

Abstract
目的
研究2例X-连锁低血磷性佝偻病(XLH)患儿及家系磷酸盐调节基因(PHEX)的突变类型,以明确其遗传学病因。
方法
回顾性分析2例XLH患者临床资料,应用高通量测序技术从基因组水平对先证者的XLH致病基因PHEX进行检测,并应用PCR-Sanger测序法对突变基因的家系分布进行验证。
结果
2例患儿均检测到PHEX基因新发突变,1例为移码突变c.931dupC,导致翻译提前终止,产生截短蛋白p.Gln311Profs*13;另1例为剪接位点突变IVS14+1G > A,导致外显子15跳跃,产生不完整的氨基酸链。2例患儿父母的基因表型均正常。
结论
c.931dupC和IVS14+1G > A是PHEX基因的两个新突变,可能是XLH新的致病性突变。
Keywords: 低血磷性佝偻病, 磷酸盐调节基因, 突变分析, 儿童
Abstract
Objective
To investigate PHEX gene mutations in 2 patients with X-linked hypophosphatemic rickets (XLH) and their families and to clarify the genetic etiology.
Methods
A retrospective analysis was performed for the clinical data of two patients with XLH. High-throughput sequencing was used to detect the PHEX gene, a pathogenic gene of XLH. PCR-Sanger sequencing was used to verify the distribution of mutations in families.
Results
Both patients had novel mutations in the PHEX gene; one patient had a frameshift mutation, c.931dupC, which caused early termination of translation and produced the truncated protein p.Gln311Profs*13; the other patient had a splice site mutation, IVS14+1G > A, which caused the skipping of exon 15 and produced an incomplete amino acid chain. Their parents had normal gene phenotypes.
Conclusions
c.931dupC and IVS14+1G > A are two novel mutations of the PHEX gene and might be the new pathogenic mutations of XLH.
Keywords: Hypophosphatemic rickets, PHEX gene, Mutation analysis, Child
X-连锁低血磷性佝偻病(X-Linked hypophos-phatemic rickets, HYP或XLH, OMIM #307800)是低血磷性佝偻病中最常见的一种,发病率约为1/20 000[1-2]。XLH症状多于1岁以后症状明显,多为X-连锁显性遗传,发病机制为磷酸盐调节基因(phosphate regulating gene with homologies to endopeptidases on the X-chromosome, PHEX)突变导致血清成纤维生长因子23(FGF23)水平升高,影响肾小管对磷的重吸收,导致尿磷排泄增多、血磷水平下降[3-4]。XLH早期表现为骨骼和牙不同程度的损害,如方颅、鸡胸、漏斗胸、肋骨串珠、肋外翻、手足镯,出牙延迟、易脱落、牙痛,直立行走后出现下肢骨软化症状:“X”或“O”型腿,骨骼疼痛、生长迟缓等[5-6];后期可出现牙脓肿、关节活动障碍[6-7]。补充磷制剂可改善其临床症状。
国内外已报道444例PHEX基因突变所致的XLH患者,多发生于外显子(约占79%),包括缺失、插入、错义及无义突变等;部分发生于内含子(约占19%),包括剪接位点突变;极少数发生于非翻译区(untranslated regions, UTR)[8-9]。XLH的基因变异具有明显的异质性,多种突变形式可引起相似的临床表现。本研究采用PCR-Sanger测序方法,分析2例低血磷性佝偻病患儿的PHEX基因,发现2种未见报道的PHEX基因新突变。
1. 资料与方法
1.1. 研究对象
先证者1,男,3岁4个月,发现“O型腿”2年。不伴骨痛。第一胎第一产、足月顺产出生,出生时身长、体重不详,出生时无窒息。母乳喂养,10月龄添加辅食,无偏食挑食。7月龄能独坐、10月龄独站、1岁2个月能独立行走。既往体健。患儿父母健康,否认近亲结婚,家族中无类似患者。体格检查:身高82 cm(-4.34 SD),体重11 kg,前额突出,无方颅,无鸡胸,无肋骨串珠,无郝氏沟,无手足镯,心肺腹部查体未见异常,“O型腿”、膝间距9 cm。实验室检查:血钙正常,血磷1.04 mmol/L(参考值:1.3~2.1 mmol/L),ALP 792.8 U/L(参考值:95~405 U/L),25(OH) D3正常,PTH正常。血气分析无异常。下肢骨片(见图 1):双膝关节骨骺端稍膨大,临时钙化带模糊。肾脏B超无异常。
1.
先证者1的髋关节、下肢X片
股骨远端及近端钙化带模糊,如箭头所示。

先证者2,女,7岁4个月,发现“X型腿”伴双下肢间断性疼痛5年。第一胎第一产、足月顺产出生,出生身长、体重不详,出生时无产伤、无窒息。喂养史、生长发育史无特殊。既往体健。患儿父母体健,否认近亲结婚,家族中无类似患者。体格检查:身高111.2 cm(-2.61 SD),体重22.5 kg,无方颅,无鸡胸,无郝氏沟,无肋串珠,无手足镯,心肺腹部查体未见明显异常,“X型腿”、踝间距12 cm。实验室检查:血钙正常,血磷0.5 mmol/L(参考值:1.3~2.1 mmol/L),ALP 428 U/L(参考值:95~405 U/L),PTH 110 pg/mL(参考值:10~69 pg/mL)。肾脏B超:双肾皮质回声稍增强,双侧肾椎体见少许钙质成分沉着。
1.2. 高通量捕获测序、突变验证及家系分析
通过重庆医科大学附属儿童医院伦理审查及患儿家属知情同意后,采集患儿及其父母空腹EDTA抗凝静脉血各2 mL,提取基因组DNA。DNA浓度 > 10 ng/μL,体积 > 100 μL;纯度应达到A260/280 > 1.8,A260/230 > 1.2。
取先证者基因组,对PHEX基因的外显子区及两侧的剪接位点序列进行高通量测序,并利用PCR-Sanger测序对先证者及家系进行突变验证和分析(中国人民解放军空军医学研究所附属医院病理检验中心完成)。引物设计根据GeneBank人类PHEX基因序列(NG_007563.2),见表 1。PCR扩增,取PCR样品用ABI遗传分析仪进行测序。阳性突变者用另一份备用样品进行复测。
1.
PHEX基因引物序列
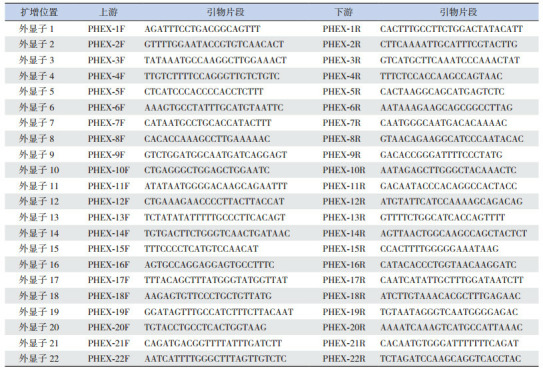
|
2. 结果
2.1. 基因测序结果
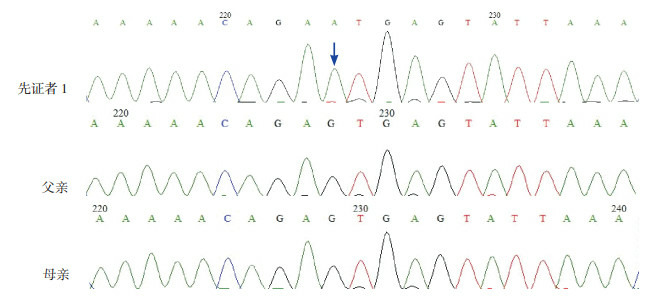
经高通量捕获测序,发现先证者1在PHEX基因第14号内含子剪切位点发生单碱基置换:IVS14+1G > A;运用PCR-Sanger测序进行突变验证及家系分析,证实患儿父母该基因位点正常。见图 2。先证者2的PHEX基因检测到1个杂合突变:位于8号外显子的移码突变c.931dupC;运用PCR-Sanger测序进行突变验证及家系分析,发现患儿父母该基因位点正常。见图 3。
2.
先证者1及家系的PHEX基因测序图
先证者1在第14号内含子发生单碱基置换:IVS14+1G > A,突变位点如箭头所示;其父母该位点正常。
3.
先证者2及家系的PHEX基因测序
先证者2在第8号外显子发生碱基插入突变:c.931dupC,突变位点如箭头所示;其父母该位点正常。

经查询Locus Database数据库,PHEX基因的c.931dupC和IVS14+1G > A两个突变均未见报道。查阅PUBMED(https://www.ncbi.nlm.nih.gov/pubmed/)和CNKI(http://www.cnki.net/)及万方(http://www.wanfangdata.com.cn/)文献数据库,均无该基因突变的文献报道。因此,本文检测到的2种PHEX基因突变为新突变。经MUTATION TASTER(www.mutationtaster.org/)生物信息学分析,2例突变均为致病性突变,但未行相关基因功能学研究。
3. 讨论
至2016年11月,PHEX database数据库已报道444例XLH患者,共计338种PHEX基因突变,其中由中国人报道的突变共计13种。PHEX基因突变中移码突变约占27%,错义突变占21%,剪接位点突变占19%。但中国人群中最多见的PHEX基因突变类型是无义突变(约占30%),其次为错义突变(约占22%),剪接位点突变约占21%。本研究2例XLH患儿检测到PHEX基因突变,分别为移码突变c.931dupC和剪接位点突变IVS14+1G > A,均为国际上首次报道的新突变。在我国已报道的XLH病例中[9-14, 16-17],PHEX基因突变以E18、E22外显子突变更为多见;而在PHEX数据库中,以E09、E22外显子突变较为常见。这提示PHEX基因突变呈散在分布,可能与种族、性别、地区等因素相关。
本研究先证者1的IVS14+1G > A突变位于14号内含子,MUTATION TASTER(www.mutationtaster.org/)生物信息学分析提示突变“GA-A-GA”为经典的剪切位点突变方式,可影响外显子的剪切,导致外显子跳跃,产生不完整的氨基酸链,合成异常蛋白。PHEX数据库中共报道了4种类型的内含子14(I14)基因突变,其中3例为碱基缺失(IVS14,c.1584+3delGAGT;IVS14+1,c.1586+1delGA;IVS14+3,c.1586+3delGAGT),另1例为同区域剪接位点碱基突变(IVS14-1,c.1587-1G > C)导致剪接位点异常[8, 18-20]。2012年Beck-Nielsen等[8]报道1例31岁的女性低血磷性佝偻病患者在PHEX基因内含子14位置上发生突变:IVS14-1,c.1587-1G > C,引起内含子保留和外显子跳跃,患者身高157 cm(-1.8 SD),表现为轻微骨骼畸形,严重牙病变,无骨折史;血磷低,1, 25(OH)2D3水平正常,PTH轻度升高,骨密度正常。本研究先证者1发生的IVS14+1G > A突变,影响了外显子剪切,可能导致外显子15的跳跃而产生不完整的氨基酸链。本研究与Beck-Nielsen等报道的患者相比,血生化表现一致,但尚无牙病变,下肢畸形较严重,身高较正常同龄儿减低。这可能与种族、年龄或性别不同有关,也可能PHEX基因突变即使发生在同一内含子不同位置,疾病的表现在也可不一致。2013年,Yue等[14]发现3例患儿分别在内含子I10(c.1174-1G > A)、I15(c.1646-2A > T)、I17(c.1768+2T > G)发生突变,这3例患儿均为男性,起病年龄0.75~5岁,2例有家族史、1例为散发,均有“O型腿”表现,无牙龈病变,与本研究的患儿表现一致。这表明基因突变的类型与表型不一定有明显的相关性。
本研究先证者2检测到PHEX基因位于8号外显子的c.931dupC移码突变:p.Gln311Profs*13,MUTATION TASTER(www.mutationtaster.org/)生物信息学分析提示,它可导致311位的氨基酸由Gln(谷氨酰胺)转变为Pro(脯氨酸),在323位出现终止密码“UGA”,编码的氨基酸序列转变为P PVRLA GLHQE GH*,导致编码氨基酸提前终止、PHEX蛋白结构及功能异常,为致病性突变。该患儿骨骼畸形明显,提示移码突变产生的截短蛋白可能导致严重的表型。目前PHEX database数据库已报道6例外显子8突变(C.871C > T,c.931C > T,c.888+2G > T,c.904A > G,c.914T > C,c.897_898del),其中5例点突变(2例错义突变、3例无义突变),1例为缺失突变[1, 18, 20-23]。目前PHEX数据库中尚无8号外显子插入突变的报道。
本研究2例XLH患儿检测到2个国际上首次报道的PHEX基因新突变,丰富了XLH的致病基因谱,有助于深入认识PHEX基因的异质性。
Biography
冉情, 女, 硕士研究生
References
- 1.Francis F, Strom TM, Hennig S, et al. Genomic organization of the human PEX gene mutated in X-linked dominant hypophosphatemic rickets. Genome Res. 1997;7(6):573–585. doi: 10.1101/gr.7.6.573. [DOI] [PubMed] [Google Scholar]
- 2.Bowe AE, Finnegan R, Jan de Beur SM, et al. FGF-23 inhibits renal tubular phosphate transport and is a PHEX substrate. Biochem Biophys Res Commun. 2001;284(4):977–981. doi: 10.1006/bbrc.2001.5084. [DOI] [PubMed] [Google Scholar]
- 3.Xia WB, Jiang Y, Li M, et al. Levels and dynamic changes of serum fibroblast growth factor 23 in hypophosphatemic rickets/osteomalacia. http://www.medscape.com/medline/abstract/20529556. Chin Med J (Eng1) 2010;123(9):1158–1162. [PubMed] [Google Scholar]
- 4.Liu S, Zhou J, Tang W, et al. Pathogenic role of Fgf23 in Hyp mice. Am J Physiol Endocrinol Metab. 2006;291(1):E38–E49. doi: 10.1152/ajpendo.00008.2006. [DOI] [PubMed] [Google Scholar]
- 5.Thakker RV, O'Riordan JL. Inherited forms of rickets and osteomalacia. Baillieres Clin Endocrinol Metab. 1988;2(1):157–191. doi: 10.1016/S0950-351X(88)80012-0. [DOI] [PubMed] [Google Scholar]
- 6.Clausmeyer S, Hesse V, Clemens PC, et al. Mutational analysis of the PHEX gene:novel point mutations and detection of large deletions by MLPA in patients with X-linked hypophosphatemic rickets. Calcif Tissue Int. 2009;85(3):211–220. doi: 10.1007/s00223-009-9260-8. [DOI] [PubMed] [Google Scholar]
- 7.Whyte MP, Schranck FW, Armamento-Villareal R. X-linked hypophosphatemia:a search for gender, race, anticipation, or parent of origin effects on disease expression in children. http://press.endocrine.org/doi/10.1210/jcem.81.11.8923863. J Clin Endocrinol Metab. 1996;81(11):4075–4080. doi: 10.1210/jcem.81.11.8923863. [DOI] [PubMed] [Google Scholar]
- 8.Beck-Nielsen SS, Brixen K, Gram J, et al. Mutational analysis of PHEX, FGF23, DMP1, SLC34a3 and CLCN5 in patients with hypophosphatemic rickets. J Hum Genet. 2012;57(7):453–458. doi: 10.1038/jhg.2012.56. [DOI] [PubMed] [Google Scholar]
- 9.宋 莹, 麻 宏伟, 黎 芳, et al. X-连锁低磷性佝偻病的基因突变分析. http://www.cjcp.org/CN/abstract/abstract13199.shtml. 中国当代儿科杂志. 2013;15(11):928–931. doi: 10.7499/j.issn.1008-8830.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 10.许莉军, 夏维波. 低血磷性佝偻病的分子遗传学研究[D]. 北京: 北京协和医学院, 2015.
- 11.王静, 金春莲. 抗维生素D佝偻病PHEX基因诊断研究[D]. 沈阳: 中国医科大学, 2007.
- 12.唐 佳, 潘 敬新, 蒋 玮莹, et al. 一例抗维生素D佝偻病的基因诊断和新突变的致病性鉴定. http://www.cnki.com.cn/Article/CJFDTOTAL-ZLYD201205033.htm 中华临床医师杂志. 2012;6(5):1226–1230. [Google Scholar]
- 13.Yuan L, Wu S, Xu H, et al. Identification of a novel PHEX mutation in a Chinese family with X-linked hypophosphatemic rickets using exome sequencing. https://www.ncbi.nlm.nih.gov/pubmed/25060345. Biol Chem. 2015;396(1):27–33. doi: 10.1515/hsz-2014-0187. [DOI] [PubMed] [Google Scholar]
- 14.Yue H, Yu JB, He JW, et al. Identification of two novel mutations in the PHEX gene in Chinese patients with hypophosphatemic rickets/osteomalacia. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0097830. PLoS One. 2013;9(5):e97830. doi: 10.1371/journal.pone.0097830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holm IA, Nelson AE, Robinson BG, et al. Mutational analysis and genotype-phenotype correlation of the PHEX gene in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab. 2001;86(8):3889–3899. doi: 10.1210/jcem.86.8.7761. [DOI] [PubMed] [Google Scholar]
- 16.Kang QL, Xu J, Zhang Z, et al. Three novel PHEX gene mutations in four Chinese families with X-linked dominant hypophosphatemic rickets. Biochem Biophys Res Commun. 2012;423(4):793–798. doi: 10.1016/j.bbrc.2012.06.042. [DOI] [PubMed] [Google Scholar]
- 17.Zhu X, Li M, Pan H, et al. Analysis of the parental origin of de novo MECP2 mutations and X chromosome inactivation in 24 sporadic patients with Rett syndrome in China. J Child Neurol. 2010;25(7):842–848. doi: 10.1177/0883073809350722. [DOI] [PubMed] [Google Scholar]
- 18.Gaucher C, Walrant-Debray O, Nguyen TM, et al. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet. 2009;125(4):401–411. doi: 10.1007/s00439-009-0631-z. [DOI] [PubMed] [Google Scholar]
- 19.Filisetti D, Ostermann G, von Bredow M, et al. Non-random distribution of mutations in the PHEX gene, and under-detected missense mutations at non-conserved residues. Eur J Hum Genet. 1999;7(5):615–619. doi: 10.1038/sj.ejhg.5200341. [DOI] [PubMed] [Google Scholar]
- 20.Holm IA, Huang X, Kunkel LM, et al. Mutational analysis of the PHEX gene in patients with X-linked hypophosphatemic rickets. http://www.endocrine-abstracts.org/ea/0021/ea0021P14.htm. Am J Hum Genet. 1997;60(4):790–797. [PMC free article] [PubMed] [Google Scholar]
- 21.Sato K, Tajima T, Nakae J, et al. Three novel PHEX gene mutations in Japanese patients with X-linked hypophosphatemic rickets. Pediatr Res. 2000;48(4):536–540. doi: 10.1203/00006450-200010000-00019. [DOI] [PubMed] [Google Scholar]
- 22.Rowe PS, Oudet CL, Francis F, et al. Distribution of mutations in the PEX gene in families with X-linked hypophosphatemic rickets (HYP) Hum Mol Genet. 1997;6(4):539–549. doi: 10.1093/hmg/6.4.539. [DOI] [PubMed] [Google Scholar]
- 23.Raeder H, Shaw N, Netelenbos C, et al. A case of X-linked hypophosphatemic rickets:complications and the therapeutic use of cinicalcet. http://www.eje-online.org/content/159/suppl_1/S101.long. Eur J Endocrinol. 2008;159(Suppl 1):S101–S105. doi: 10.1530/EJE-08-0383. [DOI] [PubMed] [Google Scholar]