

Abstract
渐进性早发性脑病是由于NAXE基因变异导致的致死性脑病。本研究报道1例渐进性早发性脑病的临床和遗传性特征。患儿,男,4岁,因行走不稳,肢体无力2年就诊。患儿及其兄(已去世)均在2岁左右起病,均表现为斜视、共济失调、肌张力降低、发育落后,且感染后多次出现呼吸衰竭。患儿NAXE基因存在新发复合杂合突变:c.255(外显子2)A > T来自患儿母亲,c.361(外显子3)G > A来自患儿父亲。NAXE基因编码NADHX和NADPHX细胞代谢产物修复所必需的差向异构酶。本病存在线粒体NAD(P)HX修复系统的缺陷,进展快,极易在感染后迅速出现呼吸衰竭。
Keywords: 渐进性早发性脑病, NAXE基因, 儿童
Abstract
Early-onset progressive encephalopathy is a lethal encephalopathy caused by NAXE gene mutations. This paper reports the clinical and genetic features of a patient with early-onset progressive encephalopathy. A 4-yearold boy admitted to the hospital had repeated walking instability and limb weakness for 2 years. The patient and his elder brother (already dead) had clinical onset at 2 years of age. Both of them showed symptoms such as strabismus, ataxia, reduced muscle tone, delayed development, and repeated respiratory failure after infection. The NAXE gene of the patient showed new compound heterozygous mutations, i.e., c.255 (exon 2) A > T from his mother and c.361 (exon 3) G>A from his father. The NAXE gene encodes an epimerase that is essential for the repair of cellular metabolites of NADHX and NADPHX. This disease is associated with a defciency of the mitochondrial NAD(P)HX repair system. Patients usually have rapid disease progression. They are also quite likely to have respiratory failure immediately after infection.
Keywords: Early-onset progressive encephalopathy, NAXE gene, Child
渐进性早发性脑病是一种非常罕见的神经系统疾患,是由于NAXE基因变异导致。临床表型包括眼部症状如眼球震颤、斜视,神经系统症状如发育落后、肌张力减退、共济失调、痉挛发作等,感染可导致患者迅速出现呼吸衰竭、危及生命。本文结合一例渐进性早发性脑病患者的临床资料并复习相关文献,探讨该病的临床及遗传学特点,以提高对本病的认识。
1. 资料与方法
1.1. 病例介绍
患儿,男4岁,因反复出现行走不稳,肢体无力2年就诊。近2年患儿反复出现站立不稳,步态蹒跚,行走时摇晃不定,多在发热,腹泻等诱因后出现,不伴有头痛、呕吐、抽搐等。患儿2岁起感染后易出现呼吸衰竭,曾4次接受呼吸机治疗。患儿因斜视于2岁行手术治疗。患儿为第二胎,足月顺产出生,无窒息史,7个月能独坐,10个月左右叫“爸爸、妈妈”,1岁2个月能独立行走。患儿哥哥2岁多出现行走不稳,因斜视于2岁半行手术治疗,3岁时因呼吸衰竭死亡。父母非近亲结婚,没有类似症状。入院时查体:体温37.8℃,心率120次/min,呼吸28次/min,发育稍落后,营养尚可,神志清楚,呼吸运动对称,未闻及干湿啰音。四肢肌张力降低,肌力4~5级,病理反射阴性,腹壁反射、提睾反射减弱。行走不稳,指鼻实验阳性,闭目难立征(+)。实验室检查:血常规正常;肝功能:谷草转氨酶(AST)56 U/L(参考值:8~40 U/L),谷丙转氨酶(ALT)76 U/L(参考值:0~40 U/L),余项正常;降钙素原5.17 ng/mL (参考值: < 0.05 ng/mL);血气分析:pH 7.33(参考值:7.35~7.45),余项正常;血乳酸、β-羟丁酸、丙酮酸、血氨正常;肌钙、肌红蛋白正常;脑脊液常规、生化未见异常。胸片:双肺炎症。腹部超声和心脏超声未见异常。脑电图、心电图正常。一次血串联质谱、尿气相色谱-质谱联用筛查提示C18-OH增高明显,考虑外长链羟基脂肪酸代谢异常可疑;复查未见明显异常。全脊髓段磁共振正常。头颅磁共振正常,出院后1年复查显示脑萎缩,见图 1。线粒体基因、线粒体核基因及线粒体呼吸链酶学检查正常。
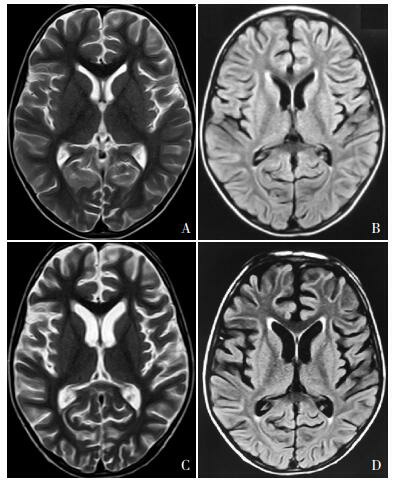
1.
头颅磁共振检查
住院时头部MRI的T2WI(A)和T2WI-FLAIR(B)显示双侧大脑半球未见脑萎缩。出院后1年复查头部MRI,T2WI(C)和T2WI-FLAIR(D)显示双侧大脑半球脑沟深宽,脑回变细,双侧脑室稍增宽。
入院后患儿迅速出现呼吸衰竭,予呼吸机辅助通气;病程中出现一次惊厥,持续约1 min,对症治疗后缓解。
1.2. 致病基因检测
取得知情同意,采集患者全血2 mL(EDTA抗凝),提取基因组DNA。参照文献与OMIM数据库,进行目标基因全外显子捕获,Illumina平台测序及数据分析。高通量测序由智因东方公司完成。并利用Sanger测序进行患者及患儿父母的突变位点验证。根据高通量测序的位点序列设计引物(见表 1),采用PCR方法进行扩增。PCR(体系为50 μL)反应条件为:95℃预变性5 min,95℃变性30 s,55℃退火30 s,72 ℃链延伸30 s,扩增30个循环;最后72℃补充延伸10 min。
1.
NAXE基因所验证位点的引物序列
| 引物名称 | 碱基序列 | 退火温度(℃) | 扩增产物大小(bp) |
| (c.361(exon3)G > A)F | CTCGTAGCCCTAGGAAGAAAGTG | 255 | 60 |
| (c.361(exon3)G > A)R | TATGGTCTATTACCGCCTGAAACC | ||
| (c.255(exon2)A > T)F | AGCTGTCTCTCCCACCCCTTT | 258 | 60 |
| (c.255(exon2)A > T)R | CACCTGCGCGACAGAGAACAC |
1.3. 突变的生物信息学分析
使用生物信息学软件进行蛋白功能预测。建议判定标准为:Provean预测值< -1.3提示有害;Polyphen2预测值> 0.8提示可能有害;Sift预测值< 0.05提示有害;M-CAP预测值> 0.025提示有害;REVEL预测值> 0.5提示有害。
2. 结果
2.1. 患者致病基因突变分析
患儿NAXE基因存在复合杂合突变,c.255(外显子2)A > T和c.361(外显子3)G > A,分别来自于父母,其父母临床表型正常。见图 2。
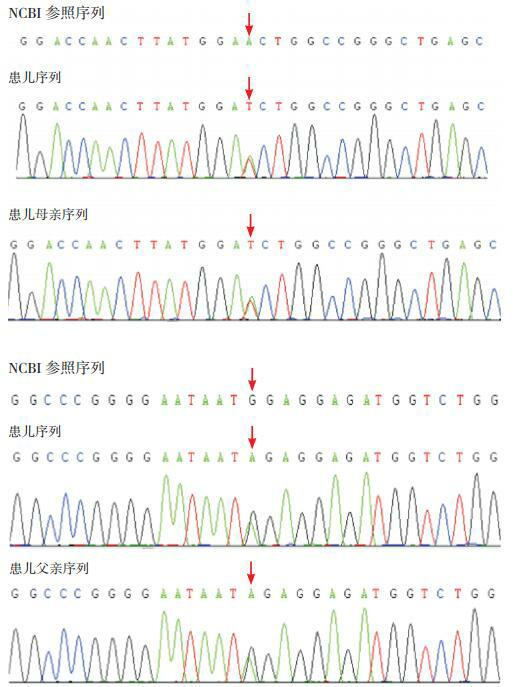
2.
患者及其父母的NAXE基因突变
患儿NAXE基因存在复合杂合突变,c.255 A>T(来自患儿母亲)和c.361 G>A(来自患儿父亲),突变位点如箭头所示。
2.2. 突变的生物信息学分析
检索文献及基因组数据库,患儿的NAXE基因突变未见收录,c.255 A > T及c.361 G > A均为错义突变。对于c.255 A > T,生物信息学软件Provean分析的预测值为-2.97(有害);Polyphen2预测值1.0(可能有害);Sift预测值为0.001(有害);M-CAP预测值0.0765(有害);REVEL预测值0.5600(有害)。c.361 G > A的Provean预测值为-7.95(有害);Polyphen2预测值1.0(可能有害);Sift预测值为0(有害);M-CAP预测值0.1965(有害);REVEL预测值0.9210(有害)。因此这2个突变均可能影响蛋白的功能,为可能的致病性突变。
2.3. 治疗
入院后予积极抗感染、控制惊厥发作、呼吸机辅助通气等对症治疗,予辅酶Q10、维生素B1、维生素B2、烟酰胺等改善内环境紊乱。患儿顺利撤除呼吸机,好转出院。出院时肌力约4~5级,出院后随访至今(2年余)患儿仍有共济失调表现,无惊厥发作。
3. 讨论
渐进性早发性脑病是一种非常罕见的常染色体隐性遗传性脑病,病情呈逐渐加重,可累及眼、呼吸系统、皮肤、肌肉软组织及神经系统等多系统[6]。临床表型包括中枢神经系统受累、眼球震颤、斜视、呼吸衰竭、皮肤红斑大疱以及气泡样损伤、肌张力低下等。中枢神经系统受累是本病重要特点,表现为急性发作的脑病,精神运动发育倒退,共济失调、震颤、四肢轻瘫,昏迷,脑水肿,脑萎缩,脑坏死,小脑水肿、脊髓病变和白质脑病等[6]。肌肉活检可发现线粒体损伤。本例患者突出表现为共济失调、肌张力低下、斜视以及反复的呼吸衰竭,临床特点与文献相符。患儿兄长也有行走不稳、斜视并且死于呼吸衰竭。反复出现的呼吸衰竭推测可能与线粒体能量代谢紊乱有关。
表 2总结了包括国外文献[6-7]报道及本研究患者的NAXE基因突变特点及临床特点。
2.
NAXE基因突变位点及方式
| 编号 | 性别 | NAXE基因突变位点 | 突变方式 | 首发症状 | 结局 |
| 1 | 男 | Mc.[177C > A]; [177C > A]; p.[Tyr59*]; [Tyr59*] | 无义突变 | 共济失调,四肢轻瘫 | 死亡 |
| 2 | 女 | c.[196C > T]; [516+1G > A]; p.[Gln66*]; [?] | 无义突变加剪切位点突变 | 肌张力降低,眼球震颤 | 死亡 |
| 3 | 男 | c.[804_807delinsA]; [804_ 807delinsA]; p.[Lys270del]; [Lys270del] | 缺失、插入 | 肌张力降低,共济失调,眼球震颤 | 死亡 |
| 4 | 男 | c.[653A > T]; [743delC]; p.[Asp218Val]; [Ala248Glufs*26] | 错义突变加移码突变 | 肌张力降低,共济失调,眼球震颤 | 死亡 |
| 5 | 男(4号患者同胞) | c.[653A > T]; [743delC]; p.[Asp218Val]; [Ala248Glufs*26] | 错义突变加移码突变 | 肌张力降低,抽搐 | 死亡 |
| 6~10 | 2男3女(5胞胎) | c. [281C > A]; (p.Ala94Asp) | 错义突变 | 肌力下降,发育倒退 | 死亡 |
| 11 | 男(本研究患者) | c.255A > T; p.E85D c.361G > A; p.G121R | 错义突变 | 斜视,共济失调 | 存活 |
患者首发症状均为共济失调、肌张力下降或发育落后,均有眼球震颤,均在病程中出现呼吸衰竭。所有患者均检测到NAXE基因突变,表现为无义突变、剪切位点突变、错义突变或移码突变;有两个家系的同胞均检测到NAXE基因突变。
渐进性早发性脑病是由于NAXE基因变异导致。烟酰胺腺嘌呤二核苷酸(还原形式,NADH;氧化形式,NAD+)和烟酰胺腺嘌呤二核苷酸磷酸盐(还原形式,NADPH;氧化形式,NADP+)是线粒体呼吸链的电子传递系统的主要氧化还原等价物,参与机体的分解代谢或合成代谢反应,同时参与抵抗活性氧的生成,人类新陈代谢所需要的大多数酶均依赖于NADH和/或NADPH。NAXE是NAD(P)HX修复系统的重要组成部分,NAXE基因编码一个差向异构酶,催化R-NAD(P)HX转化为有细胞毒性的S-NAD(P)HX,随后S-NAD(P)HX在NAXD脱氢酶作用下再转化为S-NAD(P)H,避免有毒性的S-NAD(P)HX在体内堆积[1]。NAXE基因缺陷可导致NADH脱氢酶活性抑制,毒性代谢物S-NAD HX(P)堆积,机体遭受活性氧损伤[2-5]。本例患者经高通量测序发现NAXE基因存在新发复合杂合突变,c.255(外显子2)A > T,c.361(外显子3)G > A,蛋白功能预测可能有害,确诊渐进性早发性脑病。
渐进性早发性脑病非常罕见,临床表现不具有特异性,容易误诊为急性小脑性共济失调、病毒性脑炎等。对于具有眼球震颤、斜视、反复呼吸衰竭以及中枢神经系统受累表现的患儿需要警惕本病,建议完善基因检测。渐进性早发性脑病涉及到线粒体功能障碍,预后不佳,补充NAD、烟酰胺、辅酶Q10等辅酶以增加旁路电子传递、改善线粒体氧化磷酸化功能、清除自由基等,可能改善患儿预后。
Biography
俞丹, 女, 博士, 副教授。Email:Yd540@126.com
Funding Statement
四川省科技厅重点研发项目,立项编号:2018SZ0123
References
- 1.Marbaix AY, Noël G, Detroux AM, et al. Extremely conserved ATP-or ADP-dependent enzymatic system for nicotinamide nucleotide repair. Biol Chem. 2011;286(48):41246–41252. doi: 10.1074/jbc.C111.310847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marbaix AY, Tyteca D, Niehaus TD, et al. Occurrence and subcellular distribution of the NADPHX repair system in mammals. https://www.deepdyve.com/lp/portland-press/occurrence-and-subcellular-distribution-of-the-nad-p-hx-repair-system-C5NeNpTRcG. Biochem. 2014;460(1):49–58. doi: 10.1042/BJ20131482. [DOI] [PubMed] [Google Scholar]
- 3.Prabhakar P, Laboy JI, Wang J, et al. Effect of NADH-X on cytosolic glycerol-3-phosphate dehydrogenase. Arch Biochem Biophys. 1998;360(2):195–205. doi: 10.1006/abbi.1998.0939. [DOI] [PubMed] [Google Scholar]
- 4.Kremer LS, Danhauser K, Herebian D, et al. NAXE mutations disrupt the cellular NAD(P)HX repair system and cause a lethal neurometabolic disorder of early childhood. Am J Hum Genet. 2016;99(4):894–902. doi: 10.1016/j.ajhg.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorci-Thomas MG, Thomas MJ. AIBP, NAXE, and angiogenesis:What's in a name? Circ Res. 2017;120(11):1690–1691. doi: 10.1161/CIRCRESAHA.117.311023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kremer LS, Danhauser K, Herebian D, et al. NAXE mutations disrupt the cellular NAD (P)HX repair system and cause a lethal neurometabolic disorder of early childhood. Am J Hum Genet. 2016;99(4):894–902. doi: 10.1016/j.ajhg.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spiegel R, Shaag A, Shalev S, et al. Homozygous mutation in the APOA1BP is associated with a lethal infantile leukoencephalopathy. Neurogenetics. 2016;17(3):187–190. doi: 10.1007/s10048-016-0483-3. [DOI] [PubMed] [Google Scholar]