

Abstract
8岁女性患儿,因间断咳嗽、发热3年余,再发1月余入院。近1年反复出现口腔黏膜溃疡。体格检查发现右上背部皮肤色素脱失。外院多次查血常规和免疫功能正常。多次肺部CT提示肺部感染病灶,经抗感染治疗,发热咳嗽仍反复,右肺部病变持续存在,抗结核治疗无效。多次血常规检查发现粒细胞缺乏,行基因检测,确定为RTEL1基因纯合突变所致先天性角化不良(DC)。RTEL1基因突变的先天性角化不良患者易出现肺部并发症。由于RTEL1基因序列有高度可变性,突变位点很多,突变方式多样,且可以通过常染色体显性或隐性遗传方式遗传,因此其临床表现多样,易致漏诊及误诊。对不明原因反复肺部感染的患儿,应注意口腔、皮肤和指趾甲的检查并监测血常规以排除DC。DC目前无特效治疗,出现骨髓衰竭和肺功能衰竭时,造血干细胞移植和肺移植是目前唯一的治疗方法,雄激素及其衍生物对部分病人有效,针对端粒途径的靶向药物有望给DC患者带来希望。
Keywords: 先天性角化不良, 肺部感染, 口腔溃疡, 皮肤白斑, 儿童
Abstract
An 8-year-old girl who had experienced intermittent cough and fever over a 3 year period, was admitted after experiencing a recurrence for one month. One year ago the patient experienced a recurrent oral mucosal ulcer. Physical examination showed vitiligo in the skin of the upper right back. Routine blood tests and immune function tests performed in other hospitals had shown normal results. Multiple lung CT scans showed pulmonary infection. The patient had recurrent fever and cough and persistent presence of some lesions after anti-infective therapy. The antitubercular therapy was ineffective. Routine blood tests after admission showed agranulocytosis. Gene detection was performed and she was diagnosed with dyskeratosis congenita caused by homozygous mutation in RTEL1. Patients with dyskeratosis congenita with RTEL1 gene mutation tend to develop pulmonary complications. Since RTEL1 gene sequence is highly variable with many mutation sites and patterns and can be inherited via autosomal dominant or recessive inheritance, this disease often has various clinical manifestations, which may lead to missed diagnosis or misdiagnosis. For children with unexplained recurrent pulmonary infection, examinations of the oral cavity, skin, and nails and toes should be taken and routine blood tests should be performed to exclude dyskeratosis congenita. There are no specific therapies for dyskeratosis congenita at present, and when bone marrow failure and pulmonary failure occur, hematopoietic stem cell transplantation and lung transplantation are the only therapies. Androgen and its derivatives are effective in some patients. Drugs targeting the telomere may be promising for patients with dyskeratosis congenita.
Keywords: Dyskeratosis congenita, Pulmonary infection, Oral mucosal ulcer, Skin vitiligo, Child
1. 病例介绍
患儿,女,8岁,因间断咳嗽、发热3年余,再发1月余入院。3年前因咳嗽、发热半个月第一次住院,咳嗽为阵发性,间断有白色泡沫痰,发热无规律,体温波动于38~41℃,血常规和免疫功能基本正常;肺部CT提示左上肺大片密度增高影,边缘模糊,右上肺亦可见斑片状密度增高影,考虑肺部感染;抗感染治疗后症状好转、复查胸片稍好转,出院。出院后患儿咳嗽、发热反复,每月发作2次,每次持续1周左右,抗感染治疗后可暂时缓解。出院5个月后复查肺部CT,提示左肺病灶吸收好转,右上叶大片实变影;支气管纤维镜提示支气管内膜炎;予诊断性抗结核治疗1个月,发热、咳嗽仍反复。此后近2年的时间患儿上述症状反复,多次胸片提示右肺部病变,间断予抗感染治疗。1月余前患儿再次出现咳嗽、发热,肺部CT提示右中上肺团块状、斑片状高密度灶,增强后团块状病灶增强明显,右肺门及隆凸下可见增大并明显强化的淋巴结,收住院诊治。患儿近1年反复出现口腔黏膜溃疡。既往易患呼吸道感染,6岁时行扁桃体切除术。家族中无类似病史。
入院体查:体温36.8℃,呼吸24次/min,脉搏95次/min,血压100/64 mm Hg,身高123 cm,体重23.8 kg。发育正常,营养中等,神志清楚,无特殊面容,右上背部可见一大小约3 cm×2 cm皮肤色素脱失改变(图 1),皮肤未见出血点、紫癜及瘀斑,浅表淋巴结未触及,舌右侧可见一大小约1 cm×0.5 cm溃疡灶、周边稍隆起(图 2),右肺呼吸音稍低,双肺未闻及啰音,心率95次/min,心音有力,律齐,未闻及杂音,腹软,肝脾肋下未及,肠鸣音正常。脊柱四肢、肛门外生殖器未见异常,指趾甲未见异常。辅助检查:血常规WBC 3.8× 109/L,N 0.01×109/L(0.3%),L 2.6×109/L(67.8%),E 0.4×109/L(11%),M 0.8×109/L(19.9%),Hb 115 g/L,PLT 333×109/L;肝肾功能、心肌酶、电解质、血脂分析:白蛋白33.7 g/L(参考值:40~55 g/L),肌酸激酶同工酶48.8 U/L(参考值: < 24 U/L),余项正常;体液免疫:补体C4 82.4 ng/L(参考值:120~360 ng/L),补体C3正常,IgG 24.2 g/L(参考值:7.2~16.8 g/L),IgA 4 020 mg/L(参考值:690~3 820 mg/L),IgM 3 460 mg/L(参考值:630~2 770 mg/L);细胞免疫:NK 2%(参考值:5%~27%),余项基本正常;支原体抗体1:80阳性,结核抗体-IgG阳性,PPD皮试阴性;T-SPOT阴性;血沉44 mm/h;CRP及PCT正常;G/GM实验阴性;血清铁蛋白正常;输血前检查阴性;肺部CT:双肺支气管血管束增多、紊乱,右肺上叶支气管变窄、可见团块状及斑片状密度增高影,右中肺内侧段、左下肺前基底段支气管扩张,管壁增厚,周围见斑片状及条索状密度增高影。余肺未见明显异常。肺部CT影像学讨论:右肺病变考虑(1)慢性感染性病变;(2)黏膜相关淋巴瘤。骨髓细胞学:骨髓增生活跃,粒系减少、成熟粒细胞明显减少,红系活跃,巨核系正常;外周血片:中性粒细胞缺乏,可见异形淋巴细胞。
1.
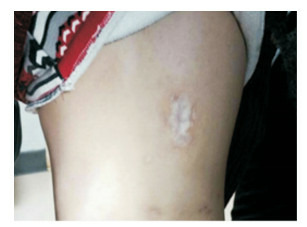
皮肤片状色素脱失
2.
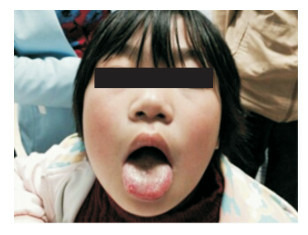
舌右侧可见溃疡,溃疡周围黏膜稍隆起
2. 诊断思维
8岁女性患儿,反复咳嗽、发热3年,抗感染治疗后症状可暂时缓解,但反复;抗结核治疗无效。多次胸片及肺部CT提示肺部病变,尤以右侧肺部病变为主;多次血常规提示中性粒细胞缺乏。本病的诊断思维从两方面考虑:反复肺部病变和粒细胞缺乏。根据肺部影像学特点需要考虑慢性感染性病变和肺黏膜相关淋巴瘤。反复发生的肺部感染常见于营养不良、先天性心脏病、免疫缺陷、呼吸道纤毛功能异常以及气管异物的患儿,发病年龄均偏小。本患儿5岁起病,无营养不良及心脏病史,多次纤维支气管镜未见异物,且病灶多发,不支持气管异物。呼吸道纤毛功能异常可表现为反复咳嗽、咯脓痰、咯血,病程进展可出现呼吸困难、发绀及杵状指,常伴有鼻窦炎及中耳炎,部分患儿有内脏转位,其影像学主要表现为双下肺支气管扩张改变[1]。本例患儿并无反复咯脓痰及鼻窦炎病史,影像学改变不支持呼吸道纤毛功能异常。患儿两次正规住院治疗,均行体液免疫及细胞免疫检查,除NK细胞偏低外未见明显异常。此外肺结核也是肺部慢性病变的常见病因,患儿经抗感染治疗,病情可暂时缓解,曾予抗结核治疗无效,不支持肺结核。肺黏膜相关淋巴瘤为低度恶性非霍奇金淋巴瘤,由于支气管黏膜在各种抗原持续刺激下,如吸烟、慢性感染以及自身免疫性疾病等,机体产生过度免疫反应,肺支气管黏膜形成黏膜相关淋巴样组织,最终发展成肺黏膜相关淋巴瘤,多见于中老年人,临床主要表现为反复咳嗽、气促及不规则发热。病变可发生于单侧肺叶,亦可累及双肺,多为肺实质病变,亦可浸润肺间质,呈境界不清的孤立性结节、肿块或斑片状影。肺部X线病灶中可见支气管充气征,部分患者因累及胸膜出现胸腔积液[2-4]。肺黏膜相关淋巴瘤的临床表现及影像学无特异性,易被误诊为肺癌、肺结核、肺部感染等疾病,对于常规抗感染及抗结核治疗效果不佳的反复肺部病变患者需警惕本病,确诊有赖于组织病理学检查。本例患儿年龄小,且肺部感染经抗感染治疗后可暂时好转,不支持肺黏膜相关淋巴瘤。粒细胞缺乏可分为先天性及获得性。本例患儿3年前粒细胞数目正常,随着病情进展,出现粒细胞减少,且伴有反复肺部感染,可能将粒细胞缺乏归结于反复感染所致,但感染所致粒细胞缺乏常见于病毒感染,而且感染控制后粒细胞可恢复正常。本例患儿除肺部感染外,无其它部位反复感染,但外周血粒细胞持续缺乏、骨髓粒系减少,不能仅以感染来解释,其它继发性引起粒细胞缺乏的因素,如免疫破坏、药物影响等也无依据,因此需要注意患儿是否存在以粒细胞减少为首发表现的遗传性骨髓衰竭性疾病,而且患儿合并有皮肤改变和反复口腔溃疡,不除外先天性角化不良(dyskeratosis congenital, DC),但症状不典型,因此进行了遗传性疾病基因筛查。
3. 进一步检查
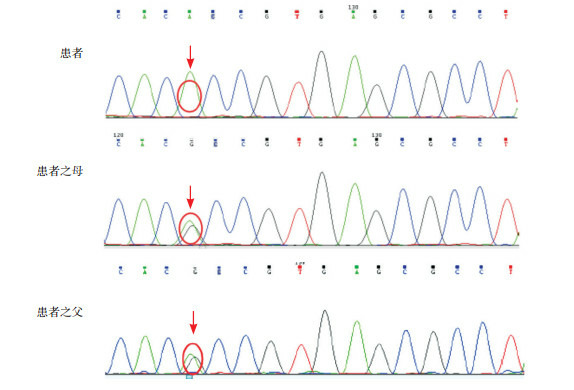
基因结果显示:患儿存在RTEL1(regulator of telomere length 1)基因纯合突变,患儿父母均为RTEL1杂合突变。见图 3。
3.
患儿及其父母RTEL1基因突变检出情况
患儿存在RTEL1基因纯合突变,患儿父母均存在RTEL1杂合突变,突变位点如箭头所示。
4. 诊断及确诊依据
诊断:先天性角化不良。依据:(1)外周血和骨髓细胞学均提示粒细胞减少;(2)反复口腔溃疡,并伴有皮肤改变;(3)不能用一般感染解释的肺部病变;(4)基因测序发现患儿存在DC致病基因之一RTEL1基因纯合突变,患儿父母存在RTEL1杂合突变。
5. 临床经过
入院后予抗感染治疗,发热、咳嗽好转。出院后间断有咳嗽,并有反复皮肤脓疱疹。出院后2个月复查中性粒细胞为0,肺部CT提示原右肺上叶、右中肺内侧段、左下肺背段及后基底段病变较前稍吸收,以右肺上叶明显,邻近肺组织扭曲,邻近支气管牵拉性扩张。此后未再行相关检查。
6. 讨论
DC是一种罕见的先天性骨髓衰竭性疾病,可累及全身各个系统,其典型的临床表现为异常皮肤色素沉着、指 (趾) 甲角化不良、口腔黏膜白斑三联征,还可有多种其它临床表现,如流泪、手掌足底多汗、脱发、结膜炎、斜视、口腔溃疡、牙齿脱落、骨质疏松、食道狭窄、肝大、肝硬化、非缺血性骨坏死等[5-6]。20%的患者可有肺部受累,表现为哮喘、肺纤维化、肝肺综合征、肺微血管异常、支气管扩张、纤维囊性发育不良、气胸、间质性肺炎、反复呼吸道感染、慢性肺炎等[7-9]。皮肤黏膜症状出现的时间差异很大,也有多种表现形式,除典型的颈部及上胸部皮肤网格样色素沉着和口腔黏膜白斑外,也可表现为反复口腔溃疡、慢性皮肤溃疡、皮肤过度角化、尿道或阴道黏膜白斑[10-11]。有些患者以骨髓衰竭或肺纤维化为首发表现,在造血干细胞移植或肺移植后逐渐出现皮肤黏膜或指趾甲的改变才考虑到DC。DC患者的骨髓增生异常综合征、急性髓系白血病、实体瘤(头颈部鳞状细胞癌和生殖器癌常见)和肺纤维化的患病风险较正常人群明显升高,是导致患者死亡的主要原因。除DC严重类型Hoyeraal-Hreidarsson综合征(HH综合征)和Revesz综合征有发育迟缓外,大部分患者的生长发育不受影响[5-6]。本病遗传方式包括X-连锁隐性遗传、常染色体显性遗传和常染色体隐性遗传。端粒及端粒酶功能缺陷是导致DC的主要原因。端粒位于染色体末端,由TTAGGG序列重复串联组成,受端粒蛋白复合物的调控,调节端粒酶活性和保护染色体完整性。端粒长度是细胞年龄、老化及复制能力的标记,随着细胞的每次分裂,端粒长度逐渐缩短。人类细胞的端粒缺陷是DNA损伤的独特形式,这种损伤的修复极其有限,病理状态下端粒异常变短可导致细胞过早衰老、死亡或绕过衰老途径,继续分裂,由于基因不稳定,最终导致肿瘤形成。端粒酶能使细胞增强其自身复制能力。多能干细胞包括造血干细胞的生命持续时间取决于端粒酶的正确功能[5]。任何导致端粒长度缺失或功能缺陷以及影响端粒酶功能的因素均可导致疾病发生[12]。目前已经发现11种基因突变可导致DC,包括ACD,CTC 1,DKC 1,NHP 2,NOP10,PARN,RTEL 1,TERC,TERT,TINF 2,和WRAP 53,这些基因突变均可导致端粒变短或功能缺陷从而出现相应的临床症状[5]。DC的临床表现有明显异质性,不同基因突变引起的疾病类型不尽相同。DKC1突变所致者多表现为经典型DC、少数表现为HH综合征,NOP 1 0、NHP 2、TCAB l突变所致者多表现为经典型DC。在同一家系中具有相同突变基因的不同成员,其外显率、疾病的严重程度以及症状出现的时间可以不同[5, 12]。
RTEL1是DNA解旋酶所必须的,对于维持端粒功能完整性起重要作用,RTEL 1基因突变导致端粒长度明显缩短。RTEL1纯合或复杂杂合突变首次在DC的严重类型HH综合征中发现,后来的研究逐渐发现RTEL1基因突变与某些脑肿瘤(如神经胶质瘤、星形细胞瘤和胶质母细胞瘤),肺部病变(特发性肺纤维化、家族性肺纤维化、间质性肺炎等)以及DC均有关[13]。RTEL1基因突变导致肺部疾病的机制尚不明确,通常认为RTEL1基因突变导致肺泡Ⅱ型上皮细胞内端粒进行性缩短,从而肺泡上皮细胞过早衰老或失去自我更新能力,但目前尚无直接证据支持这种推断。人类肺泡Ⅱ型上皮细胞的寿命及新旧更替时间不清楚,利用谱系追踪模型在小鼠的实验研究中发现:小鼠肺泡上皮细胞新旧交替发生的速度非常缓慢,因此仅用细胞分裂不足以解释肺纤维化患者中肺泡上皮细胞端粒明显缩短的现象,也不能解释影响端粒长度变化的RTEL1基因突变所致的DC患者为什么常常以肺部病变为首要表现。因此DC患者肺部病变的发生可能还有其它因素参与,其机制有待进一步研究[13-14]。RTEL1导致肺部病变的类型主要包括家族性肺纤维化、不能分类的肺纤维化、特发性肺间质性肺炎、慢性过敏性肺炎、胸膜肺实质弹力纤维增生症、伴有自身免疫性疾病的间质性肺炎、结缔组织病相关的间质纤维化等,以肺纤维化病变最为常见[15]。RTEL1基因序列有高度可变性,突变位点很多,突变方式多样,如:无义突变、剪接突变、移码突变、错义突变、短的框内缺失等,因此其临床表现多样,且具有明显异质性[16]。RTEL1突变在儿童常常以血液系统疾病为首要表现,而同一位点的突变在成年期发病则可能以肺部病变或其他系统病变为首要表现;某一位点的突变,一些患者可仅仅表现为肺纤维化;一些患者除肺部表现外,可合并肺外表现,如:骨骼畸形、肝硬化、骨质疏松等,还有些患者可以无症状长期存活[14]。本例患儿发病年龄小,系RTEL1纯合突变,临床表现不典型,除NK细胞偏低外,并无其它HH综合征的特点,如小头畸形、发育迟缓等,口腔黏膜白斑及皮肤改变也不同于典型的DC患者,而是以反复肺部感染合并局灶性纤维化及支气管扩张为主要表现,随着病情进展,逐渐出现口腔黏膜及皮肤改变以及粒细胞缺乏,提示RTEL1突变易导致肺部病变,而且遗传方式与临床表现具有异质性。
DC目前无特效治疗,骨髓衰竭、肺功能衰竭和继发恶性肿瘤是主要死亡原因。抗胸腺球蛋白(ATG)和环孢素的免疫抑制治疗对于骨髓衰竭DC患者无效。雄激素治疗对部分DC患者有效,部分患者用羟甲烯龙治疗后骨髓衰竭可暂时缓解数年之久,但其治疗过程中易出现脂质代谢紊乱和肝功能损害,明显的男性化副作用也限制了女性患者用药;达拉唑是一种雄激素的衍生物,其男性化副作用相对较轻,也试用于DC的治疗[12, 17]。目前,异基因造血干细胞移植是骨髓衰竭DC患者唯一的治愈方式。但由于DC患者存在端粒缺陷,对DNA损伤修复作用有限,移植前清髓性预处理方案中的放疗、大剂量环磷酰胺和白消安可导致DNA损伤,DC患者移植相关并发症明显增加,如移植失败、移植物抗宿主病、感染、肺纤维化、肝硬化、肝静脉阻塞等,从而使DC患者移植后的死亡率增加,生存时间缩短,移植后10年生存率为23%~27%。尽管使用氟达拉滨减低强度的预处理方案使DC患者移植成功率增加,移植相关毒副作用减少,但部分患者移植后仍可出现肺纤维化及神经系统并发症[12]。由于DC患者常常合并肺部病变,而造血干细胞移植可能加速肺部病变的进展,因此移植前后肺功能的监测至关重要[12, 18]。以肺纤维化为主要表现或造血干细胞移植后出现肺纤维化的DC患者,如果无多系统功能衰竭,可考虑行肺移植[19]。DC患者的同胞即使暂时无临床症状,仍可能存在杂合突变,因此选择移植供者时应常规进行基因筛查或端粒长度检测[12]。
7. 结语
尽管DC有典型的三联征,但由于遗传方式多样,涉及基因不同,外显率不一,临床表现有明显异质性,且可累及多系统,因此临床表现多样;而且即使典型的三联征,其症状出现的时间也不一致,易致临床漏诊及误诊。对于伴有宫内发育迟缓、小头畸形、精神发育迟滞的患儿,需警惕DC的严重类型HH综合征。儿童期出现骨髓衰竭时,最好进行基因检测排除遗传性骨髓衰竭,尤其对于有肺纤维化家族史的患儿。对于反复肺部感染患儿,治疗后病灶持续存在,不能用其它原因解释时,也需警惕DC。仔细的病史询问(反复口腔溃疡、家族中类似病史等)、体格检查(口腔、皮肤,指趾甲有无改变等)和血常规监测有助于诊断,基因检测可确诊。出现骨髓衰竭和肺功能衰竭时,造血干细胞移植和肺移植是目前唯一的治疗方法,雄激素及其衍生物对于部分病人有效,针对端粒途径的靶向药物有望给DC患者带来希望。
Biography
匡飞梅, 女, 硕士研究生
Funding Statement
国家自然科学基金(81570154;81400138)
References
- 1.申昆玲, 江载芳, 徐保平. 支气管扩张和原发性纤毛运动障碍[M]//江载芳, 申昆玲, 沈颖. 诸福棠实用儿科学上册. 第8版. 北京: 人民卫生出版社, 2015: 1300-1302.
- 2.Nicholson AG, Wotherspoon AC, Jones AL, et al. Pulmonary B-cell non-Hodgkin's lymphoma associated with autoimmune disorders:a clinic pathological review of six cases. Eur Respir J. 1996;9(10):2022–2025. doi: 10.1183/09031936.96.09102022. [DOI] [PubMed] [Google Scholar]
- 3.彭 思达, 谭 获, 黄 振倩, et al. 肺黏膜相关淋巴组织样淋巴瘤7例并临床诊断与分析. 中国肿瘤临床. 2014;41(14):922–924. doi: 10.3969/j.issn.1000-8179.20131268. [DOI] [Google Scholar]
- 4.Imai H, Sunaga N, Kaira K, et al. Clinicopathological features of patients with bronchial-associated lymphoid tissue lymphoma. Intern Med. 2009;48(5):301–306. doi: 10.2169/internalmedicine.48.1438. [DOI] [PubMed] [Google Scholar]
- 5.Savage SA. Dyskeratosis congenita[M]//Pagon RA, Adam MP, Ardinger HH, et al. Gene reviews®.[Internet]. Seattle (WA):University of Washington, Seattle; 1993-2017.
- 6.周敦华. 先天性角化不良[M]//黄绍良, 陈纯, 周敦华. 实用小儿血液病学. 北京: 人民卫生出版社, 2014: 67-69.
- 7.Verra F, Kouzan S, Saiag P, et al. Bronchoalveolar disease in dyskeratosis congenita. Eur Respir J. 1992;5(4):497–499. [PubMed] [Google Scholar]
- 8.Samuel BP, Duffner UA, Abdel-Mageed AS, et al. Pulmonary arteriovenous malformations in dyskeratosis congenita. Pediatr Dermatol. 2015;32(4):e165–166. doi: 10.1111/pde.2015.32.issue-4. [DOI] [PubMed] [Google Scholar]
- 9.Goldfarb S, Sullivan KE, Jyonouchi S. A patient with X-linked dyskeratosis congenita presenting with bronchiolitis obliterans requiring lung transplantation and immunodeficiency. Pediatr Pulmonol. 2013;48(1):91–93. doi: 10.1002/ppul.v48.1. [DOI] [PubMed] [Google Scholar]
- 10.Reimann C, Kloeckener-Gruissem B, Niemeyer CM, et al. Late manifestation of dyskeratosis congenita presenting as chronic dermal ulcer in a 37-year-old man. J Eur Acad Dermatol Venereol. 2008;22(7):897–898. doi: 10.1111/j.1468-3083.2007.02530.x. [DOI] [PubMed] [Google Scholar]
- 11.Iraji F, Jamshidi K, Pourazizi M, et al. Dyskeratosis congenita without oral involvement:A rare hereditary disease. Oman Med J. 2015;30(3):212–215. doi: 10.5001/omj.2015.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernández García MS, Teruya-Feldstein J. The diagnosis and treatment of dyskeratosis congenita:a review. http://d.scholar.cnki.net/detail/SJDP_U/SJDP14082600000050. J Blood Med. 2014;5:157–167. doi: 10.2147/JBM.S47437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vannie JB, Sarek G, Boulton SJ. RTEL1:functions of a diseaseassociated helicase. Trends Cell Biol. 2014;24(7):416–425. doi: 10.1016/j.tcb.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Kropski JA, Loyd JE. Telomeres revisited:RTEL1 variants in pulmonary fibrosis. Eur Respir J. 2015;46(2):312–314. doi: 10.1183/13993003.00710-2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Newton CA, Batra K, Torrealba J, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. 2016;48(6):1710–1720. doi: 10.1183/13993003.00308-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stanley SE, Noth I, Armanios M. What the genetics "RTEL" ing us about telomeres and pulmonary fibrosis. Am J Respir Crit Care Med. 2015;191(6):608–610. doi: 10.1164/rccm.201501-0119ED. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Islam A, Rafiq S, Kirwan M, et al. Haematological recovery in dyskeratosis congenita patients treated with danazol. Br J Haematol. 2013;162(6):854–856. doi: 10.1111/bjh.2013.162.issue-6. [DOI] [PubMed] [Google Scholar]
- 18.Barbaro P, Vedi A. Survival after hematopoietic stem cell transplant in patients with dyskeratosis congenita:Systematic review of the literature. Biol Blood Marrow Transplant. 2016;22(7):1152–1158. doi: 10.1016/j.bbmt.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Giri N, Lee R, Faro A, et al. Lung transplantation for pulmonary fibrosis in dyskeratosis congenita:Case report and systematic literature review. BMC Blood Disord. 2011;11:3. doi: 10.1186/1471-2326-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]