

Abstract
非酮性高甘氨酸血症(NKH)是由甘氨酸裂解系统缺陷引起的常染色体隐性遗传疾病,分经典型和非经典型,而非经典型表现复杂多样,诊断较为困难。该文报道1个NKH家系,父母表型正常,兄妹均非新生儿期起病,哥哥表现为难治性癫癎、严重的双侧痉挛性瘫痪和智力低下,血和脑脊液的甘氨酸浓度增高,尿甘氨酸与肌酐的比值增高,脑脊液与血甘氨酸浓度的比增高;妹妹表现为语言发育迟缓、共济失调、舞蹈病和发热诱发的精神行为异常和肌张力减退,脑脊液甘氨酸浓度增高、脑脊液与血甘氨酸浓度比增高。高通量测序提示兄妹均存在GLDC基因母源c.3006C > G(p.C1002W)错义突变和父源c.1256C > G(p.S419X)无义突变,生物学软件预测均提示致病突变。转染两种突变体GLDC基因的H293T细胞甘氨酸脱羧酶活性均有下调。NKH表型多样,二代高通量测序有利于疑似病例的确认,非经典NKH与基因突变导致的甘氨酸脱羧酶活性下调相关。
Keywords: GLDC基因, 复合杂合突变, 非酮性高甘氨酸血症, 癫癎, 语言发育迟缓, 儿童
Abstract
Nonketotic hyperglycinemia (NKH) is an autosomal recessive hereditary disease caused by a defect in the glycine cleavage system and is classified into typical and atypical NKH. Atypical NKH has complex manifestations and is difficult to diagnose in clinical practice. This article reports a family of NKH. The parents had normal phenotypes, and the older brother and the younger sister developed this disease in the neonatal period. The older brother manifested as intractable epilepsy, severe spastic diplegia, intellectual disability, an increased level of glycine in blood and cerebrospinal fluid, an increased glycine/creatinine ratio in urine, and an increased ratio of glycine concentration in cerebrospinal fluid and blood. The younger sister manifested as delayed language development, ataxia, chorea, mental and behavior disorders induced by pyrexia, hypotonia, an increased level of glycine in cerebrospinal fluid, and an increased ratio of glycine concentration in cerebrospinal fluid and blood. High-throughput sequencing found a maternal missense mutation, c.3006C > G (p.C1002W), and a paternal nonsense mutation, c.1256C > G (p.S419X), in the GLDC gene in both patients. These two mutations were thought to be pathogenic mutations by a biological software. H293T cells transfected with these two mutants of the GLDC gene had a down-regulated activity of glycine decarboxylase. NKH has various phenotypes, and high-throughput sequencing helps to make a confirmed diagnosis. Atypical NKH is associated with the downregulated activity of glycine decarboxylase caused by gene mutations.
Keywords: GLDC gene, Compound heterozygous mutation, Nonketotic hyperglycinemia, Epilepsy, Delayed language development, Child
非酮性高甘氨酸血症(nonketotic hyperglycinemia, NKH, OMIM #605899)是一组常染色体隐性遗传的代谢性疾病,又称甘氨酸脑病,发病率估计为1 : 76 000[1]。本病是由甘氨酸裂解系统活性缺乏所致的甘氨酸代谢障碍,大量甘氨酸在尿液、血清和脑脊液沉积,并通过N甲基-D-天冬氨酸(NMDA)受体过度刺激引发神经损伤[2]。甘氨酸裂解系统是4个组分构成的多酶复合物:P-蛋白(吡哆醛磷酸依赖性甘氨酸脱羧酶),H-蛋白(硫辛酸蛋白),T-蛋白(四氢叶酸依赖氨基甲基转移酶)和L-蛋白(脂酰胺脱氢酶),分别由GLDC、GCSH、AMT和GCSL基因编码。遗传学研究证实NKH主要为甘氨酸裂解系统的相关基因GLDC、GCSH或AMT突变所致,有少量研究认为也可由GLRX5、BOLA3、LIAS基因缺陷引起[3]。NKH最常见的表型为新生儿NKH,表现为出生后嗜睡、肌张力低下、肌阵挛性癫癎发作、呼吸暂停,大部分1岁以内死亡。但该疾病也存在一系列表型变异,即非经典型NKH,表现为精神运动发育迟缓、癫癎发作、共济失调、舞蹈病、语言发育落后、行为问题和热诱发的肌张力低下、精神行为异常,血清和脑脊液甘氨酸浓度升高不如新生儿NKH明显[4-5]。目前国内仅有两篇新生儿NKH报道[6-7],非经典型NKH未见报道。本研究报道一个家系中一对兄妹因GLDC复合杂合突变所致的非经典型NKH,并将相关突变转染到H293T细胞后进行蛋白表达和酶活性测定。
1. 资料与方法
1.1. 研究对象
病例1,男,6岁8个月,反复抽搐5年余就诊。9月龄起抽搐反复发作,表现为双眼凝视、呼之不应、牙关紧闭、口周发绀、肢体强直抽动,持续2~3 min不等,有时表现为肢体触电样快速抖动,每天发作10余次。诊断考虑癫癎,予以丙戊酸钠治疗,3天后患儿出现意识不清、肌张力降低,改用卡马西平、左乙拉西坦、氯硝西泮联合治疗,半年后患儿抽搐频率减少为数月1次。目前患儿不能独坐、独走,不会说话, 不会笑,不会主动抓物。患儿系第一胎第一产,足月顺产出生,出生时无窒息史。父母体健,家族中否认类似病史。体格检查:体重14 kg,头围49 cm,发育落后,营养差,神志清,反应迟钝,心、肺、腹部查体无异常,四肢肌肉萎缩、肌力4级、肌张力增高,双膝腱、跟腱反射亢进,病理反射未引出,脑膜刺激征阴性。辅助检查:尿、便常规,肝肾功能、血氨、乳酸以及脑脊液常规、生化均正常,干血斑串联质谱法分析提示氨基酸和酰基肉碱正常,头颅磁共振正常,脑电图显示广泛性癎样放电。
病例2,女,3岁5个月,病例1的妹妹。因精神行为异常、行走不稳1年余就诊。2岁时“急性上呼吸道感染”后出现烦躁不安、攻击行为、共济失调步态、四肢舞蹈样动作,以及发作性双眼斜视、上肢舞动。经抗感染及对症治疗后,患儿体温恢复正常,精神、行为异常好转。3岁1个月“急性上呼吸道感染”后再次出现烦躁不安、攻击行为,对症治疗后好转。目前患儿能独立行走,但步态不协调,可以理解简单语句,只能叫“爸爸、妈妈”。患儿系第二胎、第二产,足月顺产出生;3个月抬头,6个月独坐,17个月独立行走,3岁会叫“爸爸、妈妈”。体格检查:体重13.5 kg,头围49 cm,发育落后,营养中等,神志清,反应可,心、肺、腹部查体无异常,四肢肌力正常、肌张力偏低,直线行走不能,双膝腱、跟腱反射正常,病理反射未引出,脑膜刺激征阴性。辅助检查:血常规示淋巴细胞比例升高、余项正常;咽拭子副流感病毒阳性;血氨、血乳酸正常,尿粪常规、肝肾功能、脑脊液常规、生化均正常;干血斑串联质谱法分析提示氨基酸和酰基肉碱正常;头颅和胸腰髓磁共振正常;肌电图正常;心电图:窦性心律、室性早搏;动态心电图:窦性心动过速,频发室早,夜间出现一次2.4s的全心停搏;心脏彩超示三尖瓣轻度反流;脑电图显示广泛性、间断性、节律性δ活动。
1.2. 分子遗传学分析
取病例1外周血5 mL(EDTA抗凝),选择癫癎相关308个基因进行高通量测序(北京迈基诺公司),测序数据予以RTA软件(real-time analysis, Illumina)分析,CASAVAv1.8.2(Illumina)、BWA、GAT软件评估测序质量,获得单核苷酸变异(SNV)报告。再使用ANNOVAR、SIFT、PolyPhen-2.2.2、HGMD数据库、dbSNP数据库和1000基因组数据库进行变异注释和分析,以确认候选病原突变检测结果。对患儿突变区域进行Sanger测序验证,确定致病位点。并采集病例2及其父母的外周血5 mL(EDTA抗凝)进行Sanger测序验证。
本研究获得患儿父母知情同意。
1.3. 甘氨酸浓度检测
采集病例1血液、尿液、脑脊液各5 mL,及病例2的血液、脑脊液5 mL(尿液标本未能收集到),使用日立全自动氨基酸分析仪L8900及液相色谱法检测甘氨酸浓度(由杭州博圣生物技术公司检测)。
1.4. 蛋白免疫印迹检测GLDC蛋白
培养H293T细胞,分别转染pMCV、pMCV-GLDC,以及S419X突变或C1002W突变的pMCV-GLDC质粒,48 h后收集细胞、提取蛋白。30 μg蛋白上样,先恒压80V电泳至溴酚蓝指示剂在浓缩胶与分离胶交界处成线状,改为恒压120V至溴酚兰到凝胶底部。转膜、封闭,GLDC兔多抗(英国Abcam)4℃过夜,鼠抗兔二抗(北京中杉金桥生物技术有限公司)室温2 h反应后显影。上述实验重复3次。
1.5. 酶联免疫吸附实验检测GLDC酶活性
采用ELISA试剂盒(上海酶联生物)检测GLDC酶活性,取0.1 mL细胞上清液于反应孔中,按照试剂盒说明书进行操作,用酶标仪测定每孔在450 nm处的OD值。实验重复3次,每次检测3个复孔。
2. 结果
2.1. 患儿及父母GLDC基因测序结果
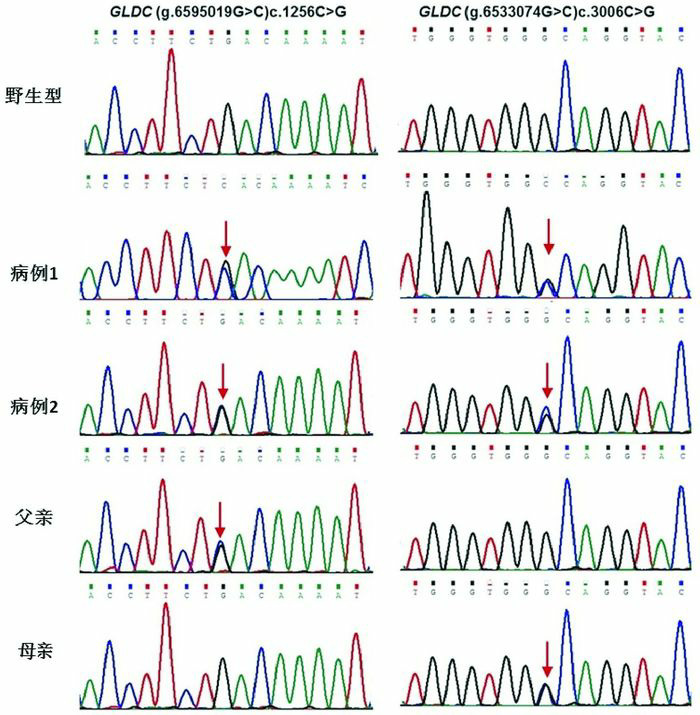
2例患儿在GLDC基因均发现突变:c.3006C > G(p.Cys1002Trp)和c.1256C > G(p.Ser419STer),均为新发突变,其中突变c.3006C > G(p.Cys1002Trp)来自母亲,突变c.1256C > G(p.Ser419STer)来自父亲,见图 1。查询致病数据库(OMIM、HGMD、Clinvar等),p.Ser419STer为无义突变,419位的丝氨酸变为终止密码子,蛋白合成提前终止,具有高致病性。p.Cys1002Trp为错义突变,PolyPhen2.2.2预测评分为1、SIFT评分为0,提示有致病性。在线生物信息软件Clustal Omega(http://www.ebi.ac.uk/Tools/msa/clustalo/)查询提示c.3006C > G(p.Cys1002Trp)在不同物种间高度保守;c.1256C > G(p.Ser419STer)在不同物种间保守度为4/7,但该突变造成蛋白合成提前终止。
1.
患儿GLDC基因测序结果及其家系验证
病例1、2检测到GLDC基因c.3006C > G(p.Cys1002Trp)和c.1256C > G(p.Ser419STer)复合杂合突变,父亲检测到GLDC基因c.1256C > G突变,母亲检测到GLDC基因c.3006C > G突变,突变位点如箭头所示。
2.2. 甘氨酸浓度分析
病例1的尿液甘氨酸浓度与肌酐的比值为1 154 μmol/mmol(参考值:64~236 μmol/mmol),血清甘氨酸浓度为751 μmol/L(参考值:0~276 μmol/L),脑脊液甘氨酸为45.3 μmol/L(参考值:1.6~19.5 μmol/L),脑脊液/血清甘氨酸之比为0.06(参考值:≤0.02)。病例2的血清甘氨酸浓度正常,脑脊液甘氨酸浓度增高(36.7 μmol/L),脑脊液/血清甘氨酸之比增高(0.13)。
2.3. GLDC蛋白表达及酶的活性
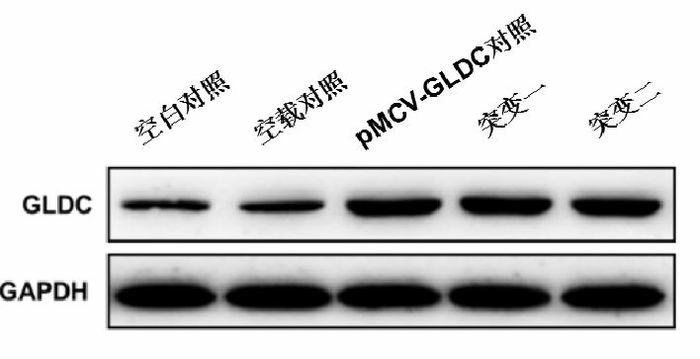
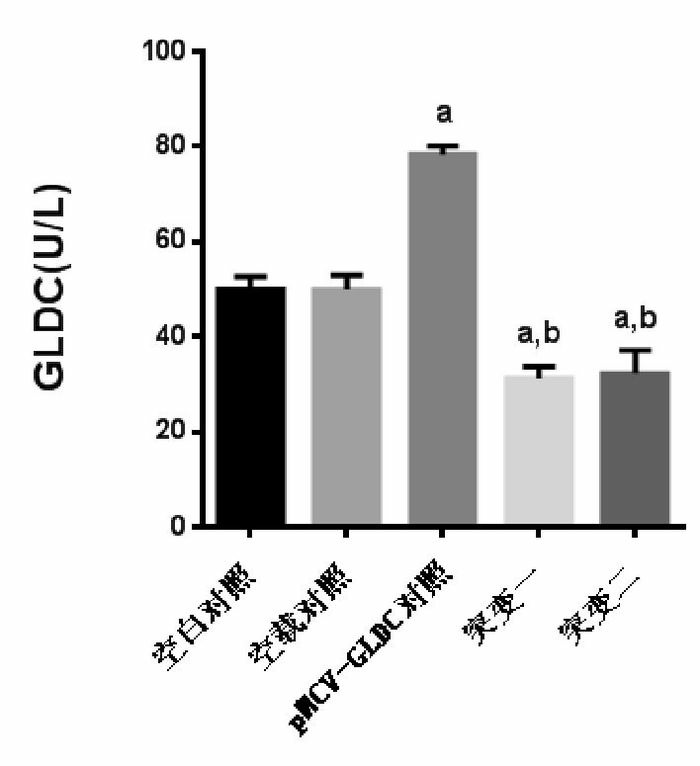
与空白对照和空载对照相比,突变一组(S419X突变)和突变二组(C1002W突变)的GLDC蛋白表达增高,酶活性降低;与pMCV-GLDC对照组相比,突变一组、突变二组的GLDC蛋白表达无明显变化,GLDC酶活性下降(P < 0.01),但突变一组和突变二组GLDC酶活性的差异无统计学意义(P > 0.05),见图 2、3。
2.
GLDC蛋白的检测
突变一和突变二的GLDC蛋白表达高于空白对照和空载对照;与pMCV-GLDC对照相比,无明显变化。
3.
GLDC酶活性的ELISA测定
a示与空白对照组及空载对照相比,P < 0.01;b示与pMCV-GLDC对照相比,P < 0.01。
3. 讨论
根据发病年龄NKH可分为新生儿型、婴儿型和迟发型。经典型NKH即新生儿型NKH,非经典型NKH包括婴儿型NKH、迟发型NKH和具有较好精神运动发育的新生儿型NKH[5]。非经典型NKH可因症状不典型,血清和脑脊液甘氨酸浓度升高不显著导致诊治延误[8]。Brenton等[4]提到尿甘氨酸浓度与肌酐的比值增高对于晚发型NKH的诊断非常重要。本研究病例1的尿甘氨酸/肌酐比值明显升高,与文献相符,遗憾的是未获得其妹妹的尿液样本。Ezgu等[9]在一个4月龄的顽固性癫癎患儿使用第二代高通量测序寻找病因,诊断速度早于其他方法。本研究2例患者均非新生儿期起病,脑脊液/血清甘氨酸之比均增高,最终通过目标基因捕获高通量测序确诊为非经典型NKH。
578个已报道的经典型NKH家系中,遗传分析确定了410个相关突变,约80%的甘氨酸脑病由GLDC基因突变引起,错义突变占了GLDC突变类型的52%,15%的患者有减毒表型[1]。晚发型NKH患儿中,大部分情况下甘氨酸裂解系统维持正常的功能,但感染可导致甘氨酸裂解系统活性进一步降低,引起共济失调、舞蹈病样动作、精神问题等非特异性脑病表现[4]。本组2例患儿,父源c.1256C > G(p.S419X)无义突变导致第419氨基酸位置上丝氨酸突变为终止密码子,改变了蛋白结构,为致病突变;母源c.3006C > G(p.C1002W)错义突变,第1002氨基酸位置上半胱氨酸变为色氨酸,蛋白预测软件分析为致病突变。本研究兄妹基因型相同,而表型并不完全一样。Korman等[10]报道GLDC基因A802V(c.2405C > T)突变的两个家系,两个家系的父母均为表型正常的基因携带者,家系A的三姐妹均为纯合突变,其中2人新生儿期出现NKH症状、1人无症状;家系B中3个含纯合突变的孩子均为严重的新生儿型NKH,2人死亡,1人遗留严重神经系统后遗症,进一步用A802V突变的GLDC基因转染COS7细胞,其甘氨酸脱羧酶活性降低至32%。Swanson等[11]也发现含有相同GLDC复合杂合突变(p.A389V/p.R515S)的两个患儿具有较大差异的临床表型。基因型相同,而临床表型不同,可能与部分突变并未导致甘氨酸脱羧酶完全失活有关,而其他遗传或非致病因子对甘氨酸脱羧酶活性可能也有影响[11]。本研究将两种突变的pMCV-GLDC转染到H293T细胞,其甘氨酸脱羧酶活性下调至野生型的39.9%和41.2%,与Korman等[10]的报道类似。
目前,NKH的标准治疗策略包括采用苯甲酸钠降低脑内甘氨酸浓度,以及受体拮抗剂右美沙芬和氯胺酮阻断NMDA受体[12-13]。Deutsch等[14]认为右美沙芬治疗减少了癫癎发作,可以改善部分患者预后。Chien等[15]则认为苯甲酸钠和右美沙芬联合对预后没有任何帮助。NKH的治疗效果与患儿体内甘氨酸脱羧酶失活程度相关。如果甘氨酸脱羧酶完全失活,即使从出生时就开始治疗,患儿仍可表现频繁的癫癎发作、智力运动发育落后和痉挛性瘫痪,甚至死亡[16];如果甘氨酸脱羧酶活性部分丧失,早期使用右美沙芬和苯甲酸钠治疗可改善预后[10, 17]。丙戊酸盐是甘氨酸裂解系统抑制剂,可导致甘氨酸浓度增高,需慎用于NKH患者[18]。本研究病例1发病初期短暂使用丙戊酸盐后病情加重可能与此有关。
总之,本研究在一个非经典型的NKH家系中发现2个新的GLDC基因突变(p.C1002W、p.S419X),体外质粒转染实验进一步证实了其致病性,本研究丰富了GLDC突变谱,为研究中国人群GLDC突变类型、遗传方式提供了一定的参考。
Biography
蒋铁甲, 男, 硕士, 主治医师
Funding Statement
浙江省省级科技项目(2012C33101)
References
- 1.Coughlin CR 2nd, Swanson MA, Kronquist K, et al. The genetic basis of classic nonketotic hyperglycinemia due to mutations in GLDC and AMT. Genet Med. 2017;19(1):104–111. doi: 10.1038/gim.2016.74. [DOI] [PubMed] [Google Scholar]
- 2.Perry TL, Urquhart N, Maclean J, et al. Nonketotic hyperglycinemia. Glycine accumulation due to absence of glycerine cleavage in brain. N Engl J Med. 1975;292(24):1269–1273. doi: 10.1056/NEJM197506122922404. [DOI] [PubMed] [Google Scholar]
- 3.Baker PR 2nd, Friederich MW, Swanson MA, et al. Variant non ketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. https://academic.oup.com/brain/article/137/2/366/282721/Variant-non-ketotic-hyperglycinemia-is-caused-by. Brain. 2014;137(Pt 2):366–379. doi: 10.1093/brain/awt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brenton JN, Rust RS. Late-onset nonketotic hyperglycinemia with a heterozygous novel point mutation of the GLDC gene. Pediatr Neurol. 2014;50(5):536–538. doi: 10.1016/j.pediatrneurol.2014.01.018. [DOI] [PubMed] [Google Scholar]
- 5.Dinopoulos A, Matsubara Y, Kure S, et al. Atypical variants of nonketotic hyperglycinemia. Mol Genet Metab. 2005;86(1-2):61–69. doi: 10.1016/j.ymgme.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 6.张 蓉, 陈 超, 曹 云, et al. 新生儿非酮症性高甘氨酸血症1例. http://www.cnki.com.cn/Article/CJFDTOTAL-GWKF201702033.htm 中国循证儿科杂志. 2009;4(2):153–155. [Google Scholar]
- 7.高 志杰, 姜 茜, 陈 倩, et al. 1个非酮性高甘氨酸血症家系的临床和分子遗传学分析. http://www.cjcp.org/CN/abstract/abstract14197.shtml. 中国当代儿科杂志. 2017;19(3):268–271. doi: 10.7499/j.issn.1008-8830.2017.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rahman S, Footitt EJ, Varadkar S, et al. Inborn errors of metabolism causing epilepsy. Dev Med Child Neurol. 2013;55(1):23–36. doi: 10.1111/dmcn.2012.55.issue-1. [DOI] [PubMed] [Google Scholar]
- 9.Ezgu F, Çiftci B, Topçu B, et al. Diagnosis of glycine encephalopathy in a pediatric patient by detection of a GLDC mutation during initial next generation DNA sequencing. Metab Brain Dis. 2014;29(1):211–213. doi: 10.1007/s11011-014-9482-y. [DOI] [PubMed] [Google Scholar]
- 10.Korman SH, Boneh A, Ichinohe A, et al. Persistent NKH with transient or absent symptoms and a homozygous GLDC mutation. Ann Neurol. 2004;56(1):139–143. doi: 10.1002/(ISSN)1531-8249. [DOI] [PubMed] [Google Scholar]
- 11.Swanson MA, Coughlin CR, Scharer GH, et al. Biochemical and molecular predictors for prognosis in nonketotic hyperglycinemia. Ann Neurol. 2015;78(4):606–618. doi: 10.1002/ana.24485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Hove JL, Vande KK, Hennermann JB, et al. Benzoate treatment and the glycine index in nonketotic hyperglycinaemia. J Inherit Metab Dis. 2005;28(5):651–663. doi: 10.1007/s10545-005-0033-x. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki Y, Kure S, Oota M, et al. Nonketotic hyperglycinemia:proposal of a diagnostic and treatment strategy. Pediatr Neurol. 2010;43(3):221–224. doi: 10.1016/j.pediatrneurol.2010.04.018. [DOI] [PubMed] [Google Scholar]
- 14.Deutsch SI, Rosse RB, Mastropaolo J. Current status of NMDA antagonist interventions in the treatment of nonketotic hyperglycinemia. http://journals.lww.com/clinicalneuropharm/Abstract/1998/03000/Current_Status_of_NMD_A_Antagonist_Interventions.1.aspx. Clin Neuropharmacol. 1998;21(2):71–79. [PubMed] [Google Scholar]
- 15.Chien YH, Hsu CC, Huang A, et al. Poor outcome for neonatal-type nonketotic hyperglycinemia treated with high-dose sodium benzoate and dextromethorphan. J Child Neurol. 2004;19(1):39–42. doi: 10.1177/08830738040190010702. [DOI] [PubMed] [Google Scholar]
- 16.Korman SH, Wexler ID, Gutman A, et al. Treatment from birth of nonketotic hyperglycinemia due to a novel GLDC mutation. Ann Neurol. 2006;59(2):411–415. doi: 10.1002/(ISSN)1531-8249. [DOI] [PubMed] [Google Scholar]
- 17.Bjoraker KJ, Swanson MA, Christodoulou J, et al. Neurodevelopmental outcome and treatment efficacy of benzoate and dextromethorphan in siblings with attenuated nonketotic hyperglycinemia. J Pediatr. 2016;170:234–239. doi: 10.1016/j.jpeds.2015.12.027. [DOI] [PubMed] [Google Scholar]
- 18.Subramanian V, Kadiyala P, Hariharan P, et al. A rare case of glycine encephalopathy unveiled by valproate therapy. J Pediatr Neurosci. 2015;10(2):143–145. doi: 10.4103/1817-1745.159200. [DOI] [PMC free article] [PubMed] [Google Scholar]