

Abstract
赖氨酸尿性蛋白耐受不良(LPI)是由SLC7A7基因突变引起的一种常染色体隐性遗传病,常常累及多个系统病变,肺部受累较为常见,儿童患者预后差。该文总结3例经SLC7A7基因分析确诊为LPI患儿的临床表现和基因突变特点。3例患儿均表现为断乳后不喜蛋白饮食、生长发育落后、贫血、肝脾肿大、骨质疏松等,均有尿乳清酸增高。2例表现有间质性肺炎、弥漫性肺间质病变。SLC7A7基因检测结果显示3例患儿中共检测出3种致病突变:c.1387delG(p.V463CfsX56)、c.1215G > A(p.W405X)和纯合c.625+1G > A。3例患儿诊断明确后,予低蛋白饮食,口服瓜氨酸100 mg/(kg·d)、蛋白琥珀酸铁4 mg/(kg·d)、葡萄糖酸钙锌10 mL/d、维生素D 400 IU/d,例3还予醋酸泼尼松5 mg/d治疗,症状和体征均有不同程度改善。LPI的氨基酸和有机酸代谢特点与尿素循环障碍较难鉴别,SLC7A7基因分析是LPI确诊的依据。
Keywords: 赖氨酸尿性蛋白耐受不良, 尿素循环障碍, 肺部疾病, SLC7A7基因, 儿童
Abstract
Lysinuric protein intolerance (LPI) is an autosomal recessive disorder caused by SLC7A7 gene mutation and often involves severe lesions in multiple systems. Lung involvement is frequently seen in children with LPI and such children tend to have a poor prognosis. This article summarizes the clinical manifestations and gene mutation characteristics of three children diagnosed with LPI by SLC7A7 gene analysis. All three children had the manifestations of aversion to protein-rich food after weaning, delayed development, anemia, hepatosplenomegaly, and osteoporosis, as well as an increase in orotic acid in urine. In addition, interstitial pneumonia and diffuse pulmonary interstitial lesions were observed in two children. SLC7A7 gene detection showed three pathogenic mutations in these children, namely c.1387delG(p.V463CfsX56), c.1215G > A(p.W405X) and homozygous c.625+1G > A. After a definite diagnosis was made, all three children were given a low-protein diet and oral administration of citrulline[100 mg/(kg·d)], iron protein succinylate[4 mg/(kg·d)], calcium and zinc gluconates oral solution (10 mL/day) and vitamin D (400 IU/day). In addition, patient 3 was given prednisone acetate (5 mg/day). The children had varying degrees of improvement in symptoms and signs. It is hard to distinguish LPI from urea cycle disorder due to the features of amino acid and organic acid metabolism in LPI, and SLC7A7 gene analysis is the basis for a definite diagnosis of LPI.
Keywords: Lysinuric protein intolerance, Urea cycle disorder, Lung disease, SLC7A7 gene, Child
赖氨酸尿性蛋白耐受不良(lysinuric protein intolerance, LPI)由Perheentupa于1965年首次报道[1],是阳离子氨基酸转运体缺陷导致尿中赖氨酸、精氨酸、鸟氨酸排泄增高,属常染色体隐性遗传。芬兰发病率最高,为1/76 000;其次日本、意大利较多报道,世界各地散发[2-4]。该病常累及呼吸、血液、肾脏和消化等多个系统,临床医生因对该病的认识不足,常常孤立分析各个系统的临床表现,进而发生误诊。该病虽然临床表现复杂,但仍有规律可循,肺间质病变常常是该病突出的临床表现及主要死因之一[5]。如出现肺间质病变并伴生长发育落后、贫血、肝脾肿大等症状和体征,同时实验室检查如血清铁蛋白、血氨、尿乳清酸等指标升高,血赖氨酸、精氨酸、鸟氨酸降低,应高度怀疑本病。尽早检测相关基因明确诊断,尽早治疗是改善患儿预后的关键。本文通过两家系3例患儿的临床及基因诊断资料分析,以加深临床医生对该病的认识。
1. 资料与方法
1.1. 研究对象
病例1:女,8岁8月。因面色苍白、发热伴加重1 d于2017年6月5日入院。患儿系第2胎第2产,足月顺产,出生体重2.15 kg,出生时有缺氧,于当地医院住院3 d(具体不详),生长发育同正常同龄儿。既往史:患儿自幼进食母乳及配方奶无明显异常,断奶后进食肉类、奶制品,易呕吐,喜食青菜和米饭。6年前外院诊断“贫血”,曾输血1次,2年前曾左侧膝部骨折。家族史:父亲39岁,体健;母亲33岁,幼时有过敏性紫癜病史;姐姐同为该病患儿(见病例2)。查体:身高110 cm(< -3 SD),体重15 kg(< -3 SD)。面色苍白,双下肢散见瘀斑,双侧颈部淋巴结肿大,左下颌腺肿痛。双肺呼吸音粗,未闻及干湿啰音。心脏查体未见异常。肝右肋下2 cm,脾左肋下5 cm,质地较硬,边界清楚。神经系统查体未见异常。辅助检查:血常规示WBC 1.8×109/L(参考值:5.0~12.0×109/L),Hb 61 g/L(参考值:110~160 g/L),PLT 81×109/L(参考值:100~300×109/L)。C反应蛋白(CRP)19.8 mg/L(参考值:0~10.0 mg/L)。肝功能:白蛋白(ALB)28.3 g/L(参考值:35.0~55.0 g/L),余正常。心肌酶:乳酸脱氢酶(LDH)1 327 IU/L(参考值:140~280 IU/L),余正常。电解质:血钙1.86 mmol/L(参考值:2.25~2.75 mmol/L),余正常。贫血七项:铁蛋白(FER)706 ng/mL(参考值:10~291 ng/mL),促红细胞素(EPO)107.87 mIU/mL(参考值:1.48~31.88 mIU/mL),叶酸(FA)7.24 ng/mL(参考值:8.86~96.24 ng/mL),可溶性转铁蛋白受体(sTFR)39.88 nmol/L(参考值:12.16~27.25 nmol/L)。血淀粉酶1 108 U/L(参考值:28~100 U/L),尿淀粉酶1 612 U/L(参考值:0~450 U/L)。血氨、血乳酸、可提取性核抗原抗体谱(ENA)、抗双链DNA抗体、抗核抗体、血管炎三项均正常。骨髓涂片示骨髓增生明显活跃,中性粒系成熟障碍伴巨幼样变,红系及巨核系未见明显异常,血小板减少,全片见少量组织细胞,偶见噬血细胞。彩超示左侧腮腺、颌下腺炎性增大,肝脏、脾脏弥漫性增大。左腕关节X线示骨质疏松。
病例2:女,11岁6月,病例1之姐姐。患儿系第1胎第1产,足月顺产,出生体重2.82 kg,出生情况可,生长发育正常。既往史:自幼不喜肉类、奶类,进食高蛋白食物后出现呕吐,平时易疲劳。查体:身高122 cm(< -2 SD),体重20.5 kg(< -3 SD)。面色苍白,心肺查体未见明显异常。肝右肋下2 cm,脾左肋下2 cm,质地较硬,边缘清楚。神经系统检查未见异常。辅助检查:血常规示WBC 3.1×109/L,Hb 103 g/L,PLT 104×109/L,余正常。肝功能:ALB 30.9 g/L,余正常。心肌酶:LDH 969 IU/L,余正常。电解质:血钙1.93 mmol/L,余正常。血脂:甘油三酯(TG)2.5 mmol/L(参考值: < 1.7 mmol/L),余正常。贫血七项:FER 1 826 ng/mL,余正常。血氨正常。左腕关节X线示骨质疏松。
病例3:女,1岁10月,因气促、纳差半年,加重伴咳嗽、发热1周于2018年2月21日入院。半年前无明显诱因出现气促,伴纳差,体重下降约2 kg,门诊多次Hb波动在65~75 g/L之间,无显著活动受限,未予特殊治疗。近1周患儿出现发热、咳嗽、气促、乏力和精神欠佳,无抽搐、无昏迷、无呕吐、无腹泻。患儿系第3胎第2产,足月顺产,出生体重3.1 kg,出生情况可。3月龄会抬头,5月龄会翻身,6月龄认人,10月龄会独坐,12月龄说话,14月龄会走。既往史无特殊。父母体健,无家族遗传病史。查体:体温38.7℃,脉搏192次/分,呼吸68次/分,血压102/70 mm Hg,身高76 cm(< -2 SD),体重9 kg(< -2 SD),神志清,精神反应差,贫血貌,口唇面色苍白,呼吸急促,三凹征(+),双肺呼吸音粗,双肺可闻及湿罗音、喘鸣音,心脏查体未见异常,腹软,肝右肋下2 cm,质软,脾左肋下3 cm,质硬。神经系统查体未见异常。双足轻度杵状趾。辅助检查:血常规示WBC 4.5×109/L,Hb 75 g/L,PLT 172×109/L,余正常。肝功能:门冬氨酸氨基转移酶(AST)129 IU/L(参考值:0~40 IU/L),ALB 28.6 g/L,余正常。心肌酶:LDH 2 244 IU/L,余正常。电解质:血钙2.11 mmol/L,余正常。血氨71.8 μmmol/L(参考值:14.0~65.0 μmmol/L)。血脂:TG 7.23 mmol/L,余正常。贫血七项:FER 7 842 ng/mL,EPO 186.87 mIU/mL,sTFR 74.31 nmol/L,余正常。乳酸正常。左腕关节X线示骨质疏松,彩超示肝、脾弥漫性增大,心脏无异常。纤维支气管镜示气管内膜炎症,取肺泡灌洗液做细胞病理,可见10%中性粒细胞,其余为组织细胞,含铁血黄素染色(-)。双肺CT示两肺纹理增多,两肺大片状模糊片絮影,双肺下叶为著,局部与相邻胸膜分界不清,双侧叶间胸膜增厚,双肺可见胸膜下线,考虑两肺弥漫性间质病变。
1.2. 遗传代谢病筛查
应用液相-串联质谱仪(API3200)行血氨基酸和酰基肉碱分析,尿有机酸分析在岛津气质联用仪(GP2020)上完成。
1.3. SLC7A7基因检测
抽取3例患儿及父母静脉血2 mL,应用DNA抽提试剂盒(GentraPuregene Blood Kit)提取全基因组DNA。送至北京金准医学检验所,应用二代测序技术检测遗传代谢病相关基因Panel(包含947个基因)寻找致病突变。高通量测序采用IDT公司的xGen Exome研究芯片,经Illumina Hiseq 2500平台完成测序,覆盖度不低于99%。找到突变位点后,用Sanger法进行验证。
1.4. 基因突变位点致病性分析
根据美国医学遗传学与基因组学学会(ACMG)遗传变异分类标准与指南[6]评判基因变异致病性。
2. 结果
2.1. 遗传代谢病筛查
例1、例3血精氨酸降低和血谷氨酰胺升高,3例尿乳清酸均升高,见表 1。
1.
三例赖氨酸尿性蛋白耐受不良患儿辅助检查资料
| 项目 | 参考值 | 例1 | 例2 | 例3 | ||||||
| 治疗前 | 治疗6月后 | 治疗1年后 | 治疗前 | 治疗后 | 治疗前 | 治疗后 | ||||
| 注:-示未测。 | ||||||||||
| 血常规 | ||||||||||
| 白细胞(×109/L) | 5.0~12.0 | 1.8 | 4.2 | 4.1 | 3.1 | 3.4 | 4.5 | 9.0 | ||
| 血红蛋白(g/L) | 110~160 | 61 | 86 | 92 | 103 | 117 | 75 | 104 | ||
| 血小板(×109/L) | 100~300 | 81 | 117 | 123 | 104 | 140 | 172 | 405 | ||
| 生化检查 | ||||||||||
| 白蛋白(g/L) | 35.0~55.0 | 25.6 | 25.7 | 23.2 | 30.9 | 31.8 | 28.6 | 30.4 | ||
| 乳酸脱氢酶(IU/L) | 140~280 | 1 327 | 420 | 258 | 969 | 1 011 | 7 842 | 1 492 | ||
| 铁蛋白(ng/mL) | 10~291 | 706 | 510 | 334 | 1 826 | 1 691 | 1 382 | 1 272 | ||
| 甘油三脂(mmol/L) | < 1.7 | 1.6 | 1.5 | 1.6 | 2.5 | 7.9 | 7.2 | 6.8 | ||
| 淀粉酶(U/L) | 28~100 | 1 108 | 114 | 168 | 128 | 116 | - | - | ||
| 血钙(mmol/L) | 2.25~2.75 | 1.86 | 1.91 | 2.06 | 1.93 | 2.14 | 2.11 | 2.18 | ||
| 血氨(μmol/L) | 14.0~65.0 | 55.2 | 42.4 | 81.8 | 45.5 | 49.4 | 71.8 | 57.2 | ||
| 遗传代谢病筛查 | ||||||||||
| 血精氨酸(μmol/L) | 1.0~70.0 | 0.8 | 1.2 | 2.5 | 2.4 | 5.9 | 0.9 | 5.0 | ||
| 血鸟氨酸(μmol/L) | 7.0~100.0 | 12.3 | 13.0 | 17.9 | 10.4 | 17.5 | 9.2 | 14.9 | ||
| 血瓜氨酸(μmol/L) | 5.5~45.0 | 15.3 | 22.1 | 20.1 | 23.1 | 36.6 | 12.1 | 62.9 | ||
| 血谷氨酰胺(μmol/L) | 1.0~50.0 | 57.6 | 28.3 | 17.6 | 38.0 | 20.2 | 45.4 | 15.1 | ||
| 尿乳清酸(mmol/molCr) | 0~2.0 | 11.7 | 0 | 0 | 4.7 | 0 | 184.2 | 0 | ||
2.2. 基因突变分析
发现例1、例2在SLC7A7基因存在c.1387delG(p.V463CfsX56)和c.1215G > A(p.W405X)复合杂合突变,分别来自父母,见图 1;例3在SLC7A7基因Intron 4处存在c.625+1G > A纯合剪接突变,分别源自父母,见图 2。c.1215G > A为新发突变,检索万方、中国知网、人类基因突变数据库(HGMD)、千人基因组和dbSNP数据库,均未见SLC7A7基因c.1215G > A突变。根据ACMG指南,c.1215G > A为无义突变(PVS1),这个变异不属于多态性变化,在人群中发生频率极低(PM2),认为c.1215G > A突变为致病性突变。
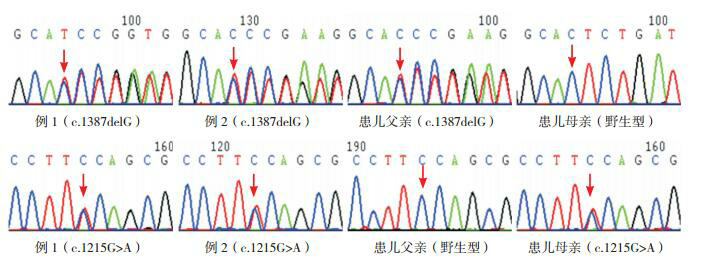
1.
患儿例1、例2(例1之姐)及其父母SLC7A7基因的Sanger测序图
红色箭头示碱基发生突变的位置。例1、例2 SLC7A7基因均检出c.1387delG和c.1215G>A复合杂合突变,父母分别携带c.1387delG、c.1215G>A杂合突变。
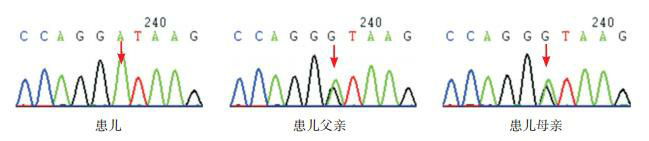
2.
患儿例3及其父母SLC7A7基因的Sanger测序图
红色箭头示碱基发生突变的位置。例3 SLC7A7基因检出c.625+1G>A纯合突变,父母分别携带c.625+1G>A杂合突变。
2.3. 治疗与随访
3例患儿诊断明确后,予低蛋白饮食,补充瓜氨酸100 mg/(kg · d)、蛋白琥珀酸铁4 mg/(kg · d)、葡萄糖酸钙锌10 mL/d、维生素D 400 IU/d。例3还予醋酸泼尼松5 mg/d治疗。例1治疗6月和1年、例2治疗6月、例3治疗3月后随访,症状体征均有不同程度改善,辅助检查见表 1。3例患儿经治疗后精氨酸和乳清酸均恢复正常。例1在治疗6月后因发热咳嗽第2次入院,诊断为肺炎,肺部CT示两肺充气不均,可见散在斑点斑片状及细条片状高密度影,主动脉弓旁可见条片状致密影,边界较清,考虑两肺感染病变(实质+间质)。治疗1年后血氨稍高至81.8 μmol/L,白蛋白偏低,淀粉酶增高,胸片示肺纹理增强,但仍偶有单、双声咳嗽。三系减少明显改善。例2治疗6月后,无肺部症状体征,仅见胸片示肺纹理增强。贫血明显改善。LDH由969 IU/L升高至1 011 IU/L,TG由2.5 mmol/L升高至7.9 mmol/L。例1、例2肝脾回缩不明显但未继续增大。例3治疗3个月后血氨降至正常,LDH、FER、TG均有不同程度降低,贫血明显改善,仍有气促,需带氧生存。2018年5月21日复查肺部CT:与2018年2月23日对比,双肺下叶病变较前好转,左肺上叶及右肺中上叶病变较前增多。
3. 讨论
LPI是一种罕见的常染色体隐性遗传代谢病,由SLC7A7基因突变导致y+LAT1蛋白缺陷,影响小肠吸收和肾小管重吸收赖氨酸、精氨酸、鸟氨酸。y+LAT1蛋白的功能是与细胞表面4F2HC(CD98)结合,负责阳性氨基酸转运。SLC7A7基因定位于14q11.2,包含11个外显子[7]。截止至2018年4月,在HGMD专业版中检索SLC7A7基因突变,共报道67种突变,包括27种错义突变、7种无义突变、6种剪切突变、微缺失和微重复分别有10种和8种、大片段缺失9种,大多为纯合突变。
本文共发现3种突变:c.1387delG、c.1215G > A、c.625+1G > A。c.1387delG杂合突变仅日本报道一例,该突变为1 387位鸟嘌呤缺失,导致第463位缬氨酸被半胱氨酸替代后发生移码,认为c.1387delG突变与致病性相关[8]。c.625+1G > A是发生在第4内含子和第3外显子拼接区的一个剪接突变,导致第4外显子缺失,该突变影响第4跨膜结构域到第3胞外结构域功能,引起y+LAT1蛋白构象改变[3]。该位点为无义突变,会造成第405位色氨酸翻译提前终止,产生截短蛋白,从而影响蛋白功能。c.1215G > A为新发错义突变,根据ACMG指南分析为致病性突变。例2与例1为姐妹关系,例1却表现出间质性肺炎和全血细胞减少等更为严重的临床表现。因此,这两例含有相同突变的姐妹临床表型具有异质性,这与以往报道的观点一致[4, 9]。
截至2018年12月,全球共有231例患者,其中儿童患者195例,我国仅有5例报道。LPI可在任何年龄发病,婴幼儿时期,大多无明显临床症状,断乳后会出现呕吐、腹泻、喂养困难等消化系统表现,常被误诊为消化不良和食物不耐受[2]。儿童期,主要表现为生长发育落后、骨质疏松、全血细胞减少、肺间质病变等。部分患儿到成年才得到诊断,主要表现为慢性肺间质病变、肾脏病变等[5, 10]。
LPI是一种多系统受累的疾病,肺部受累是其较为常见且严重的并发症。经检索发现已报道50例肺部受累患者,男34例,女16例,年龄2.9(产前~28)岁[5, 10-17]。其中儿童患者45例,大多表现为肺间质病变,较易发展为肺泡蛋白沉积症,14例患儿死亡,均在15岁前死于肺泡蛋白沉积症、肺出血等。发病初期患者可无任何呼吸系统症状,高分辨率CT可提高无症状患者的检出率[14]。据Parto报道[15],约有1/3无呼吸系统症状患者在数年后出现肺纤维化的影像学表现,肺部并发症的严重程度与影像学没有明显相关性,肺部病变出现越早,死亡率越高。近期研究表明SLC1A7/y+LAT1有抑制炎症反应的作用,当巨噬细胞和呼吸道上皮细胞SLC1A7/y+LAT1均缺陷时,会加剧LPI患者肺部炎症反应[18]。
本文3例共性的临床表现:断乳后均有拒食高蛋白饮食,出现呕吐、腹泻和贫血,生长发育明显落后,肝脾肿大,骨质疏松,实验室检查均有多项巨噬细胞活化指标异常,如外周血细胞减少,LDH、FER、TG升高等。3例均表现为乳清酸增高,例1、例3血精氨酸降低,血鸟氨酸降低不明显,血氨正常或仅轻度增高,这些指标的改变会让医生怀疑是否有鸟氨酸氨甲酰转移酶缺乏症(OTC)。但OTC患儿更多表现为神经系统的异常,血氨升高更明显,结合患儿病史和SLC7A7基因分析可明确诊断。3例患儿肺部病变的严重程度差别很大。例3在1岁4个月左右出现明显气促、咳嗽,在1岁10个月病情加重,肺CT提示弥漫性间质性肺病,肺泡灌洗液未见异常,并未发现肺泡蛋白沉积症的表现。例1确诊半年后出现间质性肺炎,1年后胸片仅表现为肺纹理增强,患儿偶有咳嗽。例2至今未出现呼吸系统症状和体征,仅胸片示肺纹理增多。3例患儿均在进一步随访观察中。
LPI以“控氨、营养、防并发症”为一般治疗原则[2]。补充瓜氨酸可纠正细胞内精氨酸缺乏,控制血氨在正常水平,并予适当蛋白,以促生长发育。本文3例患儿予瓜氨酸、维生素D、葡萄糖酸钙锌、蛋白琥珀酸铁,低蛋白饮食,例3肺间质病变重,伴有明显咳喘,予醋酸泼尼松5 mg/d,分两次口服。3例患儿经3个月~1年治疗,贫血症状明显改善,红细胞、白细胞、血小板均有回升,肝脾回缩不明显但未继续增大。但例1偶有咳嗽,例1、例2血淀粉酶仍偏高116~168(28~100 U/L),例2血脂由2.5 mmol/L升高到7.9 mmol/L。Parenti [11]曾报道两例反复发作胰腺炎合并高血脂、淀粉酶增高的患者,腹腔镜观察到胰腺纤维化和脂肪坏死。这与我们这两例的生化特点类似,本病引起高脂和高淀粉酶的机理尚不清楚,是否高脂血症导致胰脏功能受损尚需进一步观察。例3虽然咳喘等症状稍有缓解,但仍存在低氧血症,肺部影像学表现未见改善,仍表现有明显的肺间质病变。目前,肺间质病变尚无有效、安全的治疗方案。以往报道[19],粒细胞-巨噬细胞集落刺激因子(GM-CSF)雾化可以明显改善患儿的呼吸系统症状,但GM-CSF有诱发肺纤维化的危险。Santamaria[20]报道了一例合并肺泡蛋白沉积症的患儿进行肺移植,18个月后患儿肺部症状得到缓解,然而26个月后因肺泡蛋白沉积症复发死亡。
综上,LPI是一种多系统受累的疾病,通过常规实验室检测诊断困难,确定诊断需要基因检测。目前的治疗方案并不能阻止病情进展,不会改善肺间质病变,但贫血、白细胞减少、血小板减少、骨质疏松、肝脾肿大等会得到不同程度地纠正和改善。优化并尝试新的治疗方案,是提高患儿生存质量的关键。
Biography
崔冬, 男, 硕士研究生
Funding Statement
深圳三名工程项目(SZSM201812005)
References
- 1.Perheentupa J, Visakorpi JK. rotein intolerance with deficient transport of basic aminoacids. Another inborn error of metabolism. http://cn.bing.com/academic/profile?id=d736e3480fa7e1def0c49455ec8f9823&encoded=0&v=paper_preview&mkt=zh-cn. Lancet. 1965;2(7417):813–816. doi: 10.1016/s0140-6736(65)92446-3. [DOI] [PubMed] [Google Scholar]
- 2.Sebastio G, Sperandeo MP, Andria G. Lysinuric protein intolerance:reviewing concepts on a multisystem disease. http://cn.bing.com/academic/profile?id=325d961b2abb8075fea815d87a892fe6&encoded=0&v=paper_preview&mkt=zh-cn. Am J Med Genet C Semin Med Genet. 2011;157C(1):54–62. doi: 10.1002/ajmg.c.30287. [DOI] [PubMed] [Google Scholar]
- 3.Sperandeo MP, Bassi MT, Riboni M, et al. Structure of the SLC7A7 gene and mutational analysis of patients affected by lysinuric protein intolerance. Am J Hum Genet. 2000;66(1):92–99. doi: 10.1086/302700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sperandeo MP, Andria G, Sebastio G. Lysinuric protein intolerance:update and extended mutation analysis of the SLC7A7 gene. Hum Mutat. 2008;29(1):14–21. doi: 10.1002/(ISSN)1098-1004. [DOI] [PubMed] [Google Scholar]
- 5.Mauhin W, Habarou F, Gobin S, et al. Update on lysinuric protein intolerance, a multi-faceted disease retrospective cohort analysis from birth to adulthood. http://cn.bing.com/academic/profile?id=93e7e39751f8074af8303d4415313d71&encoded=0&v=paper_preview&mkt=zh-cn. Orphanet J Rare Dis. 2017;12(1):1–12. doi: 10.1186/s13023-016-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants:a joint the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torrents D, Mykkänen J, Pineda M, et al. Identification of SLC7A7, encoding y+LAT-1, as the lysinuric protein intolerance gene. Nat Genet. 1999;21(3):293–296. doi: 10.1038/6809. [DOI] [PubMed] [Google Scholar]
- 8.Shoji Y, Noguchi A, Shoji Y, et al. Five novel SLC7A7 variants and y+L gene-expression pattern in cultured lymphoblasts from Japanese patients with lysinuric protein intolerance. Hum Mutat. 2002;20(5):375–381. doi: 10.1002/(ISSN)1098-1004. [DOI] [PubMed] [Google Scholar]
- 9.Noguchi A, Nakamura K, Murayama K, et al. Clinical and genetic features of lysinuric protein intolerance in Japan. Pediatr Int. 2016;58(10):979–983. doi: 10.1111/ped.12946. [DOI] [PubMed] [Google Scholar]
- 10.DiRocco M, Garibotto G, Rossi GA, et al. Role of haematological, pulmonary and renal complications in the longterm prognosis of patients with lysinuric protein intolerance. Eur J Pediatr. 1993;152(5):437–440. doi: 10.1007/BF01955906. [DOI] [PubMed] [Google Scholar]
- 11.Parenti G, Sebastio G, Strisciuglio P, et al. Lysinuric protein intolerance characterized by bone marrow abnormalities and severe clinical course. J Pediatr. 1995;126(2):246–251. doi: 10.1016/S0022-3476(95)70552-X. [DOI] [PubMed] [Google Scholar]
- 12.Parsons H, Snyder F, Bowen T, et al. Immune complex disease consistent with systemic lupus erythematosus in a patient with lysinuric protein intolerance. J Inherit Metab Dis. 1996;19(5):627–634. doi: 10.1007/BF01799838. [DOI] [PubMed] [Google Scholar]
- 13.Gürsel T, Koçak U, Tümer L, et al. Bone marrow hemophagocytosis and immunological abnormalities in a patient with lysinuric protein intolerance. Acta Haematol. 1997;98(3):160–162. doi: 10.1159/000203612. [DOI] [PubMed] [Google Scholar]
- 14.Santamaria F, Parenti G, Guidi G, et al. Early detection of lung involvement in lysinuric protein intolerance:role of highresolution computed tomography and radioisotopic methods. Am J Respir Crit Care Med. 1996;153(2):731–735. doi: 10.1164/ajrccm.153.2.8564125. [DOI] [PubMed] [Google Scholar]
- 15.Parto K, Svedström E, Majurin ML, et al. Pulmonary manifestations in lysinuric protein intolerance. Chest. 1993;104(4):1176–1182. doi: 10.1378/chest.104.4.1176. [DOI] [PubMed] [Google Scholar]
- 16.Valimahamed-Mitha S, Berteloot L, Ducoin H, et al. Lung involvement in children with lysinuric protein intolerance. J Inherit Metab Dis. 2015;38(2):257–263. doi: 10.1007/s10545-014-9777-5. [DOI] [PubMed] [Google Scholar]
- 17.Zhang G, Cao L. New mutations in the SLC7A7 gene of two chinese sisters with lysinuric protein intolerance. Pediatr Pulmonol. 2017;52(11):E94–E96. doi: 10.1002/ppul.23760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rotoli BM, Barilli A, Visigalli R, et al. Downregulation of SLC7A7 triggers an inflammatory phenotype in human macrophages and airway epithelial cells. Front Immunol. 2018;9:508. doi: 10.3389/fimmu.2018.00508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Douda DN, Farmakovski N, Dell S, et al. SP-D counteracts GM-CSF-mediated increase of granuloma formation by alveolar macrophages in lysinuric protein intolerance. Orphanet J Rare Dis. 2009;4:29. doi: 10.1186/1750-1172-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santamaria F, Brancaccio G, Parenti G, et al. Recurrent fatal pulmonary alveolar proteinosis after heart-lung transplantation in a child with lysinuric protein intolerance. J Pediatr. 2004;145(2):268–272. doi: 10.1016/j.jpeds.2004.04.047. [DOI] [PubMed] [Google Scholar]