

Abstract
目的
探讨伴髓系恶性转化的Shwachman-Diamond综合征(SDS)患儿的临床特征及基因突变情况。
方法
采用二代测序方法检测11例伴髓系恶性转化的SDS患儿的基因突变情况,分析患儿的临床特征和基因突变谱。
结果
11例SDS患儿中,9例(82%)表现为儿童难治性血细胞减少(RCC),1例(9%)为骨髓增生异常综合征(MDS)伴原始细胞增多(MDS-EB),1例(9%)为急性髓系白血病伴MDS相关改变(AML-MRC)。发生髓系恶性转化的中位年龄为48个月(范围:7个月~14岁)。5例(45%)患儿表现为单纯的血液系统异常。所有患儿均检测到SBDS基因突变,常见的突变形式为c.258+2T > C纯合突变(5例,45%)和c.184A > T+c.258+2T > C复合杂合突变(3例,27%)。SBDS基因新变异c.634_635insAACATACCTGT+c.637_638delGA和c.8T > C分别评级为"致病的"和"可能致病的"。转化为RCC和MDS-EB/AML-MRC的患儿3年预期总体生存率分别为100%、0%(P=0.001)。
结论
SDS患儿可能以血液系统症状为唯一表现,临床应引起重视。恶性转化的类型与预后相关。
Keywords: Shwachman-Diamond综合征, 骨髓增生异常综合征, 急性髓系白血病, 儿童
Abstract
Objective
To study the clinical features and genetic mutations of children with Shwachman-Diamond syndrome (SDS) and malignant myeloid transformation.
Methods
Next-generation sequencing was used to analyze the gene mutations in 11 SDS children with malignant myeloid transformation, and their clinical features and genetic mutations were analyzed.
Results
Of the 11 children with SDS, 9 (82%) presented with refractory cytopenia of childhood (RCC), 1 (9%) had myelodysplastic syndrome with excess blasts (MDS-EB), and 1 (9%) had acute myeloid leukemia with myelodysplasia-related changes (AML-MRC). The median age of onset of malignant myeloid transformation was 48 months (ranged 7 months to 14 years). Of the 11 children, 45% had abnormalities in the hematological system alone. Mutations of the SBDS gene were detected in all 11 children, among whom 5 (45%) had c.258+2T > C homozygous mutation and 3 (27%) had c.184A > T+c.258+2T > C compound heterozygous mutation. The new mutations of the SBDS gene, c.634_635insAACATACCTGT+c.637_638delGA and c.8T > C, were rated as "pathogenic" and "possibly pathogenic" respectively. The 3-year predicted overall survival rates of children transformed to RCC and MDS-EB/AML-MRC were 100% and 0% respectively (P=0.001).
Conclusions
SDS children may have hematological system symptoms as the only manifestation, which needs to be taken seriously in clinical practice. The type of malignant transformation is associated with prognosis.
Keywords: Shwachman-Diamond syndrome, Myelodysplastic syndrome, Acute myeloid leukemia, Child
Shwachman-Diamond综合征(Shwachman-Diamond syndrome, SDS)是一种先天性骨髓衰竭性疾病(inherited bone marrow failure syndromes, IBMFS),以中性粒细胞减少、胰腺外分泌功能不全、多系统或器官畸形为特征的的常染色体隐性遗传病。SDS具有髓系恶性转化的倾向性,转化类型包括骨髓增生异常综合征(myelodysplastic syndrome, MDS)和急性髓系白血病(acute myeloid leukemia, AML)等。超过90%的SDS患者具有SBDS基因突变。SBDS基因位于7号染色体q11区,编码参与核糖体成熟的蛋白。此外,SRP54、DNAJC21和EFL1基因突变的患者也可能出现SDS的临床特征。SDS临床表型、严重程度和疾病过程在家系间和家系内差异都很大。约40%的SDS患者出现严重的血液系统并发症[1]。血液系统并发症是导致患者死亡和必须造血干细胞移植(hematopoietic stem cell transplantation, HSCT)的主要原因[2]。SDS是一种罕见病,目前国际文献累计报道约300余例,查阅国内2010年8月至2020年1月期刊论文,共报道25例,而对于伴MDS或AML的SDS患儿国内仅报道1例[3]。本研究系统报道了11例伴MDS/AML的SDS患儿的临床特征及基因突变情况。
1. 资料与方法
1.1. 研究对象
回顾性分析我院2014年1月至2019年1月确诊的11例临床或分子学诊断的伴MDS/AML的SDS患儿。SDS临床诊断标准为:同时具有血细胞减少(在至少3个月内至少有2次中性粒细胞绝对计数低于1.5×109/L或其他血细胞减少)和胰腺外分泌功能不全,或有其他支持性证据,包括骨骼畸形、行为问题、一级亲属有确诊SDS者,并排除胰腺囊性纤维化、Fanconi贫血、先天性角化不良、Diamond-Blackfan贫血等其他疾病。分子学诊断标准为:检测到SBDS基因双等位基因致病性或可疑致病性突变[2]。MDS诊断符合2016年WHO分型诊断标准中儿童MDS诊断标准[4]。AML诊断标准为骨髓/外周血中髓系原始细胞≥20%[4]。所有患儿家属均已签署知情同意,并获我院伦理委员会批准(NI2020009-EC-1)。
1.2. 临床资料的收集
收集SDS患儿的临床资料,包括年龄、性别、重要症状体征(包括发育情况、躯体畸形)、家族史以及血常规、骨髓细胞形态学、骨髓病理、染色体核型、肝肾功能和胰腺影像学结果和治疗等。
1.3. 目的基因捕获二代测序检测突变基因
所有病例均采集骨髓或外周血5 mL,EDTA抗凝,4℃保存,72 h内进行DNA提取。家系验证DNA提取自父母和/或同胞外周血DNA。设计包含目的基因区域的捕获Panel,目的区域包括SBDS、SRP54、DNAJC21、EFL1及其他IBMFS、家族性MDS/AML和MDS相关基因等564个基因的全部外显子及外显子内含子交界区。应用Illumina NextSeq 500测序平台进行NGS。靶向区域平均基因覆盖度98.1%,平均测序深度为1 000×,95%的靶向区域覆盖度 > 20×。对筛出的变异位点进行Sanger测序验证。变异的致病性判断依据美国医学遗传学与基因组学学会(American College of Medical Genetics and Genomics, ACMG)发布的序列变异解读标准和指南[5]。
1.4. 随访
所有患儿随访至2019年10月,中位随访时间为39个月(范围:4~130个月)。随访资料来源于住院病历、门诊病历。对随访期间死亡的病例,依病例记录或与患儿家属电话联系确认。
1.5. 统计学分析
采用SPSS 22.0软件进行统计学分析。计量资料以中位数(范围)表示。计数资料以例或百分比(%)表示,组间比较采用Fisher精确概率法。生存分析采用Kaplan-Meier法,采用log-rank检验进行生存率比较。P < 0.05为差异有统计学意义。总生存(overall survival, OS)期按确诊髓系恶性转化至死亡的时间或末次随访时间计算。
2. 结果
2.1. 临床特征
共纳入11例SDS患儿,男7例,女4例,首次出现SDS相关的任何体征或症状的中位年龄为出生时(范围:出生时~8岁),首次出现血细胞减少的中位年龄为29个月(范围:出生时~12岁),首次诊断为MDS/AML的中位年龄为48个月(范围:7个月~14岁)。9例患儿表现为儿童难治性血细胞减少(refractory cytopenia of childhood, RCC),1例为MDS伴原始细胞增多(MDS with excess blasts, MDS-EB),1例为AML伴MDS相关改变(acute myeloid leukemia with myelodysplasia-related changes, AML-MRC)。45%(5/11)患儿表现为单纯的血液系统异常。仅18%(2/11)患儿表现为典型中性粒细胞减少伴腹泻。27%(3/11)患儿首次出现血细胞减少即为MDS/AML,包括2例RCC,1例MDS-EB。仅患儿5有家族史,该患儿诊断为RCC,表现为全血细胞减少伴发育迟缓,而具有相同突变的同胞哥哥仅表现为腹泻。发育迟缓、心脏畸形和其他躯体畸形分别存在于45%(5/11)、18%(2/11)和18%(2/11)的患儿。27%(3/11)患儿存在胰腺影像学异常,表现为胰腺体积小于同龄人、脂肪化。11例患儿均未检测到肝肾功能异常(表 1)。
1.
11例伴髓系恶性转化的SDS患儿临床特征和基因信息
| 编号 | 性别 | 转化MDS/ AML的年龄 | SBDS基因突变位点 | 氨基酸改变 | 突变来源 | 突变形式 | ACMG致病性判定 | 转化类型 | 其他系统表型 |
| 注:[RCC]儿童难治性血细胞减少;[MDS-EB]骨髓增生异常综合征伴原始细胞增多;[AML-MRC]急性髓系白血病伴骨髓增生异常综合征相关改变。 | |||||||||
| 1 | 女 | 14岁 | c.258+2T > C | splicing | 父母 | 纯合 | 致病 | RCC | 脂肪泻、学习困难、双趾畸形、胰腺脂肪化 |
| 2 | 女 | 5岁 | c.258+2T > C | splicing | 父母 | 纯合 | 致病 | RCC | 发育迟缓 |
| 3 | 男 | 7个月 | c.258+2T > C | splicing | 父母 | 纯合 | 致病 | RCC | 无 |
| 4 | 女 | 3.8岁 | c.258+2T > C | splicing | 母源杂合 | 纯合 | 致病 | RCC | 宫内发育迟滞、发育迟缓、房间隔缺损、卵圆孔未闭 |
| 5 | 男 | 2.5岁 | c.8T > C | p.I3T | 母源杂合 | 纯合 | 可能致病 | RCC | 发育迟缓、胰腺脂肪化 |
| 6 | 男 | 6岁 | c.258+2T > C | splicing | 父源 | 复合杂合 | 致病 | RCC | 尿道下裂、卵圆孔未闭、动脉导管未闭、发育迟滞、语言发育迟缓 |
| c.634_635insAACATACC- TGT, c.637_638delGA | p.I212Kfs*16 | 母源 | |||||||
| 7 | 男 | 1岁 | c.258+2T > C | splicing | 父源 | 复合杂合 | 致病 | RCC | 无 |
| c.184A > T | p.K62X | 母源 | |||||||
| 8 | 男 | 4岁 | c.258+2T > C | splicing | 母源 | 复合杂合 | 致病 | RCC | 无 |
| c.184A > T | p.K62X | 父源 | |||||||
| 9 | 男 | 12岁 | c.258+2T > C | splicing | 母源杂合 | 纯合 | 致病 | AML-MRC | 脂肪泻、胰腺脂肪化 |
| 10 | 男 | 8岁 | c.258+2T > C | splicing | 母源 | 复合杂合 | 致病 | MDS-EB | 无 |
| 2号外显子缺失 | - | 自发突变 | |||||||
| 11 | 女 | 3岁 | c.258+2T > C | splicing | 父源 | 复合杂合 | 致病 | RCC | 无 |
| c.184A > T | p.K62X | 母源 | |||||||
确诊髓系恶性转化时SDS患儿的血液学特征见表 2,中性粒细胞减少最常见,存在于91%(10/11)的患儿。骨髓病理示11例患儿骨髓增生程度均低于相应年龄参考值。骨髓细胞形态学示低增生伴有轻度粒系、红系或巨核系的病态造血。患儿转化为MDS前的形态异常主要存在于髓系,包括成熟粒细胞低分叶及不同程度的颗粒减少。转化为MDS后的形态异常更为显著,其中髓系病态造血最为显著,包括Pelger-Huet样畸形、明显的颗粒减少及显著增大的胞浆偏碱性髓系前体细胞;此阶段患者也具有明显的红系病态造血。染色体核型分析示18%(2/11)患儿存在染色体异常,分别为inv(9)(p12q13)(患儿4)和del(20)(q11)(患儿1)。所有患儿未观察到其他系统恶性肿瘤。
2.
11例伴髓系恶性转化的SDS患儿确诊时的血液学指标特征 (n=11)
| 项目 | 例(%) |
| 中性粒细胞减少 | 10(91) |
| 重度粒细胞缺乏(< 0.2×109/L) | 3(27) |
| 粒细胞缺乏(< 0.5×109/L) | 6(55) |
| 贫血 | 9(82) |
| 重度贫血(< 60 g/L) | 6(55) |
| 中度贫血(< 90 g/L) | 2(18) |
| 血小板减少 | 7(64) |
| 血小板 < 20×1012/L | 1(9) |
2.2. 基因突变分析
11例患儿中,55%(6/11)为纯合突变,45%(5/11)为复合杂合突变。c.258+2T > C和c.184A > T分别存在于91%(10/11)、27%(3/11)的患儿(表 1)。c.258+2T > C、c.184A > T为既往报道过的致病性突变[2]。c.634_635insAACATACCTGT+c.637_638delGA(p.I212Kfs*16)为新变异。该变异为无义突变,符合PVS1;该变异在ESP数据库(http://evs.gs.washington.edu/EVS)、千人基因组数据库(http://browser.1000genomes.org)、ExAC数据库(http://exac.broadinstitute.org)正常对照人群中未发现,符合PM2;经MutationTaster(http://www.mutationtaster.org)和GERP++软件(http://mendel.stanford.edu/SidowLab/downloads/gerp/)的生物信息数据分析预测,分别提示为“有害的”、“保守的”,符合PP3;因此该突变被判定为“致病的”。c.8T > C(p.I3T)为新变异,患儿5为纯合变异,患儿哥哥具有慢性腹泻,也为该位点的纯合变异,患儿母亲携带此位点的杂合变异,符合PP1;患儿的表型和家族史对考虑的基因高度特异,符合PP4;该变异为ESP数据库、千人数据库、ExAC数据库正常对照人群中未发现的变异,符合PM2;经SIFT(https://sift.bii.a-star.edu.sg)、PolyPhen-2(http://genetics.bwh.harvard.edu/pph2)、MutationTaster和GERP++软件的生物信息数据分析预测,分别提示为“有害的”、“可能有害的”、“致病的”和“保守的”,符合PP3;该变异为错义变异,而基因的良性错义变异频率低、错义变异是疾病的普遍机制,符合PP2;因此该突变被判定为“可能致病的”。转化为AML-MRC的患儿9检测到IDH1基因c.394C > T(p.R132C)体细胞突变。不同基因型患儿的血细胞减少、染色体核型异常、死亡和非血液系统表型的发生率无显著性差异(均P > 0.05),见表 3。
3.
11例不同基因型伴髓系恶性转化的SDS患儿的临床特征比较 [例(%)]
| 项目 | c.258+2T > C纯合突变(n=5) | c.258+2T > C+ c.184A > T复合杂合突变(n=3) | 其他突变(n=3) | P值 |
| 中性粒细胞 < 0.5×109/L | 3(60) | 3(100) | 2(67) | 0.452 |
| 血红蛋白 < 90 g/L | 3(60) | 3(100) | 1(33) | 0.231 |
| 血小板 < 100×1012/L | 2(40) | 2(67) | 3(100) | 0.231 |
| 染色体核型异常 | 2(40) | 0(0) | 0(0) | 0.231 |
| 死亡 | 1(20) | 0(0) | 1(33) | 0.565 |
| 非血液系统特征表型 | 1(20) | 3(100) | 1(33) | 0.079 |
2.3. 预后分析
中性粒细胞减少的患儿在发生感染时应用人粒细胞集落刺激因子(granulocyte-colony stimulation factor, G-CSF)。无患儿接受长期G-CSF治疗或预防性治疗。2例患儿(1例RCC,1例AML-MRC)接受了HSCT治疗。所有患儿出现血细胞减少后3年预期OS率为82%(95%CI:70%~93%)。患儿转化为MDS/AML后3年预期OS率为80%(95%CI:67%~93%)。从出现血细胞减少到转化为MDS/AML的中位时间为18个月(范围:4~121个月)。转化为RCC和高危险度MDS-EB或AML的患儿3年预期OS率分别为100%和0%(P=0.001)(图 1)。转化为高危险度MDS或AML的2例患儿从确诊MDS/AML到死亡的时间分别仅4个月、5.5个月。性别、首次诊断SDS的年龄、血液学参数分布及异常染色体核型对患儿总生存均无明显影响(表 4)。
1.
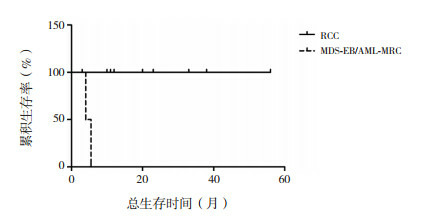
不同髓系恶性转化类型SDS患儿总生存曲线
4.
11例伴有髓系恶性转化的SDS患儿3年OS率的单因素分析
| 项目 | 例数 | 3年OS率(95%CI) | χ2值 | P值 |
| 注:[RCC]儿童难治性血细胞减少;[MDS-EB]骨髓增生异常综合征伴原始细胞增多;[AML-MRC]急性髓系白血病伴骨髓增生异常综合征相关改变。 | ||||
| 性别 | ||||
| 男 | 7 | 71%±17% | 0.928 | 0.335 |
| 女 | 4 | 100% | ||
| 首次诊断SDS年龄 | ||||
| ≤1岁 | 5 | 100% | 2.242 | 0.134 |
| > 1岁 | 6 | 60.0%±21.9% | ||
| 中性粒细胞缺乏程度 | ||||
| < 0.5×109/L | 8 | 71%±17% | 0.928 | 0.335 |
| ≥0.5×109/L | 3 | 100% | ||
| 血红蛋白浓度 | ||||
| < 90 g/L | 7 | 67%±19% | 1.465 | 0.226 |
| ≥90 g/L | 4 | 100% | ||
| 血小板 | ||||
| < 100×1012/L | 7 | 67%±19% | 1.465 | 0.226 |
| ≥100×1012/L | 4 | 100% | ||
| 染色体核型 | ||||
| 正常 | 9 | 75%±15% | 0.536 | 0.464 |
| 异常 | 2 | 100% | ||
| 转化类型 | ||||
| RCC | 9 | 100% | 11.023 | 0.001 |
| MDS-EB/AML-MRC | 2 | 0% | ||
3. 讨论
本回顾性研究报告了国内最大队列伴髓系恶性转化的SDS患儿的临床特征和基因突变情况。约20%的SDS患者以中性粒细胞 < 0.5×109/L为首次就诊原因,但仅有65%的患者同时具有典型的胰腺外分泌功能不全及骨骼畸形,因此临床中有漏诊可能[1]。一项包括1 514例接受HSCT的MDS患者的研究报道,4%的年轻患者携带SBDS基因突变,且大多数在移植前未被诊断出,提示该类患者在临床中存在被低估的可能性[6]。本研究中45%患儿表现为单纯血液系统异常,提示SDS相关基因检测在IBMFS诊断Panel中的必要性。
超过90%的SDS伴有SBDS基因突变。最常见的突变位点包括c.184A > T和c.258+2T > C[1-2, 7]。意大利SDS登记组研究结果表明c.184A > T纯合突变、c.184A > T+c.258+2T > C复合杂合突变和c.258+2T > C纯合突变最为常见,分别占56%、14%和8%[7]。本研究结果中常见的突变形式为c.258+2T > C纯合突变(45%)和c.184A > T+c.258+2T > C复合杂合突变(27%)。与既往报道一致,本研究亦未检测到SBDS基因型和表型之间的关联[7]。另外,本研究首次报道了SBDS的两个新变异,致病性突变p.I212Kfs*16和可能致病性突变p.I3T。除了SBDS,最近还有3个基因被报道具有类SDS表型,包括DNAJC21[8-9]、EFL1[10]和SRP54[11],上述基因在SDS患者中所占比例均 < 1%。本研究未检测到上述基因突变,可能上述基因突变在我国SDS患儿罕见。因此,对于疑诊SDS患儿仍首先考虑检测SBDS基因。TP53是SDS最常见的体细胞突变基因,与MDS转化风险增高和不良预后有关[6]。在1例发生AML转化的SDS患者中检测到IDH1体细胞突变,并提示可能与预后良好相关[12]。本研究中患儿9中检测到IDH1体细胞突变,一方面为这一突变可能驱动SDS的恶性转化提供了证据,另一方面该患儿预后差,提示需更多病例来明确IDH1突变对预后的影响。
SDS患者常见两种重现性细胞遗传学异常为i(7)(q10)和del(20q)[1-2],其是否与恶性转化相关尚存在争议。诸多研究表明i(7)(q10)和del(20)(q11)不增加SDS恶性转化的风险,而染色体7的非i(7q)异常,特别是单体7可能与髓系恶性转化相关[1-2, 13]。复杂核型存在于44%髓系恶性转化的SDS患者中,并且与不良预后有关[13]。本研究中除了del(20q),还检测到inv(9)(p12q13),2例患儿均表现为粒细胞缺乏,分别于14岁、45月龄时转化为RCC。而2例转化为MDS-EB/AML-MRC的患儿均为正常核型。
目前报道的SDS髓系恶性转化率差异很大,在5%到36%之间[2, 14-17]。SDS转化为MDS/AML后预后差,10年OS率仅25%[18]。2019年一项多中心回顾性研究显示中位随访4.9年,转化为MDS和AML患者的中位生存期分别为7.7年和0.99年,3年OS率分别是51%和11%[13]。本研究结果亦显示转化为MDS-EB/AML的患儿预后差,而转化为RCC的患儿可长期稳定。
SDS患者临床过程各异,如何精确预测患者预后对于治疗选择具有重要意义。本研究未发现与预后相关的因素,可能与样本数量少和患者以血液系统症状为主有关。对于转化为MDS/AML的SDS患者,HSCT虽为唯一可能治愈手段,但预后较差,诊断MDS后中位生存时间仅1.2年[6, 16],生存率仅23%[14]。对于那些患有SDS的患者,应进行每年一次的已知疾病并发症的随访,并监测其血细胞计数和骨髓转化情况[13, 16]。需注意的是,40%患者在血细胞计数稳定的情况下转化为MDS[13],因此规范监测骨髓可提高患者的长期生存。接受和不接受骨髓监测的患者3年OS率分别为62%与28% [13]。
总之,回顾性分析本中心SDS转化为MDS/AML患儿,结果显示SDS患儿可能以血液系统症状为唯一表现,临床应引起重视。c.258+2T > C和c.184A > T是最常见的突变位点,可作为基因筛查的首选。转化为MDS-EB/AML的患儿预后差,生存期短,而转化为RCC的患儿可长期稳定。进一步需要建立我国的SDS病例登记系统,建立基于证据的诊断、治疗和管理策略对于改善患儿预后至关重要。
Biography
安文彬, 女, 博士, 主治医师
Funding Statement
国家自然科学基金(81700109);协和青年科研基金&中央高校基本科研业务费专项资金(2017320024);爱佑慈善基金
References
- 1.Nelson AS, Myers KC. Diagnosis, treatment, and molecular pathology of Shwachman-Diamond Syndrome. Hematol Oncol Clin North Am. 2018;32(4):687–700. doi: 10.1016/j.hoc.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 2.Nelson A, Myers K. Shwachman-Diamond syndrome[EB/OL]. GeneReviews<sup>®</sup>, (2018-10-18)[2020-01-25]. https://www.ncbi.nlm.nih.gov/pubmed/20301722.
- 3.王 凤琦, 魏 晓楠, 邵 翠华, et al. Shwachman Diamond综合征的剪切位点突变及表型分析. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=qdyxyxb201806001 青岛大学学报(医学版) 2018;54(6):631–634. [Google Scholar]
- 4.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 5.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants:a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lindsley RC, Saber W, Mar BG, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med. 2017;376(6):536–547. doi: 10.1056/NEJMoa1611604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kuijpers TW, Alders M, Tool AT, et al. Hematologic abnormalities in Shwachman Diamond syndrome:lack of genotype-phenotype relationship. Blood. 2005;106(1):356–361. doi: 10.1182/blood-2004-11-4371. [DOI] [PubMed] [Google Scholar]
- 8.Dhanraj S, Matveev A, Li H, et al. Biallelic mutations in DNAJC21 cause Shwachman-Diamond syndrome. Blood. 2017;129(11):1557–1562. doi: 10.1182/blood-2016-08-735431. [DOI] [PubMed] [Google Scholar]
- 9.Morini J, Nacci L, Babini G, et al. Whole exome sequencing discloses heterozygous variants in the DNAJC21 and EFL1 genes but not in SRP54 in 6 out of 16 patients with Shwachman-Diamond syndrome carrying biallelic SBDS mutations. Br J Haematol. 2019;185(3):627–630. doi: 10.1111/bjh.15594. [DOI] [PubMed] [Google Scholar]
- 10.Stepensky P, Chacón-Flores M, Kim KH, et al. Mutations in EFL1, an SBDS partner, are associated with infantile pancytopenia, exocrine pancreatic insufficiency and skeletal anomalies in a Shwachman-Diamond like syndrome. J Med Genet. 2017;54(8):558–566. doi: 10.1136/jmedgenet-2016-104366. [DOI] [PubMed] [Google Scholar]
- 11.Carapito R, Konantz M, Paillard C, et al. Mutations in signal recognition particle SRP54 cause syndromic neutropenia with Shwachman-Diamond-like features. J Clin Invest. 2017;127(11):4090–4103. doi: 10.1172/JCI92876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mourad S, Bilodeau M, Roussy M, et al. IDH1 as a cooperating mutation in AML arising in the context of Shwachman-Diamond syndrome. Front Oncol. 2019;9:772. doi: 10.3389/fonc.2019.00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Myers KC, Furutani E, Weller E, et al. Clinical features and outcomes of patients with Shwachman-Diamond syndrome and myelodysplastic syndrome or acute myeloid leukaemia:a multicentre, retrospective, cohort study. Lancet Haematol. 2020;7(3):e238–e246. doi: 10.1016/S2352-3026(19)30206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Donadieu J, Fenneteau O, Beaupain B, et al. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica. 2012;97(9):1312–1319. doi: 10.3324/haematol.2011.057489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Alter BP, Giri N, Savage SA, et al. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=0e1971e8-ff4f-4a19-8787-17e89d5d3123. Haematologica. 2018;103(1):30–39. doi: 10.3324/haematol.2017.178111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenberg PS, Alter BP, Bolyard AA, et al. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006;107(12):4628–4635. doi: 10.1182/blood-2005-11-4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myers KC, Davies SM, Shimamura A. Clinical and molecular pathophysiology of Shwachman-Diamond syndrome:an update. Hematol Oncol Clin North Am. 2013;27(1):117–128. doi: 10.1016/j.hoc.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cada M, Segbefia CI, Klaassen R, et al. The impact of category, cytopathology and cytogenetics on development and progression of clonal and malignant myeloid transformation in inherited bone marrow failure syndromes. Haematologica. 2015;100(5):633–642. doi: 10.3324/haematol.2014.117457. [DOI] [PMC free article] [PubMed] [Google Scholar]