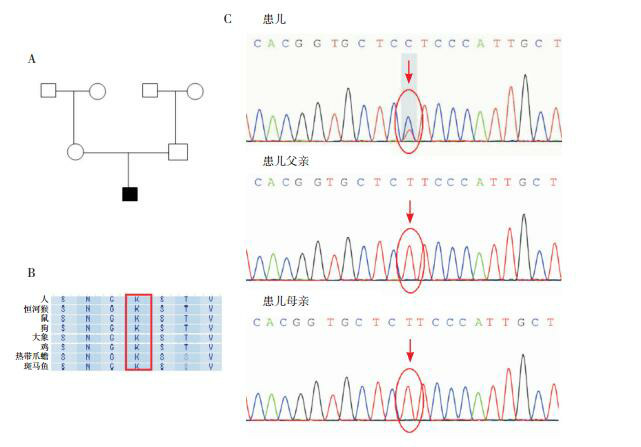
病例1携带MPZ基因c.394C > G(p.P132A)杂合变异,病例5携带MFN2基因c.326A > G(p.K109R)杂合变异。Sanger测序验证两位患儿的父母均未携带相应变异,该患儿为新发(de novo)变异。两个基因变异位点在哺乳动物中高度保守。见图 1~2。
1.
病例1家系图、Sanger测序图及保守序列分析
图A示病例1家系图,■为先证者,○为健康女性,□为健康男性;图B示MPZ蛋白第132号氨基酸位点在哺乳动物中高度保守;图C示患儿存在MPZ基因c.394C > G(p.P132A)杂合突变,父母在该位点均未携带突变(箭头所示)。

2.
病例5家系图、Sanger测序图及保守序列分析
图A示病例5家系图,■为先证者,○为健康女性,□为健康男性;图B示MFN2蛋白第109号氨基酸位点在哺乳动物中高度保守;图C示患儿存在MFN2基因c.326A > G(p.K109R)杂合突变,父母在该位点均未携带突变(箭头所示)。
应用ACMG序列变异解读标准对2个新变异进行评级。MPZ基因c.394C > G(p.P132A)杂合变异,为新发变异,无家族史,符合PS2;该变异为ESP数据库、千人数据库、ExAC数据库正常对照人群中未发现的变异,符合PM2;SIFT、PolyPhen-2、PROVEAN软件的生物信息数据分析预测提示“有害的”,MutationAssessor预测提示“致病性中等”;因此该变异被判定为“可能致病的”。MFN2基因c.326A > G(p.K109R)杂合变异,也为新发变异,无家族史,符合PS2;MFN2蛋白第107~111号氨基酸为核苷酸结合域,影响GTP的结合[8-9],符合PS3;该变异为ESP数据库、千人数据库、ExAC数据库正常对照人群中未发现的变异,符合PM2;经SIFT、PolyPhen-2、PROVEAN、MutationAssessor软件的生物信息数据分析预测,均提示为“有害的”,符合PP3;因此该突变被判定为“致病的”。见表 4。
4.
新变异的ACMG致病性分级
| 病例 | 突变基因 | SIFT | PolyPhen-2 | PROVEAN | Mutation-Assessor | ACMG致病性分级 | 致病性判定 | ||
| 强 | 中等 | 支持证据 | |||||||
| 注:-示无支持证据。 | |||||||||
| 1 | MPZ | 0.01 | 0.93 | -6.97 | 2.37 | PS2 | PM2 | - | 可能致病的 |
| 5 | MFN2 | 0 | 1.00 | -2.66 | 4.07 | PS2, PS3 | PM2 | PP3 | 致病的 |
3. 讨论
CMT是一组具有高度临床和遗传异质性的周围神经系统遗传病,诊断需结合临床分型及基因分型。二代测序技术的发展正改变着CMT基因诊断的方式。包含所有已知CMT相关基因的基因panel检测是如今最经济、最高效、最直接的基因检测方式,这能让临床医生在第一时间对所有CMT基因的突变情况有一个整体的了解。它能很好的体现基因型-表型的对应关系,为临床的治疗、随访提供可靠保障。但新变异的临床意义解读是临床工作的一个难点所在。2015年,ACMG提出了适用于所有孟德尔基因变异,包括单基因、多基因包、外显子组和基因组测序发现的遗传变异的解读标准与指南[5]。
本研究共确诊13例早发型CMT患儿,6例(46%)PMP22基因重复突变,3例(23%)MPZ基因点突变,3例(23%)MFN2基因点突变及1例(8%)NEFL基因点突变,这与之前Cornett等[10]报道的CMT患儿前三位的突变基因一致。而对于占总体CMT人群突变第二位的GJB1基因,却并非CMT患儿的主要突变基因。这是由于在脱髓鞘型中,PMP22基因重复突变、MPZ、NEFL基因突变的患者多表现为早发型;而在轴索型中,几乎所有的MFN2基因突变的患者表现为早发型,而GJB1基因突变患者无论表现为脱髓鞘型或轴索型,起病年龄均相对较晚[11]。
本研究中6例患儿起病年龄早,且正中神经MNCV < 12 m/s,符合DSS的诊断标准。突变基因包括3例MPZ基因突变和3例PMP22基因重复突变,与之前文献报道的DSS多数为MPZ及PMP22(包括整体重复及点突变)相一致[12]。近年来,DSS更被看做是一个临床综合征,尽管DSS患儿起病年龄早,且MNCV显著降低,提示着严重的脱髓鞘或者无髓鞘形成,但Gabreëls-Festen等[12]对25例DSS患者进行随访发现,仍有1/3的患者在平均年龄39.9岁时尚可独立行走1 km以上。DSS的出现并非预示着成人期的严重残疾及轮椅依赖的可能。
近年来,多项研究均提示MNCV虽能作为临床分型的主要指标,但却与疾病的进展不相平行。而cMAP尽管与起病年龄不相关,却关乎病程长短及疾病的严重程度,特别是在PMP22基因重复突变及MPZ基因突变的患者中,cMAP与远端肌力密切相关[13-14]。文中CMT1型患儿中有5人cMAP低于同龄儿,表明该型患儿除脱髓鞘病变外尚还存在轴索病变,且也预示着患儿相对预后不良。
综上,本研究依据患儿的临床表现、神经肌肉电生理特征、遗传模式、二代测序结果,结合临床分型及基因分型,采用ACMG标准对新变异致病性进行分析,共确诊早发型CMT 13例,为临床CMT的诊断确定基础。
Biography
徐佳露, 女, 博士, 主治医师
Funding Statement
国家青年科学基金(81501084)
References
- 1.Skre H. Genetic and clinical aspects of Charcot-Marie-Tooth's disease. Clin Genet. 1974;6(2):98–118. doi: 10.1111/j.1399-0004.1974.tb00638.x. [DOI] [PubMed] [Google Scholar]
- 2.Gutmann L, Shy M. Update on Charcot-Marie-Tooth disease. Curr Opin Neurol. 2015;28(5):462–467. doi: 10.1097/WCO.0000000000000237. [DOI] [PubMed] [Google Scholar]
- 3.Timmerman V, Strickland AV, Züchner S. Genetics of Charcot-Marie-Tooth (CMT) disease within the frame of the Human Genome Project success. Genes (Basel) 2014;5(1):13–32. doi: 10.3390/genes5010013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiVincenzo C, Elzinga CD, Medeiros AC, et al. The allelic spectrum of Charcot-Marie-Tooth disease in over 17, 000 individuals with neuropathy. Mol Genet Genomic Med. 2014;2(6):522–529. doi: 10.1002/mgg3.2014.2.issue-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung KW, Kim SB, Park KD, et al. Early onset severe and late-onset mild Charcot-Marie-Tooth disease with mitofusin 2(MFN2) mutations. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=HighWire000002270636. Brain. 2006;129(Pt 8):2103–2118. doi: 10.1093/brain/awl174. [DOI] [PubMed] [Google Scholar]
- 7.陈 嵘, 梁 秀龄. 腓骨肌萎缩症的临床表现和分型及遗传学. 中山医科大学学报. 1997;18(3):213–215, 255. [Google Scholar]
- 8.Santel A, Fuller MT. Control of mitochondrial morphology by a human mitofusin. J Cell Sci. 2001;114(Pt 5):867–874. doi: 10.1242/jcs.114.5.867. [DOI] [PubMed] [Google Scholar]
- 9.Rojo M, Legros F, Chateau D, et al. Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci. 2002;115(Pt 8):1663–1674. doi: 10.1242/jcs.115.8.1663. [DOI] [PubMed] [Google Scholar]
- 10.Cornett KMD, Menezes MP, Shy RR, et al. Natural history of Charcot-Marie-Tooth disease during childhood. Ann Neurol. 2017;82(3):353–359. doi: 10.1002/ana.v82.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abe A, Numakura C, Kijima K, et al. Molecular diagnosis and clinical onset of Charcot-Marie-Tooth disease in Japan. J Hum Genet. 2011;56(5):364–368. doi: 10.1038/jhg.2011.20. [DOI] [PubMed] [Google Scholar]
- 12.Gabreëls-Festen A. Dejerine-Sottas syndrome grown to maturity:overview of genetic and morphological heterogeneity and follow-up of 25 patients. J Anat. 2002;200(4):341–356. doi: 10.1046/j.1469-7580.2002.00043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim YH, Chung HK, Park KD, et al. Comparison between clinical disabilities and electrophysiological values in Charcot-Marie-Tooth 1A patients with PMP22 duplication. J Clin Neurol. 2012;8(2):139–145. doi: 10.3988/jcn.2012.8.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hattori N, Yamamoto M, Yoshihara T, et al. Demyelinating and axonal features of Charcot-Marie-Tooth disease with mutations of myelin-related proteins (PMP22, MPZ and Cx32):a clinicopathological study of 205 Japanese patients. Brain. 2003;126(Pt 1):134–151. doi: 10.1093/brain/awg012. [DOI] [PubMed] [Google Scholar]