

Abstract
患儿,男,11岁,间断乏力、行走困难6年,发作性意识不清4年;辅助检查提示严重代谢性酸中毒、低血糖、肝功能异常,CT示肝脏明显增大、呈脂肪密度影。给予补液、纠酸、纠正低血糖,以及左卡尼汀、复合维生素B、辅酶Q10等治疗,患儿持续昏迷、代谢性酸中毒及低血糖难以纠正,患儿死亡。血、尿有机酸筛查和基因检测证实为电子转运黄素蛋白脱氢酶编码基因(ETFDH)缺陷所致的迟发型戊二酸尿症Ⅱ型(GA Ⅱ c型)。GA Ⅱ c型是一种发病率很低的遗传性代谢病,易误诊误治。对于反复乏力或活动耐力下降、低血糖、肝脏明显增大伴肝功能异常的患儿,应考虑GA Ⅱ型的可能,尿有机酸分析和血串联质谱分析可提供诊断线索,ETFDH基因分析可提供确诊依据。
Keywords: 电子转运黄素蛋白脱氢酶, 戊二酸尿症, 低血糖, 肌无力, 儿童
Abstract
A boy aged 11 years was admitted due to intermittent weakness and diffculty in walking for 6 years, and hepatomegaly, glycopenia and unconsciousness for 4 years.The laboratory examinations showed severe metabolic acidosis, hypoglycemia, and abnormal liver function.CT scan showed marked liver enlargement with fat density shadow. The boy was given fluid infusion, correction of acidosis, intravenous injection of glucose, L-carnitine, compound vitamin B, and coenzyme Q10, but he was in a persistent coma and it was difficult to correct refractory metabolic acidosis and hypoglycemia.The boy died.Blood and urinary organic acid screening and gene detection confrmed that the boy had late-onset glutaric aciduria type Ⅱ (GAⅡc) caused by electron-transferring-flavoprotein dehydrogenase (ETFDH) gene defect.GAⅡc is an inherited metabolic disease with a low incidence, resulting in a high misdiagnosis rate.GAⅡc should be considered for children with recurrent weakness or reduced activity endurance, hypoglycemia, and marked liver enlargement with abnormal liver function.Urinary organic acid analysis and blood tandem mass spectrometry can help with the early diagnosis of GAⅡc, and ETFDH gene analysis helps to make a confirmed diagnosis.
Keywords: Electron-transferring-flavoprotein dehydrogenase, Glutaric aciduria, Hypoglycemia, Muscle weakness, Child
1. 病例介绍
患儿,男,11岁,间断乏力、行走困难6年,发作性意识不清4年。患儿6年前(5岁)出现活动后双下肢乏力、行走困难,偶有“腿痛”,当地疑诊重症肌无力、多发性肌炎等,口服泼尼松片(20 mg/次,Bid)、复合维生素B(1片/次,Tid)等半月余,下肢乏力症状稍改善后停药。近4年因发作性乏力、行走困难、意识不清等先后4次就诊,发现肝脏明显增大(右肋下4.0 cm)伴转氨酶增高(具体不详)、低血糖,疑诊肝糖原贮积症等遗传性代谢病,给予还原型谷胱甘肽、葡醛内酯、肌苷片、左卡尼汀、复合维生素B等治疗,症状好转但反复。半天前,患儿因意识不清就诊,查体发现肝右肋下7.0 cm,急查血糖2.1 mmol/L(参考值:3.6~6.9 mmol/L),心肌酶:AST 339 U/L(参考值:0~40 U/L),LDH 567.2 U/L(参考值:80~245 U/L),CK 1 600 U/L(参考值:0~200 U/L),以“肝大、低血糖、昏迷查因”收入院。
患儿系第3胎第3产,足月顺产出生,出生体重3.5 kg。出生史及生长发育史无异常。患儿父母体健,非近亲结婚;姐姐17岁、哥哥14岁,均体健。
入院查体:体温36.9℃,脉搏134次/min,呼吸43次/ min,血压85/49 mm Hg。体重35 kg,身高151 cm。发育正常,营养中等,鼻导管吸氧下经皮血氧饱和度72%,浅昏迷,Glasgow评分7分,面色灰暗,口周发绀,皮肤、巩膜无黄染,未见皮疹及出血点,浅表淋巴结未及肿大,双侧瞳孔等大等圆、直径2.0 mm,对光反射迟钝,口腔粘膜光滑,咽红,双侧扁桃体Ⅰ度肿大,颈软,呼吸促,三凹征阴性,双肺呼吸音对称,可闻及痰鸣音,心音低钝,律齐,心率134次/min,各瓣膜听诊区未闻及病理性杂音。腹软,肝右肋下8 cm、质韧、表面光滑,脾左肋下未触及,肠鸣音正常。无Gower征,腓肠肌无假性肥大及萎缩。四肢肌张力低,腱反射存在,双侧巴氏征阴性,克氏征及布氏征阴性,四肢末端梢凉、足背动脉搏动弱、毛细血管充盈时间4 s。
辅助检查:血气分析:pH 7.26(参考值:7.35~7.45),PaCO2 30.1 mm Hg(参考值:35~45 mm Hg),PaO2 65 mm Hg(参考值:80~100 mm Hg),HCO3-18.7 mmol/L(参考值:22~27 mmol/L),BE -10.7 mmol/L(参考值:-3~3 mmol/L),Lac 8.9 mmol/L(参考值:0~2 mmol/L);空腹血糖1.9 mmol/L(参考值:3.6~6.9 mmol/L);肝功能:ALT 288.7 U/L(参考值:0~40 U/L),AST 321.0 U/L(参考值:0~40 U/L),余项正常;血氨137.0 μmol/L(参考值:9~33 μmol/L);血常规、血沉、CRP、PCT、凝血功能、电解质、肾功能、血脂、B型钠尿肽前体、肌钙蛋白T等大致正常,甲胎蛋白、CMV-DNA、EBV-DNA无异常;乙肝病毒5项抗体均阴性,甲肝、丙肝、梅毒螺旋体、HIV抗体阴性,结核抗体阴性,腺病毒、柯萨奇病毒、流感病毒、呼吸道合胞病毒以及EB病毒抗体等均阴性。腹部彩超:肝脏增大,并弥漫性回声改变,肾脏、脾脏、胰腺、胆囊未见明显异常;腹部CT:肝脏增大,普遍呈脂肪密度,肠间隙可见脂肪密度影;胸片:两肺纹理粗,心影大小正常;心脏彩超、头颅CT平扫未见异常。
2. 诊断思维
全身型重症肌无力、肌营养不良、低钾性周期性麻痹等均可致反复发作性活动耐力下降、运动后易疲劳等症状。全身型重症肌无力(myasthenia gravis, MG)通常表现有抬头困难,行走乏力,不能久行,症状重,而且肌无力呈“晨轻暮重、休息后缓解”的特征,不伴肝脏大、低血糖等改变,与本例不符。本研究患儿自5岁开始出现反复发作的肌无力,但并无进行性加重,也无Gower征、腓肠肌假性肥大和骨骼肌萎缩等特征,CK增高也不显著,不支持典型肌营养不良(muscular dystrophy, MD)。患儿家族无类似病史、肌无力发作时血钾正常,不支持家族性低钾性周期性麻痹。
本例患儿除发作性全身乏力、活动耐力下降外,且伴意识障碍,肝脏增大、脂肪变,以及低血糖、高氨血症、代谢性酸中毒等,需考虑遗传代谢性疾病可能,如线粒体脑肌病(mitochondrial encephalomyopathy, ME)、瑞氏综合征、肝糖原贮积症(glycogen storage disease, GSD)、肉碱缺乏症、线粒体脂肪酸氧化缺陷等。
ME是一种线粒体结构或功能异常所致的多系统疾病,肌肉损害主要表现为骨骼肌不能耐受疲劳,部分患者可伴有非酮性低血糖,与本病相似;多数ME患儿伴有脑病症状如癫痫发作、共济失调、智力障碍等,本病例无类似症状,发现mtDNA缺失或大量重排可确诊。瑞氏综合征主要是线粒体代谢异常引起的肝脏为主的广泛内脏脂肪变性,表现为突出的急性脑病症状(呕吐、惊厥、意识障碍等),合并肝脏肿大、转氨酶增高、血氨、血乳酸、游离脂肪酸明显增高,以及血糖明显降低等,常有上呼吸道感染史、阿司匹林用药史等诱因。本例脑病症状不及瑞氏综合征突出,发作前无诱因,与瑞氏综合征不符。患儿反复全身乏力伴低明显血糖、肝脏增大、高氨血症、代谢性酸中毒,需要注意先天性糖代谢酶缺陷所造成的GSD可能,进一步行肾上腺素试验、胰高血糖素试验协助诊断,肝脏或肌肉组织活检发现糖代谢特异酶的缺乏可明确GSD。
其它遗传代谢性疾病如肉碱缺乏症、有机酸尿症、脂肪酸β氧化障碍等可表现为进行性肌疲劳、近端肢体肌无力、意识障碍,以及脂质沉积性肝肿大、肝功异常,低酮性低血糖、高血氨等特征,与本例患儿临床特征类相似,进一步行高效液相、气相色谱质谱联用和串联质谱分析(GC-MS/MS)、全基因组测序可辅助诊断。
3. 进一步检查
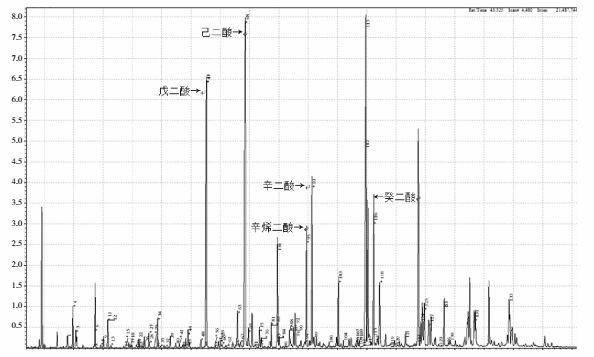
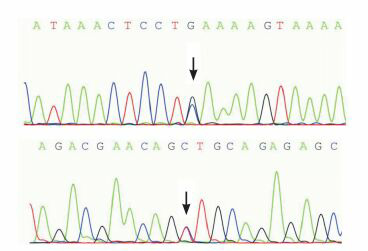
家长拒绝行肝脏、肌肉活检。肾上腺素、胰高血糖素刺激后空腹血糖升高,不支持GSD诊断。采集血、尿标本进行GC-MS/MS和基因测序(北京富佑遗传病门诊),结果:尿戊二酸、2-羟基戊二酸、3-羟基戊二酸、己二酸、辛二酸、辛烯二酸、癸二酸等明显增高(图 1),血中长链酰基肉碱(C4-C22)浓度明显增高,游离肉碱明显降低(表 1);基因测序发现电子转运黄素蛋白脱氢酶(electron-transferring-flavoprotein dehydrogenase, ETFDH)基因3号外显子的c.250G > A(chr4-159603421,p.A84T),以及12号内含子的c.1691-3C > G(chr4-159629513)两处杂合突变,见图 2。
1.
患者尿有机酸分析结果
戊二酸、己二酸、辛二酸、辛烯二酸、癸二酸明显增高。
1.
血串联质谱氨基酸及肉碱谱分析
| 检验项目 | 检测结果(μmol/L) | 参考值(μmol/L) |
| 游离肉碱 | 5.71 | 20.00~60.00 |
| 肉豆蔻酰肉碱 | 0.95 | 0~0.70 |
| 肉豆蔻烯酰肉碱 | 0.95 | 0~0.50 |
| 十八碳烯酰肉碱 | 3.46 | 0~2.80 |
| 二十碳酰肉碱 | 0.30 | 0~0.20 |
| 二十二碳酰肉碱 | 0.38 | 0~0.20 |
2.
先证者ETFDH基因测序结果
先证者ETFDH基因存在c.1691-3C > G、c.250G > A两处杂合突变(突变位点如箭头所示)。
4. 诊断及确诊依据
结合患儿临床特征、遗传代谢筛查和基因分析结果,诊断为晚发型戊二酸尿症Ⅱ型(glutaric aciduria type Ⅱ,GAⅡ型)。依据:(1)11岁年长儿,疾病呈慢性进程;(2)表现为乏力或活动耐力差、意识改变,以及肝脏增大、低血糖、代谢性酸中毒;(3)腹部CT示肝脏普遍呈脂肪密度,肠间隙可见脂肪密度影;(5)尿戊二酸增高,血游离肉碱降低、中长链酰基肉碱(C4-C22)增高;(6)ETFDH基因两处突变:c.250G > A、c.1691-3C > G,均为已报道的致病性突变。
5. 诊疗经过
入院后予气管插管机械通气,以及扩容、补液、纠酸维持内环境和血糖稳定,糖皮质激素免疫调节治疗,并予以还原性谷胱甘肽、磷酸肌酸、左卡尼汀、复合维生素B、辅酶Q10、果糖二磷酸钠、三磷酸腺苷等支持治疗,以及白醋灌肠及乳果糖降低血氨等对症治疗。3 d后患儿仍昏迷,代谢性酸中毒及低血糖难以纠正,放弃治疗后患儿死亡。
6. 讨论
GAⅡ又称多酰基辅酶A脱氢酶缺乏症(multiple acyl coenzyme A dehydrogenase deficiency, MADD),是一种常染色体隐性遗传性线粒体能量代谢障碍性疾病[1]。根据发病年龄及严重程度可分3种亚型:① GAⅡa型,新生儿期发病伴先天畸形;② GAⅡb型,新生儿期发病不伴先天畸形;③ GAⅡc型,晚发型或轻症型。GAⅡa型、Ⅱb型患者多数在新生儿早期死亡,GAⅡc型在各个年龄段均可发病,以年长儿和成人报道多见。
本病罕见,迄今尚无发病率数据,由Przyrembel等[2] 1976年首次报道,以后陆续有新生儿或成人的病例报道[3]。我国自2003年梁雁等[4]首次报道2例GAⅡc型病例,已经有超过148例的报道[5]。国内血、尿有机酸分析和串联质谱分析开展较晚,以往报道的线粒体障碍脂质沉积性肌病的病例中可能有相当一部分为GAⅡc型[6]。
GAⅡc型临床表现个体差异较大,发病前常有感染、饥饿、腹泻、高脂饮食等诱发因素。婴幼儿患者间歇性发病,表现为肌张力低下、低血糖、高氨血症、代谢性酸中毒等症状,严重者出现惊厥、意识障碍、昏迷,部分患儿有类似瑞氏综合征的表现。年长儿及成人患者起病隐匿,轻症可无症状或症状轻微,如近端肢体无力,运动不耐受或易疲劳、肌肉疼痛等,可伴有恶心、呕吐等消化道症状;重症病例急性期常有低血糖、代谢性酸中毒、高氨血症、高乳酸血症、血清酶学(ALT、AST、CK、LDH)明显增高等,部分患者甚至出现意识障碍、惊厥、昏迷,少数病例可因感染并严重代谢紊乱、脑血栓等死亡[4, 7-8]。
GAⅡc型早期症状无特异性,常以肌无力、活动耐力下降、肝肿大及肝功异常、低血糖等首诊,早期诊断较为困难。血尿有机酸分析、血肉碱谱分析可提供诊断线索,发现电子转运黄素蛋白(electron transport flavin protein, ETF)基因或ETFDH基因:编码电子转运黄素蛋白-泛醌氧化还原酶(electron transfer flowoprotein-ubiquinom oxidoreductase, ETF-QO)突变有助于确诊[9]。本例患儿5岁发病,表现为反复发作的肌无力、意识障碍,以及肝脏增大、肝功能异常、低血糖等,11岁经GC-MS/MS筛查以及基因测序确诊。需要注意的是,GAⅡ型患者在病情缓解期可无典型的有机酸改变。
ETF或ETF-QO缺陷是导致GAⅡ型的关键因素,编码ETF或ETF-QO蛋白的基因ETFA、ETFB和ETFDH分别定位于15p23~25、19q13.1和4q33,分别由12、6和13个外显子组成[10-11]。研究显示,GAⅡ型的表型可能与基因突变种类有关,如ETFA和ETFB基因突变患者往往于新生儿期发病,而ETFDH基因突变引起晚发型GAⅡ[3, 11-12]。但Grünert等[12]发现约7%的晚发型GAⅡ型病例存在ETFA和ETFB基因突变。Olsen等[10]发现新生儿期发病的GAⅡ型患者也有存在ETFDH基因突变的。本例患儿5岁起病,为晚发型GAⅡ型患者。目前已报道80多个ETFDH基因突变位点,13个外显子均已见发现突变。本研究患儿的ETFDH基因外显子及剪切位点区域的两处突变:c.250G > A(鸟嘌呤 > 腺嘌呤)和c.1691-3C > G(胞嘧啶 > 鸟嘌呤),均为已报到的致病性突变,可导致其编码的蛋白p.A84T氨基酸改变(丙氨酸 > 丝氨酸)[13-14]。
GAⅡc型目前无特效治疗方法,以饮食控制、支持对症治疗为主。(1)饮食控制:急性发作期应限制脂肪和蛋白摄入,高碳水化合物饮食或静脉输注葡萄糖以补充能量、抑制分解代谢,促进合成代谢,减少酸性代谢物产生[4, 6]。饥饿时脂肪分解代谢增加,但高蛋白饮食也可加重病情。(2)补充左旋肉碱可促进酸性代谢物排出,纠正代谢性酸中毒[9, 16]。(3)核黄素(维生素B2)治疗对部分患者有效。维生素B2是ETF、ETFDH、酰基辅酶A脱氢酶的辅酶,大剂量维生素B2可刺激缺陷脱氢酶的残余活性,明显改善GAⅡ型患者的临床症状和代谢指标[9, 14]。(4)对于伴有辅酶Q10缺乏的GAⅡ型患者,辅酶Q10与维生素B2联用可改善症状[17]。(5)糖皮质激素如泼尼松等治疗可能有效,机制与其能激活甘油三酯脂肪酶,加速脂肪分解或促进肌肉细胞摄取肉毒碱有关[18]。
GAⅡc型患者如能予以早期饮食控制及补充核黄素、肉碱等干预,多数预后良好[6, 9],也有并发严重代谢代谢紊乱、心肌炎、脑血栓等死亡的报道[7]。本例患儿病程6年,多次急性发作,未能及早诊断和饮食控制,最终死于严重代谢紊乱。因此,对不明原因的发作性肌无力、低血糖伴肝肿大及酶学异常的患者,应注意GAⅡ型可能,尽早行尿有机酸分析和血串联质谱分析,必要时行基因诊断。
Biography
崔亚杰, 男, 硕士, 主治医师
References
- 1.Frerman FE, Goodman SI. Deficiency of electron transfer flavoprotein or electron transfer flavoprotein:ubiquinone oxidoreductase in glutaric acidemia type Ⅱ fibroblasts. Proc Natl Acad Sci U S A. 1985;82(13):4517–4520. doi: 10.1073/pnas.82.13.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Przyrembel H, Wendel U, Becker K, et al. Glutaric aciduria type Ⅱ:report on a previously undescribed metabolic disorder. Clin Chim Acta. 1976;66(2):227–239. doi: 10.1016/0009-8981(76)90060-7. [DOI] [PubMed] [Google Scholar]
- 3.Yotsumoto Y, Hasegawa Y, Fukuda S, et al. Clinical and molecular investigations of Japanese cases of glutaric acidemia type 2. Mol Genet Metab. 2008;94(1):61–67. doi: 10.1016/j.ymgme.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 4.梁 雁, 刘 丽, 魏 虹, et al. 维生素B2治疗有效的晚发型戊二酸尿症Ⅱ型. 中华儿科杂志. 2003;41(12):916–920. doi: 10.3760/j.issn:0578-1310.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 5.Zhu M, Zhu X, Qi X, et al. Riboflavin-responsive multiple Acyl-CoA dehydrogenation deficiency in 13 cases, and a literature review in mainland Chinese patients. J Hum Genet. 2014;59(5):256–261. doi: 10.1038/jhg.2014.10. [DOI] [PubMed] [Google Scholar]
- 6.邢 雅智. 多种酰基辅酶A脱氢酶缺乏症的诊治进展. http://www.cnki.com.cn/Article/CJFDTOTAL-LCAK201205017.htm 国际儿科学杂志. 2010;37(5):518–521. [Google Scholar]
- 7.章 瑞南, 邱 文娟, 叶 军, et al. 多种酰基辅酶A脱氢酶缺乏症儿童与成人患者临床特点比较. http://www.cnki.com.cn/Article/CJFDTOTAL-LCAK201205017.htm 临床儿科杂志. 2012;30(5):446–449. [Google Scholar]
- 8.Taubman B, Hale DE, Kelley RI. Familial Reye-like syndrome:a presentation of medium-chain acyl-coenzyme A dehydrogenase deficiency. http://pediatrics.aappublications.org/content/79/3/382.short. Pediatrics. 1987;79(3):382–385. [PubMed] [Google Scholar]
- 9.杨 艳玲, 木村 正彦, 袁 云, et al. 戊二酸尿症Ⅱ型所致脂肪沉积性肌肉病的诊断与治疗分析. http://www.cnki.com.cn/Article/CJFDTOTAL-ZHSJ200405017.htm 中华神经科杂志. 2004;37(5):438–441. [Google Scholar]
- 10.Olsen RK, Brøner S, Sabaratnam R, et al. The ETFDH c.158A > G variation disrupts the balanced interplay of ESE-and ESS-binding proteins thereby causing missplicing and multiple Acyl-CoA dehydrogenation deficiency. Hum Mutat, 2014;35(1):86–95. doi: 10.1002/humu.2014.35.issue-1. [DOI] [PubMed] [Google Scholar]
- 11.Xi J, Wen B, Lin J, et al. Clinical features and ETFDH mutation spectrum in a cohort of 90 Chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis. 2014;37(3):399–404. doi: 10.1007/s10545-013-9671-6. [DOI] [PubMed] [Google Scholar]
- 12.Grünert SC. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orphanet J Rare Dis. 2014;9:117. doi: 10.1186/s13023-014-0117-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lan MY, Fu MH, Liu YF, et al. High frequency of ETFDH c.250G > A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin Genet. 2010;78(6):565–569. doi: 10.1111/cge.2010.78.issue-6. [DOI] [PubMed] [Google Scholar]
- 14.Wen B, Dai T, Li W, et al. Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations. J Neurol Neurosurg Psychiatry. 2010;81(2):231–236. doi: 10.1136/jnnp.2009.176404. [DOI] [PubMed] [Google Scholar]
- 15.Wang ZQ, Chen XJ, Murong SX, et al. Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G > A. J Mol Med (Berl) 2011;89(6):569–576. doi: 10.1007/s00109-011-0725-7. [DOI] [PubMed] [Google Scholar]
- 16.Mooy PD, Przyrembel H, Giesberts MA, et al. Glutaric aciduria type Ⅱ:treatment with riboflavine, carnitine and insulin. Eur J Pediatr. 1984;143(2):92–95. doi: 10.1007/BF00445792. [DOI] [PubMed] [Google Scholar]
- 17.Gempel K, Topaloglu H, Talim B, et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. https://academic.oup.com/brain/article/130/8/2037/307340/The-myopathic-form-of-coenzyme-Q10-deficiency-is. Brain. 2007;130(Pt 8):2037–2044. doi: 10.1093/brain/awm054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engel AG, Siekert RG. Lipid Storage myopathy responsive to prednisone. Arch Neurol. 1972;27(2):174–181. doi: 10.1001/archneur.1972.00490140078011. [DOI] [PubMed] [Google Scholar]