

Abstract
该文对1例婴儿型低磷酸酯酶症(HPP)患儿及其家系进行临床特点分析及碱性磷酸酯酶基因(ALPL)检测。先证者,男,5个月,多发骨骼畸形:胸骨凹陷、双侧桡骨弯曲畸形、双膝外翻畸形,伴喂养困难、体重下降、发育迟滞、反复肺炎并呼衰,血碱性磷酸酶显著降低。患儿父母、姐姐、叔父、姨母(其他家系成员未能配合)中除父母及姨母的碱性磷酸酶略低,姨母可见脊柱侧弯畸形,余均无临床表型及实验室异常。患者ALPL基因检测到来源于母亲的c.228delG突变及来源于父亲的c.407G > A复合杂合突变,其姨母携带c.228delG突变。c.407G > A突变为已报道的HPP致病突变,c.228delG为新的致病性突变。低磷酸酯酶症是由ALPL基因突变所致,ALPL基因检测是有效的诊断方法。该研究拓展了ALPL基因突变谱,为HPP的基因诊断提供了理论依据。
Keywords: 低磷酸酯酶症, ALPL基因, 家系, 儿童
Abstract
This article reported the clinical features of one child with infantile hypophosphatasia (HPP) and his pedigree information. The proband was a 5-month-old boy with multiple skeletal dysplasia (koilosternia, bending deformity of both radii, and knock-knee deformity of both knees), feeding difficulty, reduction in body weight, developmental delay, recurrent pneumonia and respiratory failure, and a significant reduction in blood alkaline phosphatase. Among his parents, sister, uncle, and aunt (other family members did not cooperate with us in the examination), his parents and aunt had a slight reduction in alkaline phosphatase and his aunt had scoliosis; there were no other clinical phenotypes or abnormal laboratory testing results. His ALPL gene mutation came from c.228delG mutation in his mother and c.407G > A compound heterozygous mutation in his father. His aunt carried c.228delG mutation. The c.407G > A mutation had been reported as the pathogenic mutation of HPP, and c.228delG mutation was a novel pathogenic mutation. Hypophosphatasia is caused by ALPL gene mutation, and ALPL gene detection is an effective diagnostic method. This study expands the mutation spectrum of ALPL gene and provides a theoretical basis for genetic diagnosis of this disease.
Keywords: Hypophosphatasia, ALPL gene, Pedigree, Child
低磷酸酯酶症(hypophosphatasia, HPP, MIM146300, 241500, 241510)是一种以骨矿化不足、血清碱性磷酸酶(alkaline phosphatase, ALP)含量及活性降低、骨特异性碱性磷酸酶(bone-specific alkaline phosphatase, BALP)活性丧失为主要临床特征的遗传代谢性骨病[1]。其遗传方式不确定,轻型的遗传方式可为常染色体隐性或显性,重型的多为常染色体隐性遗传。本病系罕见病,欧洲发病率约为1:300 000,美国报道的发病率为1:100 000[2]。根据发病年龄和骨骼病变程度,HPP分为6型:先天致死型(围生期致死)、婴儿型、儿童型、成年型、齿型及假性低磷酸酯酶症[1-3]。各型之间临床表现可有交叉。婴儿型多在1~6个月发病,多因体重不增、生长落后、颅骨软化、肋骨串珠等就诊,易被误诊为佝偻病[4]。本研究通过对1例婴儿型HPP患儿及其家系的临床特点、碱性磷酸酯酶基因(alkaline phosphatase liver/bone/kidney, ALPL)测序结果进行分析,阐述该病的遗传学发病机制,以期提高临床医生对本病的认识,加强对本病的遗传咨询和产前诊断,降低此类患儿的出生率。
1. 资料与方法
1.1. 研究对象
患儿,男,5个月。因体重不增4月余,气促、反应差5 d入院。既往因肺炎住院4次,因呼吸衰竭多次抢救治疗。患儿为第二胎第二产, 足月顺产出生,出生体重3 100 g,出生时无窒息史。人工喂养,新生儿晚期即有喂养困难,体重不增并逐渐下降。至今不会抬头、翻身。入院查体:体重2 590 g,身长56 cm。体温36.0℃,心率147次/min,呼吸59次/min。皮肤干皱,皮下脂肪消失,前囟5×5 cm,骨缝宽,枕部触诊乒乓球感。双耳位偏低,下颌小,呼吸急促、见吸气三凹征,胸骨凹陷,可见肋骨串珠,双侧桡骨弯曲畸形,双膝外翻畸形。母孕期体健,否认毒物、放射线接触史。否认近亲婚配及家族遗传病史。辅助检查:血常规WBC 13.1×109/L,余项正常;肝功能:谷丙转氨酶86 U/L(参考值9~50 U/L)、谷草转氨酶54 U/L(参考值15~45 U/L),余项正常;肌酸激酶(CK)正常;7次血钙波动在3.19~4.40 mmol/L(参考值2.08~2.60 mmol/L);5次碱性磷酸酶波动在9~17 mmol/L(参考值40~750 mmol/L);1, 25-二羟基维生素D3(VitD3)正常;甲状旁腺素3.5 pg/mL(参考值:6~80 pg/mL);降钙素22.1 pg/mL(参考值:0~18 pg/mL)。血串联质谱分析未见异常。尿气相质谱示4-羟基苯乳酸偏高。胸部X线片:支气管肺炎。左腕关节正位片:骨矿化不良,左侧尺、桡骨远端干骺端先期钙化带骨小梁模糊,并见干骺端不规则骨质缺损,未见骨化中心(见图 1)。胸腰椎正位X线片:各椎体骨质疏松、椎间隙窄、肋骨串珠及骨质密度普遍降低。
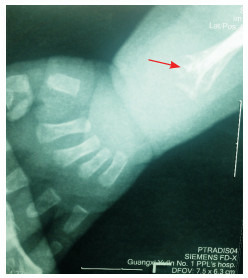
1.
患儿5月龄左腕正位片
骨矿化不良,未见骨化中心;左侧尺、桡骨远端干骺端先期钙化带骨小梁模糊,并可见干骺端不规则骨质缺损(如箭头所示)。
1.2. 家系临床资料收集
收集先证者家系3代共计10人的病史资料,以及患儿及其父母、叔父、姨母、姐姐(其他家系成员未能配合)的VitD3、PTH、降钙素、ALP、血钙等检查结果。
1.3. 目的基因扩增
获得医院医学伦理委员会批准及家属知情同意后采集先症者外周血5 mL及其父母、叔父、姨母、姐姐(其他家系成员未能配合)的外周血各2 mL。使用QiagenFlexiGene DNA Kit提取基因组DNA。http://genome.ucsc.edu/检索野生型ALPL基因序列(NM-000478)。使用引物设计软件Primer 5.0进行引物设计(见表 1),经NCBI Blast验证所有引物的特异性。反应体系(总体积25 μL):DNA样品1 μL,上、下游引物各0.5 μL,2×Taq PCR superMix 12.5 μL,ddH20 10.5 μL。PCR扩增,预变性94℃ 5 min,变性94℃ 15 s,退火60℃ 30 s,72℃延伸30 s,35个循环后予末次延伸72℃ 5 min。采用2%琼脂糖凝胶电泳检验PCR产物,溴乙锭染色。
1.
ALPL基因编码区引物序列

|
1.4. Sanger测序验证及分析
PCR产物送北京康旭医学研究所行基因测序,对先证者及其家系成员所发现的ALPL基因突变位点进行Sanger测序验证。Swiss-model在线软件预测,并用Molpobity软件评价分析组织非特异性碱性磷酸酶(tissue-nonspecific alkaline phosphatase, TNAP/TNSALP)蛋白的三级结构模型。检索PubMed、HGMD、ESP6500及SESEP实验室ALPL基因数据库,并结合相关文献行家系分析。
2. 结果
2.1. 家系的临床表现及实验室检查结果
该家系(图 2)三代共计10人,男女各5人,除先证者及其姨母有骨骼畸形(查体及脊柱X线见脊柱侧弯畸形),余均无异常临床表型。家系中收集了VitD3、PTH、降钙素、ALP、血钙等结果的成员中,患儿血钙增高、ALP明显降低,1, 25-二羟基维生素D3、PTH均正常;其父母、姨母的碱性磷酸酶略低,1, 25-二羟基维生素D3、血钙、PTH正常;患儿叔父、姐姐的血钙、ALP、1, 25-二羟基维生素D3、PTH均正常。
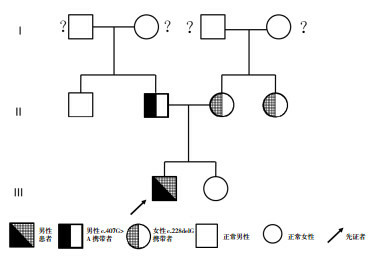
2.
患儿家系图
患者父亲为c.407G > A突变携带者,母亲及姨母为c.228delG突变携带者。?示未行基因检测。
2.2. 患儿及家系基因测序结果
该家系中患儿及其父母、叔父、姨母、姐姐(其他家系成员未能配合)进行了ALPL基因测序。患儿ALPL基因存在复合杂合突变,分别位于chr1:21887636及chr1:21889712,引起c.228delG和c.407G > A两处突变;患儿父亲携带c.407G > A突变;患儿母亲及姨母携带c.228delG突变;患儿姐姐、叔父的两突变位点均正常。见图 3。
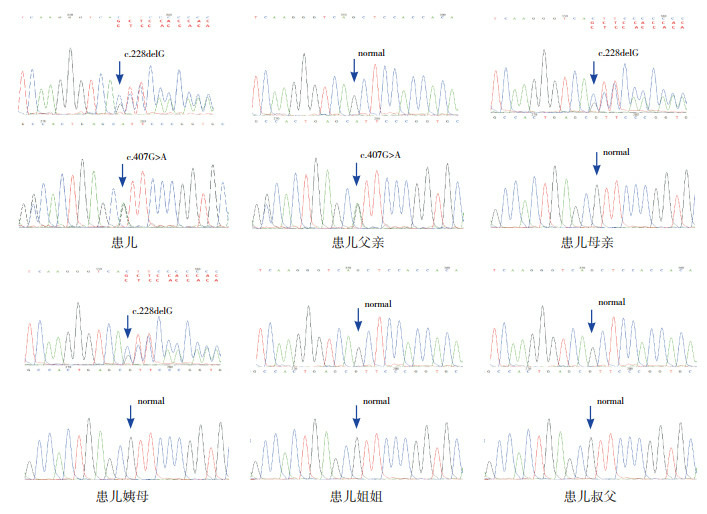
3.
患儿家系ALPL基因测序结果
患儿ALPL基因存在c.228delG(A)和c.407G > A (B) 的复合突变;患儿父亲为c.407G > A突变携带者;母亲及姨母为c.228delG突变携带者;患儿姐姐、叔父的两突变位点均正常。突变位点如箭头所示。
2.3. TNAP蛋白三级结构预测
用Swiss-model在线软件分别构建野生型及突变型TNAP蛋白三级结构模型,预测c.228delG及c.407G > A突变对TNAP蛋白结构的影响,并用Molpobity软件评价分析TNAP蛋白三级结构模型。结果显示:两突变致使TNAP蛋白A链结构发生改变,c.228delG影响一小段肽段,c.407G > A导致一个氨基酸位点改变,见图 4。
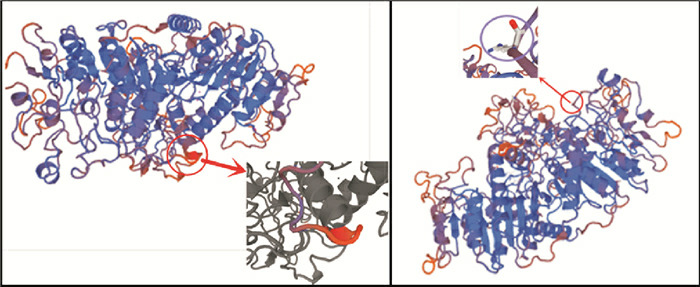
4.
TNAP蛋白质结构飘带图
红色圈内为突变影响区域。左图右下角的灰色小图为受影响区域放大图,其涂色段为c.228delG突变影响的肽段;右图左上角小图为突变影响位点的放大图,其紫色圈内为c.407G > A突变的氨基酸突变位点。
3. 讨论
HPP在1948年由Rathbun首次报道,又称Rathbun综合症[5],系因ALPL基因(又称TNS-ALP基因,编号:249;MIM-171760)突变所致。ALPL基因定位于染色体1p36-34,编码TNAP。TNAP在软骨细胞、成骨细胞、成牙本质细胞以及这些细胞分泌的基质小泡中都有表达,TNAP水解细胞外矿化抑制物无机焦磷酸盐(pyrophosphate, PPi),从而启动并促进骨组织矿化[1]。TNAP蛋白有5个重要的结构域,包括激活点、激活谷、钙结合位点、同型二聚体区及冠区,当基因突变使这些重要的结构区域发生改变时,组织非特异性碱性磷酸酶活性受到严重影响[6],进而影响骨骼矿化而致病。
HPP分为先天致死型(围生期致死型)、婴儿型、儿童型、成年型、齿型、假性低磷酸酯酶症6型。先天致死型最严重,多因死胎或出生后呼吸衰竭死亡[7]。婴儿型多在1~6个月发病,1岁左右死亡,常表现为体重不增、生长落后及类佝偻病,继发高钙血症者可致肾结石、肾损害,以及食欲差、呕吐、便秘、多饮、多尿、发热等[4, 8]。儿童型在2岁后发病,多因牙列早脱就诊,ALP显著减低,并且与疾病严重程度相关。成年型多中年以后发病,少数患者有关节、肌腱、椎间韧带钙化。牙型仅有牙冠矿化受阻表现如牙齿生长异常及恒牙早脱[2]。假性低磷酸酯酶症极罕见,临床表现及X线特点与婴儿型、儿童型、成年型相同,但ALP正常。国内报道的婴儿型HPP,除麻宏伟等报道的1例出生胎龄28周早产儿7月龄发病外,均于1~6月龄发病,均有反复的呼吸道感染、呼吸衰竭、喂养困难、营养不良等表现[9-14]。本研究先证者5月龄,新生儿晚期即出现喂养困难、体重不增并逐渐下降,具有骨矿化不全、骨骼畸形以及ALP水平降低、血钙增高等表现,符合婴儿型HPP的临床特点。
HPP临床表型的异质性及影像学表现的多样性不影响X线作为HPP临床诊断的重要依据[15]。几乎所有HPP患者的X线均可见骨矿化不足、骨质疏松、类佝偻病表现。本例患儿左腕X有骨矿化不良,骨化中心消失,干骺端先期钙化带骨小梁模糊等表现;脊柱X线显示各椎体骨质疏松、肋骨串珠、骨质密度普遍降低,符合HPP的X线特点。
ALPL基因突变是诊断HPP的金标准。目前国内已报道的婴儿型HPP患儿的突变位点包括ALPL基因第1、5、7、9、12外显子[10, 12-13]。国外HPP基因检测开展较早,报道的ALPL基因突变位点达332种,以点突变为主,大片段缺失仅9例。本研究使用二代基因测序筛查出可疑致病突变点,并用Sanger测序法验证,检测到先证者ALPL基因c.228delG和c.407G > A复合杂合突变。
c.228delG位于第4外显子,引起编码区第228号核苷酸G缺失,导致从第76号氨基酸谷氨酰胺(Gln)开始的氨基酸合成改变,并在改变后的第46个氨基酸即第112号氨基酸处终止(p.Gln76HisfsTer46),故为移码突变;该突变来源于其母,其姨母亦有此突变。c.407G > A位于第5外显子,即编码区第407号核苷酸由G变为A,导致第136号氨基酸由精氨酸(Arg)转变为组氨酸(His)(p.Arg136His);该突变来源于其父亲。患儿姐姐、叔父未见上述两种突变,且无临床症状。检索PubMed、HGMD、ESP6500及SESEP实验室的ALPL基因数据库,发现c.407G > A的致病性已有文献报道,与HPP相关[16];SIFT、MutationTaster及PolyPhen-2软件预测也提示c.407G > A导致p.Arg136His,136号氨基酸精氨酸参与蛋白质残基侧链间的相互作用。c.228delG的致病性尚未见报道。经检索1000Genomes、dbSNP数据库,该变异也不属于多态性变化,在人群中发生的频率极低。SIFT、MutationTaster及PolyPhen-2蛋白质功能预测软件提示,c.228delG导致从第76号氨基酸谷氨酰胺(Gln)开始的氨基酸合成发生改变,并在改变后的第46个氨基酸即第112号氨基酸处终止;由于110号氨基酸所处位置是酶激活位点及金属离子结合位点[17],推测突变可能影响酶与底物的结合以及蛋白功能。本研究先证者遗传了来自父亲的c.407G > A及来自母亲的c.228delG复合杂合突变,出现典型的临床症状;其父、母亲及姨母分别为c.407G > A、c.228delG突变携带者,无临床症状,故两突变在本家系中的遗传方式可能为常染色体隐性遗传,先证者同胞中基因型正常的概率为25%,如果该家系中患儿父母再次孕育,应当寻求适当的遗传学帮助,并进行产前诊断。
部分轻症HPP可自愈,病情较重者需采取多学科综合管理。血钙增高程度较重者可使用鲑降钙素治疗,并注意骨骼脱矿化所致的骨缺损及骨折的矫形管理,以及营养不良者的饮食管理和抗炎止痛药的应用。美国食品药品管理局于2013年批准了靶标酶置换药Asfotase Alfa作为先天致死型、婴儿型、儿童型HPP的治疗用药。Scott等[18]对11例3岁以下低磷酸酯酶症患儿予以24周的Asfotase Alfa治疗后,患者骨折愈合、畸形减轻、骨质重塑;并明显改善了因严重呼吸道感染所致呼吸衰竭患儿的呼吸功能;患儿的大运动、精细运动,以及认知发育、肌力均得到改善,并观察到追赶性生长。
综上,本研究通过1例婴儿型低磷酸酯酶症患儿的家系分析,总结了HPP的临床特点,发现了ALPL基因的一个新致病突变,对于提高临床医生对HPP的认识和诊断能力,以及加强对本病的遗传咨询和产前诊断、降低HPP患儿的出生率有一定的意义。
Biography
李登峰, 男, 硕士研究生, 医师
Funding Statement
广西医科大学第一附属医院科研启动基金资助项目(No.2010001)
References
- 1.Mumm S, Jones J, Finnegan P, et al. Hypophosphatasia:molecular diagnosis of Rathbun's original case. J Bone Miner Res. 2001;16(9):1724–1727. doi: 10.1359/jbmr.2001.16.9.1724. [DOI] [PubMed] [Google Scholar]
- 2.Mornet E, Yvard A, Taillandier A, et al. A molecular-based estimation of the prevalence of hypophosphatasia in the European population. Ann Hum Genet. 2011;75(3):439–445. doi: 10.1111/ahg.2011.75.issue-3. [DOI] [PubMed] [Google Scholar]
- 3.Di Mauro S, Manes T, Hessle L, et al. Kinetic characterization of hypophosphatasia mutations with physiological substrates. J Bone Miner Res. 2002;17(8):1383–1391. doi: 10.1359/jbmr.2002.17.8.1383. [DOI] [PubMed] [Google Scholar]
- 4.麻 宏伟. 表现为类佝偻病的遗传性疾病. http://www.cjcp.org/CN/abstract/abstract13198.shtml. 中国当代儿科杂志. 2013;15(11):923–927. doi: 10.7499/j.issn.1008-8830.2013.11.001. [DOI] [PubMed] [Google Scholar]
- 5.Rathbun JC. Hypophosphatasia; a new developmental anomaly. Am J Dis Child. 1948;75(6):822–831. doi: 10.1001/archpedi.1948.02030020840003. [DOI] [PubMed] [Google Scholar]
- 6.Mornet E, Stura E, Lia-Baldini AS, et al. Structural evidence for a functional role of human tissue nonspecific alkaline phosphatase in bone mineralization. J Biol Chem. 2001;276(33):31171–31178. doi: 10.1074/jbc.M102788200. [DOI] [PubMed] [Google Scholar]
- 7.Jaruratanasirikul S, Chanvitan P. Hypophosphatasia:the importance of alkaline phosphatase in bone mineralization. J Med Assoc Thai. 1999;82(12):1268–1272. [PubMed] [Google Scholar]
- 8.张 碧丽, 杨 瑄, 黄 乐, et al. 喂养困难生长发育落后高钙血症和低碱性磷酸酶血症. http://www.cjcp.org/CN/abstract/abstract11428.shtml 中国当代儿科杂志. 2001;(6):723–725. [Google Scholar]
- 9.段 泓宇, 王 一斌. 婴儿型碱性磷酸酶过少症1例报告. 临床儿科杂志. 2010;28(12):1112. doi: 10.3969/j.issn.1000-3606.2010.12.003. [DOI] [Google Scholar]
- 10.郑 雯洁, 杨 宇真, 陈 晓英, et al. 婴儿型低磷酸酶血症1例报告及文献复习. http://www.cnki.com.cn/Article/CJFDTOTAL-LCAK200902019.htm 临床儿科杂志. 2009;27(2):163–164. [Google Scholar]
- 11.麻 宏伟, 马 健. 婴儿型低碱性磷酸酶血症1例. http://www.cnki.com.cn/Article/CJFDTOTAL-XZEK200804020.htm 中国循证儿科杂志. 2008;3(4):318–319. [Google Scholar]
- 12.刘 海娟, 李 梅, 邢 小平, et al. 低磷酸酶血症一家系组织非特异性碱性磷酸酶 (TNSALP) 基因突变分析. http://www.cnki.com.cn/Article/CJFDTOTAL-JCYL201103011.htm 基础医学与临床. 2011;31(3):263–267. [Google Scholar]
- 13.赵 真, 夏 维波, 邢 小平, et al. 婴儿型低磷酸酶血症组织非特异性碱性磷酸酶基因突变检测. 中华内科杂志. 2013;52(10):824–828. doi: 10.3760/cma.j.issn.0578-1426.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 14.王 崇伟, 谢 丽尔. 婴儿型低磷酸酯酶症1例. 中华实用儿科临床杂志. 2011;26(20):1611–1612. doi: 10.3969/j.issn.1003-515X.2011.20.026. [DOI] [Google Scholar]
- 15.Berkseth KE, Tebben PJ, Drake MT, et al. Clinical spectrum of hypophosphatasia diagnosed in adults. Bone. 2013;54(1):21–27. doi: 10.1016/j.bone.2013.01.024. [DOI] [PubMed] [Google Scholar]
- 16.Taillandier A, Zurutuza L, Muller F, et al. Characterization of eleven novel mutations (M45L, R119H, 544delG, G145V, H154Y, C184Y, D289V, 862+5A, 1172delC, R411X, E459K) in the tissue-nonspecific alkaline phosphatase (TNSALP) gene in patients with severe hypophosphatasia. Mutations in brief no. 217. Online. Hum Mutat. 1999;13(2):171–172. doi: 10.1002/(ISSN)1098-1004. [DOI] [PubMed] [Google Scholar]
- 17.Le Du MH, Millan JL. Structural evidence of functional divergence in human alkaline phosphatases. J Biol Chem. 2002;277(51):49808–49814. doi: 10.1074/jbc.M207394200. [DOI] [PubMed] [Google Scholar]
- 18.Scott LJ. Asfotase Alfa:A review in paediatric-onset hypophosphatasia. Drugs. 2016;76(2):255–262. doi: 10.1007/s40265-015-0535-2. [DOI] [PubMed] [Google Scholar]