

Abstract
MECP2重复综合征(MDS)是儿科少见病,主要表现为运动发育落后、语言缺失或落后、反复感染、严重智力障碍、癫癎、孤独症样表现及婴儿早期肌张力低下等。该文3例患儿均为男孩,病例1、2首发表现为运动发育落后、语言缺失或落后,病例3首发表现为反复感染,查体均有肌张力均低下、病理征均阴性。病例1有全身强直-阵挛发作,脑电图示局灶性发作,予奥卡西平、左乙拉西坦、氯硝基安定联合抗癫癎治疗,癫癎发作控制。病例3出现过失神发作1次及点头发作3次,发作间期多次脑电图正常,未予抗癫癎治疗。3例患儿的反复感染随年龄增长得到改善,语言及智力改善不明显。微阵列比较基因组杂交技术(aCGH)检测发现3例患儿的X染色体存在MECP2基因重复,确诊为MDS。对于发育落后伴反复感染、癫癎发作的患儿,应考虑到MDS可能,早期行aCGH检测有助于诊断。
Keywords: MECP2重复综合征, 癫癎, 运动发育落后, 语言发育落后, 反复感染, 儿童
Abstract
MECP2 duplication syndrome (MDS) is a rare pediatric disease and mainly manifests as delayed motor development, language loss or delay, recurrent infection, severe intellectual disability, epilepsy, autistic symptoms, and early infantile hypotonia. In this article, the three children with this disease were all boys. Cases 1 and 2 had delayed motor development, and language loss or delay as initial manifestations, and case 3 had recurrent infection as initial manifestation. Physical examination showed hypotonia and negative pathological signs in each case. Case 1 had tonic-clonic seizures and electroencephalography showed focal seizures, for which he was given oxcarbazepine, levetiracetam, and clonazepam as the antiepileptic treatment to control seizures. Case 3 experienced one absence seizure and three head-nodding seizures with normal electroencephalographic findings during these seizures, and therefore, he was not given antiepileptic treatment. In each case, recurrent infection was improved with the increase in age, but there were no significant improvements in language or intelligence. Array-based comparative genomic hybridization (aCGH) showed MECP2 duplication in X chromosome in each case, and so they were diagnosed with MDS. MDS should be considered for children with delayed development complicated by recurrent infection and epileptic seizures, and early aCGH helps with the diagnosis of this disease.
Keywords: MECP2 duplication syndrome, Epilepsy, Delayed motor development, Delayed language development, Recurrent infection, Child
MECP2重复综合征(methyl-CpG-binding protein 2 duplication syndrome, MDS, OMIM#300260)是由MECP2基因重复突变导致的疾病,主要表现为运动发育落后、语言缺失或落后、反复感染、严重智力障碍、癫癎、孤独症样表现及婴儿早期肌张力低下等[1]。目前报道的MECP2重复综合征患者约200例;我国首例于2012年报道,至今已报道20余例,其中包括6个家系[2-6]。MECP2重复综合征是儿科少见病,容易漏诊、误诊,现将近4年收治的3例MDS患儿临床资料总结并报告如下。
1. 资料与方法
1.1. 研究对象
收集中山大学孙逸仙纪念医院儿科神经专科2012年10月至2016年10月期间确诊的3例MECP2重复综合征患儿临床资料。3例患儿均为男性,来自3个不同的家庭,确诊年龄为1岁5个月至10岁6个月,20 d至2年确诊。诊断标准参照Van Esch的研究[7]:(1)以癫癎、严重智力障碍、语言发育落后或缺失、运动发育落后、早发的肌张力低下,以及反复呼吸道感染为主要表现;(2)基因检测发现chrXq28区域存在重复片段,包含完整的MECP2基因。
1.2. 基因检测
经患儿监护人知情同意,采集患儿静脉血2 mL,行微阵列比较基因组杂交技术(array based comparative genomic hybridization, aCGH)进行基因检测,由广州金域医学检验中心完成。
2. 结果
2.1. 临床资料
病例1(就诊年龄8岁6个月)、病例2(就诊年龄1岁5个月)的首发症状均为运动、语言发育落后,均有反复上呼吸道感染。病例3(就诊年龄11个月)因反复咳嗽就诊,发现运动发育落后。3例查体仅病例1眼距较宽,肌张力均低下(病例1其后出现下肢肌张力增高),病理征均阴性。病例1于8岁8个月出现频繁癫癎发作,表现为全身强直-阵挛发作,每日发作5~6次,脑电图示局灶性发作,头颅MRI示左侧侧脑室后角小软化灶(图 1);予奥卡西平、左乙拉西坦、氯硝基安定联合抗癫癎治疗至今,现癫癎发作1~2次/月;9岁时出现特发性中枢性性早熟;随访至12岁行走仍欠稳、语言发育无明显进步。病例2仅予随访,至4岁只能咿呀发音和扶走。病例3于1岁9个月出现失神发作1次,2岁2个月出现点头发作3次,发作间期多次脑电图正常,头颅MRI示胼胝体发育不良(图 2),未予抗癫癎治疗,随访至3岁无抽搐发作,只能发单音和扶走。
1.
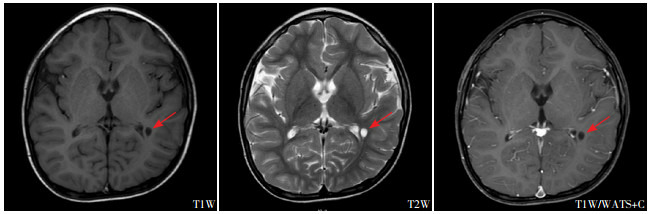
病例1头颅MRI
左侧枕叶紧邻侧脑室后角旁小软化灶(箭头所示)。
2.
病例3头颅MRI
胼胝体发育不良(箭头所示)。

病例1既往有头颅外伤史(具体不详);病例2有新生儿高胆红素血症、先天性喉软骨发育不良、缺铁性贫血病史;病例3有新生儿颅内出血、新生儿高胆红素血症、轻型β地中海贫血史。3例患儿母孕期均无特殊。
2.2. 微阵列比较基因组杂交技术检测结果
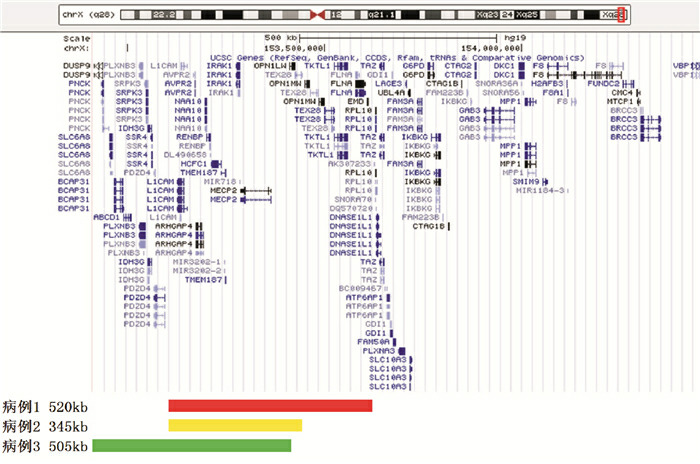
3例患儿的chrXq28区域重复片段示意图见图 3。病例1在chrXq28区域发生520 kb片段重复,arr[hg19]Xq28(153,102,572-153,622,165)×2,即患儿X染色体长臂2区8带存在2个拷贝的重复片段,重复片段定位于153,102,572-153,622,165,该片段大小为520 kb。其母未行相应检测。病例2在chrXq28区域发生345 kb片段重复,arr[hg19]Xq28(153,093,870-153,438,781)×2。其母亲为携带者:chrXq28区域发生443 kb片段重复,arr[hg19]Xq28(153,085,280-153,527,850)×3。病例3在chrXq28区域发生505 kb片段重复,arr[hg19]Xq28(152,916,853-153,421,839)×2。其母未行相应检测。
3.
3例患儿Xq28区的重复片段示意图
黑框标注的MECP2基因位于153,287,264-153,363,188,大小为75,925 bp;红色、黄色、绿色条形分别表示3例患儿重复片段的定位、大小及所包含的基因,涵盖了完整的MECP2基因。IRAK1(红色框标注)基因也存在重复拷贝。
3. 讨论
MECP2重复综合征是儿科少见病。首例MECP2重复综合征于1999年由Lubs[8]报道,当时被命名为Lubs X连锁智力障碍综合征(Lubs X-linked mental retaradation syndrome)。该综合征后来被证明为MECP2基因重复所致,2010年被命名为MECP2重复综合征(MECP2 duplication syndrome, MDS, OMIM#300260)[1]。
目前MECP2重复综合征发病率不明,约占特发性X连锁智力障碍疾病(X-linked intellectual disability, XLID)的1%,占智力障碍合并严重进展性神经系统症状的男性患儿的2%[9-10]。由于对该病的认识不足及基因检测未普及,该病的发病率可能被低估[11]。从2013年6月至今,我科300多例住院患儿行aCGH检测,诊断MECP2重复综合征3例。
MECP2基因位于X染色体长臂2区8带(Xq28),[hg19]153,287,264-153,363,188,大小为75 925 bp,有4个外显子,其侧翼序列区域富含低拷贝的重复序列(low copy-repeats, LCRs)及微同源序列,易诱发DNA双链松解,发生基因结构重排[3]。基因重排分为两种:简单重排,即重复序列为两个拷贝;复杂重排在两个拷贝之间含有正常或倒置的基因序列,或重复序列为3个拷贝及以上[12-13]。复杂重排患儿临床症状更重。本课题组3例患儿均为简单重排。
MECP2基因编码的MECP2蛋白又称甲基化CpG结合蛋白2,在脑中含量极为丰富,是神经元染色质的重要组成部分,是一个复杂的多功能核蛋白。MECP2具有抑制转录、激活转录、调节染色体构象、参与RNA剪切等多种功能[14]。研究发现,MECP2基因可以激活2 184个基因,也可抑制377个基因表达[15]。体外实验表明,海马和皮质神经元过表达MECP2蛋白时,树突分支形成减少,中枢神经系统可塑性受损,造成学习和记忆损伤[16],表现出智力及语言的障碍。另外,过表达的MECP2显著增加了海马细胞兴奋性微型突触传递[17],可导致癫癎发作。有学者发现MDS患儿的血浆F2-异构前列素水平特异性升高,与扩增的MECP2大小显著相关,推测MECP2蛋白的精确表达对稳定内环境氧化应激起关键作用,同时也与中枢神经系统氧化还原反应的平衡相关[18]。
MECP2重复综合征患儿几乎都存在IRAK1基因的重复[11]。本研究3例患儿也存在此现象(图 3)。IRAK1基因编码白介素受体相关激酶1,与MECP2基因仅有1 922 bp的距离,与反复感染可能存在某种关联[11]。但有学者认为重复表达的MECP2本身也可能引起免疫功能异常[19]。因此,重复的IRAK1与MECP2重复综合征患儿反复感染的关系仍不明确。
男性MECP2重复综合征患儿主要表现为癫癎、严重智力障碍、运动发育落后、语言缺失、反复呼吸道感染以及孤独症样表现,查体可见特殊面容、肌张力低下,头颅MRI无特征性改变,但可有胼胝体发育不良、小脑萎缩及脑室周围白质信号异常等[1, 11, 20]。MECP2重复综合征患儿的智力、语言落后并不随着年龄增长而改善;29%的MDS患儿可获得行走能力[21];超过55%的患儿2岁前曾因反复呼吸道感染住院,感染的频次随年龄增加逐渐减少。有学者认为MECP2重复综合征的癫癎发作与MECP2重复片段的大小相关[22]。超过90%的MECP2重复综合征患儿在青春期观察到癫癎发作,半数患儿每天均有发作,发作形式有强直阵挛发作、失张力发作、失神发作等,脑电图有很强的异质性[23]。本研究病例1重复片段为520 kb,于8岁6个月出现强直阵挛发作,发作次数逐渐增多,发作间期脑电图由正常演变到典型癫癎样放电;病例3重复片段为505 kb,1岁9个月出现过数次点头发作、失神发作,追踪至4岁无抽搐表现。另外,病例1在9岁出现特发性中枢性性早熟,这在以往的MECP2重复综合征患儿中未见报道。两者间的关系需进一步探讨。
MECP2重复综合征尚没有统一的诊断标准,主要根据临床表现(运动语言发育落后、反复感染、智力障碍、癫癎、孤独症样表现、肌张力低下)及基因检测确诊。由于对该病的认识不足,患儿的两个核心症状:运动、语言发育落后和反复感染容易被割裂开来,而反复感染可随着年龄增加逐渐减少,对于年长儿易被忽视,导致漏诊误诊;此外,对基因检测的不重视,亦是漏诊的一个原因。
本病尚缺乏有效治疗手段,主要采取对症、支持治疗。出现癫癎发作的患儿需及时、规范的抗癫癎治疗,一般需多药联合,但疗效欠佳。最新研究表明,通过反义寡核苷酸技术可以恢复过表达的MECP2蛋白水平,从而使症状逆转,为MECP2重复综合征的基因治疗带来希望[20]。
综上所述,对于语言、运动等全面发育落后,特别是伴反复感染、癫癎的患儿,应尽早行aCGH检测,以避免MECP2重复综合征的误诊、漏诊,对该类疾病的高危家庭提供遗传咨询及产前诊断也非常必要。
志谢
感谢上海诺华贸易有限公司在本研究病例资料收集中给予的支持。
Biography
唐丹霞, 女, 硕士研究生
References
- 1.Ramocki MB, Tavyev YJ, Peters SU. The MECP2 duplication syndrome. Am J Med Genet A. 2010;152A(5):1079–1088. doi: 10.1002/ajmg.a.v152a:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xu X, Xu Q, Zhang Y, et al. A case report of Chinese brothers with inherited MECP2-containing duplication:autism and intellectual disability, but not seizures or respiratory infections. http://bmcmedgenet.biomedcentral.com/articles/10.1186/1471-2350-13-75. BMC Med Genet. 2012;13:75. doi: 10.1186/1471-2350-13-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yi Z, Pan H, Li L, et al. Chromosome Xq28 duplication encompassing MECP2:Clinical and molecular analysis of 16 new patients from 10 families in China. Eur J Med Genet. 2016;59(6-7):347–353. doi: 10.1016/j.ejmg.2016.05.004. [DOI] [PubMed] [Google Scholar]
- 4.易 致, 潘 虹. MECP2重复综合征的研究进展. http://www.cnki.com.cn/Article/CJFDTOTAL-JKZL201602023.htm. 中华儿科杂志. 2015;53(10):792–795. [PubMed] [Google Scholar]
- 5.Lin DS, Chuang TP, Chiang MF, et al. De novo MECP2 duplication derived from paternal germ line result in dysmorphism and developmental delay. Gene. 2014;533(1):78–85. doi: 10.1016/j.gene.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Zhang Q, Zhao Y, Yang Y, et al. MECP2 duplication syndrome in a Chinese family. http://bmcmedgenet.biomedcentral.com/articles/10.1186/s12881-015-0264-0. BMC Med Genet. 2015;16:112. doi: 10.1186/s12881-015-0264-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Esch H. MECP2 duplication syndrome[M]//Pagon RA, Adam MP, Ardinger HH, et al. GeneReviews®.[Internet]. Seattle (WA):University of Washington, Seattle; 1993-2017.https://rarediseases.org/rare-diseases/mecp2-duplication-syndrome/
- 8.Lubs H, Abidi F, Bier JA, et al. XLMR syndrome characterized by multiple respiratory infections, hypertelorism, severe CNS deterioration and early death localizes to distal Xq28. Am J Med Genet. 1999;85(3):243–248. doi: 10.1002/(ISSN)1096-8628. [DOI] [PubMed] [Google Scholar]
- 9.Honda S, Hayashi S, Nakane T, et al. The incidence of hypoplasia of the corpus callosum in patients with dup (X)(q28) involving MECP2 is associated with the location of distal breakpoints. Am J Med Genet A. 2012;158A(6):1292–1303. doi: 10.1002/ajmg.a.35321. [DOI] [PubMed] [Google Scholar]
- 10.Lugtenberg D, Kleefstra T, Oudakker AR, et al. Structural variation in Xq28:MECP2 duplications in 1% of patients with unexplained XLMR and in 2% of male patients with severe encephalopathy. Eur J Hum Genet. 2009;17(4):444–453. doi: 10.1038/ejhg.2008.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Van Esch H. MECP2 duplication syndrome. http://www.mecp2duplication.com/ Mol Syndromol. 2012;2(3-5):128–136. doi: 10.1159/000329580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carvalho CM, Zhang F, Liu P, et al. Complex rearrangements in patients with duplications of MECP2 can occur by fork stalling and template switching. Hum Mol Genet. 2009;18(12):2188–2203. doi: 10.1093/hmg/ddp151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.章 清萍, 包 新华. MECP2重复综合征研究进展. http://d.g.wanfangdata.com.cn/Periodical_zhyxycx201503034.aspx. 中华医学遗传学杂志. 2015;32(3):426–429. doi: 10.3760/cma.j.issn.1003-9406.2015.03.028. [DOI] [PubMed] [Google Scholar]
- 14.Ausió J, Martínez de Paz A, Esteller M. MeCP2:the long trip from a chromatin protein to neurological disorders. Trends Mol Med. 2014;20(9):487–498. doi: 10.1016/j.molmed.2014.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Chahrour M, Jung SY, Shaw C, et al. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science. 2008;320(5880):1224–1229. doi: 10.1126/science.1153252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Na ES, Nelson ED, Kavalali ET, et al. The impact of MeCP2 loss-or gain-of-function on synaptic plasticity. Neuropsychopharmacology. 2013;38(1):212–219. doi: 10.1038/npp.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Na ES, Nelson ED, Adachi M, et al. A mouse model for MeCP2 duplication syndrome:MeCP2 overexpression impairs learning and memory and synaptic transmission. J Neurosci. 2012;32(9):3109–3117. doi: 10.1523/JNEUROSCI.6000-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Signorini C, De Felice C, Leoncini S, et al. MECP2 duplication syndrome:evidence of enhanced oxidative stress. A omparison with Rett syndrome. PLoS One. 2016;11(3):e0150101. doi: 10.1371/journal.pone.0150101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang T, Ramocki MB, Neul JL, et al. Overexpression of methyl-CpG binding protein 2 impairs T (H)1 responses. Sci Transl Med. 2012;4(163):163ra158. doi: 10.1126/scitranslmed.3004430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sztainberg Y, Chen HM, Swann JW, et al. Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. https://static1.squarespace.com/static/55325e9be4b0166f27e9ed3f/t/56cb73e2e707ebc39cec6da5/1456174078235/Zoghbi+duplication+reversal.pdf. Nature. 2015;528(7580):123–126. doi: 10.1038/nature16159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lim Z, Downs J, Wong K, et al. Expanding the clinical picture of the MECP2 duplication syndrome. Clin Genet. 2017;91(4):557–563. doi: 10.1111/cge.2017.91.issue-4. [DOI] [PubMed] [Google Scholar]
- 22.Vignoli A, Borgatti R, Peron A, et al. Electroclinical pattern in MECP2 duplication syndrome:eight new reported cases and review of literature. Epilepsia. 2012;53(7):1146–1155. doi: 10.1111/epi.2012.53.issue-7. [DOI] [PubMed] [Google Scholar]
- 23.Ramocki MB, Peters SU, Tavyev YJ, et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol. 2009;66(6):771–782. doi: 10.1002/ana.v66:6. [DOI] [PMC free article] [PubMed] [Google Scholar]