

Abstract
Background
Glioblastoma is an uncommon but highly aggressive type of brain tumour. Significant gains have been achieved in the molecular understanding and the pathogenesis of glioblastomas, however clinical improvements are difficult to obtain for many reasons. The current standard of care involves maximal safe surgical resection followed by chemoradiation and then adjuvant chemotherapy European Organisation for Research and Treatment of Cancer and the NCIC Clinical Trials Group (EORTC‐NCIC) protocol with a median survival of 14.6 months. Successive phase III international randomised controlled studies have failed to significantly demonstrate survival advantage with newer drugs.
Epidermal growth factor receptor (EGFR) is observed to be aberrant in 30% to 60% of glioblastomas. The receptor aberrancy is driven by abnormal gene amplification, receptor mutation, or both, in particular the extracellular vIII domain. EGFR abnormalities are common in solid tumours, and the advent of anti‐EGFR therapies in non‐small cell lung cancer and colorectal adenocarcinomas have greatly improved clinical outcomes. Anti‐EGFR therapies have been investigated amongst glioblastomas, however questions remain about its ongoing role in glioblastoma management. This review aimed to report on the available evidence to date and perform a systematic analysis on the risks and benefits of use of anti‐EGFR therapies in glioblastomas.
Objectives
To evaluate the efficacy and harms of anti‐EGFR therapies for glioblastoma in adults.
Search methods
We searched CENTRAL, MEDLINE, Embase, EBM Reviews databases, with supplementary handsearches to identify all available and relevant studies to 20 April 2020.
Selection criteria
All randomised controlled trials (RCTs) using anti‐EGFR therapies in adults with glioblastoma were eligible for inclusion. Anti‐EGFR therapies included tyrosine kinase inhibitors, monoclonal antibodies, or vaccines. The comparison included investigational product added to standard of care versus standard of care or placebo, or investigational product against standard of care or placebo.
Data collection and analysis
The authorship team screened the search results and recorded the extracted data for analysis. We used standard Cochrane methodology to performed quantitative meta‐analysis if two or more studies had appropriate and available data. Otherwise, we conducted a qualitative and descriptive analysis. We used the GRADE system to rate the certainty of the evidence. The analysis was performed along the two clinical settings: first‐line (after surgery) and recurrent disease (after failure of first line treatment). Where information was available, we documented overall survival, progression‐free survival, adverse events, and quality of life data from eligible studies.
Main results
The combined searches initially identified 912 records (after removal of duplicates), and further screening resulted in 19 records for full consideration. We identified nine eligible studies for inclusion in the review. There were three first‐line studies and six recurrent studies. Five studies used tyrosine kinase inhibitors (TKIs); two studies used monoclonal antibodies; and two studies used targeted vaccines. More recent studies presented greater detail in the conduct of their studies and thus had a lower risk of bias.
We observed no evidence benefit in overall survival with the use of anti‐EGFR therapy in the first‐line or recurrent setting (hazard ratio (HR) 0.89, 95% confidence interval (CI) 0.76 to 1.04; 3 RCTs, 1000 participants, moderate‐certainty evidence; and HR 0.79, 95% CI 0.51 to 1.21, 4 RCTs, 489 participants, low‐certainty evidence, respectively). All the interventions were generally well tolerated with low‐certainty evidence for lymphopenia (odds ratio (OR) 0.97, 95% CI 0.19 to 4.81; 4 RCTs, 1146 participants), neutropenia (OR 1.29, 95% CI 0.82 to 2.03; 4 RCTs, 1146 participants), and thrombocytopenia (OR 3.69, 95% CI 0.51 to 26.51; 4 RCTs, 1146 participants). A notable toxicity relates to ABT‐414, where significant ocular issues were detected.
The addition of anti‐EGFR therapy showed no evidence of an increase in progression‐free survival (PFS) in the first‐line setting (HR 0.94, 95% CI 0.81 to 1.10; 2 RCTs, 894 participants, low‐certainty evidence). In the recurrent setting, there was an increase in PFS with the use of anti‐EGFR therapy (HR 0.75, 95% CI 0.58 to 0.96, 3 RCTs, 275 participants, low‐certainty evidence). The available quality of life assessment data showed that anti‐EGFR therapies were neither detrimental or beneficial when compared to standard care (not estimable).
Authors' conclusions
In summary, there is no evidence of a demonstrable overall survival benefit with the addition of anti‐EGFR therapy in first‐line and recurrent glioblastomas. Newer drugs that are specially designed for glioblastoma targets may raise the possibility of success in this population, but data are lacking at present. Future studies should be more selective in pursuing people displaying specific EGFR targets.
Plain language summary
Drugs that target abnormal growth protein in high‐grade, aggressive brain tumours
Background
Glioblastomas are highly aggressive brain tumours. They often appear quickly with devastating effects depending on the part of the brain they are located. They often affect previously well and high functioning individuals without any ‘warning signs’. There are no known risk factors. The impact on people with glioblastomas, their family, friends, and society is highly problematic. Standard therapy involves resection of the tumour, then combined chemotherapy and radiation therapy followed by an additional six months of chemotherapy. This strategy aims only to control and contain the disease and delay its return because at present there is no cure.
Researchers have investigated and found multiple gene changes in glioblastoma tissue samples, leading to clinical trials testing new drug therapies. The protein epidermal growth factor receptor (EGFR), which normally controls cell growth, is abnormal in glioblastomas in about 30% to 60% of cases. This abnormality can lead to unrestrained cell growth, replication, and an increase in the cancer's aggressive potential. It is currently recognised that people with glioblastomas with an abnormal EGFR may have shorter survivals.
Some clinical trials with drugs targeting this protein have been conducted. This review aimed to collect all available evidence and investigate the risks and benefits for this type of therapy in glioblastomas, and in particular whether anti‐EGFR drugs can improve survival whilst remaining a tolerable therapy without side effects.
Methods
We searched medical databases for randomised controlled trials (a type of study in which participants are assigned to one of two or more treatment groups using a random method) that used anti‐EGFR therapies in people with glioblastoma up to April 2020.
Key results
Overall, no benefits were seen in improving overall survival with the use of anti‐EGFR therapy in newly diagnosed people with glioblastoma or in the recurrent setting. The use of anti‐EGFR therapies was not associated with increased side effects such as low white cells or platelet counts. There were some expected side effects including skin rashes and diarrhoea, but these were not severe and did not seem to impact participant quality of life. Anti‐EGFR therapy did not delay disease worsening in newly diagnosed people with glioblastomas but there was an improvement seen amongst those with recurrent disease.
Conclusions
At present, there is insufficient evidence to support the use of anti‐EGFR therapy in newly diagnosed or recurrent glioblastoma. Whilst the therapy is in general expected as with other anti‐EGFR therapies, significant eye side effects can arise with ABT‐414. Overall, anti‐EGFR therapies did not appear to affect quality of life. The future use of anti‐EGFR therapy in the management of glioblastoma requires more investigation. Future research should be promoted and tailored towards people with glioblastoma with known abnormal EGFR receptors.
Summary of findings
Summary of findings 1. Anti‐EGFR therapy compared to placebo or standard of care for glioblastoma in adults.
| Anti‐EGFR therapy compared to placebo or standard of care for glioblastoma in adults | |||||
| Patient or population: glioblastoma in adults Setting: Hospital (outpatients and inpatients) Intervention: anti‐EGFR therapy Comparison: placebo or standard of care | |||||
| Outcomes | № of participants (studies) | Certainty of the evidence (GRADE) | Relative effect (95% CI) | Anticipated absolute effects* (95% CI) | |
| Risk with placebo or standard of care | Risk difference with anti‐EGFR therapy | ||||
| Overall survival ‐ first‐line studies | 1000 (3 RCTs) | ⊕⊕⊕⊝ MODERATE 1 2 | HR 0.89 (0.76 to 1.04) | Study population | |
| 280 per 1000 | 26 fewer per 1000 (59 fewer to 9 more) | ||||
| Overall survival ‐ recurrent disease studies | 489 (4 RCTs) | ⊕⊕⊝⊝ LOW 3 4 5 6 | HR 0.79 (0.51 to 1.21) | Study population | |
| 250 per 1000 | 47 fewer per 1000 (114 fewer to 44 more) | ||||
| Lymphopenia | 1146 (4 RCTs) | ⊕⊕⊝⊝ LOW 7 8 | OR 0.97 (0.19 to 4.81) | Study population | |
| 97 per 1000 | 3 fewer per 1000 (77 fewer to 243 more) | ||||
| Neutropenia | 1146 (4 RCTs) | ⊕⊕⊝⊝ LOW 7 8 | OR 1.29 (0.82 to 2.03) | Study population | |
| 9 per 1000 | 3 more per 1000 (2 fewer to 9 more) | ||||
| Thrombocytopenia | 1146 (4 RCTs) | ⊕⊕⊝⊝ LOW 7 8 | OR 3.69 (0.51 to 26.51) | Study population | |
| 2 per 1000 | 5 more per 1000 (1 fewer to 44 more) | ||||
| Progression‐free survival ‐ first‐line studies | 894 (2 RCTs) | ⊕⊕⊝⊝ LOW 1 2 | HR 0.94 (0.81 to 1.10) | Study population | |
| 250 per 1000 | 22 more per 1000 (32 fewer to 75 more) | ||||
| Progression‐free survival ‐ recurrent disease studies | 275 (3 RCTs) | ⊕⊕⊝⊝ LOW 2 | HR 0.75 (0.58 to 0.96) | Study population | |
| 250 per 1000 | 56 fewer per 1000 (96 fewer to 9 fewer) | ||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; EGFR: epidermal growth factor receptor; HR: hazard ratio; OR: odds ratio; RCT: randomised controlled trial | |||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | |||||
1Lee 2015 and Westphal 2015 are both open‐label trials, hence blinding would not have been preserved. 2The three included studies all used different anti‐EGFR agents, thus affecting the directness and applicability of the evidence and requiring that it be downgraded. 3The randomisation and blinding procedures have not been clarified in Reardon 2015. van den Bent 2019 was an open‐label trial. 4High degree of heterogeneity amongst the three included studies. 5Different mechanisms of action amongst the three included studies, raising concerns about the directness of the evidence. 6Wide confidence interval, raising concerns about the precision of the outcome. 7Three of the included studies were open‐label trials, increasing the risk of bias. 8There is a degree of heterogeneity amongst these studies with regard to lines of therapy and drug mechanisms.
Background
Description of the condition
Glioblastomas are highly aggressive brain tumours that present with significant clinical challenges. Despite recent public concerns around potential causality between mobile phones and glioblastoma diagnoses, incidence rates have not increased between 1982 and 2013 (AIHW 2017; Nilsson 2017). Peak incidence occurs in the 50 to 70 yearold age group, and no specific causative agent has been identified.
Disease morbidity for glioblastomas is multifactorial. Neurological deficits may arise acutely and vary depending on tumour location. Some people develop postoperative deficits, and some have neurocognitive sequelae such as concentration and memory, or both. Others require ongoing anticonvulsant medication or corticosteroids to control cerebral oedema (Lapointe 2015). Additional morbidities include immunosuppression from chemotherapy, radiation treatment, or corticosteroids and predisposition to opportunistic infection, thrombocytopenia (reduced platelet counts), and increased risk of thrombosis (blood clots) (Qian 2016; Thaler 2013; Thaler 2014). People with glioblastoma are often premorbidly high functioning and active but subsequently become dependent on their family and friends, thus increasing psychosocial stresses that are often underestimated.
Glioblastoma treatment consists of maximal safe resection followed by concurrent chemoradiotherapy and adjuvant chemotherapy using temozolomide, resulting in a median overall survival (OS) of 14.6 months (Stupp 2005). Since 2005, there have been multiple phase III clinical trials that have added various chemotherapy combinations and monoclonal antibodies to this standard of care. However, they have all been unsuccessful in improving survival outcomes in a clinically and statistically meaningful manner (Chinot 2014; Gilbert 2014; Khasraw 2016; Stupp 2014).
The success of future trials may hinge on better participant selection with particular attention to molecular changes. Verhaak and colleagues documented the complexity of molecular changes commonly appearing in glioblastomas and suggested the identification of four molecularly distinct, clinically relevant subgroups: proneural, neural, mesenchymal, and classical (Verhaak 2010). Aldape and colleagues further investigated the importance of these molecular changes and demonstrated the need to respect these unique genetic signatures in predicting treatment sensitivity and prognosticating survival (Aldape 2015).
There have been great successes with the use of targeted therapeutic agents in other cancers. Small molecule tyrosine kinase inhibitors (TKI) targeting epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) translocations in non‐small cell lung cancer (NSCLC); v‐Raf murine sarcoma viral oncogene homolog B (BRAF) inhibition in metastatic melanoma; and anti‐EGFR antibodies in Kirsten rat sarcoma virus (KRAS) wild‐type colorectal cancers have all led to great improvements in progression‐free survival (PFS) and OS in genomically selected participants (Hauschild 2012; Karapetis 2008; Mok 2009; Shaw 2013). However, despite the discovery of many genetic alterations in glioblastoma, the successes with targeted therapy in other solid tumours have yet to be replicated in the treatment of glioblastoma. There are certainly additional factors that are unique to neuro‐oncology that need to be considered, including drug delivery through the blood–brain barrier, intratumoural heterogeneity, and variability of the genetic targets.
Description of the intervention
In normal cellular physiology, the binding of a growth factor (e.g. epidermal growth factor, EGF) to a receptor (e.g. EGFR) initiates a cascade of downstream intracellular events which regulate cell proliferation, survival, and differentiation. Overactivity of this pathway leads to uncontrolled cell growth, replication, and tumour development. This can be achieved by overexpression of the receptor, autocrine overproduction of the ligand and constitutive activation by mutations in the receptor complex (Castillo 2004).
The most frequent genetic alterations in glioblastoma are overexpression or amplification of EGFR, reported to occur in 30% to 60% of cases (Brennan 2013; Huang 2009). These EGFR abnormalities can be detected by immunohistochemistry looking for protein overexpression, fluorescence in‐situ hybridisation looking for gene amplification and polymerase chain reaction for EGFR variant III (EGFRvIII) mutation. EGFRvIII mutation is a shortened form of the gene due to loss of part of the gene (exons 2 to 7). Both EGFR overexpression and EGFRvIII can enhance glioblastoma cell growth, migration, and invasiveness (Bastien 2015; Cloughesy 2014; Haas‐Kogan 2005).
The classical genomic subtype as described by Verhaak 2010 is typically associated with EGFR amplification with a high proportion of EGFRvIII mutations. This group also has a lower median OS of 12.2 months compared to other glioblastomas in general (14.6 months) and is generally associated with an older population (over 70 years of age) (Stupp 2005; Verhaak 2010). In particular, the EGFRvIII mutant subgroup has a lower OS (less than one year) (Heimberger 2005; Shinojima 2003).
There are currently three main therapeutic methods to target EGFR overactivity: anti‐EGFR monoclonal antibodies, EGFR TKIs, and EGFR vaccines (Table 2). Anti‐EGFR monoclonal antibodies target the extracellular ligand binding domain of the receptor and block activation of the receptor and its subsequent downstream activation. EGFR TKIs target the intracellular component of the receptor associated with an activating mutation, which subsequently inhibits auto‐phosphorylation and subsequent downstream signalling. Anti‐EGFR vaccines have been designed to target the specific novel amino acid sequence arising from EGFRvIII deletion mutation and generating an immunological response.
1. Classes of anti‐EGFR therapies.
| Drug class | Description and examples |
| Anti‐EGFR monoclonal antibodies |
|
| Anti‐EGFR (tyrosine kinase inhibitors) |
|
| Anti‐EGFR vaccines |
|
EGF: epidermal growth factor; EGFR: epidermal growth factor receptor; EGFRvIII: EGFR variant III.
Anti‐EGFR vaccines and EGFR TKIs are specific to particular mutations and alterations in EGFR, whilst monoclonal antibodies target the extracellular domain of EGFR, so are effective when EGFR is amplified regardless of mutational status. Monoclonal antibodies are typically administered as intravenous injections given every one to two weeks. EGFR TKIs are typically oral tablets given daily, and EGFR vaccines are given monthly after a loading dose and via a subcutaneous route.
In this review, the experimental treatment is tested against the standard of care, which is combined chemoradiation following maximal safe resection and adjuvant chemotherapy or the best standard of care at the time of the clinical trial. This is summarised in Table 2.
How the intervention might work
The aberrant EGFR pathway is an attractive therapeutic target, and inhibition in other tumour types such as NSCLC and colorectal cancer has led to significant clinical responses. In both NSCLC and colorectal cancers, the use of EGFR TKI and monoclonal antibodies has led to improvements in OS and PFS. In NSCLC, the use of gefitinib amongst EGFR‐mutated tumours improved PFS compared to cytotoxic chemotherapy (hazard ratio 0.48, 95% confidence interval 0.36 to 0.64; P < 0.001). This was further supported in the OPTIMAL study, which used erlotinib as first‐line chemotherapy in people with EGFR‐mutated NSCLC, where the PFS improvement was 13.1 months versus 4.6 months with chemotherapy (Mok 2009; Zhou 2011). In people with colorectal cancer, those with wild‐type EGFR benefited from the use of monoclonal antibody, where OS improved with cetuximab (9.5 months with cetuximab plus best supportive care versus 4.8 months with best supportive care alone) (Karapetis 2008).
We hypothesise that anti‐EGFR therapies in EGFR‐overexpressing glioblastomas may inhibit cell proliferation and result in cell death, leading to improved survival and achieving similar results as those seen in NSCLC and colorectal cancer.
Why it is important to do this review
The purpose of this review was to find, organise, and summarise high‐level evidence in terms of the benefits and harms of anti‐EGFR therapies in people with glioblastoma to provide meaningful conclusions for clinical practice and further research. This review was driven by the encouraging results observed in phase II clinical trials involving anti‐EGFR vaccines (ACT II), where the median OS reached was 21.6 to 26 months, a significant increase compared to standard of care, which led to subsequent phase III randomised controlled trials (Sampson 2010; Schuster 2015). Targeting EGFR in glioblastoma is an active area of interest with ongoing studies in progress. Newer compounds such as depatuxizumab mafodotin (ABT‐414), an antibody–drug conjugate that can target EGFR or EGFRvIII in glioblastomas, which allows potent chemotherapy to be released inside targeted cancer cells (NCT02573324). This review will form a platform to review new data in this area as they mature.
The current standard of care for people with glioblastoma is maximum resection followed by concurrent temozolomide radiotherapy then adjuvant temozolomide, irrespective of molecular signatures. This review investigated if the addition of anti‐EGFR therapy improves outcomes in EGFR‐overexpressing glioblastomas.
Objectives
To evaluate the efficacy and harms of anti‐EGFR therapies for glioblastoma in adults.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs).
Types of participants
Adults (aged 18 years and over) with histologically confirmed glioblastoma diagnosis, either newly diagnosed or with recurrent disease.
Types of interventions
Interventions can be categorised into three groups as described by their site and mode of action against the EGFR pathway. These include anti‐EGFR monoclonal antibodies, EGFR TKIs, and anti‐EGFR vaccines. We included studies of any anti‐EGFR agents against placebo or standard of care. We included studies that combined a secondary intervention in the treatment group (such as chemotherapy or radiotherapy) if this secondary treatment was the same in the control group. The control group could receive the standard of care/active intervention (such as chemotherapy, as long as anti‐EGFR therapy was not used), placebo, or best supportive care.
In summary, the three groups would be:
anti‐EGFR monoclonal antibodies with or without chemotherapy versus placebo or standard of care with or without chemotherapy;
EGFR TKIs with or without chemotherapy versus placebo or standard of care with or without chemotherapy;
anti‐EGFR vaccine with or without standard of care versus placebo or standard of care with or without chemotherapy.
In the event there was a direct head‐to‐head comparison between two or more different anti‐EGFR therapies (with or without standard of care in either arm), we would assess this and if deemed eligible will include it in future analyses.
Types of outcome measures
We considered for evaluation any studies including at least one of the following outcomes.
Primary outcomes
Overall survival (OS): defined as time from randomisation to death from any cause.
Severe adverse events: classified according to National Cancer Institute Common Terminology Criteria for Adverse Events (NCI‐CTCAE) (NCI 2017), including percentage of treatment‐related deaths.
Secondary outcomes
Quality of life (QoL): measured against objective scales such as European Organisation for Research and Treatment of Cancer Quality of Life Core Questionnaire (EORTC QLQ‐C30; Scott 2008), European Organisation for Research and Treatment of Cancer Quality of Life Questionnaire – Brain Neoplasm (EORTC QLQ‐BN20) (Taphoorn 2010), or as defined by the trial investigators.
Progression‐free survival (PFS): defined as time from randomisation to disease progression. Disease progression may be defined by two criteria: MacDonald and Response Assessment in Neuro‐Oncology Criteria (RANO) (Macdonald 1990; Wen 2010). MacDonald's criteria was the accepted standard assessment tool in older neuro‐oncology trials until the advent of RANO criteria. RANO is now recognised as the standard response assessment tool in neuro‐oncology trials. All trials included in this review used RANO criteria for their measurements and assessments.
We presented the following outcomes in Table 1.
OS in first‐line glioblastoma studies
OS in recurrent glioblastoma studies
-
Severe adverse events
Lymphopenia
Neutropenia
Thrombocytopenia
PFS in first‐line glioblastoma studies
PFS in recurrent glioblastoma studies
Quality of life
Search methods for identification of studies
Electronic searches
The following databases were searched on 20 April 2020:
the Cochrane Central Register of Controlled Trials (CENTRAL; 2020, Issue 4), in the Cochrane Library (Appendix 1);
MEDLINE via Ovid (1946 to April week 3, 2020) (Appendix 2);
Embase via Ovid (1980 to 2020 week 16) (Appendix 3).
We applied no language restrictions to any of the searches.
Searching other resources
Two review authors (AL, MA) independently searched the following databases up to 20 February 2020 using the search strings provided by Cochrane Gynaecological, Neuro‐oncology and Orphan Cancers. These searches were also conducted in Cochrane Methodology Register, ACP Journal Club, EBM reviews, Database of Abstracts of Reviews of Effects (Ovid Technologies), and abstracts and reports from major conferences, including American Society of Clinical Oncology (ASCO), European Society of Medical Oncology (ESMO), Society of Neuro‐oncology, and European Association of Neuro‐oncology.
In addition, we searched relevant journals including Journal of Clinical Oncology, Annals of Oncology, Lancet, Lancet Oncology, New England Journal of Medicine, European Journal of Cancer, Neuro‐oncology, and Journal of Neurology and Neurosurgery.
Data collection and analysis
Two review authors (AL, MA) independently collected data and prepared the manuscript for analysis using Review Manager 5 (Review Manager 2014).
Selection of studies
We downloaded all titles and abstracts retrieved by electronic searching to a reference management database (Endnote and Covidence). Two review authors (AL, MA) independently screened the records identified from electronic and handsearches for RCTs and excluded those studies that obviously did not meet the inclusion criteria as described in Criteria for considering studies for this review. We retrieved the full‐text reports of all possibly relevant studies and assessed whether they met the inclusion criteria. We listed studies that did not meet the inclusion criteria in the Characteristics of excluded studies table with the reasons for exclusion. Any uncertainties or disagreements were resolved by discussion or by consulting a third review author (MK) if required. We identified and excluded duplicate reports and collated multiple reports of the same study so that each study, rather than each report, was the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram. We provided comprehensive details of the included studies in the Characteristics of included studies table.
We included abstracts and unpublished data only if information was available on study design and characteristics of participants, interventions, and outcomes. We contacted primary or corresponding study authors for further information and clarification to aid in this process.
Data extraction and management
Two review authors (AL, MA) independently performed data extraction. We followed Cochrane methodology as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), and used a pre‐piloted standardised data extraction form on two studies (see Appendix 4) and entered the data into Review Manager 5 for analysis (Review Manager 2014). For each eligible form, we recorded the following information: title, authors, study design, participants, setting, interventions, 'Risk of bias' items, duration of follow‐up, efficacy outcomes, QoL scores, and adverse effects. We extracted data for studies with more than one publication from the most recent publication. We highlighted short‐term adverse events if these were considered significant. We collected additional study‐related information including contact address, country, published/unpublished, language, year of publication, and sponsor of trial.
Any differences in data extraction were resolved by consensus with a third review author (MK, VH, or HW), with reference to the original article.
For time‐to‐event data (OS, PFS), we extracted hazard ratios (HRs) and 95% confidence intervals (CIs), log rank Chi2, log rank P values, numbers of events, numbers of participants per group, and medians. Where HRs were not available, we calculated them following the methods of Tierney 2007 for incorporating summary time‐to‐event data into meta‐analysis.
For dichotomous data such as adverse events, we extracted the raw data and calculate odds ratios (OR) with 95% CIs.
For continuous outcomes (QoL measures), we extracted the number of responders, mean and standard deviation (SD) in each arm to calculate the mean difference (MD) with 95% CIs. Where possible, we performed quantitative analysis on collected and calculated data. If there were insufficient data, we presented a descriptive analysis.
When possible, we extracted data for intention‐to‐treat analysis for all outcomes. We collected the time points at which outcomes were collected and reported.
Assessment of risk of bias in included studies
Two review authors (AL, MA) independently applied the 'Risk of bias' tool, resolving any differences by discussion or by appeal to a third review author (MK, VH, or HW). We judged each item at high, low, or unclear risk of bias as set out in the criteria provided by Higgins 2011 and provided a quote from the study report or a statement (or both) as justification for the judgement for each item in the 'Risk of bias' table. For attrition bias, we judged a trial to be low risk of bias if at least 80% of participants were assessed at endpoint for all outcomes. We summarised results in both a 'Risk of bias' graph and 'Risk of bias' summary. When interpreting treatment effects and meta‐analyses, we took into account the risk of bias for the studies that contribute to that outcome. Where information on risk of bias relates to unpublished data or correspondence with an investigator, we noted this in the 'Risk of bias' table.
Measures of treatment effect
We presented summary statistics for the primary endpoints (time‐to‐event data). Where HRs were not available, we calculated them following the methods of Tierney 2007 for incorporating summary time‐to‐event data into meta‐analysis. For dichotomous data such as adverse events, we extracted the incidence and total number of people evaluated and calculated for ORs. For continuous outcomes (QoL measures), we extracted data to calculate mean deviations. Where possible, we performed quantitative analysis on collected and calculated data. If there were insufficient data, we presented a descriptive analysis.
Unit of analysis issues
We have based measurement of PFS on RANO criteria (see Secondary outcomes) (Wen 2010).
Dealing with missing data
We contacted the first or corresponding author of the most recent publication in the case of missing data. We have not imputed missing data for any of the outcomes.
Assessment of heterogeneity
We have assessed heterogeneity between studies using the Cochran Q test, with a significance threshold of alpha = 0.1 and by estimation of the percentage of heterogeneity between trials that could not be ascribed to sampling variation.
In cases of substantial heterogeneity, the extra variation would have been incorporated into the analysis by using a random‐effects model. We also planned to visually inspect forest plots for heterogeneity.
We considered an I2 value of 30% or greater to represent a degree of heterogeneity worthy of further investigation. We considered the following factors as possible sources of heterogeneity:
differing clinical settings (adjuvant versus recurrent disease);
different types of anti‐EGFR therapies (as classified above);
differences in prognostic factors between studies;
study quality.
We considered these factors in the sensitivity and subgroup analyses, except in cases of differing prognostic factors.
Assessment of reporting biases
If we included 10 or more studies that investigated a particular outcome, we examined funnel plots corresponding to meta‐analysis of the outcome to assess the potential for small‐study effects such as publication bias. We planned to assess funnel plot symmetry visually, and if asymmetry was suggested, we would perform exploratory analyses to investigate it.
Data synthesis
We performed a meta‐analysis on the outcomes listed above if two or more trials of the appropriate clinical setting were available, appreciating that some statistical heterogeneity might have occurred from pooling of trials investigating different therapies. We used standard meta‐analytical techniques, employing a random‐effects model if heterogeneity in participant characteristics and treatments existed. We would group trials into first‐line or recurrent settings for analysis, as this better correlates with real‐world clinical purposes.
For time‐to‐event data, we pooled log HRs and standard error (SE) logHRs using the generic inverse variance facility of Review Manager 5 (Review Manager 2014). For dichotomous outcomes, we pooled ORs using the Mantel‐Haenszel method.
In trials with multiple treatment groups, we combined time‐to‐event outcomes by performing a separate meta‐analysis of the two‐arm HRs. Subsequently, the resulting HRs was the summary statistic for the overall trial. We followed the method described in Section 16.5 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Subgroup analysis and investigation of heterogeneity
We considered the following variables for subgroup analyses where data were available:
first‐line therapy;
recurrent disease.
Sensitivity analysis
We conducted predefined sensitivity analyses to assess the robustness of the conclusions based on studies with high or unclear risk of bias versus low risk of bias.
'Summary of findings' table and GRADE assessment of the certainty of the evidence
We have presented the overall certainty of the evidence for each outcome according to the GRADE approach, which takes into account issues not only related to internal validity (risk of bias, inconsistency, imprecision, publication bias) but also to external validity (such as directness of results) (Langendam 2013). We created a 'Summary of findings' table based on the methods described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), employing GRADEpro GDT (Table 1) (GRADEpro GDT). We used the GRADE checklist and GRADE Working Group certainty of evidence definitions (Meader 2014). We downgraded the evidence from 'high' certainty by one level for serious (or by two for very serious) concerns for each limitation.
High certainty: We are very confident that the true effect lies close to that of the estimate of the effect.
Moderate certainty: We are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different.
Low certainty: Our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect.
Very low certainty: We have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect.
Results
Description of studies
See Characteristics of included studies, Characteristics of excluded studies, Characteristics of ongoing studies
Results of the search
The literature search was supported by the Information Specialist of the Cochrane Gynaecological, Neuro‐oncology and Orphan Cancer Group, who helped to formulate the search strategies and conducted the searches in CENTRAL, MEDLINE, and Embase up to April 2020. Two review authors (AL and MA) conducted additional searches up to 20 April 2020 in Cochrane Methodology Register, ACP Journal Club, Database of Abstracts of Reviews of Effects (Ovid Technologies), and abstracts and reports from major conferences.
The main database searches identified 2879 references which were put through the Cochrane RCT Classifier. The RCT classifier is a machine learning routine that helps to distinguish between reports of RCTs (and quasi‐RCTs) and non‐RCTs. Following this step and de‐duplication, there were 908 references imported for screening using Covidence (Covidence). Additional handsearches yielded four references. Consequently, there were 912 references available for screening. Two review authors (AL and MA) independently conducted the screening procedures, identifying keywords including glioblastoma, adult, randomised, trial, EGFR (and EGFR‐related drugs – monoclonal antibodies, tyrosine kinase inhibitors, vaccines) from the title. We identified 851 references that were irrelevant. Two review authors (AL and MA) independently reviewed 61 abstracts. We excluded 42 abstracts, of which 29 abstracts had the wrong trial design, 10 were duplicate entries, 1 study was ongoing and 1 study did not have the outcomes listed as specified in our protocol. We independently reviewed the full texts of 19 records, of which 9 were found to be eligible and included in the review. This process is detailed in a PRISMA flow diagram (Figure 1).
1.
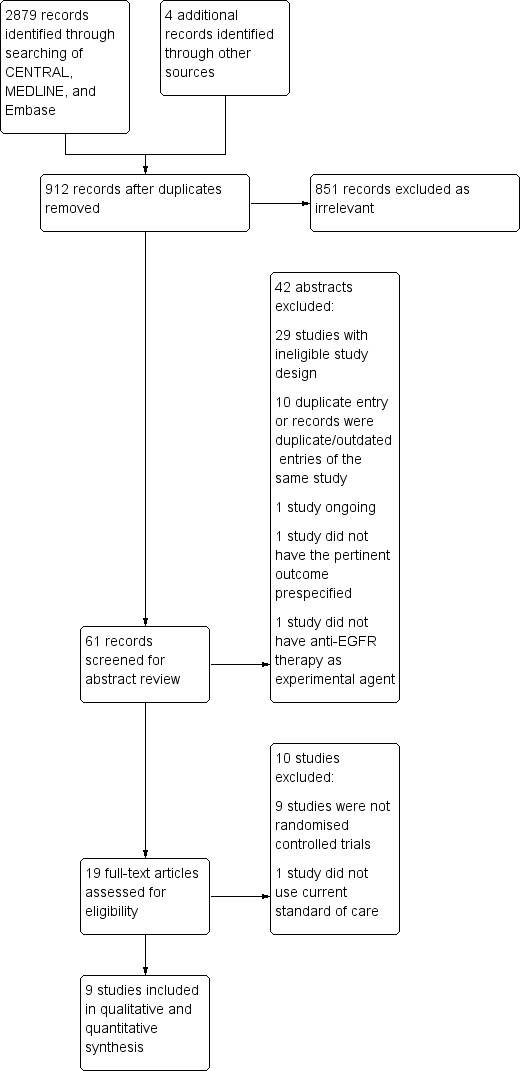
PRISMA flow diagram.
Included studies
The nine included studies can be categorised into first‐line (treatment naïve), Lee 2015; Weller 2017; Westphal 2015, and recurrent (second‐line or further setting) (Brown 2016; McNeill 2014; Reardon 2015 (ReACT); Reardon 2020; van den Bent 2009; van den Bent 2019). There were three first‐line studies and six recurrent studies investigating the role of anti‐EGFR therapies in glioblastoma. Three of the nine included studies had predefined selection for EGFR amplified or mutated glioblastomas (Reardon 2015; van den Bent 2019; Weller 2017).
The median/mean age of participants across the studies was 54 to 60 years. Three studies used OS as the primary endpoint (Lee 2015; van den Bent 2019; Weller 2017); two studies used PFS (Brown 2016; Westphal 2015); and the remaining four studies used PFS at six months (PFS6) as primary endpoint (McNeill 2014; Reardon 2015; Reardon 2020; van den Bent 2009). All of the included studies also reported secondary outcomes on OS, PFS, and toxicities. No adjustments to survival statistics were noted. However, we noted that two studies were terminated early: Brown 2016 was stopped due to cessation of the experimental drug, cediranib, by the pharmaceutical manufacturer, and Weller 2017 was closed after a second preplanned interim analysis due to futility. We also noted that Westphal 2015 presented two unplanned subgroup analyses by O6‐methylguanine–DNA methyltransferase (MGMT) methylation and EGFR statuses.
We further evaluated the included studies for risk of bias according to Cochrane guidelines (Assessment of risk of bias in included studies).
First‐line studies
Amongst the first‐line treatment studies, one study investigated the use of nimotuzumab, a monoclonal antibody (Westphal 2015); one study investigated the use of vandetanib, a multi‐TKI targeting EGFR, vascular epidermal growth factor receptor (VEGFR), and rearranged during transfection (RET) (Lee 2015); and the third study investigated the use of the EGFRvIII‐targeting vaccine rindopepimut (Weller 2017). All three studies tested their respective investigational product against the standard European Organisation for Research and Treatment of Cancer and the NCIC Clinical Trials Group (EORTC‐NCIC) protocol (Stupp 2005). The minimum Karnofsky Performance Status (KPS) was greater or equal to 60 (Karnofsky 1948). Weller 2017 specified maximal surgical resection (due to the potential risk of immunogenic flare from the vaccine), which may have inadvertently ruled out people with a poorer prognosis.
Recurrent disease studies
Amongst the recurrent treatment studies, four studies involved TKIs (Brown 2016; McNeill 2014; Reardon 2015; van den Bent 2009); one study used an antibody drug conjugate ABT‐414 (van den Bent 2019); and the remaining study investigated the use of rindopepimut vaccine (Reardon 2020). As there is no recognised standard of care in recurrent glioblastomas, the control arm was generally either cytotoxic chemotherapy, physician's choice, or conservative management. In Brown 2016, cediranib, a TKI against VEGFR, was added to gefitinib (EGFR TKI) versus cediranib alone; van den Bent 2009 investigated the use of erlotinib (EGFR TKI) versus temozolomide or carmustine; and Reardon 2015 investigated the use of afatinib (EGFR TKI) versus temozolomide versus afatinib and temozolomide in a three‐arm randomised study. These three studies had recruited participants at first recurrence only with a minimum KPS of 70. All eligible participants had received chemoradiation and adjuvant chemotherapy as per EORTC‐NCIC protocol (Stupp 2005).
In McNeill 2014, the experimental arm was vandetanib and carboplatin versus carboplatin alone. No detail was provided regarding the number of prior treatments or any previous treatments. Reardon 2020 recruited participants at first or second recurrence who were bevacizumab naïve. Participants were randomised to bevacizumab and rindopepimut versus bevacizumab plus placebo. The placebo was specially designed to generate an immune skin reaction to maintain blinding. McNeill 2014 was only available in abstract form; we contacted the corresponding authors for further information.
van den Bent 2019 investigated the use of a new antibody drug conjugate ABT‐414 (depatuxizumab mafodotin, or Depatux‐M), an anti‐EGFR monoclonal antibody drug conjugate. ABT‐414 can target both EGFR and EGFRvIII mutations and is stable in the bloodstream until it meets its target and releases anti‐microtubule agent monomethyl auristatin F (MMAF) (van den Bent 2019). This study, named INTELLANCE‐2, had three arms, with arm A using ABT‐414 only, arm B containing ABT‐414 and temozolomide, and arm C temozolomide or lomustine (depending on the time to failure from last temozolomide use).
Excluded studies
We excluded 10 studies after full‐text review. We excluded nine studies that were not randomised trials (Daugherty 2014; Hong 2012; Neyns 2009; Schuster 2015; Sepulveda 2015; Solomon 2013; van den Bent 2016; Wen 2014; Wygoda 2002). In addition, we excluded Wygoda 2006 and Solomon 2013 due to the different standard of care used; specifically they did not use temozolomide, which is now part of standard of care.
Risk of bias in included studies
The summary of the 'Risk of bias' assessment in the included studies is shown in Figure 2.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
Random sequence generation was well documented in seven of the included studies (Brown 2016; Lee 2015; Reardon 2020; van den Bent 2009; van den Bent 2019; Weller 2017; Westphal 2015), which we classified as at low risk of bias. It is unclear from the available sources how the random sequence generation was made in McNeill 2014 and Reardon 2015; we classified these two studies as at unclear risk of bias.
Four studies demonstrated adequate allocation concealment as part of their double‐blind, randomised protocol, and were therefore classified as at low risk of bias (Brown 2016; Reardon 2020; van den Bent 2009; Weller 2017). In particular, Reardon 2020 and Weller 2017 added an immunostimulant in the placebo vaccines to generate the expected skin reaction similar to rindopepimut preparations. Lee 2015, van den Bent 2019, and Westphal 2015 were open‐label studies and hence concealment could not be maintained, thus they were classified as at high risk of bias. It was unclear from the available sources how allocation concealment was preserved in McNeill 2014 and Reardon 2015, thus these two studies were classified as at unclear risk of bias.
Blinding
Three studies demonstrated evidence to support preserved blinding of participants and personnel and were assessed as at low risk of bias (Brown 2016; Reardon 2020; Weller 2017). Lee 2015, van den Bent 2019, and Westphal 2015 were open‐label studies, thus blinding would not have been preserved, and as such these studies were graded as at high risk of bias. It is unclear if blinding of participants and personnel was preserved in McNeill 2014, Reardon 2015, and van den Bent 2009, thus these studies were graded as at unclear risk of bias.
Brown 2016, van den Bent 2009, and Weller 2017 mention blinding of outcome assessors in their reports. Reardon 2015, Reardon 2020, and van den Bent 2019 mentioned that they had an independent imaging review committee, therefore we considered that these studies were at low risk of bias. It is unclear whether assessor blinding took place in Lee 2015, McNeill 2014, and Westphal 2015, thus we classified these studies as at unclear risk of bias.
Incomplete outcome data
Most of the included studies had clear flow diagrams illustrating the distribution of all trial participants and were thus classified as at low risk of bias (Brown 2016; Lee 2015; Reardon 2020; van den Bent 2009; van den Bent 2019; Weller 2017; Westphal 2015). This information was missing from McNeill 2014, as the study was available in abstract form only. Reardon 2015 had 12 participants that were not randomised after enrolment, and as this was not explained we classified the study as at high risk of bias for this domain.
Selective reporting
Most of the included studies reported on all prespecified outcomes (Brown 2016; Lee 2015; Reardon 2015; Reardon 2020; van den Bent 2009; van den Bent 2019; Weller 2017). Westphal 2015 did not report on response rates as prespecified and was thus classified as at high risk of bias. This information was missing from McNeill 2014, as the study was available in abstract form only.
Other potential sources of bias
We detected no other potential sources of bias.
Effects of interventions
See: Table 1
Primary outcomes
Overall survival
We performed meta‐analysis separately for first‐line and recurrent glioblastoma studies.
Meta‐analysis of the three first‐line studies demonstrated no reduction in the risk of death with anti‐EGFR therapies (hazard ratio (HR) 0.89, 95% confidence interval (CI) 0.76 to 1.04; 3 RCTs, 1000 participants, moderate‐certainty evidence, Analysis 1.1, Figure 3, Table 1) (Lee 2015; Weller 2017; Westphal 2015).
1.1. Analysis.

Comparison 1: Overall survival, Outcome 1: First‐line
3.

Forest plot of comparison: 1 Overall survival, outcome: 1.1 First‐line.
No heterogeneity was observed (I2 = 0%). Individually, these studies did not demonstrate a benefit with the respective experimental therapy. The median overall survival (OS) in months in the experimental arms was 16.6, 17.4, and 22.3 months for Lee 2015, Weller 2017, and Westphal 2015, respectively. These figures would be consistent with contemporary non‐EGFR‐related first‐line glioblastoma therapy studies globally (Chinot 2014; Gilbert 2014; Nabors 2015; Stupp 2014; Stupp 2017). Both Weller 2017 and Westphal 2015 demonstrated the importance of maximal safe resection in improving outcome. In Weller 2017, the subgroup with maximal safe resection reported survival above 20 months, whereas the subgroup with significant residual disease had survival around 14 to 14.8 months. Similarly, in Westphal 2015, there was an approximate difference of three to four months between residual‐ and no‐residual‐disease OS.
Four recurrent glioblastoma studies provided HRs as part of their survival data reporting (Brown 2016; Reardon 2015; Reardon 2020; van den Bent 2019). Meta‐analysis of these three studies did not show a statistically significant reduction in the risk of death with the use of anti‐EGFR therapies (HR 0.79, 95% CI 0.51 to 1.21, 4 RCTs, 489 participants, low‐certainty evidence, Analysis 1.2, Figure 4, Table 1). Significant heterogeneity was observed (I2 = 77%), which was expected given the variation in the drugs used and the lack of a standardised therapy in the recurrent setting. The included studies in this recurrent setting demonstrated a wide range of survival results in the experimental arm, from 5.6 to 12.0 months.
1.2. Analysis.

Comparison 1: Overall survival, Outcome 2: Recurrent disease
4.

Forest plot of comparison: 1 Overall survival, outcome: 1.2 Recurrent disease.
The most recent recurrent glioblastoma study reported median OS of 9.6 months in the combination ABT‐414 and temozolomide arm, 7.9 months in the ABT‐414 arm and 8.2 months in the chemotherapy arm alone (HR 0.68, 95% CI 0.48 to 0.9, P = 0.024, 260 participants) (van den Bent 2019). Reardon 2020 demonstrated a survival advantage with the combination of bevacizumab and rindopepimut vaccine against bevacizumab alone (11.0 versus 9 months) (HR 0.53, 95% CI 0.32 to 0.88, P = 0.01, 72 participants). We noted that there was a protracted delay to the full publication of this report. The remaining studies did not demonstrate any survival benefit with the use of the experimental therapy. Brown 2016 evaluated combination cediranib and gefitinib, which showed an OS difference of 1.7 months (OS 7.2 versus 5.5 months) (HR 0.68, 90% CI 0.39 to 1.19). However, this trial was terminated early due to cessation of cediranib, thus any potential survival trends must be interpreted with caution. Reardon 2015 showed no OS advantage with afatinib alone or in combination with temozolomide against temozolomide alone in a three‐arm randomised study, with OS data of 9.8 months (afatinib alone), 8.0 months (afatinib and temozolomide), and 10.6 months (temozolomide alone). McNeill 2014 reported OS data of 5.6 versus 5.2 months (vandetanib + carboplatin versus carboplatin), whilst van den Bent 2009 reported OS data of 7.7 versus 7.3 months (erlotinib versus temozolomide or carmustine).
We performed a pre‐planned sensitivity analysis between high or unclear risk of bias versus low risk of bias (Analysis 1.3). Seven studies were found to be at low risk of bias (Brown 2016; Lee 2015; Reardon 2015; Reardon 2020; van den Bent 2019; Weller 2017; Westphal 2015) and the HR for these seven studies was 0.82 (95% CI 0.71‐0.93). McNeill 2014 was the only study classified as high or unclear risk of bias and this study did not report HR for inclusion in the statistical calculations. While this sensitivity analysis demonstrated a survival benefit with the use anti‐EGFR therapy, it should be interpreted with caution given the heterogeneous nature of the patient population, the different classes of anti‐EGFR drugs used and the result was influenced heavily by one trial (Weller 2017) in particular.
1.3. Analysis.

Comparison 1: Overall survival, Outcome 3: Sensitivity analysis low risk of bias
The authorship team expressed concerns regarding comparative versus non‐comparative studies and its influence on the outcome, and thus an additional sensitivity analysis based on comparative versus non‐comparative studies was conducted (Analysis 1.4). Five of the six studies included in this primary outcome analysis were comparative RCTs, whilst Lee 2015 was a non‐comparative RCT. The HR for the five comparative RCTs was 0.89 (95% CI 0.71 to 1.12). The HR for Lee 2015 was 0.74 (95% CI 0.07 to 7.60). This did not alter the assessment for this primary outcome (Analysis 1.5). A lack of information in McNeill 2014 (the other non‐comparative study) prevented us from including this study in the statistical calculations. McNeill 2014 reported OS as 5.58 versus 5.22 months without further elaboration. This study was still in abstract form at the time of writing.
1.4. Analysis.

Comparison 1: Overall survival, Outcome 4: Sensitivity analysis for comparative and non‐comparative studies
1.5. Analysis.

Comparison 1: Overall survival, Outcome 5: Sensitivity analysis (6 studies)
Toxicities
We identified no differences between groups in the meta‐analysis of grade 3 or above toxicities amongst first‐line studies. Certain adverse events seemed to occur more frequently in the experimental arms, but these differences did not reach significance: lymphopenia (4 RCTs, 1146 participants, low‐certainty evidence, Analysis 2.1); neutropenia (4 RCTs, 1146 participants, low‐certainty evidence, Analysis 2.2); thrombocytopenia (4 RCTs, 1146 participants, low‐certainty evidence, Analysis 2.3)(Table 1).
2.1. Analysis.

Comparison 2: Toxicities of first‐line anti‐EGFR therapies ‐ grade 3 and above, Outcome 1: Lymphopenia
2.2. Analysis.

Comparison 2: Toxicities of first‐line anti‐EGFR therapies ‐ grade 3 and above, Outcome 2: Neutropenia
2.3. Analysis.

Comparison 2: Toxicities of first‐line anti‐EGFR therapies ‐ grade 3 and above, Outcome 3: Thrombocytopenia
First‐line studies
Westphal 2015 investigated the use of the monoclonal antibody nimotuzumab, and did not report significant skin toxicities as commonly seen in other anti‐EGFR monoclonal antibodies. Westphal 2015 found an increase in grade 3 or 4 toxicities (22 versus 6 events) with combination nimotuzumab and temozolomide, with increased levels of nausea and thrombocytopenia. Westphal 2015 reported three significant pulmonary embolic events in the nimotuzumab arm, with no venous thromboembolic events seen in the control group.
Lee 2015 found increased incidences of moderate to severe haematological toxicities, documenting the following toxicities of grade 3 or 4 severity in the vandetanib arm: lymphopenia 43.5% (versus 27.6% in control), leukopenia 11.6% (versus 6.9% in control), neutropenia 11.6% (versus 10.3% in control). However, these are unlikely to be of significance due to the small number of events recorded.
Weller 2017 reported mild to moderate injection site reaction relating to rindopepimut. This was a very common finding, with up to 80% noted (versus 41% in placebo). Notable but uncommon grade 3 to 4 adverse events included thrombocytopenia, fatigue, brain oedema, headaches, and seizures (all less than 10% incidence). A grade 5 pulmonary embolus was assessed as potentially related to rindopepimut. Discontinuation rates were low and due mainly to hypersensitivity from the vaccine. Importantly, the study authors did not report significant increases in immune‐related cerebral oedema or seizure.
Recurrent studies
In van den Bent 2019, significant side effects were reported in the experimental arms affecting the eyes and bone marrow. Grade 3 and 4 ocular toxicities were detected in 28.4% of participants who received ABT‐414. Only 20% of participants administered ABT‐414 did not suffer from any eye toxicities. Eye toxicities included: blurred vision, dry eye, keratitis, photophobia, and eye pain. This is a particular concern with ABT‐414 given that these issues were also encountered in the previous study by Reardon 2017. Grade 3 and 4 thrombocytopenia were also seen in 5.23% of participants who received ABT‐414.
Four recurrent studies investigated the use of oral TKIs. McNeill 2014 evaluated vandetanib, whilst the other three studies focused on specific EGFR TKI: Brown 2016 (gefitinib), Reardon 2015 (afatinib), and van den Bent 2009 (erlotinib). McNeill 2014 was similar to Lee 2015 in reporting increased incidences of moderate to severe haematological toxicities in the experimental arm. Reardon 2020 also reported mild to moderate injection site reaction relating to rindopepimut, and more detail is awaited as the data are still in abstract form. We have contacted the authors for further information.
In relation to the three specific reversible and irreversible EGFR TKI, Brown 2016 (gefitinib) found grade 3 or 4 adverse events in the combination of gefitinib and cediranib versus cediranib alone (89% versus 68%). The most frequent adverse events encountered in the combination included fatigue, hypertension, lymphopenia, anorexia, and ataxia. The study authors recorded one death from pulmonary embolus. Overall, treatment duration was longer with the combination, albeit with more dose reductions and side effects. Similarly in Reardon 2015, more adverse events were encountered with the use of afatinib both in combination with temozolomide (92.3%) or alone (85.4%) when compared to single‐agent temozolomide (56.4%). The most severe and common adverse events with the use of afatinib included fatigue, skin rash, and diarrhoea. This also contributed to a significant discontinuation (28.2%) and dose reduction rates in the combination arm (17.9%). In van den Bent 2009, erlotinib was observed to be well tolerated, and grade 3 or 4 toxicities were mainly skin related, which are commonly observed in other erlotinib trials.
In summary, toxicities associated with experimental therapies showed a slight increase in severe adverse events and adverse events when compared to the control arms.
Secondary outcomes
Progression‐free survival
We performed a meta‐analysis on two of the first‐line studies where progression‐free survival (PFS) HRs were reported (Weller 2017; Westphal 2015). There was no reduction in the risk to disease progression with the use of anti‐EGFR therapy (HR 0.94, 95% CI 0.81 to 1.10, 2 RCTs, 894 participants, low‐certainty evidence) with no significant heterogeneity observed (I2 = 0%) (Analysis 3.1, Figure 5, Table 1). These two studies found comparable PFS results ranging from 7.7 to 8.0 months. Individually, the studies did not demonstrate any PFS benefit with the addition of anti‐EGFR therapy. Westphal 2015 found a PFS of 7.7 months in their experimental arms, and Weller 2017 documented PFS 8.0 months in the rindopepimut study. These figures are slightly lower than those reported in other first‐line non‐EGFR‐related glioblastoma studies in contemporary literature such as AVAGLIO, Chinot 2014, and RTOG 0825, Gilbert 2014, where PFS was reported to be around 10.6 and 10.7 months, respectively. Lee 2015 did report a slight benefit towards vandetanib in prolonging PFS (7.7 versus 6.2 months, P = 0.6). No survival curves or HR was reported, thus this study could not be added to the meta‐analysis.
3.1. Analysis.

Comparison 3: Progression‐free survival, Outcome 1: First‐line
5.

Forest plot of comparison: 2 Progression‐free survival, outcome: 2.1 First‐line.
We performed a meta‐analysis on three of the recurrent studies (Brown 2016; Reardon 2020; van den Bent 2019), and there was a reduction in the risk to disease progression favouring anti‐EGFR therapy (HR 0.75, 95% CI 0.58 to 0.96, 3 RCTs, 275 participants, low‐certainty evidence) with no significant heterogeneity observed (I2 = 0%) (Analysis 3.2, Figure 6, Table 1). In detail, Brown 2016 documented PFS of 3.6 months versus 2.8 months in the cediranib and gefitinib arm versus the cediranib‐alone arm. With regard to recurrent glioblastoma studies, van den Bent 2019 showed a slight advantage with ABT‐414 and temozolomide with a PFS of 2.7 months (compared to 1.9 months in the ABT‐414 monotherapy and chemotherapy arms) (HR 0.77, 95% CI 0.55 to 1.07), and Reardon 2020 showed a PFS of 3.7 months in both groups (HR 0.72, 95% CI 0.43 to 1.21).
3.2. Analysis.

Comparison 3: Progression‐free survival, Outcome 2: Recurrent disease
6.

Forest plot of comparison: 3 Progression‐free survival, outcome: 3.2 Recurrent disease.
Amongst the remaining recurrent studies, the average PFS duration was similar between the experimental arm and the standard/placebo arm: 1.92 months (range 0.99 to 3.6 months). McNeill 2014 demonstrated PFS 1.7 versus 0.9 months in the vandetanib and carboplatin versus carboplatin‐alone groups, and van den Bent 2009 recorded a PFS of 1.8 months in the erlotinib group versus 2.4 months in the temozolomide or carmustine group. In Reardon 2015, there was an indication of harm in using afatinib in isolation, with a calculated HR 1.67 (0.99 months versus 1.87 months, P = 0.386), whilst the combination of afatinib and temozolomide versus temozolomide alone in the same study also showed a less favourable outcome with the addition of afatinib to temozolomide, with a calculated HR 1.31 (1.53 months versus 1.87 months, P = 0.119).
Quality of life
Four studies assessed quality of life (QoL) (Brown 2016; McNeill 2014; Weller 2017; Westphal 2015). Three studies used EORTC QLQ‐C30‐based assessment scales (Brown 2016; Weller 2017; Westphal 2015). Westphal 2015 conducted serial QoL assessments at study registration, first treatment, and then at weeks 10, 21, 33 and subsequently every 12 weeks. They found an improvement in multiple domains in favour of nimotuzumab with a maximal separation of 15 points at week 21. Weller 2017 (with the addition of MD Anderson Symptom Inventory ‐ Brain Tumor (MDASI‐BT) assessment score) found no significant differences in any of the domains between rindopepimut and placebo. Brown 2016 found that the addition of gefitinib did not have a negative impact on QoL, nor did it improve function or symptoms overall. McNeill 2014 mentioned that health‐related quality of life assessments were performed at baseline and at week 4, and changes were not predictive of time to progression or survival. More detail is awaited from publication of the full manuscript.
Discussion
Summary of main results
This review has not identified any benefit towards improved overall survival (OS) in the use of anti‐EGFR therapy in first‐line glioblastoma management (moderate‐certainty evidence, low risk of bias) (Analysis 1.1). Each of the included studies did not demonstrate a survival benefit with the use of anti‐EGFR therapy. It is disappointing that whilst anti‐EGFR therapy has proven survival benefits in most other solid tumours containing EGFR amplification or mutations, this has not been reproduced in glioblastomas (another highly EGFR‐expressing tumour) as yet. The INTELLANCE‐1 study, which is selective for EGFR amplification, mutation, or both in its participant recruitment has been suspended due to safety signals concerning lack of efficacy in May 2019 (NCT02573324).
The meta‐analysis in the recurrent setting also showed no OS benefit with the use of anti‐EGFR therapy (low‐certainty evidence, low risk of bias) (Analysis 1.2; ), despite studies by Reardon 2020 and van den Bent 2019 both observing improvements in survival. Interestingly, this survival benefit has not been translated to the first‐line setting for ABT‐414 and rindopepimut (NCT02573324; Weller 2017). The other studies did not demonstrate any improvement in OS based on the available data. It is interesting to note that in the Reardon 2015 trial, temozolomide alone outperformed either afatinib alone or in combination with temozolomide. These results held true even in a post hoc analysis where the cohort was further defined by EGFR amplification or mutation status. This would suggest that tyrosine kinase inhibitors (TKI) intrinsically are ineffective against EGFR‐drive glioblastomas.
Overall, anti‐EGFR therapies were better tolerated than expected and did not negatively impact on quality of life. In our meta‐analyses, no differences were observed between anti‐EGFR therapy and the standard of care/placebo (low‐certainty evidence, low risk of bias). The expected side effects of skin rashes and diarrhoea were generally not severe. Ocular toxicity with ABT‐414 is a particular concern given the number of participants who experienced any‐grade eye problems (van den Bent 2019). Special eye precautions have been promoted by the manufacturer to combat these problems, and frequent reviews with ophthalmologists are recommended. Deaths were recorded amongst the studies that were due to venous thromboembolic events (VTE) (second to disease progression). It is widely noted that glioblastoma has one of the highest rates of VTE amongst solid tumours (Magnus 2013; Perry 2012). It is uncertain whether EGFR‐targeted therapies would contribute to this increased risk (like anti‐angiogenic therapies), and certainly this is not recognised amongst people with non‐small cell lung cancer and colorectal cancer, where these drugs are more commonly used. It is also not known whether EGFR‐driven glioblastomas have an inherent increased risk of VTE. This may explain the poorer morbidity and mortality data often recorded in EGFR‐driven glioblastomas.
As a group, anti‐EGFR therapies did not appear to delay the time to disease progression amongst the first‐line trials (low‐certainty evidence, low risk of bias) (Lee 2015; Weller 2017; Westphal 2015), which demonstrated a progression‐free survival (PFS) duration of 6 to 8 months; this was less than that observed in both RTOG 0825 and AVAGLIO (around 10 months) (Chinot 2014; Gilbert 2014). This may be due to the ability of bevacizumab to delay/mask changes on magnetic resonance imaging (MRI) scans (Thompson 2011; Wick 2016). Whilst different radiological assessment methods were used in these trials, the differences encountered are unlikely to be explained by the assessment methods used in regard to Response Evaluation Criteria in Solid Tumours (RECIST) versus MacDonald versus RANO criteria. Amongst the included studies, only Brown 2016, van den Bent 2019, and Weller 2017 used RANO (the new accepted standard in neuro‐oncology) as the imaging assessment tool.
Surprisingly, meta‐analysis of three recurrent studies found a reduced risk of disease progression with the use of anti‐EGFR therapy (low‐certainty evidence, low risk of bias). These three studies all used different classes of EGFR drugs, and the result was largely driven by one study (van den Bent 2019), thus its importance must be interpreted with caution. Furthermore, whilst the analysis indicated a lowered risk of disease progression, this did not translate to an improvement in overall survival.
Overall completeness and applicability of evidence
The included studies were mixed in terms of the trial participants and the interventions used. We accounted for this by separating our analysis in line with the clinical situation (first‐line versus recurrent disease). Toxicities associated with experimental therapy were higher when compared to control arms, and notably ocular toxicities with ABT‐414 were particularly problematic. Most of the reported trials were not selective for people with EGFR amplification or mutation (except rindopepimut and ABT‐414 trials, where encouraging trends were observed). Further research is needed to determine if selected people with glioblastoma with known EGFR drivers respond to anti‐EGFR therapy. Use of anti‐EGFR therapy in EGFR‐driven glioblastomas may require combinations with other cytotoxic or targeted therapy agents to enhance its utility. At present, there is no evidence to support anti‐EGFR therapy in glioblastoma patients outside of clinical trial settings.
It is important to highlight the heterogeneity of the trials and the experimental drugs used. Whilst there was no significant heterogeneity observed in our meta‐analyses of first‐line studies, there was significant heterogeneity observed amongst the recurrent studies. This is to be expected given that some studies have preselected for EGFR‐amplified or ‐mutated glioblastomas (Reardon 2020; van den Bent 2019; Weller 2017), and others did not (Brown 2016; Lee 2015; McNeill 2014; Reardon 2015; van den Bent 2009; Westphal 2015). Only three of the nine included studies selected for the presence of EGFR amplification/mutation in their inclusion criteria (Reardon 2020; van den Bent 2019; Weller 2017). Reardon 2020 and Weller 2017 were investigating the use of rindopepimut vaccine. Interestingly, it was the recurrent study that showed a survival benefit (Reardon 2020), whereas the first‐line trial did not (Weller 2017). This may be attributed to some of the prespecified entry criteria in Weller 2017 (maximal surgical resection, completion of chemoradiation without progression, and maximum dexamethasone dose of 2 mg), which excluded patients with a poorer prognosis, creating a better‐than‐expected control arm. This is reflected in the whole cohort achieving medial overall survival duration of around 20 months.
The types of drugs used in these studies were also vastly different and thus contributed to the high heterogeneity and inconsistency of the results. Rindopepimut and ABT‐414 can be considered to be specially crafted towards use in glioblastomas, whilst most of the other anti‐EGFR therapies described in this review have already been commonly used in other solid tumours. The EGFR TKIs have not been able to achieve the same levels of success like that has been observed amongst non‐small cell lung cancer patients (Mok 2009). These TKIs target activating mutations in the intracellular catalytic domain of EGFR that are found in lung adenocarcinomas, whereas in glioblastomas the EGFR pathway is hyperactivated by overexpression or a mutated extracellular domain (vIII) of EGFR. ABT‐414 is designed to target the extracellular component of the receptor, and thus may explain the positive trend observed in INTELLANCE‐2 (van den Bent 2019).
Tumour heterogeneity in glioblastoma is well recognised and is increasingly seen as a contributor to the failures of targeted therapy (Eder 2014). Expression of targets or presence of mutation may vary within the tumour itself and between individuals. The development of resistance may have originated at the onset of tumour development, or indeed through evolution over time, especially when tumours are placed under stress from targeted therapy (Inda 2014). The molecular changes underlying these developments have been previously documented by Parker and colleagues (Parker 2016), and may be particularly relevant amongst EGFR‐overexpressed glioblastomas, leading to the current difficulties encountered in achieving success with anti‐EGFR therapy (Inda 2014; Sottoriva 2013). This may also help to explain the disconnect between improvements in PFS data (recurrent studies) and the lack of success in overall survival. It is likely that the experimental therapies were successful in removing the targeted population, but that ultimately led to the survival and propagation of a resistant population, thus having no impact on overall survival.
Quality of the evidence
The more recent studies were of high quality and clear and detailed in reporting their study procedures and outcomes, with a low of risk of bias (Brown 2016; Lee 2015; Reardon 2020; van den Bent 2019; Weller 2017; Westphal 2015). The remaining studies were also conducted rigorously as evidenced by the balanced nature of the randomisation. McNeill 2014 was more difficult to assess due to the lack of information and data obtained.
There is moderate degree of certainty of the overall survival trend observed amongst first‐line trials, with 1000 participants reported from 3 contributing studies. As mentioned above, these three studies all used different classes of anti‐EGFR agents, thus the applicability of these data remains uncertain and requires further validation. The other outcomes of OS (recurrent studies), toxicities, and PFS (first‐line) were not estimable, with low‐ to very low‐certainty evidence.
Potential biases in the review process
The search strategy was overseen by the Cochrane Gynaecological, Neuro‐oncology and Orphan Cancer Group to reduce the risk of introducing bias into the review process. We applied no limitations with regard to language or date of publication and made deliberate efforts to search for ongoing clinical trials. We obtained additional unpublished data through correspondence with study authors, and included this information in the review. Two review authors independently made decisions regarding study eligibility, 'Risk of bias' assessment, data collection, and grading of evidence, with any disagreements settled by a third review author.
The main bias relates to the small number of included studies, especially the older studies with smaller participant numbers and were of low or very low methodological quality, which meant that it was frequently not possible to conduct a meta‐analysis and prevented the drawing of firm conclusions regarding the clinical effectiveness of the intervention. It also meant that it was not possible to assess for publication bias. No conflicts of interest were identified for any of the study authors.
Agreements and disagreements with other studies or reviews
Given their frequency and importance in pathogenesis, EGFR alterations in glioblastomas remain an attractive target for therapies. Unfortunately, the main takeaway from this review would be the lack of survival benefit identified in the first‐line or recurrent setting with the use of anti‐EGFR therapies amongst glioblastoma patients, with significant toxicities. Other reviews have found similar results, where none of the current anti‐EGFR therapies have been truly demonstrated to be effective (An 2018; Westphal 2017), but project hope that new generations of anti‐EGFR therapies may be able to overcome past failures.
Whilst the overall results did not demonstrate survival benefit with the use of anti‐EGFR therapy, the OS data would indicate that improvements in glioblastoma care have been achieved over time, with the most recent first‐line glioblastoma studies demonstrating OS of 16 to 18 months (Chinot 2014; Gilbert 2014; Weller 2017), compared to the initial report by Stupp and colleagues in 2005, where median survival was 14.7 months (Stupp 2005).
Authors' conclusions
Implications for practice.
In summary, we have not found evidence of overall survival benefit with the use of epidermal growth factor receptor (EGFR)‐targeted therapies in glioblastoma management. EGFR‐targeted therapies were reasonably well tolerated (with special precautions required against ocular toxicities with ABT‐414). There was no evidence that they delayed disease progression in first line setting while a benefit in delaying disease progression was observed in the recurrent setting. Rindopepimut appeared promising with strong recurrent data, but no evidence of a survival benefit in the first‐line setting. Their first‐line study did achieve an overall survival of 20 months in both arms, indicating that the field has progressed from the early days of Stupp protocol and the advantage of maximal surgical resection. At present, isolated cases may still benefit from anti‐EGFR therapies, but the selection should depend on the presence of EGFR amplification or mutations as evidenced by van den Bent 2019 and Reardon 2020.
Implications for research.
Our review indicates that there are some encouraging signs from anti‐EGFR therapy in glioblastoma. The small encouraging signs identified in this report should be seen as support for further research and future studies where patient selection is driven by molecular changes and specialised drugs are designed that focus on these specific molecular targets seen in glioblastomas.
What's new
| Date | Event | Description |
|---|---|---|
| 5 August 2020 | Amended | Republished to correct review format error. |
History
Protocol first published: Issue 1, 2019 Review first published: Issue 5, 2020
| Date | Event | Description |
|---|---|---|
| 16 June 2020 | Amended | Minor typos corrected. |
| 19 May 2020 | New search has been performed | Published. |
Acknowledgements
We thank Robin Grant for clinical and editorial advice; Jo Platt for designing the search strategy; and Gail Quinn, Clare Jess, and Tracey Harrison for their contribution to the editorial process.
This project was supported by the National Institute for Health Research (NIHR), via Cochrane infrastructure funding to the Cochrane Gynaecological, Neuro‐oncology and Orphan Cancers Group. The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service, or the Department of Health.
We would like to thank the referees for their many helpful suggestions and comments, including Andrew Bryant and Juanita Lopes.
Appendices
Appendix 1. CENTRAL search strategy
#1 MeSH descriptor: [Glioblastoma] this term only #2 glioblastoma* or GBM* or GB* or astrocyt* #3 #1 or #2 #4 MeSH descriptor: [Receptor, Epidermal Growth Factor] this term only #5 EGFR* or EGF* or ERBB* or HER1* or Oncogene ERB* or ErbB‐1* or epidermal growth factor receptor* or sErbB‐1* or TGF‐alpha* or transforming growth factor alpha receptor* #6 MeSH descriptor: [Antibodies, Monoclonal] explode all trees #7 monoclonal antibod* or MAB* #8 MeSH descriptor: [Protein Kinase Inhibitors] explode all trees #9 tyrosin* near/5 (kinase* or inhibitor*) #10 PTK inhibit* or TK inhibitors* or TKI* or tyrphostins* or tyrosine phosphorylation inhibitor* or EC2* or hydroxyarl‐protein* or tyrosine* or tyrosylprotein* or phosphotransferases* or transphosphorylases* or phosphokinases* #11 nilotinib* or tasigna* or AMN107* or getfitnib* or ZD1839* or iressa* or erlotinib* or imatinib* or gleevec* or glivec* or STI‐571* #12 MeSH descriptor: [Cancer Vaccines] this term only #13 (cancer* or carcinoma* or adenocarcinoma* or neoplasm* or tumour* or tumor* or malignan* or antigen* or dendritic* or vector*) near/5 vaccin* #14 (cancer* or carcinoma* or adenocarcinoma* or neoplasm* or tumour* or tumor* or malignan* or antigen* or dendritic* or vector*) near/5 immuno* #15 rindopepimut* or CDX‐110* #16 #4 or #5 or #6 or #7 or #8 or #9 or #10 or #11 or #12 or #13 or #14 or #15 #16 #3 and #16
Appendix 2. MEDLINE Ovid search strategy
1. Glioblastoma/ 2. (glioblastoma* or GBM* or GB* or astrocyt*).mp. 3. 1 or 2 4. Receptor, Epidermal Growth Factor/ 5. (EGFR* or EGF* or ERBB* or HER1* or Oncogene ERB* or ErbB‐1* or epidermal growth factor receptor* or sErbB‐1* or TGF‐alpha* or transforming growth factor alpha receptor*).mp. 6. exp Antibodies, Monoclonal/ 7. (monoclonal antibod* or MAB*).mp. 8. exp Protein Kinase Inhibitors/ 9. (tyrosin* adj5 (kinase* or inhibitor*)).mp. 10. (PTK inhibit* or TK inhibitors* or TKI* or tyrphostins* or tyrosine phosphorylation inhibitor* or EC2* or hydroxyarl‐protein* or tyrosine* or tyrosylprotein* or phosphotransferases* or transphosphorylases* or phosphokinases*).mp. 11. (nilotinib* or tasigna* or AMN107* or getfitnib* or ZD1839* or iressa* or erlotinib* or imatinib* or gleevec* or glivec* or STI‐571*).mp. 12. Cancer Vaccines/ 13. ((cancer* or carcinoma* or adenocarcinoma* or neoplasm* or tumour* or tumor* or malignan* or antigen* or dendritic* or vector*) adj5 (vaccin* or immuno*)).mp. 14. (rindopepimut* or CDX‐110*).mp. 15. 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 16. 3 and 15 17. randomised controlled trial.pt. 18. controlled clinical trial.pt. 19. randomised.ab. 20. placebo.ab. 21. clinical trials as topic.sh. 22. randomly.ab. 23. trial.ti. 24. 17 or 18 or 19 or 20 or 21 or 22 or 23 25. (animals not (humans and animals)).sh. 26. 24 not 25 26. 16 and 26
Appendix 3. Embase Ovid search strategy
1. Glioblastoma/ 2. (glioblastoma* or GBM* or GB* or astrocyt*).mp. 3. 1 or 2 4. Receptor, Epidermal Growth Factor/ 5. (EGFR* or EGF* or ERBB* or HER1* or Oncogene ERB* or ErbB‐1* or epidermal growth factor receptor* or sErbB‐1* or TGF‐alpha* or transforming growth factor alpha receptor*).mp. 6. exp Antibodies, Monoclonal/ 7. (monoclonal antibod* or MAB*).mp. 8. exp Protein Kinase Inhibitors/ 9. (tyrosin* adj5 (kinase* or inhibitor*)).mp. 10. (PTK inhibit* or TK inhibitors* or TKI* or tyrphostins* or tyrosine phosphorylation inhibitor* or EC2* or hydroxyarl‐protein* or tyrosine* or tyrosylprotein* or phosphotransferases* or transphosphorylases* or phosphokinases*).mp. 11. (nilotinib* or tasigna* or AMN107* or getfitnib* or ZD1839* or iressa* or erlotinib* or imatinib* or gleevec* or glivec* or STI‐571*).mp. 12. Cancer Vaccines/ 13. ((cancer* or carcinoma* or adenocarcinoma* or neoplasm* or tumour* or tumor* or malignan* or antigen* or dendritic* or vector*) adj5 (vaccin* or immuno*)).mp. 14. (rindopepimut* or CDX‐110*).mp. 15. 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 16. 3 and 15 17. randomized controlled trial.pt. 18. controlled clinical trial.pt. 19. randomized.ab. 20. placebo.ab. 21. clinical trials as topic.sh. 22. randomly.ab. 23. trial.ti. 24. 17 or 18 or 19 or 20 or 21 or 22 or 23 25. (animals not (humans and animals)).sh. 26. 24 not 25 26. 16 and 26
Key: mp = title, abstract, original title, name of substance word, subject heading word, protocol supplementary concept, rare disease supplementary concept, unique identifier pt = publication type ab = abstract sh = subject heading ti = title
Appendix 4. Standardised data extraction form
| Title |
| Lead Author, Senior Author |
| Year published |
| Publication |
| Type of study |
| Trial phase |
| Intervention |
| Control |
| No. of participants |
| First‐line or recurrent disease |
| Type of participants |
| Primary outcome |
| Secondary outcome |
| Toxicity |
Data and analyses
Comparison 1. Overall survival.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 First‐line | 3 | Hazard Ratio (IV, Random, 95% CI) | 0.89 [0.76, 1.04] | |
| 1.2 Recurrent disease | 4 | Hazard Ratio (IV, Random, 95% CI) | 0.79 [0.51, 1.21] | |
| 1.3 Sensitivity analysis low risk of bias | 7 | Hazard Ratio (IV, Random, 95% CI) | 0.82 [0.71, 0.93] | |
| 1.4 Sensitivity analysis for comparative and non‐comparative studies | 6 | Hazard Ratio (IV, Random, 95% CI) | 0.89 [0.72, 1.10] | |
| 1.4.1 Comparative studies | 5 | Hazard Ratio (IV, Random, 95% CI) | 0.89 [0.71, 1.12] | |
| 1.4.2 Non‐comparative studies | 1 | Hazard Ratio (IV, Random, 95% CI) | 0.74 [0.07, 7.60] | |
| 1.5 Sensitivity analysis (6 studies) | 6 | Hazard Ratio (IV, Random, 95% CI) | 0.89 [0.72, 1.10] |
Comparison 2. Toxicities of first‐line anti‐EGFR therapies ‐ grade 3 and above.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Lymphopenia | 4 | 1146 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.19, 4.81] |
| 2.2 Neutropenia | 4 | 1146 | Odds Ratio (M‐H, Random, 95% CI) | 1.29 [0.82, 2.03] |
| 2.3 Thrombocytopenia | 4 | 1146 | Odds Ratio (M‐H, Random, 95% CI) | 3.69 [0.51, 26.51] |
| 2.4 Rash | 4 | 1146 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.14, 12.87] |
| 2.5 Diarrhoea | 5 | 1218 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.07, 6.35] |
| 2.6 Fatigue | 5 | 1218 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.18, 4.52] |
2.4. Analysis.

Comparison 2: Toxicities of first‐line anti‐EGFR therapies ‐ grade 3 and above, Outcome 4: Rash
2.5. Analysis.

Comparison 2: Toxicities of first‐line anti‐EGFR therapies ‐ grade 3 and above, Outcome 5: Diarrhoea
2.6. Analysis.

Comparison 2: Toxicities of first‐line anti‐EGFR therapies ‐ grade 3 and above, Outcome 6: Fatigue
Comparison 3. Progression‐free survival.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 First‐line | 2 | Hazard Ratio (IV, Random, 95% CI) | 0.94 [0.81, 1.10] | |
| 3.2 Recurrent disease | 3 | Hazard Ratio (IV, Random, 95% CI) | 0.75 [0.58, 0.96] | |
| 3.3 Sensitivity analysis low risk of bias PFS | 5 | Hazard Ratio (IV, Random, 95% CI) | 0.88 [0.77, 1.01] |
3.3. Analysis.

Comparison 3: Progression‐free survival, Outcome 3: Sensitivity analysis low risk of bias PFS
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Brown 2016.
| Study characteristics | ||
| Methods | Phase II multicentre, randomised, placebo‐controlled | |
| Participants | Recurrent/progressive glioblastoma patients, 38 participants in total | |
| Interventions | Cediranib and gefitinib combination versus cediranib and placebo | |
| Outcomes |
|
|
| Notes | Cediranib discontinued by AstraZeneca – trial terminated early in August 2012. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation system was managed independent of the trial management team. Registration fax from the recruiting site trial staff would use an online randomisation system to produce container numbers for the assigned treatment. |
| Allocation concealment (selection bias) | Low risk | Contents of the bottles were concealed from site staff, participants, and trial management. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not mentioned, insufficient information (likely low risk) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No missing information |
| Selective reporting (reporting bias) | Low risk | All prespecified endpoints reported. |
| Other bias | Low risk | Early termination of study due to cessation of cediranib production, decision made by manufacturer. |
Lee 2015.
| Study characteristics | ||
| Methods | Randomised 2:1, open‐label, non‐comparative, multicentre | |
| Participants | 106 randomised. First‐line glioblastomas | |
| Interventions | Vandetanib + Stupp protocol vs no vandetanib + Stupp protocol | |
| Outcomes |
|
|
| Notes | Used MacDonald criteria for radiological assessments | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “patients were randomly assigned 2:1 at registration to receive RT and temozolomide…” |
| Allocation concealment (selection bias) | High risk | Not concealed. Open‐label trial |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not concealed. Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Uncertain |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 13 patients registered but did not proceed to randomisation. |
| Selective reporting (reporting bias) | Low risk | All prespecified endpoints reported. |
| Other bias | Low risk | |
McNeill 2014.
| Study characteristics | ||
| Methods | Phase II randomised non‐comparative | |
| Participants | 66 recurrent glioblastoma patients (the trial also included 46 anaplastic astrocytoma patients) | |
| Interventions | Vandetanib and carboplatin versus carboplatin | |
| Outcomes |
Toxicity results reported on all trial participants (n = 112). SAE: vandetanib combination 37.5% vs carboplatin alone 17.85% Grade 3 thrombocytopenia (n = 14), lymphopenia (n = 12), neutropenia (n = 7), seizure (n = 5), hypertension (n = 4) |
|
| Notes | Still in abstract form only. Results have been published on ClinicalTrials.gov. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Uncertain |
| Allocation concealment (selection bias) | Unclear risk | Uncertain |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Uncertain |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Uncertain |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Uncertain |
| Selective reporting (reporting bias) | Unclear risk | Uncertain |
| Other bias | Unclear risk | Manuscript not published in full. |
Reardon 2015.
| Study characteristics | ||
| Methods | Phase I/randomised phase II, multicentre | |
| Participants | Phase II ‐ 119 recurrent glioblastoma patients | |
| Interventions | Afatinib (A) vs afatinib and temozolomide (AT) vs temozolomide (T) (1:1:1) | |
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Uncertain |
| Allocation concealment (selection bias) | Unclear risk | Uncertain |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Uncertain |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Independent review committee (ICON medical imaging) mentioned. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 12 patients were not randomised but no clear explanation. |
| Selective reporting (reporting bias) | Unclear risk | Uncertain |
| Other bias | Low risk | |
Reardon 2020.
| Study characteristics | ||
| Methods | Phase II randomised controlled trial | |
| Participants | 72 recurrent glioblastoma patients | |
| Interventions | Rindopepimut (CDX‐110) and bevacizumab versus bevacizumab + KLH control | |
| Outcomes |
|
|
| Notes | Long delay to publication ‐ 5 years from first results released in abstract form to full manuscript publication | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Prespecified randomisation list created by biostatistician. |
| Allocation concealment (selection bias) | Low risk | Participants and investigators remained blinded to treatment assignments. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Yes – double‐blind, placebo controlled Unblinded pharmacists who were otherwise uninvolved in study conduct obtained randomised treatment assignments and managed study treatment. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Independent review committee (ICON medical imaging) mentioned. Expert review committee members were otherwise independent of study conduct and were blinded to treatment allocation and investigator assessments. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None |
| Selective reporting (reporting bias) | Low risk | All prespecified endpoints reported. |
| Other bias | Low risk | |
van den Bent 2009.
| Study characteristics | ||
| Methods | Phase II randomised | |
| Participants | 110 recurrent glioblastoma patients | |
| Interventions | Erlotinib versus carmustine (if no prior temozolomide) or temozolomide | |
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Participants were randomly assigned centrally at EPRTC Data Centre either by internet or phone. |
| Allocation concealment (selection bias) | Low risk | Not mentioned but likely preserved |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Central review of imaging |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | All accounted for. |
| Selective reporting (reporting bias) | Low risk | All prespecified endpoints reported. |
| Other bias | Low risk | |
van den Bent 2019.
| Study characteristics | ||
| Methods | Phase II randomised, open‐label | |
| Participants | 260 recurrent glioblastoma patients | |
| Interventions | ABT‐414 (A) versus ABT‐414 and temozolomide (B) versus lomustine or temozolomide (C) | |
| Outcomes |
|
|
| Notes | Multiple abstracts with updates ‐ no published manuscript Comparison is between Arm A and Arm C, and Arm B and Arm C |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Assumed to be low risk as not mentioned in manuscript; evidence from study protocol |
| Allocation concealment (selection bias) | High risk | Open‐label trial |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label trial |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Central imaging review was conducted by an independent neuroradiologist. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | CONSORT diagram demonstrated that all participants were accounted for. |
| Selective reporting (reporting bias) | Low risk | None |
| Other bias | Low risk | |
Weller 2017.
| Study characteristics | ||
| Methods | Phase III randomised, double‐blind | |
| Participants | First‐line glioblastoma patient: randomisation (1:1) ‐ 195 rindopepimut and temozolomide, 210 standard of care (temozolomide) | |
| Interventions | Rindopepimut + temozolomide vs temozolomide | |
| Outcomes |
|
|
| Notes | Terminated early at second preplanned interim analysis – futility boundary crossed. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random assignment to treatment groups with a prespecified randomisation sequence with a block size of 4. |
| Allocation concealment (selection bias) | Low risk | Double‐blinded study – unblinded pharmacists obtained randomly assigned treatment assignments and managed study treatment via interactive response technology |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blinded study Placebo vaccine ‐ preloaded with immunostimulant |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Local assessors (blinded), central assessors (also blinded for PFS and ORR assessments) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | None |
| Selective reporting (reporting bias) | Unclear risk | None |
| Other bias | Low risk | |
Westphal 2015.
| Study characteristics | ||
| Methods | Phase III randomised, open‐label | |
| Participants | 142 first‐line glioblastoma patients | |
| Interventions | Nimotuzumab versus placebo | |
| Outcomes |
|
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation took place by fax after histological diagnosis of glioblastoma by local neuro‐pathological review. |
| Allocation concealment (selection bias) | High risk | Open‐label |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Open‐label |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not mentioned |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All participants accounted for. |
| Selective reporting (reporting bias) | High risk | Response rates not reported. Unplanned subgroup analyses by MGMT methylation and EGFR status presented. |
| Other bias | Low risk | |
OS: overall survival PFS: progression‐free survival PFS6: progression‐free survival at 6 months PFS12: progression‐free survival at 12 months AE: adverse events SAE: Serious adverse events ORR: overall response rate KLH: keyhole limpet hemocyanin PR: partial response SD: stable disease ITT: intention to treat QoL: quality of life MGMT: O6‐methylguanine–DNA methyltransferase EGFR: epidermal growth factor receptor
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Daugherty 2014 | Not a randomised study |
| Hong 2012 | Mixed grade III/IV, not randomised |
| Neyns 2009 | Not a randomised study |
| Schuster 2015 | Not a randomised study. Initially planned as a randomised phase II/III study, but due to voluntary attrition of first 16 participants in standard‐of‐care arm, the study was converted to open‐label, single‐arm phase II trial. |
| Sepulveda 2015 | Not a randomised study |
| Solomon 2013 | Not current standard of care |
| van den Bent 2016 | Phase I study, 3 arms but not randomised |
| Wen 2014 | Not a randomised study |
| Wygoda 2002 | Not a randomised study |
| Wygoda 2006 | Intervention not eligible |
Characteristics of ongoing studies [ordered by study ID]
NCT02573324.
| Study name | A study of ABT‐414 in subjects with newly diagnosed glioblastoma (GBM) with epidermal growth factor receptor (EGFR) amplification (INTELLANCE‐1) |
| Methods | Phase II/III randomised, placebo controlled |
| Participants | 640 first‐line EGFR‐amplified glioblastoma patients |
| Interventions | ABT‐414 vs placebo |
| Outcomes | Overall survival, progression‐free survival |
| Starting date | 7 December 2015 |
| Contact information | AbbVie Inc Study Director |
| Notes |
Differences between protocol and review
With the advent of antibody drug conjugates, this report will include this an additional class of agents within the monoclonal antibodies group. Antibody drug conjugates are compounds based on monoclonal antibodies with a linker joining up with a cytotoxic drug (or payload). The mechanism of action is similar in principle to other monoclonal antibodies. The antibody component of antibody drug conjugates will track down their target protein; this will subsequently trigger an internalisation process in the recipient cell, absorbing and releasing the cytotoxic drug/payload inside and thus leading to cell kill. This was a new drug development that was utilised in clinical trials (phase II/III) after the initial protocol for this review was published.
Another separation from the protocol relates to the addition of a sensitivity analysis regarding comparative and non comparative studies. There was disagreement within the authorship team about comparative and non‐comparative studies and the decision was to add a sensitivity analysis to confirm whether the inclusion or exclusion of Lee 2015 would alter the findings.
Contributions of authors
All authors contributed to the planning and editing of the review. AL prepared the first draft of this review; screening and searches of studies; correspondence and communication; planning of the review. MA assisted in the preparation of drafts of the review; screening and searches for studies. DC provided statistical support; addressed reviewers comments; and expertise on Cochrance reporting, editorial management. MK provided editorial reviews for the review, expertise on Cochrance reporting, and statistical advice. VH provided editorial review and supervised the planning of the review. HW provided editorial review and supervised the planning of the review.
Sources of support
Internal sources
None, Other
External sources
No sources of support supplied
Declarations of interest
AL: has received honorarium and grants from Eisai, Mundipharma, Sanofi, and Bayer for non‐glioma conditions. MA: none known. DC: has received honoraria from Novartis and Ipsen for educational activities outside the submitted work. MK: has served on AbbVie GBM advisory boards, and his institution received research grants from AbbVie and BMS to fund clinical trials in glioblastoma. VH: none known. HW: the analysis for this Cochrane Review is based on peer‐reviewed data prepared by an independent steering trials committee. My involvement in the Australian Roche advisory board was to discuss completed trial results and how the drug may be introduced into the clinic in Australian centres. My participation on the Merck Serono‐centric steering committee was to review ongoing trial recruitment and severe adverse events. None of these activities influenced the analysis of the review data or contributed to any presented/published conclusions.
Edited (no change to conclusions)
References
References to studies included in this review
Brown 2016 {published data only}
- Brown N, McBain C, Nash S, Hopkins K, Sanghera P, Saran F, et al. Multi-center randomized phase II study comparing cediranib plus gefitinib with cediranib plus placebo in subjects with recurrent/progressive glioblastoma. PLOS One 2016;11(5):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lee 2015 {published data only}
- Lee EQ, Kaley TJ, Duda DG, Schiff D, Lassman AB, Wong ET, et al. A multicenter, phase II, randomized, noncomparative clinical trial of radiation and temozolomide with or without vandetanib in newly diagnosed glioblastoma patients. Clinical Cancer Research 2015;21(16):3610-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
McNeill 2014 {published data only}
- McNeill K, Iwamoto F, Kreisl T, Sul J, Shih J, Fine H. A randomized phase II trial of vandetanib (ZD6474) in combination with carboplatin versus carboplatin alone in adults with recurrent glioblastoma. Neuro-Oncology 2014;16:v17. [Google Scholar]
Reardon 2015 {published data only}
- Reardon DA, Nabors LB, Mason WP, Perry JR, Shapiro W, Kavan P, et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro-Oncology 2015;17(3):430-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Reardon 2020 {published data only}
- Reardon DA, Desjardins A, Vredenburgh JJ, O'Rouke DM, Tran DD, Fink KL, et al. Rindopepimut with Bevacizumab for patients with relapsed EGFRvIII-expressing glioblastoma (ReACT): results of a double-blind randomized phase II trial. Clinical Cancer Research 2020;Feb 7:[Epub ahead of print]. [DOI: 10.1158/1078-0432.CCR-18-1140] [DOI] [PubMed] [Google Scholar]
van den Bent 2009 {published data only}
- den Bent MJ, Brandes AA, Rampling R, Kouwenhoven MCM, Kros JM, Carpentier AF, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. Journal of Clinical Oncology 2009;27(8):1268-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
van den Bent 2019 {published data only}
- den Bent M, Eoli M, Sepulveda JM, Smits M, Walenkamp A, Frenel J, et al. Intellance2/EORTC 1410 randomized phase II study of Depatux-M alone and with temozolomide vs temozolomide or lomustine in recurrent EGFR amplified glioblastoma. Neuro-Oncology 2019;Nov 20:pii: noz222. [DOI: 10.1093/neuonc/noz222] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weller 2017 {published data only}
- Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncology 2017;18(10):1373-85. [DOI] [PubMed] [Google Scholar]
Westphal 2015 {published data only}
- Westphal M, Heese O, Steinbach JP, Schnell O, Schackert G, Mehdorn M, et al. A randomised, open label phase III trial with nimotuzumab, an anti-epidermal growth factor receptor monoclonal antibody in the treatment of newly diagnosed adult glioblastoma. European Journal of Cancer 2015;51(4):522-32. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Daugherty 2014 {published data only}
- Daugherty LC, Fisher BJ, Morales S, Kim J, Li L, Quang TS, et al. A phase II study of surgical excision, temozolomide, radiotherapy, and anti-EGFR radioimmunotherapy (EXTRA) as adjuvant therapy in high-grade gliomas. Journal of Radiation Oncology 2014;3(4):347-53. [Google Scholar]
Hong 2012 {published data only}
- Hong J, Peng Y, Liao Y, Jiang W, Wei R, Huo L, et al. Nimotuzumab prolongs survival in patients with malignant gliomas: a phase I/II clinical study of concomitant radiochemotherapy with or without nimotuzumab. Experimental and Therapeutic Medicine 2012;4(1):151-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Neyns 2009 {published data only}
- Neyns B, Sadones J, Joosens E, Bouttens F, Verbeke L, Baurain JF, et al. Stratified phase II trial of cetuximab in patients with recurrent high-grade glioma. Annals of Oncology 2009;20(9):1596-603. [DOI] [PubMed] [Google Scholar]
Schuster 2015 {published data only}
- Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro-Oncology 2015;17(6):854-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sepulveda 2015 {published data only}
- Sepulveda JM, Vaz MA, Gil M, Reynes G, Gallego O, Bolos MV, et al. GEINO-11: A prospective multicenter, open label, phase II pilot clinical trial to evaluate safety and efficacy of Dacomitinib, a pan-HER irreversible inhibitor, in patients with recurrent glioblastoma with EGFR amplification or presence of EGFRvIII mutation. European Journal of Cancer 2015;51:S585. [Google Scholar]
Solomon 2013 {published data only}
- Solomon MT, Selva JC, Figueredo J, Vaquer J, Toledo C, Quintanal N, et al. Radiotherapy plus nimotuzumab or placebo in the treatment of high grade glioma patients: results from a randomized, double blind trial. BMC Cancer 2013;13:299. [DOI] [PMC free article] [PubMed] [Google Scholar]
van den Bent 2016 {published data only}
- Van Den Bent MJ, Gan HK, Lassman AB, Kumthekar P, Merrell R, Butowski NA, et al. Efficacy of a novel antibody-drug conjugate (ADC), ABT-414, as monotherapy in epidermal growth factor receptor (EGFR) amplified, recurrent glioblastoma (GBM). Neuro-Oncology 2016;18(18):iv44. [Google Scholar]
Wen 2014 {published data only}
- Wen PY, Chang SM, Lamborn KR, Kuhn JG, Norden AD, Cloughesy TF, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro-Oncology 2014;16(4):567-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wygoda 2002 {published data only}
- Wygoda Z, Tarnawski R, Brady L, Steplewski Z, Bazowski P, Wojtacha M, et al. Simultaneous radiotherapy and radioimmunotherapy of malignant gliomas with anti-EGFR antibody labelled with iodine 125. Nuclear Medicine Review. Central & Eastern Europe 2002;5(1):29-33. [PubMed] [Google Scholar]
Wygoda 2006 {published data only}
- Wygoda Z, Kula D, Bierzynska-Macyszyn G, Larysz D, Jarzab M, Wlaszczuk P, et al. Use of monoclonal anti-EGFR antibody in the radioimmunotherapy of malignant gliomas in the context of EGFR expression in grade III and IV tumors. Hybridoma (2005) 2006;25(3):125-32. [DOI] [PubMed] [Google Scholar]
References to ongoing studies
NCT02573324 {published data only}
- AbbVie Inc and Radiation Therapy Oncology Group. A study of ABT-414 in subjects with newly diagnosed glioblastoma with epidermal growth factor receptor amplification (Intellance 1). clinicaltrials.gov/show/NCT02573324 D.
Additional references
AIHW 2017
- Australian Institute of Health and Welfare. Cancer in Australia 2017. Cancer Series no. 101. www.aihw.gov.au/reports/cancer/cancer-in-australia-2017/contents/table-of-contents (accessed prior to 3 December 2018).
Aldape 2015
- Aldape K, Zadeh G, Mansouri S, Reifenberger G, Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathologica 2015;129(6):829-48. [DOI] [PubMed] [Google Scholar]
An 2018
- An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene 2018;37(12):1561-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bastien 2015
- Bastien JI, McNeill KA, Fine HA. Molecular characterizations of glioblastoma, targeted therapy, and clinical results to date. Cancer 2015;121(4):502-16. [DOI] [PubMed] [Google Scholar]
Brennan 2013
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155(2):462-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Castillo 2004
- Castillo L, Etienne-Grimaldi MC, Fischel JL, Formento P, Magné N, Milano G. Pharmacological background of EGFR targeting. Annals of Oncology 2004;15(7):1007-12. [DOI] [PubMed] [Google Scholar]
Chinot 2014
- Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, et al. Bevacizumab plus radiotherapy–temozolomide for newly diagnosed glioblastoma. New England Journal of Medicine 2014;370(8):709-22. [DOI] [PubMed] [Google Scholar]
Cloughesy 2014
- Cloughesy TF, Cavenee WK, Mischel PS. Glioblastoma: from molecular pathology to targeted treatment. Annual Review of Pathology 2014;9(1):1-25. [DOI] [PubMed] [Google Scholar]
Eder 2014
- Eder K, Kalman B. Molecular heterogeneity of glioblastoma and its clinical relevance. Pathology & Oncology Research 2014;20(4):777-87. [DOI] [PubMed] [Google Scholar]
Gilbert 2014
- Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. New England Journal of Medicine 2014;370(8):699-708. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015. Available at gradepro.org.
Haas‐Kogan 2005
- Haas-Kogan DA, Prados MD, Lamborn KR, Tihan T, Berger MS, Stokoe D. Biomarkers to predict response to epidermal growth factor receptor inhibitors. Cell Cycle 2005;4(10):1369-72. [DOI] [PubMed] [Google Scholar]
Hauschild 2012
- Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2012;380(9839):358-65. [DOI] [PubMed] [Google Scholar]
Heimberger 2005
- Heimberger AB, Hlatky R, Suki D, Yang D, Weinberg J, Gilbert M, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clinical Cancer Research 2005;11(4):1462. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Huang 2009
- Huang PH, Xu AM, White FM. Oncogenic EGFR signaling networks in glioma. Science Signaling 2009;2(87):re6. [DOI] [PubMed] [Google Scholar]
Inda 2014
- Inda MM, Bonavia R, Seoane J. Glioblastoma multiforme: a look inside its heterogeneous nature. Cancers 2014;6(1):226-39. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Karapetis 2008
- Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. New England Journal of Medicine 2008;359(17):1757-65. [DOI] [PubMed] [Google Scholar]
Karnofsky 1948
- Karnofsky DA, Burchenal JH. The evaluation of chemotherapeutic agents against neoplastic disease. In: MacLeod CM, editors(s). Evaluation of Chemotherapeutic Agents. New York (NY): Columbia University Press, 1949:196. [Google Scholar]
Khasraw 2016
- Khasraw M, Lee A, McCowatt S, Kerestes Z, Buyse ME, Back M, et al. Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy in patients with glioblastoma and unmethylated MGMT gene promoter in ExCentric, an open-label phase II trial. Journal of Neuro-oncology 2016;128(1):163-71. [DOI] [PubMed] [Google Scholar]
Langendam 2013
- Langendam MW, Akl EA, Dahm P, Glasziou P, Guyatt G, Schunemann HJ. Assessing and presenting summaries of evidence in Cochrane Reviews. Systematic Reviews 2013;23(2):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lapointe 2015
- Lapointe S, Florescu M, Nguyen DK, Djeffal C, Bélanger K. Prophylactic anticonvulsants for gliomas: a seven-year retrospective analysis. Neuro-Oncology Practice 2015;2(4):192-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Macdonald 1990
- Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. Journal of Clinical Oncology 1990;8(7):1277-80. [DOI] [PubMed] [Google Scholar]
Magnus 2013
- Magnus N, D'Asti E, Garnier D, Meehan B, Rak J. Brain neoplasms and coagulation. Seminars in Thrombosis & Hemostasis 2013;39(8):881-95. [DOI] [PubMed] [Google Scholar]
Meader 2014
- Meader N, King K, Llewellyn A, Norman G, Brown J, Rodgers M, et al. A checklist designed to aid consistency and reproducibility of GRADE assessments: development and pilot validation. Systematic Reviews 2014;3:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mok 2009
- Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin–paclitaxel in pulmonary adenocarcinoma. New England Journal of Medicine 2009;361(10):947-57. [DOI] [PubMed] [Google Scholar]
Nabors 2015
- Nabors LB, Fink KL, Mikkelsen T, Grujicic D, Tarnawski R, Nam DH, et al. Two cilengitide regimens in combination with standard treatment for patients with newly diagnosed glioblastoma and unmethylated MGMT gene promoter: results of the open-label, controlled, randomized phase II CORE study. Neuro-Oncology 2015;17(5):708-17. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCI 2017
- National Cancer Institute. Common Terminology Criteria for Adverse Events (CTCAE) v5.0, November 2017. https://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/CTCAE_v5_Quick_Reference_8.5x11.pdf (accessed 20 Feb 2020).
Nilsson 2017
- Nilsson J, Holgersson G, Carlsson T, Henriksson R, Bergström S, Bergqvist M. Incidence trends in high-grade primary brain tumors in males and females. Oncology Letters 2017;13(4):2831-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Parker 2016
- Parker NR, Hudson AL, Khong P, Parkinson JF, Dwight T, Ikin RJ, et al. Intratumoral heterogeneity identified at the epigenetic, genetic and transcriptional level in glioblastoma. Scientific Reports 2016;6:22477. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Perry 2012
- Perry JR. Thromboembolic disease in patients with high-grade glioma. Neuro-Oncology 2012;14(suppl 4):iv73-iv80. [DOI] [PMC free article] [PubMed] [Google Scholar]
Qian 2016
- Qian C, Yan H, Hu X, Zhang W, Liu H. Increased risk of venous thromboembolism in patients with brain tumors: a systematic review and meta-analysis. Thrombosis Research 2016;137:58-63. [DOI] [PubMed] [Google Scholar]
Reardon 2017
- Reardon DA, Lassman AB, den Bent M, Kumthekar P, Merrell R, Scott AM, et al. Efficacy and safety results of ABT-414 in combination with radiation and temozolomide in newly diagnosed glioblastoma. Neuro-Oncology 2017;19(7):965-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Sampson 2010
- Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. Journal of Clinical Oncology 2010;28(31):4722-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Scott 2008
- Scott NW, Fayers PM, Aaronson NK, Bottomley A, Graeff A, Groenvold M, et al, on behalf of the EORTC Quality of Life Group. EORTC QLQ-C30 reference values, 2008. www.eortc.org/app/uploads/sites/2/2018/02/reference_values_manual2008.pdf (accessed prior to 3 December 2018).
Shaw 2013
- Shaw AT, Kim DW, Nakagawa K, Seto T, Crinó L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. New England Journal of Medicine 2013;368(25):2385-94. [DOI] [PubMed] [Google Scholar]
Shinojima 2003
- Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Research 2003;63(20):6962. [PubMed] [Google Scholar]
Sottoriva 2013
- Sottoriva A, Spiteri I, Piccirillo SG, Touloumis A, Collins VP, Marioni JC, et al. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proceedings of the National Academy of Sciences of the United States of America 2013;110(10):4009-14. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Stupp 2014
- Stupp R, Hegi ME, Gorlia T, Erridge SC, Perry J, Hong YK, et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncology 2014;15(10):1100-8. [DOI] [PubMed] [Google Scholar]
Stupp 2005
- Stupp R, Mason WP, den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. New England Journal of Medicine 2005;352(10):987-96. [DOI] [PubMed] [Google Scholar]
Stupp 2017
- Stupp R, Taillibert S, Kanner A, Read W, Steinberg D, Lhermitte B, et al. Effect of tumor-treating fields plus maintenance temozolomide vs maintenance temozolomide alone on survival in patients with glioblastoma: A randomized clinical trial. JAMA 2017;318(23):2306-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Taphoorn 2010
- Taphoorn MJ, Claassens L, Aaronson NK, Coens C, Mauer M, Osoba D, et al. An international validation study of the EORTC brain cancer module (EORTC QLQ-BN20) for assessing health-related quality of life and symptoms in brain cancer patients. European Journal of Cancer 2010;46(6):1033-40. [DOI] [PubMed] [Google Scholar]
Thaler 2013
- Thaler J, Preusser M, Ay C, Kaider A, Marosi C, Zielinski C, et al. Intratumoral tissue factor expression and risk of venous thromboembolism in brain tumor patients. Thrombosis Research 2013;131(2):162-5. [DOI] [PubMed] [Google Scholar]
Thaler 2014
- Thaler J, Ay C, Kaider A, Reitter EM, Haselbock J, Mannhalter C, et al. Biomarkers predictive of venous thromboembolism in patients with newly diagnosed high-grade gliomas. Neuro-oncology 2014;16(12):1645-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Thompson 2011
- Thompson Eric M, Frenkel Eugene P, Neuwelt Edward A. The paradoxical effect of bevacizumab in the therapy of malignant gliomas. Neurology 2011;76(1):87-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 2007;8(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Verhaak 2010
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010;17(1):98-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wen 2010
- Wen PY, Macdonald DR, Reardon DA, Cloughesy TF, Sorensen AG, Galanis E, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. Journal of Clinical Oncology 2010;28(11):1963-72. [DOI] [PubMed] [Google Scholar]
Westphal 2017
- Westphal M, Maire CL, Lamszus K. EGFR as a target for glioblastoma treatment: an unfulfilled promise. CNS drugs 2017;31(9):723-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wick 2016
- Wick W, Chinot OL, Bendszus M, Mason W, Henriksson R, Saran F, et al. Evaluation of pseudoprogression rates and tumor progression patterns in a phase III trial of bevacizumab plus radiotherapy/temozolomide for newly diagnosed glioblastoma. Neuro-Oncology 2016;18(10):1434-41. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhou 2011
- Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncology 2011;12(8):735-42. [DOI] [PubMed] [Google Scholar]