

Abstract
对22例临床拟诊断为杜氏肌营养不良症(DMD)患儿的家系临床资料进行分析,探讨二代测序(NGS)技术在DMD患者分子诊断中的临床应用价值。先证者同时送检遗传性神经肌肉病相关panel行NGS检测和Dystrophin基因多重连接探针扩增术(MLPA)检测,分析两种方法检出的Dystrophin基因外显子缺失/重复突变结果,Dystrophin基因点突变经Sanger测序验证。通过NGS测序,22例先证者均检出Dystrophin基因突变,其中外显子缺失/重复型突变14例,点突变及微小变异8例。MLPA检测结果与NGS检测结果一致。Sanger测序结果显示NGS检测出的点突变及微小变异是正确的。1例错义突变c.6290G>T,1例无义突变c.3487C>T,4例微小缺失导致的移码突变c.1208delG、c.7497_7506delGGTGGGTGAC、c.9421_9422delAA、c.8910_8913delTCTC在人类基因突变数据库中均未见报道,为Dystrophin基因新突变。该研究结果显示NGS技术可全面检测Dystrophin基因外显子缺失/重复、点突变、微小缺失和内含子突变等变异,在DMD患者分子诊断中的临床应用有一定价值,值得推广。
Keywords: 杜氏肌营养不良症, 分子诊断, 二代测序技术, 多重连接探针扩增术, Sanger测序, 儿童
Abstract
The purpose of this study is to analyze the family's clinical data of 22 children who were given an intended clinical diagnosis of Duchenne muscular dystrophy (DMD), and to explore the clinical value of next-generation sequencing (NGS) in the molecular diagnosis of DMD. The probands were simultaneously tested by NGS for a gene panel associated with hereditary neuromuscular disease and multiplex ligation-dependent probe amplifcation (MLPA) for the Dystrophin gene. The exon deletion/repetition mutations of the Dystrophin gene determined by both methods were compared and the point mutations of the Dystrophin gene were verifed by Sanger sequencing. Dystrophin gene mutations were found in all the 22 probands, including 14 exon deletion/repetition mutations and 8 point mutations/minor variations. The results of MLPA detection were consistent with those of NGS. The results of Sanger sequencing showed that the point mutations and minor variations determined by NGS were correct. One missense mutation (c.6290G>T), 1 nonsense mutation (c.3487C>T) and 4 minor deletion-induced frameshift mutations (c.1208delG, c.7497_7506delGGTGGGTGAC, c.9421_9422delAA and c.8910_8913delTCTC) had not been reported in the Human Gene Mutation Database, and thus were considered as novel mutations of the Dystrophin gene. The results of this study showed that NGS can detect variations in the Dystrophin gene, including exon deletion/repetition, point mutation, minor deletion and intron mutation. Therefore, NGS is of certain clinical value in the molecular diagnosis of DMD and is worthy of recommendation.
Keywords: Duchenne muscular dystrophy, Molecular diagnosis, Next-generation sequencing, Multiplex ligationdependent probe amplifcation, Sanger sequencing, Child
杜氏肌营养不良症(Duchenne muscular dystrophy, DMD)是一种致死性的X连锁隐性遗传性疾病,由基因Dystrophin突变所致,男性活婴的发病率为1/3 500[1]。该病是儿童期最常见的肌肉病,一般3~5岁隐匿发病,9~12岁丧失站立和行走能力,多于20岁左右因心、肺功能衰竭而死亡[2]。Dystrophin基因是人体最大的基因之一,突变类型复杂多样,包含外显子(大片段)缺失/重复、单碱基点突变、微小缺失/插入、少量复杂突变和内含子突变等,导致DMD患者的基因诊断方法很难统一[3-4]。以往常用的分子诊断方法有:多重连接探针扩增术(multiplex ligation-dependent probe amplification, MLPA)、基因芯片、多重聚合酶链反应、逆转录PCR、短串联重复序列连锁分析、荧光原位杂交及引物原位标记技术等,各有优缺点,单选一种技术方法常会存在漏检发生[5]。目前,二代测序(next-generation sequencing, NGS)技术是唯一可以同时检测所有突变类型的平台[6]。刘敏娟等[7]于2012年初步探讨了NGS对DMD患者基因检测临床应用的可行性,6例患者成功检测到Dystrophin基因点突变、缺失和重复,但该技术尚未广泛应用于DMD的基因诊断。本研究中22例先证者同时送检遗传性神经肌肉病相关panel和Dystrophin基因的MLPA检测,比较分析两种方法检出的Dystrophin基因外显子缺失/重复结果,采用Sanger测序验证Dystrophin基因点突变,旨在进一步分析NGS技术在DMD基因诊断中的临床应用价值。
1. 资料与方法
1.1. 研究对象
2017年5月至2018年7月郑州大学第一附属医院儿科门诊就诊的22例临床拟诊断为DMD的男性患儿纳入研究。年龄50 d至11岁,均无神经肌肉病及其他遗传性疾病家族史。其中,因发现血清肌酶升高就诊4例;双下肢肌无力,行走姿势或步态异常,走路、跑步易摔倒就诊9例;行走费力、上楼和蹲下立起困难或不能就诊8例;另有1例因运动及智力发育落后,先不会爬、后不能独立站立就诊。22例患儿家属均同意行致病基因检测并签署了知情同意书。本研究已获得郑州大学第一附属医院伦理委员会批准。
1.2. 基因组DNA的提取
抽取患儿及其父母、兄弟姐妹外周血各2 mL,置于乙二胺四乙酸抗凝管中,冷链运送至武汉康圣达医学检验所有限公司,按照标准流程提取基因组DNA。
1.3. NGS检测及数据分析
选择遗传性神经肌肉病相关panel(检测基因594个)检测。采用安捷伦SureSelect Human All Exon V6定制试剂盒进行全部外显子区域及外显子-内含子交界处DNA捕获并富集,Illumina HiSeq平台进行测序。测序片段通过Burrows-Wheeler Aligner(BWA)软件(版本:0.7.9a)和UCSChg19人类参考基因组进行比对,比对结果采用Picard(版本:1.115)软件去除重复序列。
采用Genome Analysis Tool Kit(GATK,v3.2)软件进行变异检测,包括碱基质量分数校正,InDels位置和质量分数校正,SNVs和InDels变异发现和分型[8-9]。基于同批次一致性背景库和bed区域的测序深度计算基因外显子的拷贝数变异(copy number variation, CNV)[10-11]。CNV分析主要分两步:一是计算每个测序目标的测序深度,在每个样本中,剔除所有中位测序深度 < 20或 > 4 000、长度 < 20 bp或 > 2 000 bp、映射系数 < 0.9、GC含量 < 20%或 > 80%的区域,采用k=5的k均值聚类算法创建一个参考集;二是均一化和CNV分析,采用默认参数的CNVkit工具对测序数据深度均一化和CNV分析,在均一化阶段,利用参考集去除系统误差和偏差,提高信噪比。
运用基因检测智能操作系统(Genetic Testing Intelligent Execution System)进行变异注释,SNPEFF软件(hg19, GRCh37)进行变异解读,其中功能性的编码区和剪切位点的变异将进入下一步分析。通过ClinVar、在线人类孟德尔遗传(On-line Mendelian Inheritance in Man, OMIM)和人类基因突变数据库(Human Gene Mutation Database, HGMD)等查找变异位点或疑似致病突变的收录情况,进行基因突变致病的分析等。
1.4. MLPA检测
采用SALSAP034和P035(MRC-Holland,荷兰)试剂盒,按照说明书步骤,将变性后基因组DNA与SALSA MLPA探针杂交18 h,加入连接酶和缓冲液进行连接反应,经PCR扩增后用ABI3500XL基因分析仪(Applied Biosystems, 美国)分析,所得数据用Gene Marker v2.2.0软件分析。每次检测都包括2个健康对照样本,通过计算健康对照人群平均扩增峰值,对所扩增峰值进行标准化处理,分析得出MLPA分析结果图。
1.5. PCR扩增及Sanger测序
通过NGS检测出先证者为基因点突变及微小缺失时,先证者及家属选择PCR扩增Sanger测序验证。根据NGS分析结果,使用NCBI中Primer-BLAST在线软件(https://www.ncbi.nlm.nih.gov/tools/primer-blast/)设计目标区域的扩增引物。以家系成员DNA为模板,使用TaqDNA聚合酶扩增。1%琼脂糖凝胶电泳检测PCR产物,纯化的PCR产物在全自动测序仪ABI377(Applied Biosystems, 美国)上进行双向测序,测序结果用Chromas(version2.23)软件分析,并在NCBI中与正常序列(NM_004006.2)比对分析。
2. 结果
2.1. NGS检测结果
通过NGS测序,22例先证者均检出Dystrophin基因变异,检出率为100%:外显子(大片段)缺失突变13例(59%),外显子(大片段)重复突变1例(5%),无义突变2例(9%),错义突变1例(5%),微小缺失4例(18%),内含子突变1例(5%)(图 1)。患者均为半合子突变。通过在HGMD检索,本研究检测出的14例Dystrophin基因外显子(大片段)缺失/重复均已收录,1例无义突变c.10171C > T(p.Arg3391Stop)和1例内含子突变c.31+36947G > A已收录;1例错义突变c.6290G > T(p.Gly2097Val),1例无义突变c.3487C > T(p.Gln1163Stop),4例微小缺失导致的移码突变c.1208delG(p.Gly403fs)、c.7497_7506delGGTGGGTGAC(p.Met2499fs)、c.9421_9422delAA(p.Asn3141fs)、c.8910_8913delTCTC(p.Ser2970fs)在HGMD均未见报道,为新突变。SIFT软件和PolyPhen软件对c.6290G > T错义突变致病性评估分别为:SIFT预测为有害,PolyPhen预测为可能有害,综合分析该错义突变可能产生基因损伤效应,有致病性可能。
1.
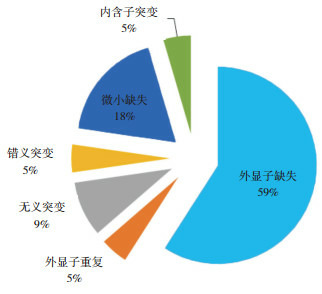
22个家系的Dystrophin突变类型分布图
2.2. MLPA检测结果
22例先证者通过MLPA检测,检出13例外显子缺失突变,1例外显子重复突变,检测结果与NGS测序结果完全一致。8例先证者母亲进行验证,2例为杂合突变携带者,6例为野生型(表 1)。
1.
22例DMD家系基因检测结果
| 编号 | 先证者NGS结果 | MLPA结果* | Sanger验证结果# | HGMD收录 |
| 注:*MLPA结果中的“-”表示先证者MLPA结果为阴性;#Sanger验证结果中的“-”表示先证者未进行Sanger验证。 | ||||
| 1 | 外显子49-52半合子缺失 | 先证者阳性,母亲野生型 | - | 有 |
| 2 | c.6290G > T(p.Gly2097Val)半合子突变 | - | 先证者阳性,母亲及妹妹野生型 | 无 |
| 3 | c.10171C > T(p.Arg3391Stop)半合子突变 | - | 先证者阳性,母亲及姐姐杂合突变 | 有 |
| 4 | 外显子45-61半合子重复 | 先证者阳性,母亲杂合重复 | - | 有 |
| 5 | c.1208delG(p.Gly403fs)半合子突变 | - | 先证者阳性,母亲杂合突变, 弟弟半合子突变 | 无 |
| 6 | 外显子45-54半合子缺失 | 先证者阳性,母亲未验证 | - | 有 |
| 7 | 外显子46半合子缺失 | 先证者阳性,母亲未验证 | - | 有 |
| 8 | c.7497_7506delGGTGGGTGAC(p.Met2499fs)半合子突变 | - | 先证者阳性,母亲及姐姐杂合突变 | 无 |
| 9 | c.3487C > T(p.Gln1163Stop)半合子突变 | - | 先证者阳性,母亲杂合突变 | 无 |
| 10 | c.9421_9422delAA(p.Asn3141fs)半合子突变 | - | 先证者阳性,母亲杂合突变 | 无 |
| 11 | 外显子61半合子缺失 | 先证者阳性,母亲杂合缺失 | - | 有 |
| 12 | 外显子22-44半合子缺失 | 先证者阳性,母亲野生型 | - | 有 |
| 13 | 外显子46-59半合子缺失 | 先证者阳性,母亲野生型 | - | 有 |
| 14 | c.31+36947G > A半合子突变(内含子突变) | - | 先证者阳性,母亲杂合突变 | 有 |
| 15 | 外显子45-52半合子缺失 | 先证者阳性,母亲未验证 | - | 有 |
| 16 | c.8910_8913delTCTC(p.Ser2970fs)半合子突变 | - | 先证者阳性,母亲及妹妹杂合突变 | 无 |
| 17 | 外显子10-11半合子缺失 | 先证者阳性,母亲野生型 | - | 有 |
| 18 | 外显子3-17半合子缺失 | 先证者阳性,母亲野生型 | - | 有 |
| 19 | 外显子50半合子缺失 | 先证者阳性,母亲未验证 | - | 有 |
| 20 | 外显子46-52半合子缺失 | 先证者阳性,母亲野生型 | - | 有 |
| 21 | 外显子12-15半合子缺失 | 先证者阳性,母亲未验证 | - | 有 |
| 22 | 外显子49-50半合子缺失 | 先证者阳性,母亲未验证 | - | 有 |
2.3. PCR扩增Sanger测序验证点突变
8例先证者Dystrophin基因点突变经PCR扩增Sanger测序验证均为阳性,证实NGS检测结果是准确的。8例先证者母亲进行验证,7例为杂合突变携带者,1例为野生型;2例先证者姐姐及1例先证者妹妹为杂合突变携带者;1例先证者弟弟为半合子突变(表 1)。
3. 讨论
DMD是由位于Xp21.2的基因Dystrophin缺陷所致,是一种严重的、致死性神经肌肉病,患者多在20岁左右死于心、肺等功能衰竭。然而,本病迄今为止仍缺乏有效的治疗方法[1-2],高效、准确的基因诊断和携带者筛查是有效降低DMD携带者患儿出生的关键。目前,临床上常见的DMD基因诊断方法是采用MLPA方法检测[12],检测结果阴性患者再选择NGS检测点突变和其他微小变异[13]。本研究中,22例疑似DMD先证者同时送检遗传性神经肌肉病相关panel进行NGS检测和Dystrophin基因MLPA检测,检测结果证实NGS检出的Dystrophin基因外显子缺失和重复与MLPA检测结果完全一致,点突变/微小缺失经Sanger测序验证结果准确无误,进一步说明NGS技术是可以全面检测复杂突变类型的Dystrophin基因变异,可在临床中广泛推广和应用。
DMD患儿在婴幼儿期的临床表型难以与其他肌营养不良区分,如强直性肌营养不良、肢带型肌营养不良等。针对婴幼儿期的患者,如果直接选择MLPA方法检测Dystrophin基因外显子缺失/重复,存在漏检点突变和微小变异的可能,也存在漏检其他肌营养不良疾病的可能。因此,本研究选择遗传性神经肌肉病相关panel进行NGS检测致病基因,虽未检测到其他致病基因的突变,但22例先证者的阳性率为100%,检出8例Dystrophin基因点突变/微小变异,14例Dystrophin基因大片段缺失/重复。
DMD患者中大约70%是由Dystrophin基因外显子缺失/重复造成,其余由Dystrophin基因点突变(无义/错误)、微小缺失/插入导致[3-4]。王凤羽等[14]和王军等[15]报道的MLPA检测DMD患者的阳性率均在72%左右,其他患者临床未确诊。为了避免漏检,白莹等[13]报道的研究中,先采用MLPA检测先证者外显子缺失/重复,对MLPA检测阴性的先证者应用NGS检测点突变情况,应用PCR扩增Sanger测序对点突变验证。该研究时间为2010年3月至2014年12月,此期间应用NGS检测DMD患者的基因变异情况是昂贵的,所以推荐的DMD基因诊断流程是以MLPA方法为基础。随着NGS测序成本的逐年下降,临床接受度提高。本研究中22例先证者同时送检遗传性神经肌肉病相关panel和Dystrophin基因MLPA检测,NGS检测出的Dystrophin基因外显子缺失、外显子重复、微小缺失、单碱基点突变和内含子突变的比例分别为59%、5%、18%、14%和5%,与已报道文献结果一致[16-17],经MLPA和Sanger家系验证分析,无假阳性和假阴性结果存在。本研究中,NGS检测的外显子拷贝数变异是基于同批次一致性背景库和bed区域的测序深度计算的,与MLPA检测结果一致。本研究检测出Dystrophin基因内含子突变1例c.31+36947G > A,属于外显子-内含子交界处,NGS技术也会漏检部分内含子突变,采用全基因组测序则可避免,但费用昂贵,难以在临床应用。
本研究通过对先证者母亲进行携带筛查发现,Dystrophin基因新发突变率高达44%(7/16)。有研究报道,对于新发突变DMD患者的母亲,外周血没有检测出为携带者的不能排除存在生殖嵌合体,建议对其再生育胎儿进行产前基因诊断[13]。鉴于Dystrophin基因新发突变率和突变特点,推荐选择NGS进行Dystrophin基因产前诊断。
本研究22例先证者同时送检遗传性神经肌肉病相关panel和Dystrophin基因的MLPA检测,结果显示NGS技术在Dystrophin基因诊断中的临床应用是值得推广的。NGS测序应用于Dystrophin基因产前诊断有一定临床应用价值,但尚需进行大样本量Dystrophin基因突变分析。本研究中发现的6例新突变丰富了Dystrophin基因突变谱。
Biography
田培超, 男, 博士, 副主任医师, 副教授。Email:tpczzu@126.com
References
- 1.Birnkrant DJ, Bushby K, Bann CM, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1:diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17(3):251–267. doi: 10.1016/S1474-4422(18)30024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freund AA, Scola RH, Arndt RC, et al. Duchenne and Becker muscular dystrophy:a molecular and immunohistochemical approach. Arq Neuropsiquiatr. 2007;65(1):73–76. doi: 10.1590/S0004-282X2007000100016. [DOI] [PubMed] [Google Scholar]
- 3.陈 远春, 代 英, 钟 敏. 3例杜氏肌营养不良家系基因诊断策略探讨. 重庆医学. 2016;45(7):926–928. doi: 10.3969/j.issn.1671-8348.2016.07.020. [DOI] [Google Scholar]
- 4.Aartsma-Rus A, Van Deutekom JC, Fokkema IF, et al. Entries in the Leiden Duchenne muscular dystrophy mutation database:an overview of mutation types and paradoxical cases that confrm the reading-frame rule. Muscle Nerve. 2006;34(2):135–144. doi: 10.1002/(ISSN)1097-4598. [DOI] [PubMed] [Google Scholar]
- 5.王 晶, 贺 勇. 杜氏肌营养不良分子诊断技术的研究进展. 国际检验医学杂志. 2014;35(10):1307–1309. doi: 10.3969/j.issn.1673-4130.2014.10.032. [DOI] [Google Scholar]
- 6.Lim BC, Lee S, Shin JY, et al. Genetic diagnosis of Duchenne and Becker muscular dystrophy using next-generation sequencing technology:comprehensive mutational search in a single platform. J Med Genet. 2011;48(11):731–736. doi: 10.1136/jmedgenet-2011-100133. [DOI] [PubMed] [Google Scholar]
- 7.刘 敏娟, 谢 敏, 毛 君, et al. 第2代测序技术在假肥大型肌营养不良基因诊断中的应用. 中华医学遗传学杂志. 2012;29(3):249–254. doi: 10.3760/cma.j.issn.1003-9406.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 8.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit:a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet. 2011;43(5):491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krumm N, Sudmant PH, Ko A, et al. Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22(8):1525–1532. doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fowler A, Mahamdallie S, Ruark E, et al. Accurate clinical detection of exon copy number variants in a targeted NGS panel using DECoN. Wellcome Open Res. 2016;1:20. doi: 10.12688/wellcomeopenres. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.殷 玉洁, 黄 玉萍, 陆 超, et al. 6例杜氏肌营养不良回顾性分析. http://www.zgddek.com/CN/abstract/abstract14244.shtml. 中国当代儿科杂志. 2017;19(4):405–409. doi: 10.7499/j.issn.1008-8830.2017.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.白 莹, 李 双, 宗 亚楠, et al. 杜氏/贝氏肌营养不良症433个家系的基因突变分析. 中华医学杂志. 2016;96(16):1261–1269. doi: 10.3760/cma.j.issn.0376-2491.2016.16.008. [DOI] [Google Scholar]
- 14.王 凤羽, 孙 伟伟, 李 聪敏, et al. 河南省杜氏肌营养不良患者dystrophin基因突变的MLPA检测. 郑州大学学报(医学版) 2011;46(6):842–846. doi: 10.3969/j.issn.1671-6825.2011.06.009. [DOI] [Google Scholar]
- 15.王 军, 彭 武, 胡 雪姣, et al. 中国西南地区170例杜氏/贝氏肌营养不良症dystrophin基因变异谱分析. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=hxykdxxb201602019 四川大学学报(医学版) 2016;47(2):232–237. [Google Scholar]
- 16.Takeshima Y, Yagi M, Okizuka Y, et al. Mutation spectrum of the dystrophin gene in 442 Duchenne/Becker muscular dystrophy cases from one Japanese referral center. J Hum Genet. 2010;55(6):379–388. doi: 10.1038/jhg.2010.49. [DOI] [PubMed] [Google Scholar]
- 17.Juan-Mateu J, Gonzalez-Quereda L, Rodriguez MJ, et al. DMD mutations in 576 dystrophinopathy families:a step forward in genotype-phenotype correlations. PLoS One. 2015;10(8):e0135189. doi: 10.1371/journal.pone.0135189. [DOI] [PMC free article] [PubMed] [Google Scholar]