

Abstract
Background
Convalescent plasma and hyperimmune immunoglobulin may reduce mortality in patients with viral respiratory diseases, and are currently being investigated in trials as potential therapy for coronavirus disease 2019 (COVID‐19). A thorough understanding of the current body of evidence regarding the benefits and risks is required.
Objectives
To continually assess, as more evidence becomes available, whether convalescent plasma or hyperimmune immunoglobulin transfusion is effective and safe in treatment of people with COVID‐19.
Search methods
We searched the World Health Organization (WHO) COVID‐19 Global Research Database, MEDLINE, Embase, Cochrane COVID‐19 Study Register, Centers for Disease Control and Prevention COVID‐19 Research Article Database and trial registries to identify completed and ongoing studies on 4 June 2020.
Selection criteria
We followed standard Cochrane methodology.
We included studies evaluating convalescent plasma or hyperimmune immunoglobulin for people with COVID‐19, irrespective of study design, disease severity, age, gender or ethnicity.
We excluded studies including populations with other coronavirus diseases (severe acute respiratory syndrome (SARS) or Middle East respiratory syndrome (MERS)) and studies evaluating standard immunoglobulin.
Data collection and analysis
We followed standard Cochrane methodology.
To assess bias in included studies, we used the Cochrane 'Risk of bias' tool for randomised controlled trials (RCTs), the Risk of Bias in Non‐randomised Studies ‐ of Interventions (ROBINS‐I) tool for controlled non‐randomised studies of interventions (NRSIs), and the assessment criteria for observational studies, provided by Cochrane Childhood Cancer for non‐controlled NRSIs.
Main results
This is the first living update of our review. We included 20 studies (1 RCT, 3 controlled NRSIs, 16 non‐controlled NRSIs) with 5443 participants, of whom 5211 received convalescent plasma, and identified a further 98 ongoing studies evaluating convalescent plasma or hyperimmune immunoglobulin, of which 50 are randomised. We did not identify any completed studies evaluating hyperimmune immunoglobulin.
Overall risk of bias of included studies was high, due to study design, type of participants, and other previous or concurrent treatments.
Effectiveness of convalescent plasma for people with COVID‐19
We included results from four controlled studies (1 RCT (stopped early) with 103 participants, of whom 52 received convalescent plasma; and 3 controlled NRSIs with 236 participants, of whom 55 received convalescent plasma) to assess effectiveness of convalescent plasma. Control groups received standard care at time of treatment without convalescent plasma.
All‐cause mortality at hospital discharge (1 controlled NRSI, 21 participants)
We are very uncertain whether convalescent plasma has any effect on all‐cause mortality at hospital discharge (risk ratio (RR) 0.89, 95% confidence interval (CI) 0.61 to 1.31; very low‐certainty evidence).
Time to death (1 RCT, 103 participants; 1 controlled NRSI, 195 participants)
We are very uncertain whether convalescent plasma prolongs time to death (RCT: hazard ratio (HR) 0.74, 95% CI 0.30 to 1.82; controlled NRSI: HR 0.46, 95% CI 0.22 to 0.96; very low‐certainty evidence).
Improvement of clinical symptoms, assessed by need for respiratory support (1 RCT, 103 participants; 1 controlled NRSI, 195 participants)
We are very uncertain whether convalescent plasma has any effect on improvement of clinical symptoms at seven days (RCT: RR 0.98, 95% CI 0.30 to 3.19), 14 days (RCT: RR 1.85, 95% CI 0.91 to 3.77; controlled NRSI: RR 1.08, 95% CI 0.91 to 1.29), and 28 days (RCT: RR 1.20, 95% CI 0.80 to 1.81; very low‐certainty evidence).
Quality of life
No studies reported this outcome.
Safety of convalescent plasma for people with COVID‐19
We included results from 1 RCT, 3 controlled NRSIs and 10 non‐controlled NRSIs assessing safety of convalescent plasma. Reporting of adverse events and serious adverse events was variable. The controlled studies reported on adverse events and serious adverse events only in participants receiving convalescent plasma. The duration of follow‐up varied. Some, but not all, studies included death as a serious adverse event.
Grade 3 or 4 adverse events (13 studies, 201 participants)
The studies did not report the grade of adverse events. Thirteen studies (201 participants) reported on adverse events of possible grade 3 or 4 severity. The majority of these adverse events were allergic or respiratory events. We are very uncertain whether or not convalescent plasma therapy affects the risk of moderate to severe adverse events (very low‐certainty evidence).
Serious adverse events (14 studies, 5201 participants)
Fourteen studies (5201 participants) reported on serious adverse events. The majority of participants were from one non‐controlled NRSI (5000 participants), which reported only on serious adverse events limited to the first four hours after convalescent plasma transfusion. This study included death as a serious adverse event; they reported 15 deaths, four of which they classified as potentially, probably or definitely related to transfusion. Other serious adverse events reported in all studies were predominantly allergic or respiratory in nature, including anaphylaxis, transfusion‐associated dyspnoea, and transfusion‐related acute lung injury (TRALI). We are very uncertain whether or not convalescent plasma affects the number of serious adverse events.
Authors' conclusions
We are very uncertain whether convalescent plasma is beneficial for people admitted to hospital with COVID‐19. For safety outcomes we also included non‐controlled NRSIs. There was limited information regarding adverse events. Of the controlled studies, none reported on this outcome in the control group. There is only very low‐certainty evidence for safety of convalescent plasma for COVID‐19.
While major efforts to conduct research on COVID‐19 are being made, problems with recruiting the anticipated number of participants into these studies are conceivable. The early termination of the first RCT investigating convalescent plasma, and the multitude of studies registered in the past months illustrate this. It is therefore necessary to critically assess the design of these registered studies, and well‐designed studies should be prioritised. Other considerations for these studies are the need to report outcomes for all study arms in the same way, and the importance of maintaining comparability in terms of co‐interventions administered in all study arms.
There are 98 ongoing studies evaluating convalescent plasma and hyperimmune immunoglobulin, of which 50 are RCTs. This is the first living update of the review, and we will continue to update this review periodically. These updates may show different results to those reported here.
Plain language summary
Plasma from people who have recovered from COVID‐19 to treat individuals with COVID‐19
Coronavirus disease 2019 (COVID‐19) is a highly infectious respiratory illness caused by a newly recognised type of coronavirus. People infected with this virus may not show signs of the disease, others may develop symptoms, including fever, cough, shortness of breath and sore throat. In some people the infection is more severe and can cause breathing difficulties, leading to hospitalisation, admission to intensive care or death. Currently, no vaccine or specific treatment is available.
People who have recovered from COVID‐19 develop natural defences to the disease in their blood (antibodies). Antibodies are found in part of the blood called plasma. Plasma from blood donated from recovered patients, which contains COVID‐19 antibodies, can be used to make two preparations. Firstly, convalescent plasma, which is plasma that contains these antibodies. Secondly, hyperimmune immunoglobulin, which is more concentrated, and therefore contains more antibodies.
Convalescent plasma and hyperimmune immunoglobulin have been used successfully to treat other respiratory viruses. These treatments (given by a drip or injection) are generally well‐tolerated, but unwanted effects can occur.
What did we want to find?
We wanted to know whether plasma from people who have recovered from COVID‐19 is an effective treatment for people with COVID‐19, and whether this treatment causes any unwanted effects. We are continually updating this review as more evidence becomes available.
Our methods
On 4 June 2020 we searched major medical databases for clinical studies on treatment with convalescent plasma or hyperimmune immunoglobulin for people with COVID‐19. Studies could be conducted anywhere in the world and include participants of any age, gender or ethnicity, with mild, moderate or severe COVID‐19.
Key results
We included 20 completed studies with 5443 participants; 5211 participants received convalescent plasma. We found one randomised controlled trial ((RCT) 103 participants; 52 participants received convalescent plasma). RCTs are clinical studies where people are randomly allocated to receive the treatment (intervention group) or to receive a different treatment or no treatment (control group). RCTs produce the best evidence. We found three controlled non‐randomised studies of interventions ((controlled NRSIs) 236 participants; 55 participants received convalescent plasma). These controlled NRSIs did not randomly allocate participants but did include a control group of participants who did not receive convalescent plasma. The remaining 16 studies (5201 participants) were not randomised and did not include a control group (non‐controlled NRSIs) but provided information about unwanted effects of convalescent plasma.
To assess whether convalescent plasma is an effective treatment for COVID‐19, we evaluated results from the RCT and three controlled NRSIs. The control groups received standard care at the time of treatment without convalescent plasma. There was not enough evidence to determine whether or not convalescent plasma affected the risk of death due to any cause at hospital discharge, time to death, or need for breathing support.
To assess whether convalescent plasma causes unwanted effects, we also evaluated the 16 non‐controlled NRSIs (5201 participants). We identified some serious unwanted effects, which could be related to convalescent plasma, including death, allergic reactions or respiratory complications. We are very uncertain whether or not convalescent plasma affects the number of serious unwanted events.
None of the included studies reported effects on quality of life.
Certainty of the evidence
Our certainty (confidence) in the evidence was very limited because there was only one randomised study and most studies did not use reliable methods to measure their results. Furthermore, participants received various treatments alongside convalescent plasma, and some had underlying health problems.
Conclusion
We are very uncertain whether plasma from people who have recovered from COVID‐19 is an effective treatment for people hospitalised with COVID‐19. We are very uncertain whether or not convalescent plasma affects the number of serious harms. These findings could be related to the natural progression of the disease, other treatments that the participants received, or to convalescent plasma. Our searches found 98 ongoing studies evaluating convalescent plasma and hyperimmune immunoglobulin, of which 50 are randomised. This is the first living update of our review, and we will continue to update this review with results from completed studies.
Summary of findings
Background
Description of the condition
The clinical syndrome coronavirus disease 2019 (COVID‐19) is a new, rapidly emerging zoonotic infectious disease caused by severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2; WHO 2020a). On 11 March 2020, the World Health Organization (WHO) declared the current COVID‐19 outbreak a pandemic, with the outbreak resulting in more than 11.5 million cases and over 535,000 deaths worldwide as of 7 July 2020 (WHO 2020b; WHO 2020c). Although there are similarities with historic coronavirus epidemics, with severe acute respiratory syndrome (SARS) and Middle East respiratory syndrome (MERS) responsible for 813 and 858 deaths respectively, the scale and impact of the COVID‐19 pandemic presents unprecedented challenges to health facilities and healthcare workers all over the world (WHO 2007; WHO 2019).
With a preliminary hospitalisation rate of 12.3 patients per 100,000 population in the USA, COVID‐19 has taken a toll on healthcare capacity, and especially on intensive care unit (ICU) capacity (CDC 2020a). Early reports of the case fatality rate suggest that it ranges between 0.7% and 4%, with higher rates also reported (WHO 2020a; WHO 2020c). However, these numbers should be interpreted with great care due to the data pertaining to the early emergency response, which due to shortage of test kits has led to selective testing of people with severe disease, underreporting of cases and delays from confirmation of a case to time of death (Kim 2020). The median incubation period of SARS‐CoV‐2 was reported to be five days, with 97.5% of cases developing symptoms within 11.5 days of infection (Lauer 2020). Common signs and symptoms can include fever, dry cough, fatigue and sputum production (WHO 2020a). Other, less commonly reported signs and symptoms are shortness of breath, sore throat, headache, myalgia or arthralgia, chills, nausea or vomiting, nasal congestion, diarrhoea, haemoptysis and conjunctival congestion (WHO 2020a). Of the reported cases, 80% are estimated to have a mild or asymptomatic course of infection, and an estimated 5% of cases are admitted to the ICU with acute respiratory distress syndrome (ARDS), septic shock or multiple organ failure, or both (Team 2020; WHO 2020a). A risk factor for developing infection and progressing to severe disease is old age, with people aged over 80 years at highest risk of mortality. Other risk factors are cardiovascular disease, obesity, hypertension, diabetes, chronic respiratory disease, cancer and compromised immune status (Chen 2020a; Huang 2020; Liang 2020; WHO 2020a; Wu 2020a).
SARS‐CoV‐2 is a positive‐sense, single‐stranded RNA (ribonucleic acid) virus with a large genome. Although not much is known about the specific mechanisms underlying severe disease in COVID‐19, there are indications that the virus is capable of inducing an excessive immune reaction in the host, with highly activated but decreased numbers of CD4+ and CD8+ T cells detected in the peripheral blood of people with COVID‐19 (Xu 2020). Early reports also showed that people critically ill with COVID‐19 frequently exhibit a hypercoagulable state and endothelial inflammation, which is hypothesised to lead to the high burden of thromboembolic events seen in this population (Driggin 2020). Preliminary reports into the pathophysiology of SARS‐CoV‐2 have further indicated that the observed decrease in human angiotensin‐converting enzyme 2 (ACE2) activity may play a role in causing the rapid deterioration of patient lung function (Tolouian 2020; Van de Veerdonk 2020). ACE2 is a protein that functions as the receptor facilitating entry of SARS‐CoV‐2 into the host cell, and is most abundant on type II alveolar cells in the lungs.
Description of the intervention
Convalescent plasma, convalescent serum and hyperimmune immunoglobulin prepared from convalescent plasma, are interventions that have been used in the past to treat conditions when no vaccine or pharmacological interventions were available. Diphtheria, pneumococcal pneumonia, hepatitis A and B, mumps, polio, measles and rabies are conditions where convalescent plasma has been shown to be effective (Eibl 2008).
A systematic review has shown that convalescent plasma may have clinical benefit for people with influenza and SARS (Mair‐Jenkins 2015). This systematic review included observational studies and randomised controlled trials (RCTs) investigating the use of convalescent plasma, serum or hyperimmune immunoglobulin for treating severe acute respiratory infections of laboratory‐confirmed or suspected viral aetiology, and included investigations with patients of any age and sex. Control interventions consisted of sham, or placebo, therapy and no therapy. The authors concluded that, although the included studies were generally small and of low quality, with a moderate to high risk of bias, the use of convalescent plasma may reduce mortality and appears safe (Mair‐Jenkins 2015). The authors also suggested that the effectiveness of convalescent plasma in reducing hospital length of stay is dependent on early administration of the therapy, and use as prophylaxis is more likely to be beneficial than treating severe disease. However, the optimal timing and dosage of convalescent plasma therapy is unknown.
There is conflicting evidence about the effect of convalescent plasma or hyperimmune immunoglobulin for treating severe acute respiratory infections. Studies investigating the effectiveness of hyperimmune immunoglobulin for influenza have been contradictory, with some RCTs showing effectiveness (Hung 2013), whereas others show no benefit (Beigel 2017; Beigel 2019; Davey 2019).
Although convalescent plasma is generally thought to be a safe and well‐tolerated therapy, adverse events can occur. Limited information is available about specific adverse events related to convalescent plasma therapy, but symptoms that have been reported are similar to those for other types of plasma blood components, including fever or chills, allergic reactions, and transfusion‐related acute lung injury (TRALI; Beigel 2019; Chun 2016; Luke 2006). Furthermore, the transfer of coagulation factors present in plasma products is potentially harmful for people with COVID‐19, who are already at an increased risk of thromboembolic events (Driggin 2020). Plasma transfusions are also known to cause transfusion‐associated circulatory overload (TACO). TACO and TRALI are especially important to consider, because COVID‐19 patients with comorbidities, who might be eligible for experimental treatment with convalescent plasma therapy, are at an increased risk of these adverse events. There are risk‐mitigation strategies that can be implemented to prevent TRALI. These include limiting donations from female donors, especially those with a history of pregnancy, and screening of donors for antibodies that are implicated in TRALI (Otrock 2017). In addition to the aforementioned adverse events, transfusion‐transmitted infections, red blood cell alloimmunisation and haemolytic transfusion reactions have also been described following plasma transfusion, although they are less common (Pandey 2012). Pathogen inactivation can be implemented to decrease the risk of transmitting infections by transfusion (Rock 2011).
When compared to convalescent plasma, hyperimmune immunoglobulin has the advantage of preventing transfer of potentially harmful coagulation factors that are present in plasma products. The amount and antibody concentration can be more accurately dosed compared to convalescent plasma, and hyperimmune immunoglobulin can be prepared in a consistent manner (Hung 2013). Not many studies have reported on adverse events of hyperimmune immunoglobulin, but the safety profile of standard intravenous immunoglobulin is known and the adverse events reported here are also likely to occur in hyperimmune immunoglobulin therapy. Common adverse events of intravenous immunoglobulin that occur immediately after administration are: infusion site pain; swelling and erythema; and immediate systemic reactions, such as head and body aches, chills and fever (Stiehm 2013). Other, less common early adverse reactions to immunoglobulin therapy are pulmonary complications, such as pulmonary embolism, pulmonary oedema and pleural effusion, with TRALI also reported (Baudel 2020; Stiehm 2013). Anaphylactic and anaphylactoid reactions to immunoglobulin therapy are rare (Brennan 2003; Stiehm 2013). Delayed adverse events of immunoglobulin therapy, which occur within hours to days of initiation of immunoglobulin therapy, are persistent headaches (common), aseptic meningitis, renal failure, thromboembolic events, and haemolytic reactions (Sekul 1994; Stiehm 2013). Transmission of infectious agents has been described after administration of intravenous immunoglobulin, but this risk is considered to be low (Stiehm 2013). Other, severe adverse events that occur late after administration are lung disease, enteritis and dermatological disorders (Stiehm 2013).
A theoretical risk related to virus‐specific antibodies, which are transferred with convalescent plasma and hyperimmune immunoglobulin administration, is antibody‐dependent enhancement of infection (Morens 1994). Here, virus‐binding antibodies facilitate the entry and replication of virus particles into monocytes, macrophages and granulocytic cells and thereby increase the risk of more severe disease in the infected host. Although antibody‐dependent enhancement has not been demonstrated in COVID‐19, it has been seen with previous coronavirus infections when the antibodies given targeted a different serotype of the virus (Wan 2020; Wang 2014). A mechanism for antibody‐dependent enhancement in COVID‐19 has recently been proposed, with non‐neutralising antibodies to variable S domains potentially enabling an alternative infection pathway via Fc receptor‐mediated uptake (Ricke 2020). Antibody‐dependent enhancement is therefore a potentially harmful consequence of convalescent plasma and hyperimmune immunoglobulin therapy for COVID‐19.
In summary, the benefits of the intervention, both for convalescent plasma or hyperimmune immunoglobulin, should be carefully considered in view of the risks of adverse events.
How the intervention might work
Convalescent plasma contains pathogen‐specific neutralising antibodies, which can neutralise viral particles, and treatment with convalescent plasma or hyperimmune immunoglobulins confers passive immunity to recipients. The duration of conferred protection can differ depending on the timing of administration, ranging from weeks to months after treatment (Casadevall 2020a).
By neutralising SARS‐CoV‐2 particles, early treatment with convalescent plasma is postulated to increase the patient’s own capacity to clear the initial inoculum (Casadevall 2020a; Robbins 1995). This could lead to a reduction in mortality and fewer hospitalised patients progressing to the ICU. Furthermore, convalescent plasma may reduce the length of ICU stay in critically ill patients (Mair‐Jenkins 2015), thus helping to lift pressure from global healthcare systems and increasing ICU capacity.
Preliminary evidence in humans and rhesus macaques has shown that reinfection with SARS‐CoV‐2 is not likely, with most (but not all) patients who recovered from COVID‐19 producing sufficient amounts of neutralising antibodies to protect against reinfection (Bao 2020a; Wu 2020b). This implies that convalescent plasma from people who have recovered from SARS‐CoV‐2 infection is capable of conferring passive immunity. A recently reported case series also indicated sufficient neutralising antibody titres in convalescent plasma to neutralise SARS‐CoV‐2 in five COVID‐19 patients, who all recovered after treatment (Shen 2020). It is important to note, however, that research in other coronavirus species has shown that immunity may not be long‐lasting, with two to three years of protection estimated from work with SARS and MERS (Mo 2006; Payne 2016). Furthermore, there are indications that the severity of infection has an impact on antibody titres, with less severe disease leading to lower neutralising antibody response in people with SARS and COVID‐19 (Ho 2005; Zhao 2020a).
Why it is important to do this review
There is a clear, urgent need for more information to guide clinical decision‐making for COVID‐19 patients. Pharmacological treatment options are being investigated in many ongoing trials, with currently only treatment of dexamethasone proven to be effective in reducing mortality (Horby 2020), and remdesivir shown to reduce time to recovery (Beigel 2020). Current treatment further consists of supportive care with extracorporeal membrane oxygenation in severe cases and oxygen supply in mild cases (CDC 2020b; WHO 2020d). Despite these treatments, people hospitalised with COVID‐19 are still at a high risk of mortality. A vaccine could aid in inducing immunity in the population and preventing transmission to those who are at risk for severe disease, but no vaccine is currently available, although multiple candidate vaccines are in development. Until these vaccines are available and distributed, convalescent plasma is a potential therapy for COVID‐19 patients. Convalescent plasma, and hyperimmune immunoglobulin to a certain extent, can be prepared and made rapidly available by blood banks and hospitals when enough potential donors have recovered from the infection, using readily available materials and methods (Bloch 2020). However, its safety and efficacy are not well characterised, and there are costs associated with pursuing the use of convalescent plasma for treatment of COVID‐19.
A multitude of clinical trials investigating the safety and effectiveness of convalescent plasma or hyperimmune immunoglobulins have been announced, and their results will need to be interpreted with care. Thus, there needs to be a thorough understanding of the current body of evidence regarding the use of convalescent plasma for people with COVID‐19, and an extensive review of the available literature is required.
Objectives
To continually assess, as more evidence becomes available, whether convalescent plasma or hyperimmune immunoglobulin transfusion is effective and safe in the treatment of people with COVID‐19.
Methods
Criteria for considering studies for this review
Types of studies
The protocol for this review was registered with the Center for Open Science (Piechotta 2020).
To assess the benefits and safety of convalescent plasma therapy for COVID‐19 we included randomised controlled trials (RCTs), as such studies, if performed appropriately, give the best evidence for experimental therapies in highly controlled therapeutic settings. For RCT data, we used the methods recommended by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2019a), as specified in the description of the methods.
In case of insufficient evidence available from RCTs, we had planned to include prospective controlled non‐randomised studies of interventions (NRSIs), including quasi‐randomised controlled trials (e.g. assignment to treatment by alternation or by date of birth), controlled before‐and‐after (CBA) studies, and interrupted time series (ITS) studies. We had planned to use the methods proposed in the Cochrane Handbook for Systematic Reviews of Interventions for the inclusion of controlled NRSIs in systematic reviews (Reeves 2019).
As planned at the protocol stage, we further included retrospective controlled NRSIs, because of insufficient evidence (very low‐certainty evidence or no evidence) available from RCTs and prospective controlled NRSIs and adapted the methods for the inclusion of controlled NRSIs in systematic reviews as specified by the Cochrane Handbook for Systematic Reviews of Interventions (Reeves 2019).
The evidence that we found from the RCT was at unclear or high risk of bias and at serious risk of bias for the controlled NRSIs, and none of the studies reported safety data for the control arm. So we also included safety data from prospective and retrospective non‐controlled NRSIs, for example, case series (please see Differences between protocol and review), and followed the methodology as specified in the protocol (Piechotta 2020).
We followed the suggestions specified in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2019a), as far as possible, and applied the methodology outlined in the following sections. We considered studies including one or more participant(s) with coronavirus disease 2019 (COVID‐19).
We included full‐text publications, abstract publications, and results published in trials registries, if sufficient information was available on study design, characteristics of participants, interventions and outcomes. We did not apply any limitation with respect to the length of follow‐up.
Types of participants
We included individuals with a confirmed diagnosis of COVID‐19, with no age, gender or ethnicity restrictions.
We excluded studies including populations with other coronavirus diseases (severe acute respiratory syndrome (SARS) or Middle East respiratory syndrome (MERS)). We also excluded studies including populations with mixed viral diseases (e.g. influenza), unless the trial authors provided subgroup data for people with COVID‐19.
Types of interventions
We included the following interventions.
Convalescent plasma from people who had recovered from SARS‐CoV‐2 infection
Hyperimmune immunoglobulin therapy
We did not include studies on standard immunoglobulin.
We included the following comparisons for studies with a control arm.
Convalescent plasma therapy versus control treatment, for example, drug treatments (including but not limited to hydroxychloroquine, remdesivir), standard immunoglobulin. Co‐interventions are allowed, but must be comparable between intervention groups.
We had planned to additionally include the following comparisons for studies with a control arm, but did not identify any studies.
Convalescent plasma versus standard care or placebo
Convalescent plasma therapy versus hyperimmune immunoglobulin
Hyperimmune immunoglobulin versus standard care or placebo
Hyperimmune immunoglobulin versus control treatment, for example, drug treatments (including but not limited to hydroxychloroquine, remdesivir). Co‐interventions are allowed, but must be comparable between intervention groups.
Types of outcome measures
We evaluated core outcomes as pre‐defined by the Core Outcome Measures in Effectiveness Trials Initiative for COVID‐19 patients (COMET 2020).
Primary outcomes
Effectiveness of convalescent plasma for people with COVID‐19
All‐cause mortality at hospital discharge
Time to death
Secondary outcomes
Effectiveness of convalescent plasma for people with COVID‐19
-
Improvement of clinical symptoms, assessed by need for respiratory support at up to 7 days; 8 to 15 days; 16 to 30 days:
oxygen by mask or nasal prongs
oxygen by non‐invasive ventilation (NIV) or high‐flow
intubation and mechanical ventilation
mechanical ventilation plus high‐flow oxygen
extracorporeal membrane oxygenation (ECMO)
30‐day and 90‐day mortality
Time to discharge from hospital
Admission to the intensive care unit (ICU)
Length of stay on the ICU
Quality of life, assessed with standardised scales (e.g. WHOQOL‐100) at up to 7 days; up to 30 days, and longest follow‐up available
Safety of convalescent plasma for people with COVID‐19
Number of participants with grade 3 and grade 4 adverse events, including potential relationship between intervention and adverse reaction (e.g. transfusion‐related acute lung injury (TRALI), transfusion‐transmitted infection, transfusion‐associated circulatory overload (TACO), transfusion‐associated dyspnoea (TAD), acute transfusion reactions)
Number of participants with serious adverse events
Timing of outcome measurement
For time‐to‐event outcomes, such as mortality, discharge from hospital, and improvement of clinical symptoms, we included outcome measures representing the longest follow‐up time available.
We included all other outcome categories for the observational periods that the study publications reported. We included those adverse events occurring during active treatment and had planned to include long‐term adverse events as well. If sufficient data had been available, we planned to group the measurement time points of eligible outcomes, for example, adverse events and serious adverse events, into those measured directly after treatment (up to seven days after treatment), medium‐term outcomes (15 days after treatment) and longer‐term outcomes (over 30 days after treatment).
Search methods for identification of studies
We carry out weekly searches for completed and ongoing studies in all languages in order to limit language bias.
Electronic searches
We designed and tested search strategies for electronic databases according to methods suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Lefebvre 2019), CD developed them and Cochrane Haematology's Information Specialist (IM) peer reviewed them. In this emerging field, we expected that at least the abstract would be in English. If studies were published in other languages than those our review team could accommodate (English, Dutch, German, French, Italian, Malay and Spanish), we involved Cochrane TaskExchange to identify people within Cochrane to translate these studies.
As publication bias might influence all subsequent analyses and conclusions, we searched all potential relevant trials registries in detail to detect ongoing as well as completed studies, but not yet published studies. Nowadays, it is mandatory to provide results at least in the trials registry. In case results were not published elsewhere, we had planned to extract and analyse these data. However, no outcome data have yet been added to the trials registries.
We searched the following databases and sources, from 1 January 2019 to 4 June 2020.
-
Databases of medical literature
MEDLINE (Ovid, 23 April to 4 June 2020), Appendix 1
Embase (Ovid, 23 April to 4 June 2020), Appendix 2
PubMed (for epublications ahead of print only; searched 4 June 2020), Appendix 3
Center for Disease Control and Prevention COVID‐19 Research Article Database (www.cdc.gov/library/researchguides/2019novelcoronavirus/databasesjournals.html; downloaded 4 June 2020), Appendix 4
Cochrane COVID‐19 Study Register (covid-19.cochrane.org; searched 4 June 2020), Appendix 5
-
Trials registries and registry platforms to identify ongoing studies and results of completed studies
ClinicalTrials.gov ‐ COVID‐19 subset (included in Cochrane COVID‐19 Study Register)
WHO International Clinical Trials Registry Platform (ICTRP) ‐ COVID‐19 subset (included in Cochrane COVID‐19 Study Register)
Searching other resources
We handsearched the reference lists of all identified studies, relevant review articles and current treatment guidelines for further literature; and
contacted experts in the field, drug manufacturers and regulatory agencies in order to retrieve information on unpublished studies.
Data collection and analysis
Selection of studies
Two out of four review authors (SJV, KLC, VP, NS) independently screened the results of the search strategies for eligibility for this review by reading the abstracts using Covidence software. We coded the abstracts as either 'retrieve' or 'do not retrieve'. In the case of disagreement or if it was unclear whether we should retrieve the abstract or not, we obtained the full‐text publication for further discussion. Two review authors assessed the full‐text articles of selected studies. If the two review authors were unable to reach a consensus, they consulted a third review author to reach a final decision.
We documented the study selection process in a flow chart, as recommended in the PRISMA statement (Moher 2009), and show the total numbers of retrieved references and the numbers of included and excluded studies. We list all articles that we excluded after full‐text assessment and the reasons for their exclusion in the Characteristics of excluded studies table.
Data extraction and management
One review author (NS, SJV, KLC, or VP) performed all data extractions and assessments. Two other review authors (NS, SJV, KLC, or VP) verified the accuracy and (where applicable) the plausibility of extractions and assessment.
Two review authors (VP or NS) independently assessed eligible studies obtained in the process of study selection (as described above) for methodological quality and risk of bias. If the review authors were unable to reach a consensus, we consulted a third review author (SJV or KLC).
One review author (NS, SJV, KLC, or VP) extracted data using a customised data extraction form developed in Microsoft Excel (Microsoft Corporation 2018); please see Differences between protocol and review). Another review author (NS, SJV, KLC, or VP) verified the accuracy and (where applicable) the plausibility of extractions and assessment. We conducted data extraction according to the guidelines proposed by Cochrane (Li 2019). If the review authors were unable to reach a consensus, we consulted a third review author.
We collated multiple reports of one study so that the study, and not the report, is the unit of analysis.
We extracted the following information.
General information: author, title, source, publication date, country, language, duplicate publications
Quality assessment: study design, confounding, definition of risk estimates, selection bias, attrition bias, detection bias, reporting bias
Study characteristics: trial design, setting and dates, source of participants, inclusion/exclusion criteria, comparability of groups, treatment cross‐overs, compliance with assigned treatment, length of follow‐up
Participant characteristics: age, gender, ethnicity, number of participants recruited/allocated/evaluated, disease, severity of disease, additional diagnoses, previous treatments (e.g. experimental drug therapies, oxygen therapy, ventilation), whether the donors were tested by nasal swabs or whether the plasma was tested
-
Interventions: convalescent plasma therapy or hyperimmune immunoglobulin therapy, concomitant therapy, duration of follow‐up, donors' disease severity, how donations were tested for neutralising antibody
For studies including a control group: comparator (type)
-
Outcomes
-
Effectiveness of convalescent plasma for people with COVID‐19:
all‐cause mortality at hospital discharge
time to death
improvement of clinical symptoms, assessed through need for respiratory support at up to 7 days; 8 to 15 days; 16 to 30 days
30‐day and 90‐day mortality
time to discharge from hospital
admission to the ICU
length of stay on the ICU
-
Safety of convalescent plasma for people with COVID‐19:
number of participants with grade 3 and grade 4 adverse events, including potential relationship between intervention and adverse reaction (e.g. TRALI, TACO, TAD, acute transfusion reactions)
number of participants with serious adverse events
-
Assessment of risk of bias in included studies
Randomised controlled trials
Two review authors (VP, NS) independently assessed risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (please see Differences between protocol and review), with any disagreements resolved by discussion (Higgins 2011). In order to rate the certainty of the evidence, we assessed risk of bias per outcome rather than per study only. We completed a 'Risk of bias' table for each included study using the 'Risk of bias' tool in Review Manager Web. Risk of bias judgements in RCTs are 'high', 'unclear' or 'low'.
Controlled non‐randomised studies of interventions
As reported above, we had planned to include controlled non‐randomised studies of intervention (NRSI) trials if there was insufficient evidence from RCTs.
Two review authors (VP, NS) independently assessed eligible studies for methodological quality and risk of bias (using the Risk Of Bias in Non‐randomised Studies ‐ of Interventions (ROBINS‐I) tool; Sterne 2016). The quality assessment strongly depends upon information on the design, conduct and analysis of the trial. The two review authors resolved any disagreements regarding quality assessments by discussion, and in case of discrepancies among their judgements, or inability to reach consensus, we had planned to consult a third review author until consensus could be reached. We asked the Cochrane Editorial and Methods Department (Theresa Moore) to review our judgements for reasonability. The categories for 'Risk of bias' judgements for controlled NRSIs using ROBINS‐I are 'low risk', 'moderate risk', 'serious risk' and 'critical risk' of bias.
We assessed the following domains of bias.
Bias due to confounding
Bias in selection of participants into the study
Bias in classification of interventions
Bias due to deviations from intended interventions
Bias due to missing data
Bias in measurement of outcomes
Bias in selection of the reported result
For every criterion we made a judgement using one of five response options.
Yes
Probably yes
Probably no
No
No information
Non‐controlled non‐randomised studies of interventions
As specified in the Types of studies section, the evidence that we found from the RCT was at unclear or high risk of bias and at serious risk of bias for the controlled NRSIs, and none of the studies reported safety data for the control arm. So we also included safety data from prospective and retrospective non‐controlled NRSIs.
Because we only included safety data from non‐controlled NRSIs, we only assessed methodological quality and risk of bias for studies reporting any safety data.
Two review authors (VP, NS) assessed eligible studies for methodological quality and risk of bias (using the 'Risk of bias' assessment criteria for observational studies tool provided by Cochrane Childhood Cancer (see Table 4; Mulder 2019). We performed and presented any 'Risk of bias' judgements per outcome per study.
1. 'Risk of bias' assessment criteria for observational studies.
| Heading | Internal validity | External validity |
| Study group |
Selection bias (representative: yes/no)
or
|
Reporting bias (well defined: yes/no)
and
|
| Follow‐up |
Attrition bias (adequate: yes/no)
or
|
Reporting bias (well defined: yes/no)
|
| Outcome |
Detection bias (blind: yes/no)
|
Reporting bias (well defined: yes/no)
|
| Risk estimation |
Confounding (adjustment for other factors: yes/no)
|
Analyses (well defined: yes/no)
|
The quality assessment strongly depends upon information on the design, conduct and analysis of the study. The two review authors (VP, NS) resolved any disagreements regarding the quality assessments by discussion; in case of disagreement they would have consulted a third review author (SJV or KLC).
We assessed the following domains of bias.
-
Internal validity
Unrepresentative study group (selection bias)
Incomplete outcome assessment/follow‐up (attrition bias)
Outcome assessors unblinded to investigated determinant (detection bias)
Important prognostic factors or follow‐up not taken adequately into account (confounding)
-
External validity
Poorly defined study group (reporting bias)
Poorly defined follow‐up (reporting bias)
Poorly defined outcome (reporting bias)
Poorly defined risk estimates (analyses)
For every criterion, risk of bias judgements are 'high', 'unclear' or 'low'.
We used the Risk‐of‐bias VISualization tool (robvis) to generate risk of bias summary figures (McGuinness 2020).
Measures of treatment effect
Randomised controlled trials
For continuous outcomes, we had planned to record the mean, standard deviation and total number of participants in both the treatment and control groups. For dichotomous outcomes, we had planned to record the number of events and total number of participants in both the treatment and control groups.
For continuous outcomes using the same scale we had planned to perform analyses using the mean difference (MD) with 95% confidence intervals (CIs). For continuous outcomes measured with different scales we had planned to perform analyses using the standardised mean difference (SMD). For interpreting SMDs, we had planned to re‐express SMDs in the original units of a particular scale with the most clinical relevance and impact.
If available, we extracted and reported hazard ratios (HRs) for time‐to‐event outcomes (time to death). If HRs were not available, we made every effort to estimate the HR as accurately as possible using the available data and a purpose‐built method based on the Parmar and Tierney approach (Parmar 1998; Tierney 2007). If sufficient studies had provided HRs, we planned to use HRs rather than risk ratios (RRs) or MDs in a meta‐analysis.
For dichotomous outcomes, we had planned to report the pooled RR with a 95% CI (Deeks 2019). If the number of observed events had been small (less than 5% of sample per group), and if studies had balanced treatment groups, we planned to report the Peto odds ratio (OR) with 95% CI (Deeks 2019).
Controlled non‐randomised studies of interventions
For dichotomous outcomes, if available, we had planned to extract and report the RR with a 95% CI from statistical analyses adjusting for baseline differences (such as Poisson regressions or logistic regressions) or the ratio of RRs (i.e. the RR post‐intervention/RR pre‐intervention).
For continuous variables, if available, we had planned to extract and report the absolute change from a statistical analysis adjusting for baseline differences (such as regression models, mixed models or hierarchical models), or the relative change adjusted for baseline differences in the outcome measures (i.e. the absolute post‐intervention difference between the intervention and control groups, as well as the absolute pre‐intervention difference between the intervention and control groups/the post‐intervention level in the control group; EPOC 2017).
Non‐controlled non‐randomised studies of interventions
For non‐controlled NRSIs we did not carry out an analysis using quantitative data from indirect controls, as we are aware of the difficulties of indirect comparisons of participant groups with varying baseline characteristics, especially in the absence of individual patient data. Because authors of non‐controlled NRSIs, often discuss their findings using information from other intervention and observational studies as implicit controls, we discussed our findings extensively in the context of what is known about the outcome of 'comparable' patients receiving other experimental treatments but not convalescent plasma therapy or hyperimmune immunoglobulin therapy. We did not meta‐analyse the data but provided information from individual studies within tables.
Unit of analysis issues
We did not combine any data from different study designs. Meta‐analysis was not appropriate for the identified controlled NRSIs because of critical risk of bias. Meta‐analysis was also not appropriate for the non‐controlled NRSIs as described above. We reported and presented results narratively, instead.
Please refer to Appendix 6 for information regarding how we had planned to combine studies with multiple treatment groups.
Dealing with missing data
Chapter 6 of the Cochrane Handbook for Systematic Reviews of Interventions suggests a number of potential sources for missing data, which we needed to take into account: at study level, at outcome level and at summary data level (Higgins 2019b). In the first instance, it is of the utmost importance to differentiate between data 'missing at random' and 'not missing at random'.
We requested missing data from the study authors. We contacted four principal investigators from included studies (Duan 2020; Li 2020; Liu 2020; Zeng 2020). We received one response (Liu 2020), stating that the authors were not able to provide additional data for this version of the review. We contacted seven principal investigators from ongoing studies, which were planned to be completed (ChiCTR2000030010; ChiCTR2000030039; ChiCTR2000030179; ChiCTR2000030627; NCT04264858; NCT04345991; NCT04376788), but did not receive any responses. As we have not pooled any data at this point, we did not have to make any assumptions. If, for updates of this review, data are still missing, we will have to make explicit assumptions of any methods the included studies used. For example, we will assume that the data were missing at random or we will assume that missing values had a particular value, such as a poor outcome.
Assessment of heterogeneity
We did not combine any data from different study designs. Meta‐analysis was not appropriate for the identified controlled NRSIs because of critical risk of bias. Meta‐analysis was also not appropriate for the non‐controlled NRSIs as described above. We reported and presented results narratively, instead.
Please refer to Appendix 6 for information regarding how we had planned to assess heterogeneity.
Assessment of reporting biases
As mentioned above, we searched trials registries to identify completed studies that have not been published elsewhere, to minimise or determine publication bias. We included studies irrespective of their publication status as recommended in Cochrane Handbook for Systematic Reviews of Interventions (McKenzie 2019).
In an update of this review, we intend to explore potential publication bias by generating a funnel plot and statistically testing this by conducting a linear regression test (Sterne 2019), for meta‐analyses involving at least 10 studies. We will consider P < 0.1 as significant for this test.
Data synthesis
Please refer to Appendix 6 for information regarding how we had planned to synthesise data from RCTs and controlled NRSIs.
We did not meta‐analyse data from non‐controlled NRSIs, as there might be no additional benefit in meta‐analysing data without a control group. We reported outcome data of each included trial within tables.
As data did not allow quantitative assessment, we presented outcome data individually per study within tables.
Subgroup analysis and investigation of heterogeneity
Considering the currently available evidence, any analyses were inappropriate for this version of the review. We therefore plan to perform subgroup analyses of the following characteristics in an update of this review.
Age of participants (divided into applicable age groups, e.g. children; 18 to 65 years, 65 years and older)
Severity of condition
Pre‐existing conditions (diabetes, respiratory disease, hypertension, immunosuppression)
We will use the tests for interaction to test for differences between subgroup results.
Sensitivity analysis
Considering the currently available evidence, any analyses were inappropriate for this version of the review. We will perform only one sensitivity analysis for the following in an update of this review.
'Risk of bias' assessment components (low risk of bias versus high risk of bias)
To assess the influence of study quality on an outcome, we will perform sensitivity analyses per outcome, comparing studies with at least one domain of high risk of bias to those without high risk of bias.
Influence of completed, but not published studies
Influence of premature termination of studies
Summary of findings and assessment of the certainty of the evidence
We used the GRADE approach to assess the certainty of the evidence for the following outcomes (please find the rationale for the amendment of graded outcomes in the Differences between protocol and review).
All‐cause mortality at hospital discharge
Time to death
-
Clinical improvement (assessed by need for respiratory support) at the following time points
7 days post‐convalescent plasma transfusion
15 days post‐convalescent plasma transfusion
30 days post‐convalescent plasma transfusion
Quality of life
Grade 3 or 4 adverse events
Serious adverse events
We used GRADEpro GDT software to create a 'Summary of findings' table, as suggested in the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2019a). We assessed the certainty of the evidence for non‐controlled NRSIs as reported in the GRADE guidance 3, starting from low‐certainty evidence (Balshem 2011). As we used the ROBINS‐I tool to assess risk of bias for controlled NRSIs, we followed GRADE guidance 18, starting from high‐certainty evidence with the opportunity to downgrade by three points for critical risk of bias (Schünemann 2019b). For time‐to‐event outcomes we calculated absolute effects at specific time points as recommended in the GRADE guidance 27 (Skoetz 2020). We phrased the findings and certainty of the evidence as suggested in the informative statement guidance (Santesso 2020).
Results
Description of studies
Results of the search
For this update, we identified 1856 new records, in addition to the 1267 potentially relevant records from the first version (altogether 3123 references). After removing duplicates, we screened 1678 new records for this update (altogether 2717 records) based on their titles and abstracts, and we excluded 2535 records that did not meet the prespecified inclusion criteria. We evaluated the remaining 182 records and screened the full texts, or, if these were not available, abstract publications or trials registry entries. See Figure 1 for the study flow diagram (Moher 2009).
1.

Study flow diagram
We identified 118 potentially eligible studies within 120 citations: 20 completed studies (22 records) (Ahn 2020; Anderson 2020; Bao 2020b; Duan 2020; Jin 2020; Joyner 2020; Kong 2020; Li 2020; Liu 2020; Pei 2020; Perotti 2020; Salazar 2020; Shen 2020; Tan 2020; Yang 2020; Ye 2020; Zeng 2020; Zhang 2020a; Zhang 2020b; Çınar 2020), and 98 ongoing studies (see 'Ongoing studies' below).
Included studies
We included 20 studies describing 5443 participants in this review, of whom 5211 received convalescent plasma (Ahn 2020; Anderson 2020; Bao 2020b; Çınar 2020; Duan 2020; Jin 2020; Joyner 2020; Kong 2020; Li 2020; Liu 2020; Pei 2020; Perotti 2020; Salazar 2020; Shen 2020; Tan 2020; Yang 2020; Ye 2020; Zeng 2020; Zhang 2020a; Zhang 2020b).
Design and sample size
Efficacy outcomes
We evaluated efficacy and safety outcomes from controlled studies: one RCT (Li 2020; 103 participants of whom 52 received convalescent plasma) and three controlled NRSIs (Duan 2020; Liu 2020; Zeng 2020; 236 participants of whom 55 received convalescent plasma).
Safety outcomes
For safety outcomes, we also evaluated non‐controlled NRSIs. However, six non‐controlled NRSIs (case reports or case series) did not report whether they evaluated adverse events and are therefore not considered in further analyses (Anderson 2020; Bao 2020b; Kong 2020; Shen 2020; Yang 2020; Çınar 2020). We extracted safety data from 14 studies with 5201 participants who received convalescent plasma (Ahn 2020; Duan 2020; Jin 2020; Joyner 2020; Li 2020; Liu 2020; Pei 2020; Perotti 2020; Salazar 2020; Tan 2020; Ye 2020; Zeng 2020; Zhang 2020a; Zhang 2020b). Of the 10 non‐controlled NRSIs, one is an ongoing expanded access study (Joyner 2020; NCT04338360), reporting data for 5000 participants. Because it only reports on the first 5000 participants and meanwhile (as of 09 July 2020; US Covid Plasma 2020) enrolled 48,125 participants of which 31,497 received convalescent plasma, we also kept this record as an ongoing study. Perotti 2020 was prospectively registered and analysed 46 participants. The remaining non‐controlled NRSIs were not prospectively registered and reported data for one to 25 participants (Ahn 2020; Jin 2020; Pei 2020; Salazar 2020; Tan 2020; Ye 2020; Zhang 2020a; Zhang 2020b).
Setting
The one RCT and two controlled NRSIs originated from China (Duan 2020; Li 2020; Zeng 2020), and one controlled NRSIs originated from the USA (Liu 2020). Of the 10 additionally included non‐controlled NRSIs that we analysed for safety outcomes, six originated from China (Jin 2020; Pei 2020; Tan 2020; Ye 2020; Zhang 2020a; Zhang 2020b), two originated from the USA (Joyner 2020; Salazar 2020), one from South Korea (Ahn 2020), and one from Italy (Perotti 2020).
Participants
The RCT by Li 2020 and the controlled NRSIs study by Liu 2020 included participants with clinical symptoms meeting the definitions of severe or life‐threatening disease. Duan 2020 transfused convalescent plasma in severely ill individuals. The controlled NRSIs study by Zeng 2020 evaluated critically ill individuals, admitted to ICU.
The majority of the additional studies evaluated for safety outcomes transfused convalescent plasma in critically ill individuals (Ahn 2020; Jin 2020; Joyner 2020; Pei 2020; Perotti 2020; Salazar 2020; Ye 2020; Zhang 2020a; Zhang 2020b). Two of these studies included at least one or more participants with moderate disease severity (Jin 2020; Pei 2020), and one of these studies included one participant with mild disease severity (Ye 2020). One study described one hospitalised participant with moderate disease severity (Tan 2020).
Interventions
All included completed studies evaluated convalescent plasma. We did not identify any completed studies evaluating hyperimmune immunoglobulin (IgG).
In all of the controlled studies evaluated for efficacy and safety outcomes, the dose and volume of convalescent plasma transfused varied. Li 2020 randomised participants into two groups ‐ the convalescent plasma group received one or more doses of 4 mL/kg to 13 mL/kg per recipient body weight with a median volume of 200 mL (interquartile range (IQR) 200 mL to 300 mL) transfused alongside standard therapy (which included antivirals, antibiotics, standard immunoglobulin, Chinese herbal medications, steroids, interferon) and the control group received standard therapy without convalescent plasma. Only convalescent plasma units with an receptor‐binding domain (RBD) of S protein (S‐RBD)–specific IgG titre of at least 1:640, correlating to serum antibody neutralisation titre of 1:80, were used for the study.
Duan 2020 transfused one dose of 200 mL of convalescent plasma alongside standard therapy (which included antivirals, antibiotics, antifungals, steroids) and compared to historic controls matched for age, gender and disease severity who received standard therapy. They evaluated neutralising activity against SARS‐CoV‐2 in these plasma units by classical plaque reduction test using a recently isolated viral strain with an antibody cut‐off titre of over 1:160.
Liu 2020 was a matched cohort study that retrospectively compared 39 participants, who were transfused two doses of 250 mL of convalescent plasma alongside standard therapy (which included antivirals, antibiotics, steroids, stem cells, hydroxychloroquine and immunomodulatory agents) to matched controls using a propensity score. They performed calendar period matching on the following variables: administration of hydroxychloroquine and azithromycin; intubation status and duration; length of hospital stay; and oxygen requirement on the day of transfusion. They matched control patients to plasma recipients by length of stay prior to transfusion and measured antibody titre using a two‐step Spike protein‐directed ELISA (enzyme‐linked immunosorbent assay) with a target anti‐spike titre of at least 1:320 dilution.
Zeng 2020 was a matched cohort study that transfused six participants one to two doses of convalescent plasma (median 300 mL each dose, range 200mL to 600 mL) alongside standard therapy (which included antivirals, antibiotics, steroids, hydroxychloroquine) and compared this group retrospectively to matched controls. Gold immunochromatography for SARS‐CoV‐2 IgM and IgG tests were performed using blood samples, however they did not report any antibody titres.
In the non‐controlled NRSIs that we evaluated for safety outcomes, dose and volume of plasma also varied greatly. The total volume of convalescent plasma transfused varied between 200 mL and 2400 mL, with participants receiving between one to eight doses of plasma. Five studies reported antibody titres (Jin 2020; Pei 2020; Perotti 2020; Salazar 2020; Zhang 2020b). Two studies reported neutralising antibody titres (Jin 2020; Perotti 2020).
Of the above studies, only eight reported some information on plasma donors (Ahn 2020; Jin 2020; Li 2020; Pei 2020; Perotti 2020; Salazar 2020; Ye 2020; Zhang 2020b). Six studies reported the gender of donors ‐ of these, five included both male and female donors (Li 2020; Pei 2020; Perotti 2020; Salazar 2020; Zhang 2020b), but most of these studies excluded prior pregnancy or tested for HLA and/or HNA antibodies except for Zhang 2020b. However, Pei 2020 included one female donor with a previous history of pregnancy.
Some studies provided information on previously reported symptoms and disease severity of convalescent plasma donors (Ahn 2020; Duan 2020; Salazar 2020; Zhang 2020b). Ahn 2020 reported that the two included donors had been admitted to hospital with fever, cough and pneumonia. Duan 2020 reported that donors had been admitted to hospital, but no other information on severity of illness was available. Salazar 2020 reported that all donors were symptomatic. Zhang 2020b reported that all six donors had fever and cough during the course of disease and were admitted to the hospital.
In the seven studies that reported assessment of donor recovery, all donors were symptom‐free and completely recovered from coronavirus disease 2019 (COVID‐19) prior to donating plasma (Ahn 2020; Duan 2020; Li 2020; Pei 2020; Salazar 2020; Ye 2020; Zhang 2020b). Seven studies specified that donors had a negative SARS‐CoV‐2 reverse transcription polymerase chain reaction (RT‐PCR) test prior to convalescent plasma donation (Duan 2020; Jin 2020; Li 2020; Perotti 2020; Salazar 2020; Ye 2020; Zhang 2020b). It was not always clear on what kind of specimen the RT‐PCR test had been performed; three studies reported that the tests were performed on upper respiratory tract swabs (Li 2020; Perotti 2020; Zhang 2020b), one study reported that the test was performed on sputum (Duan 2020), whereas three did not report information on the origin of the donor sample (Jin 2020; Salazar 2020; Ye 2020). Ye 2020 and Zhang 2020b reported that an RT‐PCR test had also been performed on the convalescent plasma product, in addition to RT‐PCR testing of the donor.
Outcomes
We evaluated efficacy and safety outcomes in one RCT and three controlled NRSIs. In Li 2020, the primary outcome was time to clinical improvement within 28 days, defined as patient discharged alive or reduction of 2 points on a 6‐point disease severity scale ranging from 1 (discharge) to 6 (death). Secondary outcomes were 28‐day mortality, time to hospital discharge and clearance of viral PCR results within 72 hours.
In Duan 2020, primary outcomes were safety. Secondary outcomes included improvement of clinical symptoms, radiological and laboratory parameters within three days of transfusion. In Liu 2020, primary outcomes reported were supplemental oxygen requirements and survival at days 1, 7, 14 post‐transfusion. In Zeng 2020, the primary outcome was survival and secondary outcomes were clearance of viral PCR and radiological improvement.
We evaluated safety outcomes in all studies that reported these outcomes. Twelve studies reported assessment of adverse events of possibly grade 3 or grade 4 severity (Ahn 2020; Duan 2020; Jin 2020; Li 2020; Liu 2020; Pei 2020; Perotti 2020; Salazar 2020; Tan 2020; Ye 2020; Zeng 2020; Zhang 2020b). Zhang 2020a reported that no adverse event had been observed for one of their participants. It was unclear whether the other participants experienced any adverse events.
Fourteen studies (5201 participants) assessed and reported serious adverse events (Ahn 2020; Duan 2020; Jin 2020; Joyner 2020; Li 2020; Liu 2020; Pei 2020; Perotti 2020; Salazar 2020; Tan 2020; Ye 2020; Zeng 2020; Zhang 2020a; Zhang 2020b).
Please refer to the Characteristics of included studies for more detailed information.
Ongoing studies
Of the 98 ongoing studies, six are expanded access studies from the USA (NCT04338360; NCT04358211; NCT04360486; NCT04363034; NCT04372368; NCT04374370). For the NCT04338360 study, safety data of 5000 participants have been reported (Joyner 2020). However, as Joyner 2020 only reports on the first 5000 participants, and 48,125 participants (of whom 31,497 received convalescent plasma), have been enrolled in the study as of 9 July 2020 (US Covid Plasma 2020), we decided to treat this record as an ongoing study.
50 are RCTs (ChiCTR2000030010; ChiCTR2000030179; ChiCTR2000030627; ChiCTR2000030702; ChiCTR2000030929; EUCTR2020‐001310‐38; IRCT20200310046736N1; IRCT20200404046948N1; IRCT20200409047007N1; IRCT20200413047056N1; NCT04332835; NCT04333251; NCT04342182; NCT04344535; NCT04345289; NCT04345523; NCT04345991; NCT04346446; NCT04348656; NCT04355767; NCT04356534; NCT04358783; NCT04359810; NCT04361253; NCT04362176; NCT04364737; NCT04366245; NCT04372979; NCT04373460; NCT04374487; NCT04374526; NCT04375098; NCT04376788; NCT04377568; NCT04380935; NCT04381858; NCT04381936; NCT04383535; NCT04385043; NCT04385186; NCT04385199; NCT04388410; NCT04390503; NCT04391101; NCT04392414; NCT04393727; NCT04395170; NCT04397757; NCT04403477; NCT04405310).
Of these, 28 are expected to be completed in 2020 (ChiCTR2000030010; ChiCTR2000030179; ChiCTR2000030627; ChiCTR2000030702; ChiCTR2000030929; IRCT20200310046736N1; IRCT20200404046948N1; IRCT20200409047007N1; IRCT20200413047056N1; NCT04332835; NCT04342182; NCT04345523; NCT04345991; NCT04346446; NCT04348656; NCT04356534; NCT04376788; NCT04380935; NCT04381858; NCT04383535; NCT04385186; NCT04385199; NCT04388410; NCT04392414; NCT04393727; NCT04397757; NCT04403477; NCT04405310), and plan to evaluate between 15 and 1200 participants. Of these studies, five RCTs were scheduled to be completed by the time of writing (ChiCTR2000030010; ChiCTR2000030179; ChiCTR2000030627; NCT04345991; NCT04376788), but results are not published yet and study investigators did not reply to our requests.
Four further, large RCTs are planned to be completed in 2021: NCT04344535 and NCT04362176, each randomising 500 participants, NCT04345289, evaluating 1500 participants and NCT04381936 randomising 12,000 participants to six different treatment options (lopinavir‐ritonavir, corticosteroid, hydroxychloroquine, azithromycin, tocilizumab and convalescent plasma).
Please refer to Characteristics of ongoing studies and to Table 5 for more detailed information.
2. Summary: design and planned completion date of ongoing studies.
| Study ID | Title | Link | Design | Planned number of participants | Planned completion date | Completed/terminated | Results available | Other study ID |
| ChiCTR2000029850 | Efficacy and safety of convalescent plasma treatment for severe patients with novel coronavirus pneumonia (COVID‐19): a prospective cohort study | www.chictr.org.cn/showproj.aspx?proj=49533 | Controlled NRSI | 20 | 15 February 2022 | |||
| ChiCTR2000030010 | A randomized, double‐blind, parallel‐controlled, trial to evaluate the efficacy and safety of anti‐SARS‐CoV‐2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia patients (COVID‐19) | www.chictr.org.cn/showproj.aspx?proj=49777 | RCT | 100 | 31 May 2020 | |||
| ChiCTR2000030039 | Clinical study for infusing convalescent plasma to treat patients with new coronavirus pneumonia (COVID‐19) | www.chictr.org.cn/showproj.aspx?proj=49544 | Controlled NRSI | 60 | 1 February 2020 | |||
| ChiCTR2000030179 | Experimental study of novel coronavirus pneumonia rehabilitation plasma therapy severe novel coronavirus pneumonia (COVID‐19) | www.chictr.org.cn/showproj.aspx?proj=50059 | RCT | 100 | 24 February 2020 | |||
| ChiCTR2000030627 | Study on the application of convalescent plasma therapy in severe COVID‐19 | www.chictr.org.cn/showproj.aspx?proj=50727 | RCT | 30 | 30 May 2020 | |||
| ChiCTR2000030702 | Convalescent plasma for the treatment of common COVID‐19: a prospective RCT | www.chictr.org.cn/showproj.aspx?proj=50537 | RCT | 30 | 15 August 2020 | |||
| ChiCTR2000030929 | A randomized, double‐blind, parallel‐controlled trial to evaluate the efficacy and safety of anti‐SARS‐CoV‐2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia (COVID‐19) | www.chictr.org.cn/showproj.aspx?proj=50696 | RCT | 30 | 16 June 2020 | |||
| ChiCTR2000031501 | The efficacy of convalescent plasma in patients with critical novel coronavirus pneumonia (COVID‐19): a pragmatic, prospective cohort study | www.chictr.org.cn/showproj.aspx?proj=50254 | Controlled NRSI | 20 | 17 July 2020 | |||
| EUCTR2020‐001310‐38 | A randomized, prospective, open label clinical trial on the use of convalescent plasma compared to best supportive care in patients with severe COVID‐19 | www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2020-001310-38 | RCT | 106 | NR | |||
| IRCT20151228025732N53 | Therapeutic effects of plasma of recovered people from COVID‐19 on hospitalized patients with this disease | en.irct.ir/trial/46931 | Controlled NRSI | 12 | 20 June 2020 | |||
| IRCT20200310046736N1 | Comparison of the therapeutic effect of convalescent plasma and plasma‐derived immunoglobulin‐enriched solution on COVID‐19 patients | en.irct.ir/trial/46424 | RCT | 45 | 24 July 2020 | |||
| IRCT20200325046860N1 | Convalescent plasma therapy for COVID‐19 patients | en.irct.ir/trial/46759 | Non‐controlled NRSI | 200 | 20 August 2020 | |||
| IRCT20200404046948N1 | Efficacy and safety of convalescent plasma in the treatment of COVID‐19 | en.irct.ir/trial/46973 | RCT | 60 | 20 June 2020 | |||
| IRCT20200409047007N1 | Effect of COVID 19 survivors plasma in COVID 19 patients with ARDS | en.irct.ir/trial/47058 | RCT | 32 | 15 August 2020 | |||
| IRCT20200413047056N1 | Comparison between the efficacy of intravenous immunoglobulin and convalescent plasma in COVID‐19 | en.irct.ir/trial/47212 | RCT | 15 | 19 June 2020 | |||
| NCT04264858 | An exploratory clinical study on the treatment of acute severe 2019‐nCoV pneumonia with immunoglobulin from cured 2019‐nCoV pneumonia patients | clinicaltrials.gov/show/NCT04264858 | Non‐controlled NRSI | 10 | 31 May 2020 | ChiCTR2000030841 | ||
| NCT04292340 | The efficacy and safety of anti‐SARS‐CoV‐2 inactivated convalescent plasma in the treatment of novel coronavirus pneumonia patient (COVID‐19): an observational study | clinicaltrials.gov/show/NCT04292340 | Non‐controlled NRSI | 15 | 31 July 2020 | |||
| NCT04327349 | Investigating effect of convalescent plasma on COVID‐19 patients outcome: a clinical trial | clinicaltrials.gov/show/NCT04327349 | Non‐controlled NRSI | 30 | 30 September 2020 | |||
| NCT04332380 | Convalescent plasma for patients with COVID‐19: a pilot study | clinicaltrials.gov/show/NCT04332380 | Non‐controlled NRSI | 10 | 31 December 2020 | |||
| NCT04332835 | Convalescent plasma for patients with COVID‐19: a randomized, open label, parallel, controlled clinical study | clinicaltrials.gov/show/NCT04332835 | RCT | 80 | 31 December 2020 | |||
| NCT04333251 | Evaluating convalescent plasma to decrease coronavirus associated complications. A phase I study comparing the efficacy and safety of high‐titer anti‐Sars‐CoV‐2 plasma vs best supportive care in hospitalized patients with interstitial pneumonia due to COVID‐19 | clinicaltrials.gov/show/NCT04333251 | RCT | 115 | 31 December 2022 | |||
| NCT04333355 | Phase 1 study to evaluate the safety of convalescent plasma as an adjuvant therapy in patients with SARS‐CoV‐2 infection | clinicaltrials.gov/show/NCT04333355 | Non‐controlled NRSI | 20 | 30 Apr 2021 | |||
| NCT04338360 | Expanded access to convalescent plasma for the treatment of patients with COVID‐19 | ClinicalTrials.gov/show/NCT04338360 | Expanded access | NR | NR | Preprint, subset of data | ||
| NCT04340050 | COVID‐19 convalescent plasma | ClinicalTrials.gov/show/NCT04340050 | Non‐controlled NRSI | 10 | 31 December 2021 | |||
| NCT04342182 | Convalescent plasma as therapy for Covid‐19 severe SARS‐CoV‐2 disease (CONCOVID Study) (ConCoVid‐19 | ClinicalTrials.gov/show/NCT04342182 | RCT | 426 | 1 July 2020 | |||
| NCT04343261 | Convalescent plasma in the treatment of COVID 19 | ClinicalTrials.gov/show/NCT04343261 | Non‐controlled NRSI | 15 | 1 April 2021 | |||
| NCT04343755 | Convalescent plasma as treatment for hospitalized subjects with COVID‐19 infection | ClinicalTrials.gov/show/NCT04343755 | Non‐controlled NRSI | 55 | 1 April 2021 | |||
| NCT04344535 | Convalescent plasma vs. standard plasma for COVID‐19 | ClinicalTrials.gov/show/NCT04344535 | RCT | 500 | 31 August 2021 | |||
| NCT04345289 | Efficacy and safety of novel treatment options for adults with COVID‐19 pneumonia (CCAP) | ClinicalTrials.gov/show/NCT04345289 | RCT | 1500 | 15 June 2021 | EUCTR2020‐001367‐88 | ||
| NCT04345523 | Convalescent plasma therapy vs. SOC for the treatment of COVID19 in hospitalized patients (ConPlas‐19) | ClinicalTrials.gov/show/NCT04345523 | RCT | 278 | 1 July 2020 | |||
| NCT04345679 | Anti COVID‐19 convalescent plasma therapy | ClinicalTrials.gov/show/NCT04345679 | Non‐controlled NRSI | 20 | 1 April 2021 | |||
| NCT04345991 | Efficacy of convalescent plasma to treat COVID‐19 patients, a nested trial in the CORIMUNO‐19 cohort | ClinicalTrials.gov/show/NCT04345991 | RCT | 120 | 1 June 2020 | |||
| NCT04346446 | Efficacy of convalescent plasma therapy in severely sick COVID‐19 patients | ClinicalTrials.gov/show/NCT04346446 | RCT | 20 | 20 June 2020 | |||
| NCT04346589 | Convalescent antibodies infusion in critically ill COVID 19 patients | ClinicalTrials.gov/ct2/show/NCT04346589 | Non‐controlled NRSI | 10 | 1 July 2020 | |||
| NCT04347681 | Potential efficacy of convalescent plasma to treat severe COVID‐19 and patients at high risk of developing severe COVID‐19 | ClinicalTrials.gov/show/NCT04347681 | Non‐controlled NRSI | 40 | 11 April 2021 | |||
| NCT04348656 | Convalescent plasma for hospitalized adults with COVID‐19 respiratory illness (CONCOR‐1) | ClinicalTrials.gov/show/NCT04348656 | RCT | 1200 | 31 December 2020 | |||
| NCT04348877 | Plasma rich antibodies from recovered patients from COVID19 | ClinicalTrials.gov/show/NCT04348877 | Non‐controlled NRSI | 20 | 1 December 2020 | |||
| NCT04352751 | Experimental use of convalescent plasma for passive immunization in current COVID‐19 pandemic in Pakistan in 2020 | ClinicalTrials.gov/show/NCT04352751 | Non‐controlled NRSI | 2000 | 1 April 2021 | |||
| NCT04353206 | Convalescent plasma in ICU patients with COVID‐19‐induced respiratory failure | ClinicalTrials.gov/show/NCT04353206 | Non‐controlled NRSI | 90 | 1 May 2021 | |||
| NCT04354831 | A study evaluating the efficacy and safety of high‐titer anti‐SARS‐CoV‐2 plasma in hospitalized patients with COVID‐19 infection | ClinicalTrials.gov/ct2/show/NCT04354831 | Non‐controlled NRSI | 106 | 1 May 2023 | |||
| NCT04355767 | Convalescent plasma vs. placebo in emergency room patients with COVID‐19 | ClinicalTrials.gov/ct2/show/NCT04355767 | RCT | 206 | 1 December 2022 | |||
| NCT04355897 | CoVID‐19 plasma in treatment of COVID‐19 patients | ClinicalTrials.gov/ct2/show/NCT04355897 | Non‐controlled NRSI | 100 | 1 August 2020 | |||
| NCT04356482 | Convalescent plasma for ill patients by COVID‐19 | ClinicalTrials.gov/show/NCT04356482 | Non‐controlled NRSI | 90 | 1 December 2020 | |||
| NCT04356534 | Convalescent plasma trial in COVID ‐19 patients | ClinicalTrials.gov/show/NCT04356534 | RCT | 40 | 30 June 2020 | |||
| NCT04357106 | COPLA study: treatment of severe forms of coronavirus infection with convalescent plasma | ClinicalTrials.gov/show/NCT04357106 | Non‐controlled NRSI | 10 | 1 August 2020 | |||
| NCT04358211 | Expanded access to convalescent plasma to treat and prevent pulmonary complications associated with COVID‐19 | ClinicalTrials.gov/show/NCT04358211 | Expanded access | NR | NR | |||
| NCT04358783 | Convalescent plasma compared to the best available therapy for the treatment of SARS‐CoV‐2 pneumonia | ClinicalTrials.gov/show/NCT04358783 | RCT | 30 | 30 May 2020 | |||
| NCT04359810 | Plasma therapy of COVID‐19 in critically ill patients | ClinicalTrials.gov/show/NCT04359810 | RCT | 105 | 1 April 2021 | |||
| NCT04360486 | Treatment of COVID‐19 with Anti‐Sars‐CoV‐2 convalescent plasma (ASCoV2CP) | ClinicalTrials.gov/show/NCT04360486 | Expanded access | NR | NR | |||
| NCT04361253 | Evaluation of SARS‐CoV‐2 (COVID‐19) antibody‐containing plasma therapy | ClinicalTrials.gov/show/NCT04361253 | RCT | 220 | 1 December 2021 | |||
| NCT04362176 | Passive immunity trial of Nashville II | ClinicalTrials.gov/show/NCT04362176 | RCT | 500 | 1 April 2021 | |||
| NCT04363034 | Arkansas expanded access COVID‐19 convalescent plasma treatment program | ClinicalTrials.gov/ct2/show/NCT04363034 | Expanded access | NR | NR | |||
| NCT04364737 | Convalescent plasma to limit COVID‐19 complications in hospitalized patients | ClinicalTrials.gov/show/NCT04364737 | RCT | 300 | 30 April 2023 | |||
| NCT04365439 | Convalescent plasma for COVID‐19 | ClinicalTrials.gov/show/NCT04365439 | Non‐controlled NRSI | 10 | 30 June 2020 | |||
| NCT04366245 | Clinical trial to evaluate the efficacy of treatment with hyperimmune plasma obtained from convalescent antibodies of COVID‐19 infection | ClinicalTrials.gov/show/NCT04366245 | RCT | 72 | 1 December 2021 | |||
| NCT04372368 | Convalescent plasma for the treatment of patients with COVID‐19 | ClinicalTrials.gov/show/NCT04372368 | Expanded access | NR | NR | |||
| NCT04372979 | Efficacy of convalescent plasma therapy in the early care of COVID‐19 patients | ClinicalTrials.gov/show/NCT04372979 | RCT | 80 | 1 May 2021 | |||
| NCT04373460 | Convalescent plasma to limit SARS‐CoV‐2 associated complications | ClinicalTrials.gov/show/NCT04373460 | RCT | 1344 | 31 January 2023 | |||
| NCT04374370 | SARSCoV2 (COVID‐19) convalescent plasma (CP) expanded access protocol (EAP) | ClinicalTrials.gov/show/NCT04374370 | Expanded access | NR | NR | |||
| NCT04374487 | A phase II, open label, RCT to assess the safety and efficacy of convalescent plasma to limit COVID‐19 associated complications | ClinicalTrials.gov/show/NCT04374487 | RCT | 100 | 9 May 2021 | |||
| NCT04374526 | Early transfusion of convalescent plasma in elderly COVID‐19 patients to prevent disease progression | ClinicalTrials.gov/show/NCT04374526 | RCT | 182 | 30 June 2021 | |||
| NCT04374565 | Convalescent plasma for treatment of COVID‐19 patients with pneumonia | ClinicalTrials.gov/show/NCT04374565 | Non‐controlled NRSI | 29 | 5 April 2021 | |||
| NCT04375098 | Efficacy and safety of early COVID‐19 convalescent plasma in patients admitted for COVID‐19 infection | ClinicalTrials.gov/show/NCT04375098 | RCT | 30 | 1 December 2021 | |||
| NCT04376034 | Convalescent plasma collection and treatment in pediatrics and adults | ClinicalTrials.gov/show/NCT04376034 | Non‐controlled NRSI | 240 | 30 Mar 2021 | |||
| NCT04376788 | Exchange transfusion versus plasma from convalescent patients with methylene blue in patients with COVID‐19 | ClinicalTrials.gov/show/NCT04376788 | RCT | 15 | 1 June 2020 | |||
| NCT04377568 | Efficacy of human coronavirus‐immune convalescent plasma for the treatment of COVID‐19 disease in hospitalized children | ClinicalTrials.gov/show/NCT04377568 | RCT | 100 | 1 May 2022 | |||
| NCT04377672 | Human convalescent plasma for high risk children exposed or infected with SARS‐CoV‐2 | ClinicalTrials.gov/show/NCT04377672 | Non‐controlled NRSI | 30 | 18 May 2022 | |||
| NCT04380935 | Effectiveness and safety of convalescent plasma therapy on COVID‐19 patients with acute respiratory distress syndrome | ClinicalTrials.gov/show/NCT04380935 | RCT | 60 | 31 August 2020 | |||
| NCT04381858 | Convalescent plasma vs human immunoglobulin to treat COVID‐19 pneumonia | ClinicalTrials.gov/show/NCT04381858 | RCT | 500 | 30 September 2020 | |||
| NCT04381936 | Randomised evaluation of COVID‐19 therapy (RECOVERY) | ClinicalTrials.gov/ct2/show/NCT04381936 | RCT | 12000 | 30 June 2021 | ISRCTN50189673 | ||
| NCT04383535 | Convalescent plasma and placebo for the treatment of COVID‐19 severe pneumonia | ClinicalTrials.gov/show/NCT04383535 | RCT | 333 | 20 August 2020 | |||
| NCT04383548 | Clinical study for efficacy of anti‐corona VS2 immunoglobulins prepared from COVID19 convalescent plasma prepared by VIPS mini‐pool IVIG medical devices in prevention of SARS‐CoV‐2 infection in high risk groups as well as treatment of early cases of COVID | ClinicalTrials.gov/show/NCT04383548 | Non‐controlled NRSI | 100 | 1 January 2021 | |||
| NCT04384497 | Convalescent plasma for treatment of COVID‐19: an exploratory dose identifying study | ClinicaltTrials.gov/show/NCT04384497 | Non‐controlled NRSI | 50 | 1 December 2020 | |||
| NCT04384588 | COVID19‐convalescent plasma for treating patients with active symptomatic COVID 19 infection (FALP‐COVID) | ClinicalTrials.gov/show/NCT04384588 | Controlled NRSI | 400 | 6 April 2021 | |||
| NCT04385043 | Hyperimmune plasma in patients with COVID‐19 severe infection | ClinicalTrials.gov/show/NCT04385043 | RCT | 400 | 15 May 2021 | |||
| NCT04385186 | Inactivated convalescent plasma as a therapeutic alternative in patients with CoViD‐19 | ClinicalTrials.gov/show/NCT04385186 | RCT | 60 | 30 November 2020 | |||
| NCT04385199 | Convalescent plasma for patients with COVID‐19 | ClinicalTrials.gov/show/NCT04385199 | RCT | 30 | 1 August 2020 | |||
| NCT04388410 | Safety and efficacy of convalescent plasma transfusion for patients with SARS‐CoV‐2 infection | ClinicalTrials.gov/show/NCT04388410 | RCT | 250 | 31 December 2020 | |||
| NCT04388527 | COVID‐19 convalescent plasma for mechanically ventilated population | ClinicalTrials.gov/show/NCT04388527 | Non‐controlled NRSI | 50 | 30 September 2020 | |||
| NCT04389710 | Convalescent plasma for the treatment of COVID‐19 | ClinicalTrials.gov/show/NCT04389710 | Non‐controlled NRSI | 100 | 14 April 2021 | |||
| NCT04389944 | Amotosalen‐ultraviolet a pathogen‐inactivated convalescent plasma in addition to best supportive care and antiviral therapy on clinical deterioration in adults presenting with moderate to severe COVID‐19 | ClinicalTrials.gov/show/NCT04389944 | Non‐controlled NRSI | 15 | 30 June 2020 | |||
| NCT04390178 | Convalescent plasma as treatment for acute coronavirus disease (COVID‐19) | ClinicalTrials.gov/show/NCT04390178 | Non‐controlled NRSI | 10 | 1 December 2020 | |||
| NCT04390503 | Convalescent plasma for COVID‐19 close contacts | ClinicalTrials.gov/ct2/show/NCT04390503 | RCT | 200 | 1 April 2021 | |||
| NCT04391101 | Convalescent plasma for the treatment of severe SARS‐CoV‐2 (COVID‐19) | ClinicalTrials.gov/show/NCT04391101 | RCT | 231 | 31 December 2021 | |||
| NCT04392232 | A phase 2 study of COVID 19 convalescent plasma in high risk patients with COVID 19 infection | ClinicalTrials.gov/show/NCT04392232 | Non‐controlled NRSI | 100 | 31 December 2020 | |||
| NCT04392414 | Hyperimmune convalescent plasma in moderate and severe COVID‐19 disease | ClinicalTrials.gov/show/NCT04392414 | RCT | 60 | 15 September 2020 | |||
| NCT04393727 | Transfusion of convalescent plasma for the early treatment of pneumonia due to SARSCoV2 | ClinicalTrials.gov/show/NCT04393727 | RCT | 126 | 30 August 2020 | |||
| NCT04395170 | Convalescent plasma compared to anti‐COVID‐19 human immunoglobulin and standard treatment (TE) in hospitalized patients | ClinicalTrials.gov/show/NCT04395170 | RCT | 75 | 1 June 2021 | |||
| NCT04397523 | Efficacy and safety of COVID‐19 convalescent plasma | ClinicalTrials.gov/show/NCT04397523 | Non‐controlled NRSI | 20 | 29 April 2021 | |||
| NCT04397757 | COVID‐19 convalescent plasma for the treatment of hospitalized patients with pneumonia caused by SARS‐CoV‐2 | ClinicalTrials.gov/show/NCT04397757 | RCT | 80 | 13 November 2020 | |||
| NCT04403477 | Convalescent plasma therapy in severe COVID‐19 infection | ClinicalTrials.gov/show/NCT04403477 | RCT | 20 | 30 October 2020 | |||
| NCT04404634 | Convalescent plasma to limit coronavirus associated complications | ClinicalTrials.gov/show/NCT04404634 | RCT | 300 | 31 January 2023 | |||
| NCT04405310 | Convalescent plasma of Covid‐19 to treat SARS‐COV‐2 a randomized double blind 2 center trial | ClinicalTrials.gov/show/NCT04405310 | RCT | 80 | 20 July 2020 | |||
| NCT04407208 | Convalescent plasma therapy in patients with COVID‐19 | ClinicalTrials.gov/show/NCT04407208 | Non‐controlled NRSI | 10 | 1 August 2020 | |||
| NCT04408040 | Use of convalescent plasma for COVID‐19 | ClinicalTrials.gov/show/NCT04408040 | Non‐controlled NRSI | 700 | 1 June 2022 | |||
| NCT04408209 | Convalescent plasma for the treatment of patients with severe COVID‐19 infection | ClinicalTrials.gov/show/NCT04408209 | Non‐controlled NRSI | 60 | 15 September 2021 | |||
| NCT04412486 | COVID‐19 convalescent plasma (CCP) transfusion | ClinicalTrials.gov/show/NCT04412486 | Non‐controlled NRSI | 100 | 31 May 2022 | |||
| U1111‐1251‐9286 | Effect of convalescent plasma in patients with severe COVID‐19 | www.ensaiosclinicos.gov.br/rg/RBR-4vm3yy/ | Non‐controlled NRSI | 20 | 31 May 2022 | |||
| NR: not reported; RCT: randomised controlled trial; NRSI: non‐randomised study of intervention | ||||||||
Excluded studies
We excluded 62 references that did not match our inclusion criteria.
Thirty‐two were a review of the literature, an editorial, letter or an opinion (Alzoughool 2020; Barone 2020; Bloch 2020; Cao 2020b; Casadevall 2020a; Casadevall 2020b; Chen 2020a; Datta 2020; Dzik 2020; Fleming 2020; Hammarström 2020; Jawhara 2020; Kesici 2020; Khanna 2020; Knudson 2020; Kominers 2020; Kumar 2020; Lanza 2020; Pawar 2020; Roback 2020; Rubin 2020; Seghatchian 2020; Sheridan 2020; Syal 2020; Tanne 2020; Lancet Haematology 2020; Tiberghien 2020; Wong 2020; Yoo 2020; Zeng 2020a; Zhao 2020b; Zhu 2020).
Sixteen studies were performed with an intervention other than convalescent plasma or hyperimmune immunoglobulin (Cao 2020a; Chen 2020b; Chen 2020c; Díez 2020; Hu 2020; ISRCTN86534580; Jiang 2020; Lin 2020; NCT04261426; NCT04344379; NCT04350580; NCT04368013; Robbiani 2020; Shi 2020; Xie 2020; de Assis 2020).
Four studies were cancelled by the investigator before recruiting participants into the study (ChiCTR2000030312; ChiCTR2000030381; ChiCTR2000030442; NCT04325672).
Four studies pertained to feasibility of collection of convalescent plasma only (Budhai 2020; NCT04344015; NCT04344977; NCT04360278).
Three studies reported standard operating procedure related to plasma donation (Brasil Ministerio 2020; Franchini 2020; Ministerio de Salud 2020).
Two references were in Chinese (Qiu 2020; Tu 2020). Both were translated and assessed by Rujan Shrestha and Ya‐Ying Wang via Cochrane TaskExchange. The papers reported on a generalised collection of information about the COVID‐19 infection of two participants relating to aetiology, pathology, symptoms, clinical presentation and some generalised pharmacological treatment methods.
One study included an irrelevant participant population (participants exposed to COVID‐19; NCT04323800).
Risk of bias in included studies
Risk of bias in randomised controlled trials
We assessed methodological quality and risk of bias for one study (Li 2020), using the 'Risk of bias' tool recommended in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
Overall judgement
Overall, we rated the risk of bias to be unclear for mortality outcomes, and outcomes assessing improvement of clinical symptoms (efficacy outcomes), and to be high for safety outcomes. The full judgement for the study per category is presented in Figure 2 and the support for judgement in Appendix 7.
2.
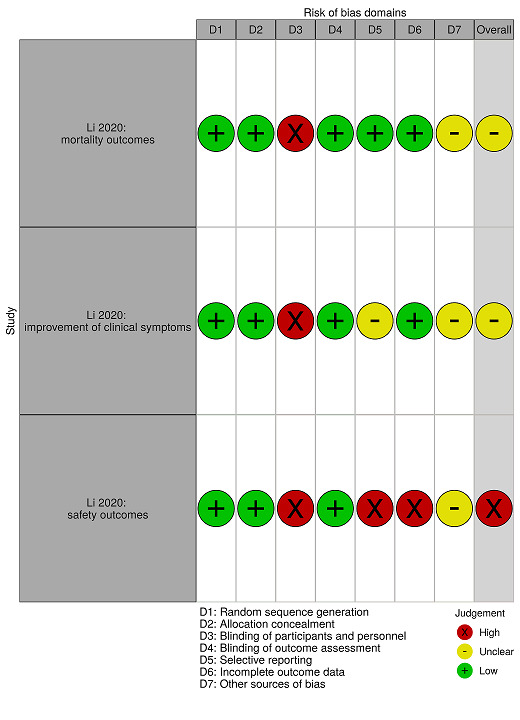
Risk of bias summary for randomised controlled trial
Allocation (selection bias)
We judged the risk of attrition bias to be low, as random sequence generation and allocation concealment was described in detail.
Blinding (performance bias and detection bias)
We judged the risk of performance bias to be high, because the trial was not masked for participants and personnel.
We judged the risk of detection bias to be low, because the study authors reported that the outcome assessors were blinded to the study group allocation.
Selective reporting (reporting bias)
We judged the risk of reporting bias to be low for mortality outcomes because the study authors reported that they determined mortality outcomes at the protocol stage.
We judged the risk of reporting bias to be unclear for outcomes addressing improvement of clinical symptoms, because the comparisons at days 7, 14, and 28 were added as a post hoc analysis.
We judged the risk of reporting bias to be high for safety outcomes, because the study authors only reported transfusion‐related adverse events.
Incomplete outcome data (attrition bias)
We judged the risk of attrition bias to be low for mortality outcomes and outcomes addressing improvement of clinical symptoms, because the study authors reported results for the intention‐to‐treat population.
We judged the risk of attrition bias to be high for safety outcomes, because the study authors reported safety data for the intervention group only.
Other potential sources of bias
The trial was terminated early because no participants could be enrolled to the trial after the containment of the epidemic in Wuhan, China. We are unclear about the potential bias of this pre‐termination.
In addition, we noticed that effect estimates, which are indicated as odds ratios (ORs) in the primary study, are in fact risk ratios (RRs). We are unclear about the potential bias of this incorrect use.
Risk of bias in controlled non‐randomised studies of interventions
We assessed methodological quality and risk of bias for three studies (Duan 2020; Liu 2020; Zeng 2020), using the Risk Of Bias in Non‐randomised Studies ‐ of Interventions (ROBINS‐I) tool (Sterne 2016).
Overall bias
Overall, we rated the risk of bias within and across studies to be critical for all assessed outcomes. Studies are too problematic to provide any useful evidence, however better evidence is not yet available. We present the full judgement per trial and category, including the support for judgement, in Appendix 8; and the 'Risk of bias' summary in Figure 3.
3.
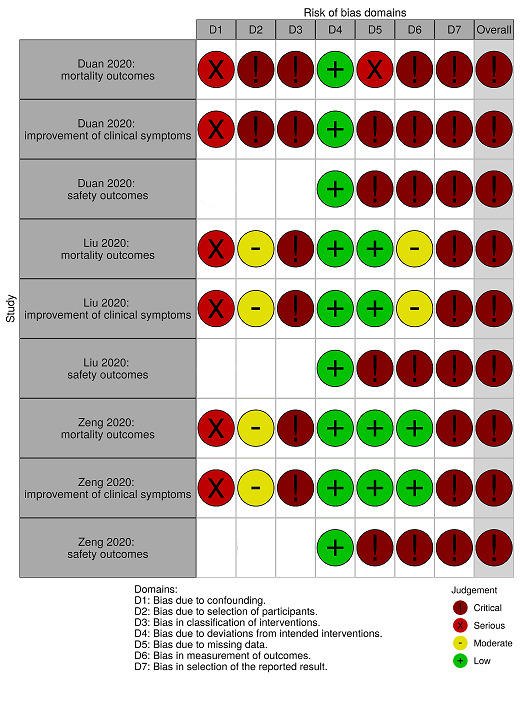
'Risk of bias' summary for controlled non‐randomised studies of interventions
Bias due to confounding
We judged the risk of bias due to confounding to be serious for all studies for mortality outcomes and outcomes addressing improvement of clinical symptoms. Duan 2020 adjusted for age, gender, and disease severity, but did not adjust for important confounding factors, including co‐morbidities, previous treatments and time of disease onset. Liu 2020 adjusted for antiviral treatments, intubation status and duration, length of hospital stay, and oxygen requirement on the day of transfusion, but did not adjust for important confounding factors including age and gender. Zeng 2020 did not adjust for any confounding factors.
Assessment of risk of bias due to confounding was not applicable for all studies for safety outcomes, because they reported adverse events for the intervention group only; either after plasma transfusion or transfusion‐related events only.
Bias in selection of participants into the study
We judged the risk of bias in selection of participants into the study to be critical for Duan 2020 for mortality outcomes and outcomes addressing improvement of clinical symptoms. The study included a small sample size and it was unclear how they selected participants into the intervention group, and for how long they followed up participants of the historical control group.
We judged the risk of bias in selection of participants into the study to be moderate for Liu 2020 and Zeng 2020 for mortality outcomes and outcomes addressing improvement of clinical symptoms. In Liu 2020, selection into the study may have been related to intervention and outcome, but the study authors used appropriate methods to adjust for the selection bias. Zeng 2020 performed allocation to intervention and control group based on donor availability.
Assessment of risk of bias in selection of participants into the study was not applicable for all studies for safety outcomes, because they reported adverse events for the intervention group only; either after plasma transfusion or transfusion‐related events only.
Bias in classification of interventions
We judged the risk of bias in classification of interventions to be critical for all studies for mortality outcomes and outcomes addressing improvement of clinical symptoms, because they assigned participants to the control group retrospectively, and knowledge of patient outcomes at the time of assignment to the control group could have had a major impact on the selection and classification of interventions.
Assessment of risk of bias in classification of interventions was not applicable for all studies for safety outcomes, because they reported adverse events for the intervention group only; either after plasma transfusion or transfusion‐related events only.
Bias due to deviations from intended interventions
We judged the risk of bias due to deviations from intended intervention to be low for all studies and all outcomes, because all assessed participants received the intended interventions.
Bias due to missing data
We judged the risk of bias due to missing data to be serious for Duan 2020 for mortality outcomes, because they reported mortality for participants in the intervention group until day 3 of follow‐up, and it was unclear how long they followed the control group. We judged the risk of bias due to missing data to be low for Liu 2020 and Zeng 2020 for mortality outcomes, because results were reasonably complete.
We judged the risk of bias due to missing data to be critical for Duan 2020 for outcomes addressing improvement of clinical symptoms, because they did not report how long they followed the control group and they did not assess clinical status in terms of respiratory support. We judged the risk of bias due to missing data to be low for Liu 2020 and Zeng 2020 for outcomes addressing improvement of clinical symptoms, because all participants who were still alive had been discharged by the end of follow‐up.
We judged the risk of bias due to missing data to be critical for all studies for safety outcomes, because studies did not report safety data for the control group.
Bias in measurement of outcomes
We judged the risk of bias in measurement of outcomes to be critical for Duan 2020 for mortality outcomes and outcomes addressing improvement of clinical symptoms, because it was unclear whether the follow‐up was comparable between groups.
We judged the risk of bias in measurement of outcomes to be moderate for Liu 2020 for mortality outcomes and outcomes addressing improvement of clinical symptoms, because median follow‐ups were comparable between groups. However, outcome assessors were not blinded to the intervention and the study was performed retrospectively.
We judged the risk of bias in measurement of outcomes to be low for Zeng 2020 for mortality outcomes, and outcomes addressing improvement of clinical symptoms, because all participants were followed until death or discharge.
We judged the risk of bias in measurement of outcomes to be critical for all studies for safety outcomes, because safety data were not reported for the control group.
Bias in selection of the reported results
We judged the risk of bias in selection of the reported results to be critical for all studies and all outcomes, because all studies were performed retrospectively, and the selection of all reported results are likely biased.
Risk of bias in non‐controlled non‐randomised studies of interventions (for safety assessment)
We assessed methodological quality and risk of bias for 10 non‐controlled NRSIs (Ahn 2020; Jin 2020; Joyner 2020; Pei 2020; Perotti 2020; Salazar 2020; Tan 2020; Ye 2020; Zhang 2020a; Zhang 2020b), using the 'Risk of bias' assessment criteria tool for observational studies provided by Cochrane Childhood Cancer (see Table 4; Mulder 2019). We only assessed risk of bias for safety outcomes. We therefore only assessed risk of bias for those non‐controlled NRSIs that reported safety data.
Overall judgement
In addition to the high risk of bias due to the non‐randomised and non‐controlled study design, we rated the overall risk of bias within and across studies to be high. We present the full judgement per trial and category in Figure 4 and the support for judgement in Appendix 9.
4.
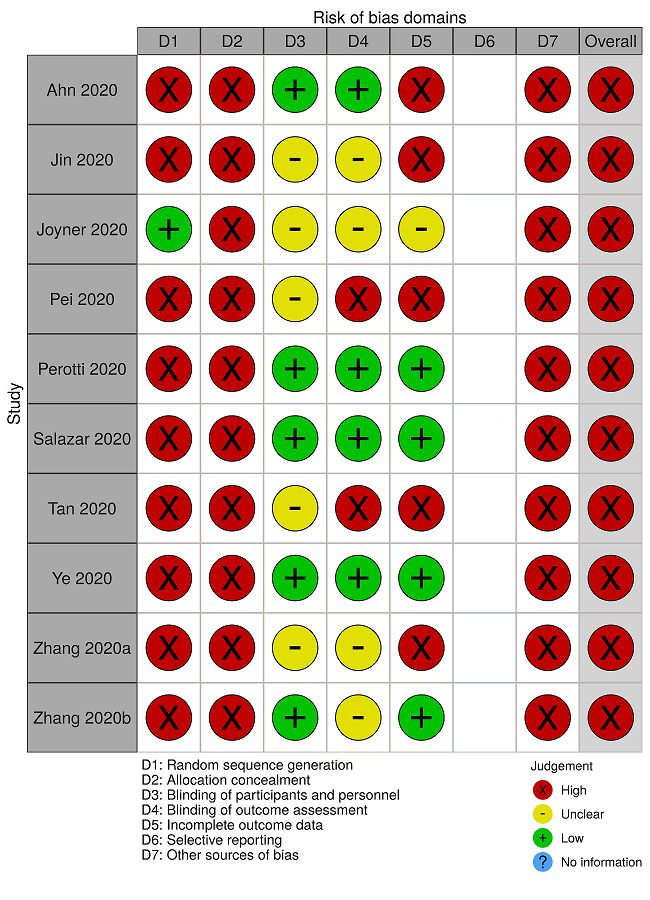
'Risk of bias' summary for non‐controlled non‐randomised studies of interventions (assessing safety data only)
Allocation (selection bias)
Except for one study (Joyner 2020), all studies were at high risk of selection bias. We considered study groups not to be representative, as all studies included low numbers of participants (1 to 46 participants) with no control groups.
We judged risk of selection bias to be low for Joyner 2020 because of the prospective study design, the large population size, and the fact that the first 5000 enrolled participants were considered in this interim analysis.
Blinding (performance bias and detection bias)
All studies were unblinded and therefore at high risk of performance and detection bias.
Incomplete outcome data (attrition bias)
We assessed attrition bias in terms of whether studies (equally) assessed outcomes for all participants.
We judged the risk of attrition bias to be low for five studies (Ahn 2020; Perotti 2020; Salazar 2020; Ye 2020; Zhang 2020b), because they assessed and reported adverse events and symptoms for all participants.
We judged the risk of attrition bias to be unclear for the other five studies (Jin 2020; Joyner 2020; Pei 2020; Tan 2020; Zhang 2020a), because it was unclear whether they had assessed adverse events for all participants or whether they had selectively reported outcomes. Jin 2020 assessed the outcome for all participants, however the observation period was unclear. Joyner 2020 reported preliminary results only and only reported serious adverse events over a four‐hour observation period. Pei 2020 reported one serious adverse event occurring in one participant, however did not report whether they had assessed or observed other adverse events. Tan 2020 reported that their participant experienced moderate fever after the transfusion, however did not report whether other adverse events occurred. Zhang 2020a described that they had observed no adverse events for one of their participants after plasma transfusion, but did not provide any information regarding the occurrence of adverse events for the other participants. They stated in the conclusions that they had not observed any serious adverse events.
Selective reporting (reporting bias)
We assessed reporting bias in terms of whether the study group and intervention were well‐defined and whether the outcomes were equally reported for all participants and the length of follow‐up was mentioned.
Well‐defined study group and intervention
We judged the risk of reporting bias to be low for four studies (Ahn 2020; Perotti 2020; Salazar 2020; Ye 2020), because both the study population and intervention were well described.
Jin 2020, Joyner 2020, and Zhang 2020a described the study population, but reported only limited information on the intervention. Zhang 2020b provided clear information on the intervention, but scarcely described the participant. We therefore judged the risk of reporting bias to be unclear for these four studies.
We judged the risk of bias to be high for two studies (Pei 2020; Tan 2020), which only reported limited information on the study population and the intervention. However, Pei 2020 was a preprint only, and claimed that the participant characteristics would be provided in the supplementary material once published.
Well‐defined outcomes
We judged the risk of reporting bias to be low for four studies (Perotti 2020; Salazar 2020; Ye 2020; Zhang 2020b), because the observation period and results were reported for all participants.
We judged the risk of reporting bias to be unclear for Joyner 2020 because only serious adverse events were reported and results are preliminary.
We judged the risk of reporting bias to be high for the other five studies (Ahn 2020; Jin 2020; Pei 2020; Tan 2020; Zhang 2020a), because it was unclear whether adverse events had been equally assessed for all participants or whether outcomes were selectively reported. Pei 2020 reported one serious adverse event occurring in one participant, however did not report whether they had assessed or observed other adverse events. Tan 2020 reported that their participant experienced moderate fever after the transfusion, however did not report whether other adverse events occurred. Zhang 2020a described they had not observed any adverse events for one of their participants after plasma transfusion, but did not provide any information regarding the occurrence of adverse events for the other participants. They stated in the conclusions that they had not observed any serious adverse events.
Other potential sources of bias
We further considered confounding and poorly‐defined risk estimates as potential sources of bias.
Confounding
All studies were at high risk of confounding because none of the studies adjusted for confounding factors, including concomitant treatments.
Poorly‐defined risk estimates
None of the studies performed any analyses.
Effects of interventions
See: Table 1
Summary of findings 1. Convalescent plasma for people with COVID‐19.
| Convlescent plasma for people with COVID‐19 | ||||||
|
Patients or population: people with COVID‐19 Settings: inpatient Intervention: convalescent plasma transfusion Comparison: no convalescent plasma transfusion | ||||||
| Outcomes and study design | Anticipated absolute effects (95% CI) | Relative effect (95% CI) | No of participants (studies) | Certainty of the evidence | Comments | |
| Control group risk (without convalescent plasma)a | Risk with convalescent plasma | |||||
| All‐cause mortality at hospital discharge | ||||||
| Randomised controlled trials | NA | NA | NA | 0 | NA | |
| Controlled non‐randomised studies of interventions | 933 per 1000 |
830 per 1000 (569 to 1000) |
RR 0.89 (95% CI 0.61 to 1.31) | 21 (1 study) | ⊕⊝⊝⊝ Very lowb,c |
|
| Time to death | ||||||
| Randomised controlled trials (follow‐up 28 days) | 240 per 1000 dead |
184 per 1000 (79 to 393) |
HR 0.74 (95% CI 0.30 to 1.82) | 103 (1 study) | ⊕⊝⊝⊝ Very Lowc,d |
|
| Controlled non‐randomised studies of interventions (follow‐up 11 days) | 243 per 1000 dead |
120 per 1000 (59 to 235) |
HR 0.46, 95% CI 0.22 to 0.96 | 195 (1 study) | ⊕⊝⊝⊝ Very lowb,e |
|
|
Improvement of clinical symptoms, assessed by need for respiratory support Follow‐up: 7 days | ||||||
| Randomised controlled trials at day 7 |
98 per 1000 |
96 per 1000 (29 to 312) |
RR 0.98 (95% CI 0.30 to 3.19) | 103 (1 study) | ⊕⊝⊝⊝ Very Lowc,d |
|
| Controlled non‐randomised studies of interventions | NA | NA | NA | 0 | NA | None of the identified studies reported this outcome. |
|
Improvement of clinical symptoms, assessed by need for respiratory support Follow‐up: 15 days | ||||||
| Randomised controlled trials at day 14 | 176 per 1000 |
326 per 1000 (160 to 663) |
RR 1.85 (95% CI 0.91 to 3.77) | 103 (1 study) | ⊕⊝⊝⊝ Very Lowc,d |
|
| Controlled non‐randomised studies of interventions | 756 per 1000 |
817 per 1000 (688 to 975) |
RR 1.08 (95% CI 0.91 to 1.29) | 195 (1 study) | ⊕⊝⊝⊝ Very lowb,c |
|
|
Improvement of clinical symptoms, assessed by need for respiratory support Follow‐up: 30 days | ||||||
| Randomised controlled trials at day 28 | 431 per 1000 |
523 per 1000 (344 to 780) |
RR 1.20 (95% CI 0.80 to 1.81) | 103 (1 study) | ⊕⊝⊝⊝ Very Lowc,d |
|
| Controlled non‐randomised studies of interventions | NA | NA | NA | 0 | NA | None of the identified studies reported this outcome. |
| Quality of Life | ||||||
| Randomised controlled trials | NA | NA | NA | 0 | NA | None of the identified studies reported this outcome. |
| Controlled non‐randomised studies of interventions | NA | NA | NA | 0 | NA | None of the identified studies reported this outcome. |
| Grade 3 or 4 adverse eventsf | ||||||
| Randomised controlled trials | NR | NR in a comparative design | NA | NA | NA | All included controlled trials reported safety data for the intervention group only, so we included the results here under non‐controlled non‐randomised studies of interventions. |
| Controlled non‐randomised studies of interventions | NR | NR in a comparative design | NA | NA | NA | |
| Non‐controlled non‐randomised studies of interventions | NA | NA | NA | 201 (13 studies) | ⊕⊝⊝⊝ Very lowg,h |
We were unable to summarise numerical data in any meaningful way. We have provided an overview of the reported adverse events for each study in Table 2. Studies did not report the grade of adverse events. The majority of these adverse events were allergic or respiratory events. |
| Serious adverse events | ||||||
| Randomised controlled trials | NR | NR in a comparative design | NA | NA | NA | All included controlled trials reported safety data for the intervention group only, so we included the results here under non‐controlled non‐randomised studies of interventions. |
| Controlled non‐randomised studies of interventions | NR | NR in a comparative design | NA | NA | NA | |
| Non‐controlled non‐randomised studies of interventions | NA | NA | NA | 5201 (14 studies) | ⊕⊝⊝⊝ Very low g,h |
We were unable to summarise numerical data in any meaningful way. An overview of the reported serious adverse events is provided in Table 3 per study. The majority of participants were from one non‐controlled non‐randomised study of intervention (5000 participants), which reported only on serious adverse events limited to the first four hours after convalescent plasma transfusion. This study included death as a serious adverse event; they reported 15 deaths, four of which they classified as potentially, probably or definitely related to transfusion. Other serious adverse events reported in all studies were predominantly allergic or respiratory in nature, including: anaphylaxis; transfusion‐associated dyspnoea; and transfusion‐related acute lung injury (TRALI). |
|
GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. CI: confidence interval; HR: hazard ratio; NA: not available; NR: not recorded; RR: risk ratio | ||||||
aControl group risk extracted from included studies. bRisk of bias within this study is critical, so we downgraded three points for risk of bias. cWe downgraded two points for imprecision because of the very small information size and results including both potential benefit and potential harm. dRisk of bias within this study and for this outcome is unclear, so we downgraded one point for risk of bias. eWe downgraded one point for imprecision because of the very small information size. fWe assume these adverse events are grade 3‐4; not all of the studies reported grading of adverse events. gRisk of bias across studies is high for this outcome, so we downgraded one point for risk of bias. hWe included intervention arms of controlled studies and non‐controlled studies only, so we started assessment from low‐certainty evidence and did not summarise outcome data across studies.
In Table 1, we present certainty of the evidence for our prioritised outcomes (please see 'Summary of findings and assessment of the certainty of the evidence' in Data synthesis).
Effectiveness of convalescent plasma for people with COVID‐19
All‐cause mortality at hospital discharge
Randomised controlled trials
Li 2020 reported 28‐day mortality. As not all participants had been discharged at the end of follow‐up (28 days), we could not analyse all‐cause mortality at hospital discharge.
Controlled non‐randomised studies of interventions
All three controlled NRSIs (236 participants) reported mortality data for the intervention and control group (Duan 2020; Liu 2020; Zeng 2020). However, we were able to evaluate all‐cause mortality at hospital discharge for Zeng 2020 only (21 participants), as not all participants had been discharged at the end of follow‐up in Liu 2020 and hospital discharge was unclear for Duan 2020 (end of follow‐up three days after transfusion).
Zeng 2020 reported that five out of six participants in the intervention group died, and that 14 out of 15 participants in the control group died. One participant from each group was discharged (RR* 0.89, 95% CI 0.61 to 1.31; very low‐certainty evidence). The evidence is very uncertain whether there is a difference between patients receiving convalescent plasma or not.
*We calculated the effect estimate with the reported outcome data. We did not adjust for any confounding factors.
Time to death
Randomised controlled trials
Li 2020 (103 participants) suggests that compared to the control group, convalescent plasma may prolong time from randomisation to death but the evidence is very uncertain (HR* 0.74, 95% CI 0.30 to 1.82; very low‐certainty evidence).
*The study authors calculated effect estimates. HRs were calculated using unadjusted Cox proportional hazards models.
Subgroup analysis: severity of disease
The study authors reported subgroup analyses for participants with severe disease and participants with life‐threatening disease. No participant with severe disease died in the convalescent plasma arm, therefore the study authors could not calculate a HR. For participants with life‐threatening disease, the evidence is uncertain whether convalescent plasma therapy prolongs time to death (HR* 0.86, 95% CI 0.34 to 2.41).
*The study authors calculated effect estimates. HRs were calculated using Cox proportional hazards models adjusted for disease severity.
Controlled non‐randomised studies of interventions
Liu 2020 (195 participants) reported time to death after a median follow‐up time of 11 days for the convalescent plasma group and nine days for the control group. Convalescent plasma may prolong time to death, but the evidence is very uncertain (HR* 0.46, 95% CI 0.22 to 0.96; very low‐certainty evidence).
*We calculated the effect estimate with the reported outcome data using the Parmar and Tierney approach (Tierney 2007), as described in the Measures of treatment effect. We did not adjust for any confounding factors, however the study population was a 1:4 matched‐sample and adjusted for duration of symptoms prior to admission, therapeutic anticoagulant, broad‐spectrum antibiotics, and antivirals.
Subgroup analysis: severity of disease
We identified a significant subgroup difference (test for interaction P = 0.05) for non‐intubated participants, favouring the convalescent plasma transfusion arm (HR* 0.19, 95% CI 0.05 to 0.72) and no evidence of a difference for participants who were intubated (HR* 1.24, 95% CI 0.33 to 4.67).
*The study authors calculated effect estimates (Liu 2020). HRs were calculated for a 1:4 matched‐sample and adjusted for duration of symptoms prior to admission, therapeutic anticoagulant, broad‐spectrum antibiotics, and antivirals
Improvement of clinical symptoms (assessed by need for respiratory support)
Randomised controlled trials
Li 2020 reported this outcome for 103 participants. However, the definition of the outcome differed from the one we used. The study authors defined clinical improvement as discharged or a reduction of 2 points on a 6‐point disease severity scale:
6 points death
5 points hospitalisation plus extracorporeal membrane oxygenation (ECMO) or invasive mechanical ventilation
4 points hospitalisation plus noninvasive ventilation or high‐flow supplemental oxygen
3 points hospitalisation plus supplemental oxygen (not high‐flow or noninvasive ventilation)
2 points hospitalisation with no supplemental oxygen
1 point hospital discharge
Improvement of clinical symptoms was reported at days 7, 14, and 28 (see Table 2). The study authors added a post hoc analysis to compare the rates of improvement at these days.
3. Improvement of clinical symptoms (assessed by need for respiratory support) .
| Study | Number of participants | Baseline | At day 7 | At day 15 | Up to day 30 | From baseline to longest follow‐up | ||||||
| Intervention group | Control group | Intervention group | Control group | Intervention group | Control group | Intervention group | Control group | Intervention group | Control group | Intervention group | Control group | |
| Randomised controlled trials (RCTs) | ||||||||||||
| Li 2020 | 52 | 51 |
|
|
5/52 improved (9.6%) | 5/51 improved (9.8%) | 17/52 improved (32.7%) | 9/51 improved (17.6%) | 27/52 improved (51.9%) | 22/51 improved (43.1%) | After 28 days: 27/52 improved (51.9%) |
After 28 days: 22/51 improved (43.1%) |
| Controlled non‐randomised studies of interventions (NRSIs) | ||||||||||||
| Duan 2020 | 10 | 10 |
|
NR |
3/8 participants with clinical improvement
|
NR | NR | NR | NR | NR | Longest follow‐up: 3 days after transfusion (3/8 participants with clinical improvement)
|
NR |
| Liu 2020 | 39 | 156 |
|
|
NR | NR |
|
|
NR | NR | Median follow‐up time was 11 (1 to 28) days
|
Median follow‐up time was 9 (0 to 31) days
|
| NR | NR |
|
|
NR | NR | |||||||
| Zeng 2020 | 6 | 15 |
|
|
NR | NR |
NR |
NR | NR | NR |
Longest follow‐up: NR
|
Longest follow‐up: NR
|
|
NR |
|
NR |
|
NR |
|||||||
| CP: convalescent plasma; ECMO: extracorporeal membrane oxygenation; NIV: non‐invasive ventilation; NR: not reported | ||||||||||||
The evidence is very uncertain for the effect of convalescent plasma on clinical symptoms, assessed by need for respiratory support at day 7 (RR* 0.98, 95% CI 0.30 to 3.19; very low‐certainty evidence). Convalescent plasma transfusion may increase improvement of clinical symptoms, as assessed by need for respiratory support at 14 days (RR* 1.85, 95% CI 0.91 to 3.77; very low‐certainty evidence), and 28 days (RR* 1.20, 0.80 to 1.81; very low‐certainty evidence), but the evidence is very uncertain.
*We calculated the effect estimate with the reported outcome data (see Analysis 1.1; Analysis 1.2; Analysis 1.3). We did not adjust for any confounding factors.
1.1. Analysis.

Comparison 1: Results from RCT, Outcome 1: Improvement of clinical symptoms at up to 7 days (assessed by need for respiratory support)
1.2. Analysis.

Comparison 1: Results from RCT, Outcome 2: Improvement of clinical symptoms at 8 to 15 days (assessed by need for respiratory support)
1.3. Analysis.

Comparison 1: Results from RCT, Outcome 3: Improvement of clinical symptoms at 16 to 30 days (assessed by need for respiratory support)
Li 2020 expressed effect estimates as odds ratios (ORs), therefore we recalculated relative effects as RRs. We noticed, that our calculation arrived at the same numerical values, and therefore highlight that effect estimates that are indicated as ORs by Li 2020 are in fact RRs.
Subgroup analysis: severity of disease
We did not find any evidence for subgroup differences amongst participants with severe or life‐threatening disease for the three reported time points (at 7, 14 and 28 days, see Analysis 1.4; Analysis 1.5; Analysis 1.6, respectively).
1.4. Analysis.

Comparison 1: Results from RCT, Outcome 4: Improvement of clinical symptoms at up to 7 days (assessed by need for respiratory support): subgroup severity of disease
1.5. Analysis.

Comparison 1: Results from RCT, Outcome 5: Improvement of clinical symptoms at 8 to 15 days (assessed by need for respiratory support): subgroup severity of disease
1.6. Analysis.

Comparison 1: Results from RCT, Outcome 6: Improvement of clinical symptoms at 16 to 30 days (assessed by need for respiratory support): subgroup severity of disease
Controlled non‐randomised studies of interventions
All controlled NRSIs reported this outcome (please see Table 2), however reporting differed across studies.
Liu 2020 assessed improvement of clinical symptoms, as assessed by need for respiratory support at day 14, including 39 participants in the intervention group and 156 participants in the control group. Zeng 2020 reported that one out of six participants in the intervention group and one out of 15 participants in the control group had improved and were discharged at the latest point of follow‐up (time point unclear). It was unclear whether they still needed any respiratory support after discharge. Five out of six participants in the intervention group and 14 out of 15 in the control group had died.
Duan 2020 reported improvement of clinical symptoms, as assessed by need for respiratory support only for the intervention group. They reported a decreased need for respiratory support in four out of 10 participants within three days of convalescent plasma transfusion. One other participant was reported to require only intermittent oxygenation after previously receiving continuous low‐flow oxygenation via nasal cannula. The study also reported on two individuals who required no respiratory support preceding convalescent plasma therapy. No information on improvement of clinical symptoms for other time points was available.
30‐day and 90‐day mortality
Randomised controlled trials
Li 2020 reported no significant difference in 28‐day mortality between both groups (RR* 0.65, 95% CI 0.29 to 1.46). The study authors did not evaluate 90‐day mortality.
*We calculated the effect estimate with the reported outcome data (see Analysis 1.7). We did not adjust for any confounding factors.
1.7. Analysis.

Comparison 1: Results from RCT, Outcome 7: 30‐day mortality
Li 2020 expressed effect estimates as ORs, therefore we recalculated relative effects as RRs. We noticed, that our calculation arrived at the same numerical values, and therefore highlight that effect estimates, which are indicated as ORs by Li 2020, are in fact RRs.
Controlled non‐randomised studies of interventions
None of the controlled NRSIs reported 28‐day mortality.
Time to discharge from hospital
Randomised controlled trials
Li 2020 provided clear criteria for hospital discharge and assessed this outcome by day 28:
body temperature returned to normal for longer than three days
respiratory symptoms improved without the need for oxygen support
two consecutive, negative PCR test results from nasopharyngeal swabs at least 24 hours apart
The median time from randomisation to discharge in the convalescent plasma group was 28 days (IQR 13 to indeterminate) and was not determinable (IQR 19 to indeterminate) in the control group. The results show that compared to standard therapy alone, convalescent plasma therapy may slightly reduce time to discharge (HR* 1.61, 95% CI 0.88 to 2.93).
*The study authors calculated effect estimates. HRs were calculated using unadjusted Cox proportional hazards models.
Controlled non‐randomised studies of interventions
Time to discharge from hospital was not reported for any of the controlled NRSIs. Duan 2020, Liu 2020 and Zeng 2020 reported how many participants had been discharged at longest follow‐up available (0 to 31 days), without any evidence of a difference between the participants in the convalescent plasma transfusion group and the control group (Duan 2020: RR* 7.00, 95% CI 0.41 to 120.16; Liu 2020: RR* 1.08, 95% CI 0.86 to 1.35; Zeng 2020: RR* 2.50, 95% CI 0.18 to 33.83).
*We calculated the effect estimate with the reported outcome data (see Analysis 2.1). We did not adjust for any confounding factors.
2.1. Analysis.

Comparison 2: Results from controlled NRSIs, Outcome 1: Time to discharge from hospital
Admission to the ICU
None of the controlled studies reported this outcome.
Length of stay on the ICU
None of the controlled studies reported this outcome.
Quality of life
None of the controlled studies reported this outcome.
Safety of convalescent plasma for people with COVID‐19
For safety outcomes we included data from RCTs, controlled NRSIs, and non‐controlled NRSIs. As the controlled studies reported adverse events or serious adverse events for participants receiving convalescent plasma only, all studies are listed together.
Number of participants with grade 3 and grade 4 adverse events
Thirteen studies (201 participants) reported assessment of adverse events of possibly grade 3 or grade 4 severity (Ahn 2020; Duan 2020; Jin 2020; Li 2020; Liu 2020; Pei 2020; Perotti 2020; Salazar 2020; Tan 2020; Ye 2020; Zeng 2020; Zhang 2020a; Zhang 2020b). However, Zhang 2020a only reported that they had observed no adverse events for one of their participants; it was unclear whether the other three participants did or did not experience any adverse events. Twelve studies therefore reported the presence or absence of adverse events for all participants receiving convalescent plasma.
Four studies reported the occurrence of adverse events that were possibly grade 3 or 4 severity but they did not report the degree of severity (see Table 3).
4. Adverse events: grade 3 or 4.
| Study | Number of participants | Grade 3 or 4 adverse eventsa |
| Ahn 2020 | 2 | 0 |
| Duan 2020b | 10 (convalescent plasma group) | 0 |
| Jin 2020 | 6 | 0 |
| Li 2020 | 52 (convalescent plasma group) | 3 (in 2 participants)
|
| Liu 2020 | 39 (convalescent plasma group) | 0 |
| Pei 2020 | 3 | 1 (anaphylactic shock) |
| Perotti 2020 | 46 | 5 (in 4 participants)
|
| Salazar 2020c | 25 | 0 |
| Tan 2020 | 1 | 1 (fever) |
| Ye 2020 | 6 | 0 |
| Zeng 2020 | 6 (convalescent plasma group) | 0 |
| Zhang 2020ad | 4 | 0 |
| Zhang 2020b | 1 | 0 |
aWe assume that these adverse events were grade 3 or 4, but the studies did not specify the degree of severity. bOne participant with evanescent red face (grade unclear). cOne participant with morbilliform rash one day post‐transfusion that lasted for several days (grade unclear). dAssessment of adverse events only reported for one individual. Unclear information provided for the other three participants.
Li 2020 (52 participants, intervention arm of the RCT only) mentioned that one participant experienced chills and rashes within two hours of convalescent plasma transfusion, which they classified as non‐severe allergic transfusion reaction and also a probable non‐severe febrile non‐haemolytic transfusion reaction. The participant recovered fully after treatment with dexamethasone and promethazine. In addition, there was one non‐severe allergic transfusion reaction classified as a serious adverse event (further described below).
One non‐controlled NRSI (Perotti 2020, 46 participants), reported five events in four participants, including chills and fever, urticaria, one anaphylaxis, one possible transfusion‐related acute lung injury (TRALI) and one subsegmental pulmonary embolus (but relation unlikely/excluded).
Tan 2020, a case study, reported that the participant experienced moderate fever (38.9 °C) after convalescent plasma transfusion.
One of the three participants in Pei 2020 had severe anaphylactic shock after receiving 30 mL of plasma from a female donor with a history of pregnancy.
Nine studies (reporting on 99 participants) reported no adverse events that were possibly of grade 3 or grade 4 severity.
Reporting of adverse events was variable across these studies. In the controlled studies, there was reporting on adverse events only in participants receiving convalescent plasma, with no reporting in the control group. The duration of follow‐up for observation of adverse events varied across all studies. In addition, it was difficult to ascertain whether some of the adverse events were related to convalescent plasma transfusion, or due to underlying disease or other treatments, or both.
The evidence is very low certainty and none of the studies reported this outcome for any control group.
Number of participants with serious adverse events
Fourteen studies (5201 participants) assessed serious adverse events (Ahn 2020; Duan 2020; Jin 2020; Joyner 2020; Li 2020; Liu 2020; Pei 2020; Perotti 2020; Salazar 2020; Tan 2020; Ye 2020; Zeng 2020; Zhang 2020a; Zhang 2020b).
Four studies reported on the occurrence of serious adverse events (Joyner 2020; Li 2020; Pei 2020; Perotti 2020), see Table 6.
5. Serious adverse events .
| Study | Number of participants | Serious adverse events |
| Ahn 2020 | 2 | 0 |
| Duan 2020 | 10 (convalescent plasma group) | 0 |
| Jin 2020 | 6 | 0 |
| Joyner 2020 | 5000 | Within 4 hours after transfusion
|
| Li 2020 | 52 (convalescent plasma group) | 1 possible severe transfusion‐associated dyspnoea (patient had "shortness of breath, cyanosis, and severe dyspnoea within 6 hours of transfusion. The participant was given dexamethasone, aminophylline, and other supportive care immediately and gradually improved after 2 hours)." |
| Liu 2020 | 39 (convalescent plasma group) | 0 |
| Pei 2020 | 3 | 1 (anaphylactic shock) |
| Perotti 2020 | 46 | 3
|
| Salazar 2020 | 25 | 0 |
| Tan 2020 | 1 | 0 |
| Ye 2020 | 6 | 0 |
| Zeng 2020 | 6 (convalescent plasma group) | 0 |
| Zhang 2020a | 4 | 0 |
| Zhang 2020b | 1 | 0 |
| TACO: transfusion‐associated circulatory overload; TRALI: transfusion‐related acute lung injury | ||
Joyner 2020 reported data for 5000 participants from an ongoing US FDA (Food and Drug Administration) Expanded Access Programme. The study authors evaluated the incidence of serious adverse events in the first four hours after convalescent plasma transfusion only. Fifteen participants (0.3%) died; the study authors classified four of these deaths as potentially, probably, or definitely related to the convalescent plasma transfusion (0.1%). Eleven TRALIs (0.2%), seven TACOs (0.1%), and three severe allergic reactions (0.1%) occurred, all of them related to the plasma transfusion.
Li 2020 (52 participants, intervention arm from the included RCT) mentioned that one participant suffered from shortness of breath, cyanosis, and severe dyspnoea within six hours of convalescent plasma transfusion, which they classified as possible severe transfusion‐associated dyspnoea. After medical treatment the symptoms gradually improved over two hours.
Three serious events occurred in the single‐arm study by Perotti 2020 (46 participants): anaphylaxis/hypersensitivity, TRALI (relation possible) and subsegmental pulmonary embolism (but relation is considered to be unlikely/excluded).
One participant in Pei 2020 (3 participants) experienced a serious adverse event. As described above, this individual had severe anaphylactic shock after receiving convalescent plasma from a female donor with a history of pregnancy.
No serious adverse events occurred in 10 studies (100 participants).
Reporting of serious adverse events was variable across these studies. In the controlled studies, there was reporting on serious adverse events in participants receiving convalescent plasma only with no reporting in the control group. The duration of follow‐up for observation of serious adverse events varied across all studies. Some, but not all, studies included death as a serious adverse event. In addition, it was difficult to ascertain whether some of the adverse events were related to convalescent plasma transfusion, or due to underlying disease or other treatments, or both.
We are very uncertain whether or not convalescent plasma affects the number of serious adverse events. The evidence is very low certainty.
Discussion
Summary of main results
The aim of this review was to assess the effectiveness and safety of convalescent plasma and hyperimmune immunoglobulin in the treatment of coronavirus disease 2019 (COVID‐19). This is the first living update of our review.
We identified one randomised controlled trial (RCT) (which was stopped early), three controlled non‐randomised studies of interventions (NRSIs), and 16 non‐controlled NRSIs (for safety outcomes only). These studies evaluated 5443 participants, of whom 5211 received convalescent plasma. We identified a further 98 ongoing studies evaluating convalescent plasma or hyperimmune immunoglobulin, of which 50 are randomised.
Risk of bias
The risk of bias of the included RCT was unclear for efficacy outcomes and high for safety outcomes. All controlled NRSIs were at an overall critical risk of bias.
For safety outcomes, we also included and assessed non‐controlled NRSIs in addition to the controlled studies. As six non‐controlled NRSIs did not report safety data, we included 10 non‐controlled NRSIs for safety outcomes. The overall risk of bias of the 10 assessed non‐controlled NRSIs was also high.
Effectiveness of convalescent plasma for people with COVID‐19
We do not know whether the following results are related to the underlying natural history of the disease, other concomitant treatment, or convalescent plasma. We only included results from controlled studies to assess effectiveness of convalescent plasma. We rated all outcomes as very low certainty, and we were unable to pool data across studies. We present results per study.
All‐cause mortality at hospital discharge
We could not analyse results from the RCT as not all participants had been discharged at the end of follow‐up (28 days). We included one controlled NRSI (reporting on 21 participants) to assess this outcome. We are very uncertain whether convalescent plasma has any effect on all‐cause mortality at hospital discharge.
Time to death
We included one RCT (103 participants) and one controlled NRSI (195 participants) to assess this outcome. Convalescent plasma may prolong time from randomisation or start of treatment to death compared to the control group, but the evidence is very uncertain.
Improvement of clinical symptoms (as assessed by need for respiratory support)
We included one RCT (reporting on 103 participants) and one controlled NRSI (reporting on 195 participants) to assess this outcome. We are very uncertain whether convalescent plasma has any effect on the improvement of clinical symptoms at seven days, 14 days, and 28 days (very low‐certainty evidence). Two other controlled NRSIs (reporting on 20 and 21 participants, respectively) reported clinical improvement for the intervention group only, and the observation period was unclear.
Quality of life
None of the included studies reported this outcome.
Safety of convalescent plasma for people with COVID‐19
We included results from RCTs, controlled NRSIs, and non‐controlled NRSIs to assess the safety of convalescent plasma. Only 14 out of the 20 studies reported safety outcomes; six non‐controlled NRSIs did not report safety outcomes. Reporting of adverse events and serious adverse events was variable across these studies. In the RCT and controlled NRSIs, there was reporting on adverse events and serious adverse events only in participants receiving convalescent plasma, with no reporting of these outcomes in the control group. The duration of follow‐up for observation of adverse events and serious adverse events varied across all studies. Some, but not all, studies included death as a serious adverse event. In addition, it was difficult to ascertain whether some of these events were related to convalescent plasma transfusion or due to underlying disease and/or other treatments.
Adverse events
The grade of adverse events after convalescent plasma transfusion was not reported. Thirteen studies (201 participants) reported on adverse events (of possible grade 3 or 4 severity). The majority of these adverse events comprised allergic or respiratory events. We are very uncertain whether or not convalescent plasma therapy affects the risk of moderate to severe adverse events (very low‐certainty evidence).
Serious adverse events
Fourteen studies (5201 participants) reported on serious adverse events. The majority of participants were from one non‐controlled NRSI (5000 participants), which reported only on serious adverse events limited to the first four hours after convalescent plasma transfusion. This study included death as a serious adverse event. They reported 15 deaths, four of which they classified as potentially, probably or definitely related to transfusion. Other serious adverse events reported in all studies also predominantly comprised allergic or respiratory adverse events, which include anaphylaxis, transfusion‐associated dyspnoea and TRALI. We are very uncertain whether or not convalescent plasma affects the number of serious adverse events.
Overall completeness and applicability of evidence
We identified one RCT (that was stopped early), three controlled NRSIs, and 16 non‐controlled NRSIs, evaluating convalescent plasma in adults, most with severe COVID‐19. These studies included 5443 participants (ranging from 1 to 5000 participants), of whom 5211 received convalescent plasma. Most of these participants had also received different treatment options, either solely or in combination. These included antivirals, antifungals or antibiotics, corticosteroids, hydroxychloroquine and respiratory support (ECMO, mechanical ventilation or oxygen). For effectiveness of convalescent plasma therapy, we included controlled studies only (4 studies, 339 participants). In the three controlled NRSIs, selection of the control groups is only briefly reported and relevant confounding factors were not considered in the analyses (e.g. age, gender, severity of disease, co‐morbidities).
None of the controlled studies reported adverse events for the control arm. One large, non‐controlled NRSI (5000 participants) provided serious adverse events data, which occurred within the first four hours after convalescent plasma transfusion. The evidence for grade 3 and 4 adverse events is very uncertain, as adverse events were inconsistently reported across study designs.
We identified 98 ongoing studies, six are expanded access studies from the USA, and 50 are RCTs. Of these studies, five RCTs were planned to be completed already (ChiCTR2000030010; ChiCTR2000030179; ChiCTR2000030627; NCT04345991; NCT04376788), but results are not published yet and study investigators did not reply to our requests for additional information. An additional 23 RCTs are planned to be completed in 2020. The publication of the results of these studies will necessitate an update of this review. The conclusions of the updated review could differ from those of the present review, and may allow for a better judgement regarding the effectiveness and safety of convalescent plasma therapy.
Certainty of the evidence
It is important to note that the outcome measures are heterogeneous with wide variation in reporting across the included studies.
We identified one unblinded RCT, which was stopped early because there were no more eligible participants due to containment of the epidemic in Wuhan, China (Li 2020). It is unclear to what extent this pre‐termination may bias the results of the study. The certainty of the evidence in the reported outcomes was further reduced because of the very small information size and results including both potential benefit and potential harm for convalescent plasma therapy.
We identified three controlled NRSIs (Duan 2020; Liu 2020; Zeng 2020), which were all at critical risk of bias. None of these studies provided results for the same outcome, however, even if they had, we would not have meta‐analysed the results because of this critical risk of bias. The certainty of the evidence in the reported outcomes was further reduced because of the very small information size and results mostly including both potential benefit and potential harm for convalescent plasma therapy.
Because all included controlled studies report safety data only for the intervention group, we considered the results in a similar way to the non‐controlled NRSIs. We were unable to pool numerical data in any meaningful way and therefore reported results separately per study. The evidence is of very low certainty and without a control group, the outcome could not be considered in context.
Potential biases in the review process
To avoid potential bias in the review, we had planned to include the best available evidence. However, as COVID‐19 is a novel disease, results from large RCTs are not yet available. In fact, we could only identify one RCT, three controlled NRSIs, and 16 non‐controlled NRSIs. To increase the informative value of our review, we are tracking all registered trials and will continually update this review as more evidence becomes available. As explained above, the only RCT was stopped early due to the containment of the epidemic in Wuhan, China, leading to the enrolment of fewer participants than planned and consequently a lower power to detect an effect. We anticipate the lower numbers of people hospitalised with COVID‐19 and eligible for inclusion will also be a concern for other, ongoing studies that are not international. There are currently still many new trials being registered in registries, as can be seen from the additional 28 RCTs added to the list of ongoing studies in this update of the review.
Two experienced Information Specialists developed a sensitive search strategy, to identify all ongoing and completed studies. We searched all relevant databases and trials registries, and two review authors conducted all review steps independently and in duplicate. We are confident that we identified all relevant published and ongoing studies and will monitor them closely in the future. However, it is unclear whether ongoing studies will be completed before the global containment of the pandemic.
Another consideration for this rapidly evolving field is the availability of preprint articles that have not yet undergone peer review. In this review, we also included such preprints. However, we are aware of the potentially lower quality of these publications, and that results could change once the peer‐reviewed journal publications are available.
Although we have very limited confidence in the available evidence, we are not aware of any deficiencies in our review process. However, we are certain that the results are likely to be substantially different and conclusions may change as soon as peer‐reviewed high‐certainty evidence becomes available.
Agreements and disagreements with other studies or reviews
This systematic review identified very low‐certainty evidence on the safety and effectiveness of convalescent plasma for people with COVID‐19.
A recent systematic review and meta‐analysis found low‐certainty evidence for the use of convalescent plasma for treating people with infections with different aetiologies (Mair‐Jenkins 2015). The authors reported a systematic review and meta‐analysis of the literature on the use of convalescent plasma and hyperimmune immunoglobulin in treating severe acute respiratory infections of viral aetiology, and found that this treatment is likely to be both safe and effective in preventing mortality. The study identified a 75% reduction in the odds of mortality in their exploratory post hoc meta‐analysis across all viral aetiologies. The studies included in this review were performed with people treated with convalescent plasma for severe acute respiratory syndrome (SARS) and influenza. The limited number of identified studies and the low quality of included, mainly non‐controlled NRSIs restricted the authors’ ability to analyse extensively the risks and benefits of convalescent plasma therapy. Recommendations from the authors were to investigate the use of convalescent plasma and hyperimmune immunoglobulin in large, well‐designed clinical trials or other formal evaluations to obtain better‐certainty evidence, and to evaluate the optimal treatment regimen.
Results from several large RCTs on the use of convalescent plasma and hyperimmune immunoglobulin in treating severe influenza have recently been made public (Beigel 2017; Beigel 2019; Davey 2019; Hung 2013). However, the results from these studies are inconsistent, with some studies showing a beneficial effect of convalescent plasma for treating people with severe influenza, whereas other studies show no benefit. The studies were well designed and reported in detail the timing of the intervention and relevant outcomes. One study reported effectiveness of hyperimmune immunoglobulin, but only in a post hoc analysis of a subgroup of participants treated within five days of symptom onset (Hung 2013). In a different study, for the subgroup analysis of people with influenza B, the effect of hyperimmune immunoglobulin also resulted in a demonstrable clinical and virological benefit (Davey 2019). Different mechanisms in the human immune system and their role in responding to different circulating influenza strains might further explain why the results of clinical trials of convalescent plasma and hyperimmune immunoglobulin for influenza varied (Davey 2019). Influenza A immunity is reported to carry over to the next years, known as heterosubtypic immunity (Kreijtz 2011), and the current outbreak of COVID‐19 can, in that sense, not be compared with seasonal influenza. Notwithstanding these differences, which might explain why the aforementioned influenza studies were not successful in clearly demonstrating benefit, the possibility of a null effect of convalescent plasma over a suitable comparator cannot be ruled out with the currently available evidence on COVID‐19.
The adverse events associated with plasma transfusions are well characterised. Critically ill patients receiving plasma transfusions have an especially high risk of TACO, which is the leading cause of transfusion‐related mortality (Pandey 2012). Many countries have now introduced risk mitigation strategies to decrease the risk of TRALI. In the UK in 2018 there was only one confirmed case of TRALI.
In this systematic review of the literature, which mainly identified studies that included people with COVID‐19 with severe or critical illness, we identified a small proportion of participants experiencing any grade 3 or 4 adverse event, or serious adverse event. With the information available at this moment from published trials registry entries, it is apparent that the majority of clinical trials are enrolling people with COVID‐19 who have progressed to moderate or severe disease. Despite there being some evidence from other infectious diseases that early therapy might be more effective (Mair‐Jenkins 2015), targeting this population is justifiable given the evident lack of effective interventions for COVID‐19. The population that is eligible for treatment in these trials with convalescent plasma is potentially at high risk of transfusion reactions, and when treating critically ill people with COVID‐19, their status should be carefully monitored.
Authors' conclusions
Implications for practice.
The currently available evidence on the safety and effectiveness of convalescent plasma and hyperimmune immunoglobulin for treatment of people hospitalised with COVID‐19 is of very low certainty. Thus, any conclusions that are drawn based on these data are of limited value and these conclusions are subject to change as more reliable results become available. For the primary outcomes, there was not enough evidence to determine whether or not convalescent plasma affected the risk of all‐cause mortality at hospital discharge, time to death or improvement of clinical symptoms, assessed by the need for respiratory support. Other outcomes that were reported in a subset of the included studies were length of stay on the intensive care unit (ICU) and time to discharge from hospital, but reporting of these outcomes was not complete. None of the studies assessed quality of life. Most studies assessed the risks of the intervention, but reporting was heterogeneous. More thorough investigations, preferably well‐designed clinical trials, are needed in order to assess the benefits and risks of convalescent plasma therapy for people with COVID‐19.
Implications for research.
For the first version of the living systematic review investigating the use of convalescent plasma or hyperimmune immunoglobulin for people with COVID‐19, we included data from one small randomised controlled trial (RCT). The recruitment rate of this RCT was lower than expected, leading to the enrolment of far fewer participants than planned and consequently a lower power to detect an effect. The study authors noted this as one of the limitations of their study. We anticipate the currently decreasing number of people with COVID‐19 in some countries being eligible for inclusion, and head to head studies evaluating other potential beneficial drugs to treat COVID‐19 will also be a concern for other, ongoing studies. There are currently still many new studies being registered in trials registries, as can be identified from the list of ongoing studies in this review.
In addition to the notion that there are potentially too few eligible participants for all these studies, the importance of good study design should be stressed. We identified many ongoing, single‐arm intervention studies and expanded access registrations, whereas there urgently needs to be good‐quality evidence on the use of convalescent plasma for COVID‐19. This evidence should ideally be from RCTs with an appropriate control arm and preferably with a blinded design. The importance of reporting outcomes consistently for all study arms, and ensuring comparability of study arms in terms of co‐interventions, cannot be overstated. Although the numbers of infected individuals are declining, there remains the possibility of a second wave in the near future, and therefore careful consideration of study design is warranted.
Another consideration for research in this rapidly evolving field is the availability of preprint articles that have not yet undergone peer review. In this review, the status of publications has been included in the Characteristics of included studies table. However, it is important to continue to be aware of the potentially lower quality of these publications.
What's new
| Date | Event | Description |
|---|---|---|
| 3 June 2020 | New citation required and conclusions have changed | We included results from one RCT and three controlled NRSIs and added further safety data from non‐controlled NRSIs. |
| 31 May 2020 | New search has been performed | We included eight new studies. |
History
Review first published: Issue 5, 2020
Acknowledgements
This review was published in collaboration with the Cochrane Editorial and Methods Department. We particularly thank Sarah Hodgkinson (Associate Editor, Cochrane Editorial and Methods Department), Analysis of Review Group Output (ARGO) for their comments on the Abstract, Clare Dooley (Managing Editor), and Denise Mitchell (Copy Editor) for their excellent support. Thanks also to the Cochrane Editorial and Methods Department team, for their valuable comments on the review and timely management of the editorial process.
We thank Robin Featherstone (Information Specialist, Cochrane Editorial and Methods Department) for commenting on the search strategy and Gerald Gartlehner and Adrienne Stevens for their advice on rapid review methodology. We thank Susan J Brunskill for her support in identifying included and ongoing studies. We thank Theresa Moore (Methodology Editor, Editorial and Methods Department) for reviewing our assessment for the non‐randomised controlled trials with ROBINS‐I.
We thank all external peer reviewers who read and commented on this review. We thank Miquel Lozano (MD, PhD Clinic University Hospital, University of Barcelona, Spain), and Dr Michael James Ankcorn (Department of Virology, Northern General Hospital, Sheffield Teaching Hospitals NHS Foundation Trust, UK), who greatly helped to improve this review.
We thank Rujan Shrestha and Ya‐Ying Wang for translating and assessing articles in Chinese language for us via Cochrane TaskExchange.
Appendices
Appendix 1. Search strategy MEDLINE
1. Coronavirus Infections/
2. Coronavirus/
3. "Betacoronavirus"/
4. ((corona* or corono*) adj1 (virus* or viral* or virinae*)).tw,kf.
5. (coronavirus* or coronovirus* or coron?virinae* or "2019‐nCoV" or 2019nCoV or 2019‐CoV or nCoV2019 or "nCoV‐2019" or "COVID‐19" or COVID19 or "CORVID‐19" or CORVID19 or "WN‐CoV" or WNCoV or "HCoV‐19" or HCoV19 or CoV or "2019 novel*" or Ncov or "n‐cov" or "SARS‐CoV‐2" or "SARSCoV‐2" or "SARSCoV2" or "SARS‐CoV2" or SARSCov19 or "SARS‐Cov19" or "SARSCov‐19" or "SARS‐Cov‐19" or SARSr‐cov or Ncovor or Ncorona* or Ncorono* or NcovWuhan* or NcovHubei* or NcovChina* or NcovChinese* or Wuhan virus* or novel CoV or CoV 2 or CoV2 or betacoron?vir*).tw,kf.
6. (((respiratory* adj2 (acute* or symptom* or disease* or illness* or condition*)) or "sea‐food market*" or "seafood market*" or "food market*" or "foodmarket*") adj10 (Wuhan* or Hubei* or China* or Chinese* or Huanan*)).tw,kf.
7. ((outbreak* or wildlife* or wild‐life or pandemic* or epidemic*) adj3 (Wuhan* or Hubei* or China* or Chinese* or Huanan*)).tw,kf.
8. (anti‐flu* or anti‐influenza* or antiflu* or antinfluenza*).tw,kf.
9. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8
10. Plasma/
11. Immunoglobulins/
12. Immunoglobulins, Intravenous/
13. Immune Sera/
14. ((convalesc* or recovered or cured or rehabilitat* or survivor* or survived or virus‐positive or virus neutrali* or virus inactivated or antibod* or high‐titre* or high‐titer*) adj6 (plasma or blood or serum or sera)).mp.
15. ((plasma adj1 therap*) or gamma‐globulin* or "γ‐Globulin" or hyper‐Ig).tw,kf.
16. ((hyperimmune or hyper‐immune or high‐dos*) adj3 (plasma or immunoglobulin* or IVIG* or immune globulin* or globulin* or IgG)).tw,kf.
17. (plasma adj5 (immun* or antibod* or exchange* or donor* or donat* or transfus* or infus*)).mp.
18. ((convalesc* or recovered or cured or rehabilitat* or survivor* or survived or virus‐positive or virus inactivated or antibody‐positive) adj5 (donor* or donat*)).mp.
19. (((serum or sera) adj2 (therap* or treatment*)) or serotherap* or sero‐therap*).tw,kf.
20. 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19
21. 9 and 20
22. Covid‐19 Serotherapy.px
23. (Flu‐IVIG or ((anti‐flu* or anti‐influenza* or antiflu* or antinfluenza*) adj5 plasma)).mp.
24. 21 or 22 or 23
25. (exp Animals/ or exp Animal Experimentation/ or exp Models, Animal/) not Humans/
26. 24 not 25
27. limit 26 to yr="2019 ‐Current"
Appendix 2. Search strategy Embase
# Searches
1. "Coronavirus Infections"/ or "Coronavirus Infection"/
2. Coronavirinae/ or Coronavirus/ or exp Betacoronavirus/
3. ((corona* or corono*) adj1 (virus* or viral* or virinae*)).tw,kw.
4. (coronavirus* or coronovirus* or coron?virinae* or "2019‐nCoV" or 2019nCoV or 2019‐CoV or nCoV2019 or "nCoV‐2019" or "COVID‐19" or COVID19 or "CORVID‐19" or CORVID19 or "WN‐CoV" or WNCoV or "HCoV‐19" or HCoV19 or CoV or "2019 novel*" or Ncov or "n‐cov" or "SARS‐CoV‐2" or "SARSCoV‐2" or "SARSCoV2" or "SARS‐CoV2" or SARSCov19 or "SARS‐Cov19" or "SARSCov‐19" or "SARS‐Cov‐19" or SARSr‐cov or Ncovor or Ncorona* or Ncorono* or NcovWuhan* or NcovHubei* or NcovChina* or NcovChinese* or Wuhan virus* or novel CoV or CoV 2 or CoV2 or betacoron?vir*).tw,kw.
5. (((respiratory* adj2 (acute* or symptom* or disease* or illness* or condition*)) or "sea‐food market*" or "seafood market*" or "food market*" or "foodmarket*") adj10 (Wuhan* or Hubei* or China* or Chinese* or Huanan*)).tw,kw.
6. ((outbreak* or wildlife* or wild‐life* or pandemic* or epidemic*) adj3 (Wuhan* or Hubei* or China* or Chinese* or Huanan*)).tw,kw.
7. (anti‐flu* or anti‐influenza* or antiflu* or antifluenza*).tw,kw.
8. or/1‐7
9. Plasma Transfusion/
10. exp Immunoglobulin/
11. ((convalesc* or recovered or cured or survivor* or survived or rehabilitat* or virus‐positive or virus‐neutrali* or virusinactived or antibody‐rich or high‐tire* or high‐titer*) adj6 (plasma or blood or serum or sera)).mp.
12. ((plasma adj1 therap*) or gamma‐globulin or "y‐Globulin" or hyper‐lg).tw,kw.
13. (plasma adj5 (immun* or antibod* or exchange* or donor* or donat* or transfus* or infus*)).mp.
14. ((convalesc* or recovered or cured or survivor* or rehabilitat* or survived or virus‐positive or virus inactived or antibody‐positive) adj5 (donor* or donat*)).mp.
15. (plasma adj5 (immun* or antibod* or exchange* or donor* or donat* or transfus* or infus*)).mp.
16. ((hyperimmune or hyper‐immune or high‐dos*) adj3 (plasma or immunoglobulin* or IVIG* or immune globulin* or globulin* or IgG)).tw,kw.
17. (plasma adj5 (immun* or antibod* or exchange* or donor* or donat* or transfus* or infus*)).mp.
18 ((convalesc* or recovered or cured or rehabilitat* or survivor* or survived or virus‐positive or virus inactivated or antibody‐positive) adj5 (donor* or donat*)).mp.
19. (((serum or sera) adj2 (therap* or treatment*)) or serotherap* or sero‐therap*).tw,kw.
20. or/9‐19
21. (Flu‐IVIG or ((anti‐flu* or antiflu*) adj5 plasma)).mp.
22. (8 and 20) or 21
23. (exp animal/ or nonhuman/) not exp human/
24. a nimal experiment/ not (human experiment/ or human/)
25. 23 or 24
26. 22 not 25
Appendix 3. Search strategy PubMed
#1 (corona‐virus* OR corono‐virus* OR coronavirus* OR coronovirus* OR coronavirinae* OR coronovirinae* OR betacoronavirus OR Wuhan* OR Hubei* OR Huanan OR "2019 nCoV" OR 2019nCoV OR 2019 CoV OR nCoV2019 OR "nCoV 2019" OR "COVID 19" OR COVID19 OR "CORVID 19" OR CORVID19 OR "WN CoV" OR WNCoV OR "HCoV 19" OR HCoV19 OR CoV OR "2019 novel*" OR Ncov OR "n cov" OR "SARS CoV 2" OR "SARSCoV 2" OR "SARS‐CoV‐2" OR "SARSCoV‐2" OR "SARSCoV2" OR "SARS CoV2" OR „SARS‐Cov2“ OR SARSCov19 OR "SARS Cov19" OR "SARSCov 19" OR "SARS Cov 19" OR Ncovor OR Ncorona* OR Ncorono* OR NcovWuhan* OR NcovHubei* OR NcovChina* OR NcovChinese* OR novel CoV OR CoV2 OR SARSr‐cov)v 19" OR "SARS Cov 19" OR Ncovor OR Ncorona* OR Ncorono* OR NcovWuhan* OR NcovHubei* OR NcovChina* OR NcovChinese* OR SARSr‐cov)
#2 (((respiratory* AND (acute* OR symptom* OR disease OR diseases OR diseased OR illness* OR condition*)) OR "seafood market*" OR "sea food market*" OR "food market*" OR "foodmarket*") AND (Wuhan* OR Hubei* OR China OR "China’s" OR Chinese* OR Huanan*))
#3 ((outbreak* OR wildlife* OR wild‐life* OR pandemic* OR epidemic*) AND (China OR "China’s" OR Chinese* OR Huanan* OR Wuhan OR Hubei*))
#4 (anti‐flu* OR anti‐influenza* OR antiflu* OR antinfluenza*)
#5 #1 OR #2 OR #3 OR #4
#7 ((convalesc*[TIAB] OR recovered[TIAB] OR cured[TIAB] OR survivor*[TIAB] OR survived[TIAB] OR virus‐positive[TIAB] OR virus‐neutrali*[TIAB] OR "virus inactivated"[TIAB] OR antibod*[TIAB] OR high‐titre*[TIAB] OR high‐titer*) AND (plasma[TIAB] OR blood[TIAB] OR donor*[TIAB] OR donat*[TIAB]))
#8 ("therapeutic plasma" OR "plasma therapy" OR "immune plasma" OR "plasma exchange" OR gamma‐globulin* or "γ‐Globulin" or hyper‐Ig)
#9 (plasma[TI] AND (immun*[TIAB] OR transfus*[TIAB] OR infus*[TIAB]))
#10 ((hyperimmune OR hyper‐immune OR high‐dos*) AND (plasma OR immunoglobulin* OR IVIG* OR immune globulin* OR globulin*))
#11 #7 OR #8 OR #9 OR #10
#12 #6 AND #11
#13 (Flu‐IVIG OR ((anti‐flu* or anti‐influenza* or antiflu* or antinfluenza*) AND plasma))
#14 #12 OR #13
#15 (publisher[sb] OR inprocess[sb] OR pubmednotmedline[sb]) #16 #13 AND #15: Publication date from 2019/11/01 to present
Appendix 4. Search strategy CDC COVID‐19 Database (for searching in Endnote)
Any Field: plasma or hyperimmune or hyper‐immune or IVIG or immunoglobulin* or immune‐globulin* or globulin* or gamma‐globulin or γ‐Globulin or hyper‐Ig or serum or convalesc* or sera or donor or donat* or sero* or flu‐IVIG or antiflu* or anti‐flu*
Appendix 5. Search strategy Cochrane COVID‐19 Study Register
plasma OR hyperimmune OR hyper‐immune OR IVIG OR immunoglobulin OR globulin OR gamma‐globulin OR γ‐Globulin OR hyper‐Ig OR serum OR sera OR donor OR donation OR sero* OR flu‐IVIG OR antiflu* OR anti‐flu
Appendix 6. Planned methodology for randomised controlled trials (RCTs) and non‐randomised studies of interventions (NRSIs)
Data extraction and management
Assessment of risk of bias in included studies
Randomised controlled trials
We had planned to use the Risk of Bias 2.0 (RoB 2) tool to analyse the risk of bias in the underlying study results (Sterne 2019). Of interest for this review was the effect of the assignment to the intervention (the intention‐to‐treat (ITT) effect) and we would have performed all assessments with RoB 2 on this effect. The outcomes that we would have addressed are those specified for inclusion in Table 1. Accordingly, the outcomes had been prioritised according to the Core Outcome Measures in Effectiveness Trials Initiative for Covid‐19 patients (COMET 2020).
One review author would have assessed the risk of bias for each study result. A second review author would have verified the accuracy and the plausibility. In case of discrepancies among their judgements or inability to reach consensus, we had planned to consult a third review author to reach a final decision. We would have assessed the following types of bias as outlined in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2019a).
Bias arising from the randomisation process
Bias due to deviations from the intended interventions
Bias due to missing outcome data
Bias in measurement of the outcome
Bias in selection of the reported result
To address these types of bias we had planned to use the signalling questions recommended in RoB 2 and make a judgement using the following options:
'yes': if there is firm evidence that the question is fulfilled in the study (i.e. the study is at low or high risk of bias for the given the direction of the question);
'probably yes': a judgement has been made that the question is fulfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question);
'no': if there is firm evidence that the question is unfilled in the study (i.e. the study is at low or high risk of bias for the given the direction of the question);
'probably no': a judgement has been made that the question is unfilled in the study (i.e. the study is at low or high risk of bias given the direction of the question);
'no information' if the study report does not provide sufficient information to allow any judgement.
We had planned to use the algorithms proposed by RoB 2 to assign each domain one of the following levels of bias:
low risk of bias;
some concerns;
high risk of bias.
Subsequently we had planned to derive a 'Risk of bias' rating for each prespecified outcome in each study in accordance with the following suggestions.
'Low risk of bias': the trial is judged to be at low risk of bias for all domains for this result.
'Some concerns': the trial is judged to raise some concerns in at least one domain for this result, but not to be at high risk of bias for any domain.
'High risk of bias': the trial is judged to be at high risk of bias in at least one domain for the result or the trial is judged to have some concerns for multiple domains in a way that substantially lowers confidence in the results.
Data synthesis
If the clinical and methodological characteristics of individual studies were sufficiently homogeneous, we had planned to pool the data in meta‐analysis. We had planned to perform analyses according to the recommendations of the Cochrane Handbook for Systematic Reviews of Interventions (Deeks 2019). We would not have conducted meta‐analyses that involved both RCTs and controlled NRSIs. We had planned to conduct separate meta‐analyses for each comparison.
We had planned to use the Review Manager Web software for analyses (Review Manager Web). One review author would have entered the data into the software, and a second review author would have checked the data for accuracy.
We had planned to use the random‐effects model for all analyses as we anticipate that true effects will be related but will not be the same for included studies. If we could not perform a meta‐analysis, we had planned to comment on the results as a narrative with the results from all studies presented in tables.
For RCTs, when meta‐analysis had been feasible, we had planned to use the random‐effects model for pooling the data. For binary outcomes, we had planned to base the estimation of the between‐study variance using the Mantel‐Haenszel method. We had planned to use the inverse variance method for continuous outcomes, outcomes that include data from cluster‐RCTs, or outcomes where HRs are available. We planned to explore heterogeneity above 80% with subgroup analyses. If we could not find a cause for the heterogeneity then we had planned not to perform a meta‐analysis, but comment on the results as a narrative with the results from all studies presented in tables.
If a meta‐analysis had been feasible for controlled NRSIs we had planned to analyse the different types of studies separately. We had planned to only analyse outcomes with adjusted effect estimates if these were adjusted for the same factors using the inverse‐variance method as recommended in Chapter 24 of the Cochrane Handbook for Systematic Reviews of Interventions (Reeves 2019).
Appendix 7. 'Risk of bias' assessment of randomised controlled trials (RCTs), using RoB 1.0
We assessed methodological quality and risk of bias using the 'Risk of bias' tool recommended in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
| 'Risk of bias' assessment of Li 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Mortality Clinical improvement Adverse events |
Low | Quote: "Patients were randomly assigned via computer‐generated random numbering (1:1) to receive standard treatment coupled with convalescent plasma transfusion or standard treatment alone (control group) (Figure 1). The randomization was stratified based on the severity of COVID‐19 (severe or life‐threatening) and a randomization schedule was generated using block randomization with block size of 4 for each type of COVID‐19 by SAS software." |
| Allocation concealment (selection bias) | Mortality Clinical improvement Adverse events |
Low | Quote: "This random number will connect the subject to the designated treatment group (experimental group or control group) for treatment. [...] Staff responsible for randomization will only be responsible for the assignment of random groups and will not be involved in any specific trial operations." |
| Blinding of participants and personnel (performance bias) | Mortality Clinical improvement Adverse events |
High | Quote: "open‐label" Co‐interventions not balanced across arms |
| Blinding of outcome assessment (detection bias) | Mortality | Low | Quote: "To avoid assessment bias, the evaluation of clinical outcomes was performed by an investigator who was blind to the study group allocation." |
| Clinical improvement Adverse events |
Low | Quote: "To avoid assessment bias, the evaluation of clinical outcomes was performed by an investigator who was blind to the study group allocation." | |
| Selective reporting (reporting bias) | Mortality | Low | Reported as determined at protocol stage |
| Clinical improvement | Unclear | Quote: "A post hoc analysis was added to compare rates of improvement at days 7, 14, and 28." | |
| Adverse events | High | Only transfusion‐related adverse events reported | |
| Incomplete outcome data (attrition bias) | Mortality | Low | ITT population reported |
| Clinical improvement | Low | ITT population reported | |
| Adverse events | High | No safety data for control group available | |
| Other bias | Mortality Clinical improvement Adverse events |
Unclear | Quote: "Due to the containment of the COVID‐19 epidemic in Wuhan, China, the numbers of patients with COVID‐19 decreased in late March 2020. [...] The trial was terminated early after 103 of a planned 200 patients were enrolled." The study expressed effect estimates as odds ratios. Therefore we recalculated relative effects as risk ratios. We noticed that our calculation arrived at the same numerical values, and therefore highlight that effect estimates, which are indicated as odds ratios in the primary study, are in fact risk ratios. |
Appendix 8. 'Risk of bias' assessment of controlled non‐randomised studies of interventions (NRSIs), using ROBINS‐I
We assessed methodological quality and risk of bias using the Risk Of Bias in Non‐randomised Studies ‐ of Interventions (ROBINS‐I) tool (Sterne 2016).
| 'Risk of bias' assessment of Duan 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Bias due to confounding | Mortality Clinical improvement |
Serious | Quote: "Historic control group was formed by random selection of 10 patients from the cohort treated in the same hospitals and matched by age, gender, and severity of the diseases to the 10 cases in our trial." Not adjusted for co‐morbidities, previous treatments, time of disease onset, etc. |
| Adverse events | Not applicable | Paper only reports transfusion‐related adverse events for intervention group | |
| Bias in selection of participants into the study | Mortality Clinical improvement |
Critical | Small sample size, unclear how participants were selected into intervention group, unclear how long participants of historical control group were followed |
| Adverse events | Not applicable | Paper only reports transfusion‐related adverse events for intervention group | |
| Bias in classification of interventions | Mortality Clinical improvement |
Critical | Assignment to control group was done retrospectively. Treatment details of control group are not provided, and it is unclear whether patients were treated during the same period or at a time when less was known about the disease and the treatment options. Knowledge of patients' outcomes at the time of assignment to the control group could have had a major impact on the selection. |
| Adverse events | Not applicable | Paper only reports transfusion‐related adverse events for intervention group | |
| Bias due to deviations from intended interventions | Mortality Clinical improvement Adverse events |
Low | All participants received the intended intervention. |
| Bias due to missing data | Mortality | Serious | Mortality is reported for participants in intervention group until day 3 of follow‐up. Unclear how long control group was followed and how clinical status was assessed |
| Clinical improvement | Critical | Unclear how long control group was followed and clinical status in terms of respiratory support was not assessed | |
| Adverse events | Critical | No safety data for control group reported | |
| Bias in measurement of outcomes | Mortality | Critical | Unclear whether follow‐up was comparable between groups |
| Clinical improvement | Critical | Clinical course is reported for participants in intervention group until day 3 of follow‐up | |
| Adverse events | Critical | Only transfusion‐related adverse events reported | |
| Bias in selection of the reported results | Mortality Clinical improvement |
Critical | Study was registered as single‐arm trial and control group was retrospectively selected |
| Adverse events | Critical | Observation period unclear; only transfusion‐related adverse events assessed and reported | |
| Overall bias | Mortality | Critical | The study is too problematic to provide any useful evidence, however better evidence is yet insufficient. |
| Clinical improvement | Critical | The study is too problematic to provide any useful evidence, however better evidence is yet insufficient. | |
| Adverse events | Critical | The study is too problematic to provide any useful evidence, however better evidence is yet insufficient. | |
| Risk of bias assessment of Liu 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Bias due to confounding | Mortality Clinical improvement |
Serious | Only adjusted for hydroxychloroquine and azithromycin, intubation status and duration, length of hospital stay, and oxygen requirement on the day of transfusion. Not adjusted for e.g. age and gender |
| Adverse events | Not applicable | Paper only reports transfusion‐related adverse events for intervention group | |
| Bias in selection of participants into the study | Mortality Clinical improvement |
Moderate | Selection into the study may have been related to intervention and outcome, but the study authors used appropriate methods to adjust for the selection bias. Quote: "propensity score‐matched analysis using The Mount Sinai Hospital’s COVID‐19 confirmed patient pool from the same calendar period (24 March 2020 105 to 8 April 2020). A logistic regression was fit to predict the potential for plasma therapy based on time series data obtained at baseline upon admission, prior to transfusion, and the day of 107 transfusion." |
| Adverse events | Not applicable | Paper only reports transfusion‐related adverse events for intervention group | |
| Bias in classification of interventions | Mortality Clinical improvement |
Critical | Assignment to control group was done retrospectively. Treatment details of control group are not provided. Knowledge of participants' outcomes at the time of assignment to the control group could have had a major impact on the selection. |
| Adverse events | Not applicable | Paper only reports transfusion‐related adverse events for intervention group | |
| Bias due to deviations from intended interventions | Mortality Clinical improvement |
Low | All participants received the intended intervention. Most common co‐interventions (hydroxychloroquine and azithromycin, intubation status and duration, length of hospital stay, and oxygen requirement on the day of transfusion) were propensity score‐matched. Other co‐interventions were administered too infrequently to enforce exact matching |
| Adverse events | |||
| Bias due to missing data | Mortality Clinical improvement |
Low | Data were reasonably complete |
| Adverse events | Critical | No safety data for control group available | |
| Bias in measurement of outcomes | Mortality Clinical improvement |
Moderate | Median follow‐up comparable between groups. However, outcome assessors were not blinded to intervention and the study was performed retrospectively. |
| Adverse events | Critical | Only transfusion‐related adverse events reported | |
| Bias in selection of the reported results | Mortality Clinical improvement |
Critical | Retrospective study; selection of all reported results are likely biased |
| Adverse events | Critical | Observation period unclear, non‐occurrence of transfusion‐related adverse events only reported in discussion section | |
| Overall bias | Mortality | Critical | The study is too problematic to provide any useful evidence, however better evidence is yet insufficient. |
| Clinical improvement | Critical | The study is too problematic to provide any useful evidence, however better evidence is yet insufficient. | |
| Adverse events | Critical | The study is too problematic to provide any useful evidence, however better evidence is yet insufficient. | |
| 'Risk of bias' assessment of Zeng 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Bias due to confounding | Mortality Clinical improvement |
Serious | Not adjusted for confounding factors |
| Adverse events | Not applicable | Paper only reports adverse events after plasma transfusion for intervention group | |
| Bias in selection of participants into the study | Mortality Clinical improvement |
Moderate | Allocation to intervention and control group based on donor‐availability Quote: "A total of 21 contemporaneous critically ill patients with COVID‐19 were enrolled in the current study (Table 1), and all of them required intensive care unit admission. Six of the patients received convalescent plasma treatment based on the limited availability of convalescent plasma and ABO compatibility. Five of 6 patients in the convalescent plasma treatment group and 11 of 15 in the non–convalescent plasma treatment (control) group were male." |
| Adverse events | Not applicable | Paper only reports adverse events after plasma transfusion for intervention group | |
| Bias in classification of interventions | Mortality Clinical improvement |
Critical | Retrospective study design. Despite missingness of donors, unclear how control group was selected. Treatment details of control group are provided, but knowledge of participants' outcomes at the time of assignment to the control group could have had a major impact on the selection. |
| Adverse events | Not applicable | Paper only reports adverse events after plasma transfusion for intervention group | |
| Bias due to deviations from intended interventions | Mortality Clinical improvement |
Low | All participants received intended intervention. Co‐interventions (e.g. antiviral therapy, traditional Chinese medicine, etc.) seem to be balanced across treatment groups. |
| Adverse events | Low | All assessed participants received intended intervention | |
| Bias due to missing data | Mortality | Low | Data were reasonably complete |
| Clinical improvement | Low | Living participants discharged | |
| Adverse events | Critical | No safety data for control group available | |
| Bias in measurement of outcomes | Mortality Clinical improvement |
Low | Follow‐up until death or discharge |
| Adverse events | Critical | Only adverse events after plasma transfusion reported | |
| Bias in selection of the reported results | Mortality Clinical improvement |
Critical | Retrospective study; selection of all reported results are likely biased |
| Adverse events | Critical | Retrospective study; selection of all reported results are likely biased; only transfusion‐related adverse events reported; no safety data for control group available | |
| Overall bias | Mortality | Critical | The study is too problematic to provide any useful evidence, however better evidence is yet insufficient. |
| Clinical improvement | Critical | The study is too problematic to provide any useful evidence, however better evidence is yet insufficient. | |
| Adverse events | Critical | The study is too problematic to provide any useful evidence, however better evidence is yet insufficient. | |
Appendix 9. 'Risk of bias' assessment of non‐controlled non‐randomised studies of interventions (NRSIs), using the 'Risk of bias' assessment criteria for observational studies tool provided by Cochrane Childhood Cancer
We assessed methodological quality and risk of bias using the 'Risk of bias' assessment criteria for observational studies tool provided by Cochrane Childhood Cancer (see Table 4; Mulder 2019).
| 'Risk of bias' assessment of Ahn 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Not available | High | 2 participants only |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | High | Not blinded, awareness of intervention can bias assessment of subjective outcomes |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Low | Assessed and reported for both cases |
| Well‐defined study group (reporting bias) | Not available | Low | Population and intervention are well described |
| Well‐defined outcome (reporting bias) | Adverse events | High | No adverse reaction occurred after the administration of convalescent plasma in both cases. Observation period not reported |
| Well‐defined risk estimates (analyses) | Adverse events | Not applicable | No analyses performed |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Not available | High | Not adjusted for confounding factors |
| 'Risk of bias' assessment of Jin 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Not available | High | 6 of 146 COVID‐19 patients in Guizhou Jiangjunshan Hospital who received convalescent plasma therapy included in report |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | High | Not blinded, awareness of intervention can bias assessment of subjective outcomes |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Unclear | Assessed for all participants over study period, observation period unclear |
| Well‐defined study group (reporting bias) | Not available | Unclear | Study population well described, but intervention scarcely described |
| Well‐defined outcome (reporting bias) | Adverse events | High | Reported for all participants, but observation period unclear |
| Well‐defined risk estimates (analyses) | Adverse events | Not applicable | No analyses performed |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Not available | High | Not adjusted for confounding, results only reported for 6 of 146 participants receiving convalescent plasma |
| 'Risk of bias' assessment of Joyner 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Not available | Low | Large population size, prospective study, interim analysis of first 5000 patients |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | High | Not blinded, awareness of intervention can bias assessment of subjective outcomes |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Unclear | Preliminary results; only serious adverse events assessed, 4‐h follow‐up |
| Well‐defined study group (reporting bias) | Adverse events | Unclear | Study population well described, intervention scarcely described |
| Well‐defined outcome (reporting bias) | Adverse events | Unclear | Preliminary results; serious adverse events only |
| Well‐defined risk estimates (analyses) | Adverse events | Not available | No analyses performed |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Not available | High | Not adjusted for confounding factors |
| 'Risk of bias' assessment of Pei 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Not available | High | 3 participants only |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | High | Not blinded, awareness of intervention can bias assessment of subjective outcomes |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Unclear | Serious adverse events reported for 1 participant, not reported whether other participants experienced any adverse events |
| Well‐defined study group (reporting bias) | Not available | High | Study population and intervention insufficiently described |
| Well‐defined outcome (reporting bias) | Adverse events | High | Observation period not described |
| Well‐defined risk estimates (analyses) | Adverse events | Not available | No analyses performed |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Not available | High | Comorbidities and disease presentation and course not clearly reported; not adjusted for confounding factors |
| 'Risk of bias' assessment of Perotti 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Not available | High | 46 participants only |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | High | Not blinded, but awareness of intervention can bias assessment of subjective outcomes |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Low | Assessed and reported for all participants, 7‐day follow‐up |
| Well‐defined study group (reporting bias) | Not available | Low | Study population and intervention well described |
| Well‐defined outcome (reporting bias) | Adverse events | Low | Assessed and reported for all participants, 7‐day follow‐up |
| Well‐defined risk estimates (analyses) | Adverse events | Not available | No analyses performed |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Not available | High | Not adjusted for confounding factors |
| 'Risk of bias' assessment of Salazar 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Not available | High | 25 participants only |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | High | Not blinded, but awareness of intervention can bias assessment of subjective outcomes |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Low | All participants observed for occurrence of adverse events |
| Well‐defined study group (reporting bias) | Not available | Low | Study population and intervention well described |
| Well‐defined outcome (reporting bias) | Adverse events | Low | All observed adverse events described |
| Well‐defined risk estimates (analyses) | Adverse events | Not available | No analyses performed |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Not available | High | Not adjusted for confounding factors |
| 'Risk of bias' assessment of Tan 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Not available | High | 1 participant only |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | High | Not blinded, but awareness of intervention can bias assessment of subjective outcomes |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Unclear | Only fever reported, not reported whether other adverse events occurred |
| Well‐defined study group (reporting bias) | Not available | High | 1 participant only, not much information (e.g. age, comorbidities, clinical symptoms), intervention not described in detail |
| Well‐defined outcome (reporting bias) | Adverse events | High | Only fever reported, not reported whether other adverse events occurred |
| Well‐defined risk estimates (analyses) | Adverse events | Not available | No analyses performed |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Not available | High | Not adjusted for confounding factors |
| 'Risk of bias' assessment of Ye 2020 | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Not available | High | 6 participants only |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | High | Not blinded, but awareness of intervention can bias assessment of subjective outcomes |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Low | None occurred (3‐day follow‐up) |
| Well‐defined study group (reporting bias) | Not available | Low | Study population and intervention well described |
| Well‐defined outcome (reporting bias) | Adverse events | Low | 3‐day follow‐up |
| Well‐defined risk estimates (analyses) | Adverse events | Not available | No analyses performed |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Not available | High | Not adjusted for confounding factors |
| 'Risk of bias' assessment of Zhang 2020a | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Not available | High | 4 participants only |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | High | Not blinded, but awareness of intervention can bias assessment of subjective outcomes |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Unclear | Clinical course reported, but not whether adverse events occurred |
| Well‐defined study group (reporting bias) | Not available | Unclear | Study group well described but not intervention |
| Well‐defined outcome (reporting bias) | Adverse events | High | Not described in detail, unclear whether adverse events occurred |
| Well‐defined risk estimates (analyses) | Adverse events | Not available | No analyses performed |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Not available | High | Not adjusted for confounding factors |
| 'Risk of bias' assessment of Zhang 2020b | |||
| Domain | Assessed outcomes | Authors' judgement | Support for judgement |
| Representative study group (selection bias) | Not available | High | 1 participant only |
| Outcome detectors blinded to intervention (detection bias) | Adverse events | High | Not blinded, awareness of intervention can bias assessment of subjective outcomes |
| Complete outcome assessment/follow‐up (attrition bias) | Adverse events | Low | Reported that no transfusion‐related acute lung injuries were observed |
| Well‐defined study group (reporting bias) | Not available | Unclear | Participant not described in detail |
| Well‐defined outcome (reporting bias) | Adverse events | Low | No adverse events occurred |
| Well‐defined risk estimates (analyses) | Adverse events | Not available | No analyses performed |
| Important prognostic factors or follow‐up taken adequately into account (confounding) | Not available | High | Not adjusted for confounding factors |
Data and analyses
Comparison 1. Results from RCT.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Improvement of clinical symptoms at up to 7 days (assessed by need for respiratory support) | 1 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.30, 3.19] |
| 1.2 Improvement of clinical symptoms at 8 to 15 days (assessed by need for respiratory support) | 1 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 1.85 [0.91, 3.77] |
| 1.3 Improvement of clinical symptoms at 16 to 30 days (assessed by need for respiratory support) | 1 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 1.20 [0.80, 1.81] |
| 1.4 Improvement of clinical symptoms at up to 7 days (assessed by need for respiratory support): subgroup severity of disease | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.4.1 Severe disease | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 0.72 [0.18, 2.85] |
| 1.4.2 Life‐threatening disease | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 2.00 [0.19, 20.86] |
| 1.5 Improvement of clinical symptoms at 8 to 15 days (assessed by need for respiratory support): subgroup severity of disease | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.5.1 Severe disease | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 2.23 [1.05, 4.76] |
| 1.5.2 Life‐threatening disease | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.22, 4.55] |
| 1.6 Improvement of clinical symptoms at 16 to 30 days (assessed by need for respiratory support): subgroup severity of disease | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.6.1 Severe disease | 1 | 45 | Risk Ratio (M‐H, Random, 95% CI) | 1.34 [0.98, 1.83] |
| 1.6.2 Life‐threatening disease | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.33, 2.24] |
| 1.7 30‐day mortality | 1 | 101 | Risk Ratio (M‐H, Random, 95% CI) | 0.65 [0.29, 1.46] |
Comparison 2. Results from controlled NRSIs.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Time to discharge from hospital | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ahn 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Anderson 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Bao 2020b.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Duan 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Jin 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Joyner 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Kong 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Li 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Liu 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Pei 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants | (Preprint only, participant characteristics will be described in the supplementary material; not accessible yet)
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Perotti 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Salazar 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Shen 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Tan 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Yang 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Ye 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Zeng 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Zhang 2020a.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Zhang 2020b.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
Çınar 2020.
| Study characteristics | ||
| Methods |
|
|
| Participants |
|
|
| Interventions |
|
|
| Outcomes |
|
|
| Notes |
|
|
AE: adverse event; ALT: alanine aminotransferase; ARDS: acute respiratory distress syndrome; AST: aspartate transaminase; COI: conflict of interest; COPD: chronic obstructive pulmonary disease; CP: convalescent plasma; CPAP: continuous positive airway pressure; CPK: creatine phosphokinase; CRP: C‐reactive protein; CT: computed tomography; ECMO: extracorporeal membrane oxygenation; ELISA: enzyme‐linked immunosorbent assay; FiO2: fractional inspired oxygen; GI: gastrointestinal; HBV/HCV: hepatitis B/C; HLA: human leukocyte antigen; HNA: human neutrophil antigen; ICU: intensive care unit; IgA (B/G/M): immunoglobulin A (B/G/M); IL‐6: interleukin‐6; IQR: interquartile range; IRB: Institutional Review Board; IV: intravenous; IVIG: intravenous immunoglobulin; LDH: lactate dehydrogenase; MODS: multiple organ dysfunction syndrome; NR: not reported; OD: optical density; OSA: obstructive sleep apnoea; PaO2: arterial blood oxygen partial pressure; PCR: polymerase chain reaction; QoL: quality of life; RBD: receptor binding domain; RCT: randomised controlled trial; RNA: ribonucleic acid; RT‐PCR: reverse transcription polymerase chain reaction; SAE: serious adverse event; SARS: severe acute respiratory syndrome; SARS‐CoV‐2: severe acute respiratory syndrome coronavirus 2; TACO: transfusion‐associated circulatory overload; TAD: transfusion‐associated dyspnoea; TRALI: transfusion‐related acute lung injury
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Alzoughool 2020 | Review |
| Barone 2020 | Review |
| Bloch 2020 | Review |
| Brasil Ministerio 2020 | Standard operating procedure |
| Budhai 2020 | Feasibility of plasma collection only |
| Cao 2020a | Ineligible intervention |
| Cao 2020b | Review |
| Casadevall 2020a | Review |
| Casadevall 2020b | Editorial |
| Chen 2020a | Review |
| Chen 2020b | Ineligible intervention |
| Chen 2020c | Ineligible intervention |
| ChiCTR2000030312 | Study cancelled before starting recruitment |
| ChiCTR2000030381 | Study cancelled before starting recruitment |
| ChiCTR2000030442 | Study cancelled before starting recruitment |
| Datta 2020 | Review |
| de Assis 2020 | Ineligible indication |
| Dzik 2020 | Review |
| Díez 2020 | Ineligible intervention |
| Fleming 2020 | Letter |
| Franchini 2020 | Standard operating procedure |
| Hammarström 2020 | Review |
| Hu 2020 | Ineligible intervention |
| ISRCTN86534580 | Ineligible intervention |
| Jawhara 2020 | Review |
| Jiang 2020 | Ineligible intervention |
| Kesici 2020 | Letter |
| Khanna 2020 | Review |
| Knudson 2020 | Letter |
| Kominers 2020 | Review |
| Kumar 2020 | Review |
| Lancet Haematology 2020 | Editorial |
| Lanza 2020 | Letter |
| Lin 2020 | Ineligible intervention |
| Ministerio de Salud 2020 | Standard operating procedure |
| NCT04261426 | Ineligible intervention |
| NCT04323800 | Ineligible participant population (participants exposed to COVID‐19) |
| NCT04325672 | Study cancelled before starting recruitment |
| NCT04344015 | Feasibility of plasma collection only |
| NCT04344379 | Ineligible intervention |
| NCT04344977 | Feasibility of plasma collection only |
| NCT04350580 | Ineligible intervention |
| NCT04360278 | Feasibility of plasma collection only |
| NCT04368013 | Ineligible intervention |
| Pawar 2020 | Review |
| Qiu 2020 | No use of convalescent plasma. Reporting on generalised collection of information about Covid‐19 infection relating to aetiology, pathology, symptoms, clinical presentation and some generalised pharmacological treatment methods. The papers do not contain patient data that we required. Article translated by Rujan Shrestha and Ya‐Ying Wang via Cochrane TaskExchange |
| Roback 2020 | Review |
| Robbiani 2020 | Ineligible intervention |
| Rubin 2020 | Review |
| Seghatchian 2020 | Review |
| Sheridan 2020 | Review |
| Shi 2020 | Ineligible intervention |
| Syal 2020 | Review |
| Tanne 2020 | Review |
| Tiberghien 2020 | Review |
| Tu 2020 | No use of convalescent plasma. Reporting on generalised collection of information about Covid‐19 infection relating to aetiology, pathology, symptoms, clinical presentation and some generalised pharmacological treatment methods. The papers do not contain patient data that we required. Article translated by Rujan Shrestha and Ya‐Ying Wang via Cochrane TaskExchange |
| Wong 2020 | Review |
| Xie 2020 | Ineligible intervention |
| Yoo 2020 | Review |
| Zeng 2020a | Letter |
| Zhao 2020b | Review |
| Zhu 2020 | Letter |
Characteristics of ongoing studies [ordered by study ID]
ChiCTR2000029850.
| Study name | Study on convalescent plasma treatment for severe patients with novel coronavirus pneumonia (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 15 February 2020 |
| Contact information | Liang Yu The First Affiliated Hospital of Zhejiang University, State Key Laboratory for Diagnosis and Treatment of Infectious Diseases, National Clinical Research Center for Infectious Disease, 79 Qingchun Road, Shangcheng District, Hangzhou, Zhejiang, 310003, yu‐liang@zju.edu.cn Xiaowei Xu 79 Qingchun Road, Shangcheng District, Hangzhou, Zhejiang, China, 310003, xxw69@126.com |
| Notes |
|
ChiCTR2000030010.
| Study name | A randomized, double‐blind, parallel‐controlled, trial to evaluate the efficacy and safety of anti‐SARS‐CoV‐2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia patients (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 19 February 2020 |
| Contact information | Liu Ying Wuhan Jinyintan Hospital (Wuhan Infectious Diseases Hospital) , 1 Yintan Road, Dongxihu District, Wuhan, Hubei, China , 430023, whsjytyy_gcp@163.com Zhang Dingyu 1 Yintan Road, Dongxihu District, Wuhan, Hubei, China, 430023, 1813886398@qq.com |
| Notes |
|
ChiCTR2000030039.
| Study name | Clinical study for infusing convalescent plasma to treat patients with new coronavirus pneumonia (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 February 2020 |
| Contact information | Liping Wang Affiliated Hospital of Xuzhou Medical University, 9 Kunpeng Road, Gulou District, Xuzhou, Jiangsu, 163wangliping@163.com China Xuebing Yan 9 Kunpeng Road, Gulou District, Xuzhou, Jiangsu, China, yxbxuzhou@126.com |
| Notes |
|
ChiCTR2000030179.
| Study name | Experimental study of novel coronavirus pneumonia rehabilitation plasma therapy severe novel coronavirus pneumonia (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 24 February 2020 |
| Contact information | Liu Wei The First Affiliated Hospital of Nanchang University, 17 Yongwai Main Street, Nanchang, Jiangxi, China, 330006, cdyfyliuwei@163.com Le Aiping 17 Yongwai Main Street, Nanchang, Jiangxi, China, 330006, leaiping@126.com |
| Notes |
|
ChiCTR2000030627.
| Study name | Study for using the healed novel coronavirus pneumonia (COVID‐19) patients plasma in the treatment of severe critical cases |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 February 2020 |
| Contact information | Guojun Zhang The First Affiliated Hospital of Zhengzhou University, 1 Jianshe Road East, Zhengzhou, He'nan, China, zlgj‐001@126.com Guojun Zhang 1 Jianshe Road East, Zhengzhou, He'nan, China, zlgj‐001@126.com |
| Notes |
|
ChiCTR2000030702.
| Study name | Plasma of the convalescent in the treatment of novel coronavirus pneumonia (COVID‐19) common patient: a prospective clinical trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 15 February 2020 |
| Contact information | Liu Zhong Institute of Blood Transfusion, Chinese Academy of Medical Sciences, 26 Huacai Road, Chenghua District, Chengdu, Sichuan, China, 610000, Liuz@ibt.pumc.edu.cn Cao Bin 2 Yinghua Street East, Chaoyang District, Beijing, China, 100029, caobin_ben@163.com |
| Notes |
|
ChiCTR2000030929.
| Study name | A randomized, double‐blind, parallel‐controlled trial to evaluate the efficacy and safety of anti‐SARS‐CoV‐2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 17 March 2020 |
| Contact information | Lianghao Zhang 11443556@qq.com Sinopharm Wuhan Blood Products Co., Ltd. 1 Golden Industrial Park Road, Zhengdian, Jiangxia District, Wuhan, Hubei, China |
| Notes |
|
ChiCTR2000031501.
| Study name | The efficacy of convalescent plasma in patients with critical novel coronavirus pneumonia (COVID‐19): a pragmatic, prospective cohort study |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 17 March 2020 |
| Contact information | Weiqin LI liweiqindr@vip.163.com Eastern Theater General Hospital 305 Zhongshandong road, Xuanwu district, Nanjing, Jiangsu, China |
| Notes |
|
EUCTR2020‐001310‐38.
| Study name | A randomized, prospective, open label clinical trial on the use of convalescent plasma compared to best supportive care in patients with severe COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions | Interventions
|
| Outcomes |
|
| Starting date | 6 April 2020 |
| Contact information | Sixten Körper, IKT Ulm, 89081 Ulm, Germany; s.koerper@blutspende.de |
| Notes |
|
IRCT20151228025732N53.
| Study name | Evaluation of the therapeutic effects of convalescent plasma (CP) of recovered people from COVID‐19 in improving clinical and laboratory symptoms of hospitalised patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 April 2020 |
| Contact information | Alireza Emadi Semnan University of Medical Sciences, Semnan, Iran +98 23 3345 1336 are20935@semums.ac.ir |
| Notes |
|
IRCT20200310046736N1.
| Study name | Comparison of the therapeutic effect of convalescent plasma and plasma‐derived immunoglobulin‐enriched solution on COVID‐19 patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 24 March 2020 |
| Contact information | Parastoo Moradi Choghakabodi, Iran (Islamic Republic of); parastoomoradi40@yahoo.com |
| Notes |
|
IRCT20200325046860N1.
| Study name | Evaluation of convalescent plasma therapy in the treatment of patients with COVID‐19 disease |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 15 March 2020 |
| Contact information | Hassan Abolghasemi +98 21 8126 3166 h.abolghasemi.ha@gmail.com |
| Notes |
|
IRCT20200404046948N1.
| Study name | Randomized, parallel‐controlled and multi‐center clinical study evaluating the efficacy and safety of convalescent plasma, in the treatment of patients with severe SARS‐CoV‐2 infection (COVID‐19) |
| Methods |
|
| Participants | Participants
|
| Interventions |
|
| Outcomes |
|
| Starting date | 13 April 2020 |
| Contact information | Ramin Hamidi Farahani, Artesh University of Medical Sciences, Tehran, Iran; Amir.salarian@gmail.com |
| Notes |
|
IRCT20200409047007N1.
| Study name | The effect of plasma administration of COVID‐19 survivors in patients with acute respiratory distress syndrome due to COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 13 April 2020 |
| Contact information | Dr Mohsen Seddigh Shamsi, Mashhad University of Medical Sciences, Department of Internal Medicine, Taqi Abad Square, Mashhad, Iran |
| Notes |
|
IRCT20200413047056N1.
| Study name | Comparison between the efficacy of intravenous immunoglobulin and convalescent plasma in improving the condition of patients with COVID‐19: a randomized clinical trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 18 April 2020 |
| Contact information | Malihe Zangoue, Birjand University of Medical Sciences, Birjadn, Iran; mzangoue@yahoo.com |
| Notes |
|
NCT04264858.
| Study name | Treatment of acute severe 2019‐nCoV pneumonia with immunoglobulin from cured patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 17 March 2020 |
| Contact information | Xiang Cheng Department of Cardiology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology Wuhan, Hubei, China, 430022 |
| Notes |
|
NCT04292340.
| Study name | The efficacy and safety of anti‐SARS‐CoV‐2 inactivated convalescent plasma in the treatment of novel coronavirus pneumonia patient (COVID‐19): an observational study |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 February 2020 |
| Contact information | Hongzhou Lu, Ph.D+86‐021‐37990333 ext 3222 luhongzhou@fudan.edu.cn Shanghai Public Health Clinical Center Shanghai, Shanghai, China, 201508 |
| Notes |
|
NCT04327349.
| Study name | Investigating effect of convalescent plasma on COVID‐19 patients outcome: a clinical trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 28 March 2020 |
| Contact information | NR |
| Notes |
|
NCT04332380.
| Study name | Convalescent plasma for patients with COVID‐19: a pilot study (CP‐COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 April 2020 |
| Contact information | Juan M Anaya Cabrera, MD, PhD ; +57 321 233 9828; anayajm@gmail.com Manuel E Rojas Quintana, MD, MSc; +57 315 459 9951; manuel_9316@hotmail.com |
| Notes |
|
NCT04332835.
| Study name | Convalescent plasma for patients with COVID‐19: a randomized, open label, parallel, controlled clinical study (CP‐COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 May 2020 |
| Contact information | Juan M Anaya Cabrera, MD, PhD; +57 321 233 9828; anayajm@gmail.com Manuel E Rojas Quintana, MD, MSc; +57 315 459 9951; manuel_9316@hotmail.com |
| Notes |
|
NCT04333251.
| Study name | Evaluating convalescent plasma to decrease coronavirus associated complications. A phase I study comparing the efficacy and safety of high‐titer anti‐SARS‐CoV‐2 plasma vs best supportive care in hospitalized patients with interstitial pneumonia due to COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 April 2020 |
| Contact information | NR |
| Notes |
|
NCT04333355.
| Study name | Phase 1 study to evaluate the safety of convalescent plasma as an adjuvant therapy in patients with SARS‐CoV‐2 infection |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 15 April 2020 |
| Contact information | Servando Cardona‐Huerta, MD., Ph.D; +5218112121946; servandocardona@tec.mx Sylvia De la Rosa, MD; +5218111832730; sylvia.delarosa@tec.mx |
| Notes |
|
NCT04338360.
| Study name | Expanded access to convalescent plasma for the treatment of patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | NR |
| Contact information | Michael Joyner, MD; 507‐255‐4288; USCOVIDplasma@mayo.edu |
| Notes |
|
NCT04340050.
| Study name | Pilot study for use of convalescent plasma collected from patients recovered from COVID‐19 disease for transfusion as an empiric treatment during the 2020 pandemic at the University of Chicago Medical Center |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 10 April 2020 |
| Contact information | Maria Lucia Madariaga, MD; 773‐270‐2004; mlmadariaga@bsd.uchicago.edu |
| Notes |
|
NCT04342182.
| Study name | Convalescent plasma therapy from recovered COVID‐19 patients as therapy for hospitalized patients with COVID‐19 (CONCOVID Study) (ConCoVid‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 8 April 2020 |
| Contact information | Bart Rijnders, MD, PhD+31107033510; b.rijnders@erasmusmc.nl |
| Notes |
|
NCT04343261.
| Study name | Convalescent plasma in the treatment of COVID 19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 10 April 2020 |
| Contact information | Contact: Latha Dulipsingh, MD860‐714‐4402; Latha.Dulipsingh@trinityhealthofne.org |
| Notes |
|
NCT04343755.
| Study name | Phase IIa study exploring the safety and efficacy of convalescent plasma from recovered COVID‐19 donors collected by plasmapheresis as treatment for hospitalized subjects with COVID‐19 infection |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 9 April 2020 |
| Contact information |
|
| Notes |
|
NCT04344535.
| Study name | Convalescent plasma to reduce complications associated with COVID‐19 infection: a randomized trial comparing the efficacy and safety of high‐titre anti‐SARS‐CoV‐2 plasma vs. standard plasma in hospitalized patients with COVID‐19 infection |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 8 April 2020 |
| Contact information | Contact information not shared Responsible party: Elliott Bennett‐Guerrero, Professor of Anesthesiology, Stony Brook University |
| Notes |
|
NCT04345289.
| Study name | Efficacy and safety of novel treatment options for adults with COVID‐19 pneumonia. A double‐blinded, randomized, multi‐stage, 6‐armed placebo‐controlled trial in the framework of an adaptive trial platform |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 April 2020 |
| Contact information | Contact: Thomas Benfield, MD, DMSc+45 38622302 thomas.lars.benfield@regionh.dk |
| Notes |
|
NCT04345523.
| Study name | Multi‐center, randomized clinical trial of convalescent plasma therapy versus standard of care for the treatment of COVID‐19 in hospitalized patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 3 April 2020 |
| Contact information | Cristina Avendaño Solá, MD, PhD +34 91 191 64 79 cavendano@salud.madrid.org |
| Notes |
|
NCT04345679.
| Study name | Anti COVID‐19 convalescent plasma therapy |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 14 April 2020 |
| Contact information |
|
| Notes |
|
NCT04345991.
| Study name | Cohort multiple randomized controlled trials open‐label of immune modulatory drugs and other treatments in covid‐19 patients ‐ CORIMUNO‐CORIPLASM: efficacy of convalescent plasma to treat SARS‐CoV2 infected patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 14 April 2020 |
| Contact information | Karine LACOMBE, PU‐PH +33 149283196 karine.lacombe2@aphp.fr |
| Notes |
|
NCT04346446.
| Study name | Efficacy of convalescent plasma therapy in severely sick COVID‐19 patients: a pilot randomized controlled trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 14 April 2020 |
| Contact information | Dr Meenu Bajpai, MD, Institute of Liver and Biliary Sciences, India mailto:meenubajpai%40hotmail.com?subject=NCT04346446, ILBS‐COVID‐02, Efficacy of Convalescent Plasma Therapy in Severely Sick COVID‐19 Patients |
| Notes |
|
NCT04346589.
| Study name | A pilot study to explore the efficacy and safety of rescue therapy with antibodies from convalescent patients obtained with double ‐filtration plasmapheresis (DFPP) and infused in critically ill ventilated patients with coronavirus disease 2019 (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | April 2020 |
| Contact information | Piero Luigi Ruggenenti, MD; 0039 035 267 ext 3814; pruggenenti@asst-pg23.it |
| Notes |
|
NCT04347681.
| Study name | A national collaborative multicenter phase II study for potential efficacy of convalescent plasma to treat severe COVID‐19 and patients at high risk of developing severe COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 12 April 2020 |
| Contact information | Hani AL‐Hashmi, MD; 00966564773377; hanih.hashmi@kfsh.med.sa Mahammad Awadallah, MSc; 00966545032312; mahammad.awadalla@kfsh.med.sa |
| Notes |
|
NCT04348656.
| Study name | A randomized open‐label trial of CONvalenscent plasma for hospitalized adults with acute COVID‐19 respiratory illness (CONCOR‐1) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 27 April 2020 |
| Contact information | Donald M Arnold, MD, McMaster University, Hamilton, Canada arnold@mcmaster.ca |
| Notes |
|
NCT04348877.
| Study name | Plasma rich antibodies from recovered patients from COVID19 (PRA‐001) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 April 2020 |
| Contact information | Hossam Fahmy, Professor of Faculty of Medicine, Ain Shams University |
| Notes |
|
NCT04352751.
| Study name | Experimental use of convalescent plasma for passive immunization in current COVID‐19 pandemic in Pakistan in 2020 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | April 2020 |
| Contact information | Contact: Dr. Arshi Naz, PhD,Diplab; 00923232234376; labarshi@yahoo.com Contact: Dr. Neeta Maheshwary, MBBS M.Phil; 00923208247773; drneeta@hiltonpharma.com |
| Notes |
|
NCT04353206.
| Study name | A feasibility study assessing the safety of multiple doses of anti‐SARS‐CoV‐2 plasma in mechanically ventilated intubated patients with respiratory failure due to COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | May 2020 |
| Contact information | Noah Merin, MD PhD; 310‐423‐1160; Noah.Merin@cshs.org David Hager, MD PhD; dhager1@jhmi.edu |
| Notes |
|
NCT04354831.
| Study name | An open label, phase 2 study evaluating the efficacy and safety of high‐titre anti‐SARS‐CoV‐2 plasma in hospitalized patients with COVID‐19 infection |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 May 2020 |
| Contact information | Mary Beth Graham, MD, Medical College of Wisconsin, USA mailto:mbgraham%40mcw.edu?subject=NCT04354831, PRO00037712, A Study Evaluating the Efficacy and Safety of High‐Titer Anti‐SARS‐CoV‐2 Plasma in Hospitalized Patients With COVID‐19 Infection |
| Notes |
|
NCT04355767.
| Study name | Convalescent plasma to limit coronavirus associated complications: a randomized double‐blind, phase 2 study comparing the efficacy and safety of high‐titer anti‐SARS‐CoV‐2 plasma vs. placebo in emergency room patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | May 2020 |
| Contact information | Study team; 650‐724‐7186; jcunning@stanford.edu |
| Notes |
|
NCT04355897.
| Study name | CoVID‐19 plasma in treatment of COVID‐19 patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | NR |
| Contact information | Dean J Kereiakes, MD; 513‐585‐1777; lindnermd@thechristhospital.com |
| Notes |
|
NCT04356482.
| Study name | Determination of the dose and effectiveness of convalescent plasma in severely and very severely ill patients by COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | May 2020 |
| Contact information | Luis M Villela, MD; +526624756529; luisvillela@yahoo.com Diego Espinoza, MD; +526623862375; dr.espinoza.peralta@gmail.com |
| Notes |
|
NCT04356534.
| Study name | Use of convalescent plasma therapy for COVID‐19 patients with hypoxia: a prospective randomized trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 19 April 2020 |
| Contact information | Manaf Al Qahtani, Dr. Royal College of Surgeons in Ireland ‐ Bahrain; mailto:mqahtani%40rcsi‐mub.com?subject=NCT04356534, BDF/R&REC/2020‐423, Convalescent Plasma Trial in COVID ‐19 Patients |
| Notes |
|
NCT04357106.
| Study name | COPLA Study: treatment of severe forms of coronavirus infection with convalescent plasma |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 13 April 2020 |
| Contact information | Juan Carlos Olivares‐Gazca, MD, MPH; 2222438100; jolivares@hsctmexico.com José Manuel Priesca‐Marin, MD; 2222438100; mpriesca@hsctmexico.com |
| Notes |
|
NCT04358211.
| Study name | Expanded access to convalescent plasma to treat and prevent pulmonary complications associated with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | April 3 2020 |
| Contact information | Nakhle Saba, MD nsaba@tulane.edu Tulane Medical CenterAvailable New Orleans, Louisiana, USA, 70112 |
| Notes |
|
NCT04358783.
| Study name | Phase II, randomized, double‐blind, controlled clinical trial evaluating the efficacy and safety of plasma from patients cured of COVID‐19 compared to the best available therapy in subjects with SARS‐CoV‐2 pneumonia |
| Methods |
Clinical trial comparing convalescent plasma to BAT for the treatment of severely ill and critically ill patient with COVID‐19 Patients with SARS‐CoV‐2 PCR‐confirmed infection with pulmonary infiltrates and hypoxaemia will be screened and invited to participate |
| Participants |
Patients with SARS‐CoV‐2 PCR‐confirmed infection with pulmonary infiltrates and hypoxaemia will be screened and invited to participate |
| Interventions |
|
| Outcomes |
|
| Starting date | 27 April 2020 |
| Contact information | Contact: Eduardo Pérez Alba, MD +52 8117998705 md.eduardo.perez@gmail.com Contact: Laura Marina Nuzzolo Shihadeh, MD +52 8112773423 laura.nuzzolo@gmail.com Hospital Universitario José E. Gonzalez, UANL, Mexico |
| Notes |
|
NCT04359810.
| Study name | A phase 2, randomized clinical trial to evaluate the efficacy and safety of human anti‐SARS‐CoV‐2 convalescent plasma in severely ill adults with COVID‐19 |
| Methods |
Number of centres: NR Intervention model description: a total of 105 eligible participants will be randomised in a 2:1 ratio to receive either CP qualitatively positive for SARS‐CoV‐2 antibody (anti‐SARS‐CoV‐2 plasma) or non‐CP fresh frozen (control plasma) |
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 21 April 2020 |
| Contact information | Contact: Max O'Donnell, MD 212‐305‐5794 mo2130@cumc.columbia.edu Contact: Andrew Eisenberger, MD 212‐305‐0983 abe6@cumc.columbia.edu Columbia University Irving Medical Center/NYPRecruiting New York, New York, USA, 10032 |
| Notes |
|
NCT04360486.
| Study name | Expanded access protocol for the treatment of coronavirus disease 2019 (COVID‐19) with anti‐SARS‐CoV‐2 convalescent plasma (ASCoV2CP) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 24 April 2020 |
| Contact information | Andrew P Cap, MS, MD, PhD, FACP andrew.p.cap.mil@mail.mil U.S. Army Medical Research and Development Command |
| Notes |
|
NCT04361253.
| Study name | A prospective, randomized, double‐masked, placebo‐controlled trial of high‐titer COVID‐19 convalescent plasma (HT‐CCP) for the treatment of hospitalized patients with COVID‐19 of moderate severity |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 30 April 2020 |
| Contact information | Richard Kaufman, MD 617‐732‐5232 rmkaufman@bwh.harvard.edu Karina Oganezova 6177328624koganezova@bwh.harvard.edu Brigham and Women's Hospital, Boston, Massachusetts, USA, 02115 |
| Notes |
|
NCT04362176.
| Study name | A randomized, controlled clinical trial to test the safety and efficacy of convalescent donor plasma to treat COVID‐19 in hospitalized adults |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 24 April 2020 |
| Contact information | Amanda J Bistran‐Hall (615) 875‐8531 amanda.j.bistran-hall@vumc.org Principal Investigator: Todd Rice, MD Vanderbilt University Medical Center Vanderbilt University Medical Center Nashville, Tennessee, USA, 37203 |
| Notes |
|
NCT04363034.
| Study name | Arkansas expanded access COVID‐19 convalescent plasma treatment program |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 27 April 2020 |
| Contact information | Danielle Evans (501) 526‐7906 DEvans@uams.edu David Avery (501) 214‐2101 daavery@uams.edu University of Arkansas |
| Notes |
|
NCT04364737.
| Study name | Convalescent plasma to limit coronavirus associated complications: a randomized blinded phase 2 study comparing the efficacy and safety of anti‐SARS‐CoV2 plasma to placebo in COVID‐19 hospitalized patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 17 April 2020 |
| Contact information | Mila B Ortigoza, MD, PhD Mila.Ortigoza@nyulangone.org Michelle Chang Michelle.Chang3@nyulangone.org
|
| Notes |
|
NCT04365439.
| Study name | Convalescent plasma for the treatment of moderate‐severe COVID‐19: a proof‐of‐principle study |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 27 April 2020 |
| Contact information | Contact: Enos Bernasconi, M.D. +41 91 811 60 22 enos.bernasconi@eoc.ch Contact: Beatrice Bernasconi +41 91 811 60 21 beatrice.bernasconi@eoc.ch Ente Ospedaliero Cantonale, Bellinzona Principal Investigator: Stefano Fontana, M.D. Servizio Trasfusionale, Lugano |
| Notes |
|
NCT04366245.
| Study name | Phase I / II multicentre, randomized and controlled clinical trial to evaluate the efficacy of treatment with hyperimmune plasma obtained from convalescent antibodies of COVID‐19 infection |
| Methods |
Inclusion criteria:
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 23 April 2020 |
| Contact information | Ana Cardesa Gil 697 95 69 41 ext 0034 ana.cardesa@juntadeandalucia.es Hospital Unversitario Virgen Macarena, Sevilla, Spain, 41009 |
| Notes |
|
NCT04372368.
| Study name | Convalescent plasma for the treatment of patients with COVID‐19 |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | NR |
| Contact information | Contact: John D Beckham, MD303‐724‐4927 David.beckham@cuanschutz.edu |
| Notes | Recruitment status: available Prospective completion date: NR Sponsor/funding: University of Colorado, Denver, Investigators Principal Investigator: John D Beckham, MD University of Colorado Denver, Anschutz Medical Campus |
NCT04372979.
| Study name | Evaluation of efficacy of COVID‐19 convalescent plasma versus standard plasma in the early care of COVID‐19 patients hospitalized outside intensive care units |
| Methods | Trial design: triple‐blinded, parallel, clinical RCT Sample size: 80 Setting: inpatient Country: France Language: translated to English Number of centres: at least 4 |
| Participants | Inclusion criteria:
OR 1 of the biological criteria :
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | May 2020 |
| Contact information | Contact: Christophe MARTINAUD, PU PH +33 141467241christophe.martinaud@intradef.gouv.fr Contact: Christophe RENARD +33 140514103christophe1.renard@intradef.gouv.fr |
| Notes | Recruitment status: not yet recruiting Prospective completion date: October 2020 Sponsor/funding: Direction Centrale du Service de Santé des Armées, University Hospital, Grenoble; Investigators Study Director:Hervé FOEHRENBACHDirection Centrale du Service de Santé des Armées (DCSSA), Study Director:Catherine VERRETService de Santé des Armées‐Direction de la Formation de la Recherche et de l'Innovation, Principal Investigator:Christophe MARTINAUDCentre de Transfusion Sanguine des Armées, Principal Investigator:Jean‐Luc BOSSONStatistical and methodological investigator ‐ Laboratoire TIMC UMR 5525 CNRS Equipe Themas |
NCT04373460.
| Study name | Comparison of the efficacy and safety of human coronavirus immune plasma (HCIP) vs. control (SARS‐CoV‐2 non‐immune) plasma among outpatients with symptomatic COVID‐19 |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 19 May 2020 |
| Contact information | David J Sullivan, MD 410‐502‐2522 dsulliv7@jhmi.edu, David Sullivan, MD 410‐502‐2522 dsulliv7@jhmi.edu |
| Notes | Recruitment status: not yet recruiting Prospective completion date: 21 December 2022 Sponsor/funding: Johns Hopkins University, State of Maryland, Bloomberg Foundation, Principal Investigator: David J Sullivan, MD The Johns Hopkins University |
NCT04374370.
| Study name | Severe acute respiratory syndrome coronavirus 2 of the genus betacoronavirus (SARSCoV2) convalescent plasma (CP) expanded access protocol (EAP) |
| Methods |
|
| Participants | Inclusion criteria:
Severe disease is defined as:
Life‐threatening disease is defined as:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | NR |
| Contact information | Contact: Chris Ensor, Pharm D 413.519.7056 Chris.Ensor@AdventHealth.com |
| Notes | Recruitment status: available Prospective completion date: NR Sponsor/funding: AdventHealth Orlando, Available: Orlando, Florida, United States, 32803, Principal Investigator: Eduardo Oliveira, MD AdventHealth |
NCT04374487.
| Study name | A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma to limit COVID‐19 associated complications |
| Methods |
|
| Participants | Inclusion criteria
Or in case of severe or immediately life‐threatening COVID‐19, for example:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 9 May 2020 |
| Contact information | Sangeeta Pathak, MBBS, Diploma 9873081647 sangeeta.pathak@maxhealthcare.com Sandeep Budhiraja, MRCP, FACP 9810262954 sbudhiraja@maxhealthcare.com |
| Notes | Recruitment status: not yet recruiting Prospective completion date: 9 May 2021 Sponsor/funding: Max Healthcare Insititute Limited, Investigators Principal Investigator: Sangeeta Pathak, MBBS, Diploma Max Super Speciality Hospital, Saket (DDF) |
NCT04374526.
| Study name | Early transfusIon of COVID‐19 convalescent plasma in elderly COVID‐19 patients to prevent disease progression |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 27 May 2020 |
| Contact information | Raffaele Landolfi, Prof. 06 30154435 ext +39 raffaele.landolfi@unicatt.it Luciana Teofili, Prof. 06 30154180 ext +39 luciana.teofili@unicatt.it |
| Notes | Recruitment status: recruiting Prospective completion date: 30 June 2021 Sponsor/funding: Fondazione Policlinico Universitario Agostino Gemelli IRCCS |
NCT04374565.
| Study name | Efficacy and safety of high‐titer anti‐SARS‐CoV‐2 (COVID19) convalescent plasma for hospitalized patients with infection due to COVID‐19 to decrease complications: a phase II trial |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 5 May 2020 |
| Contact information | Kristen M Petros De Guex, MA 434) 924‐5059 KMP6F@hscmail.mcc.virginia.edu William B Harrington, MPH 434‐409‐5060 wh7fd@hscmail.mcc.virginia.edu |
| Notes | Recruitment status: recruiting Prospective completion date: 5 April 2021 Sponsor/funding: |
NCT04375098.
| Study name | Efficacy and safety of early anti‐SARS‐COV‐2 convalescent plasma in patients admitted for COVID‐19 infection: a randomized phase II trial |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 4 May 2020 |
| Contact information | Contact: Maria Elvira Balcells, MD +562 23543508 ebalcells@uc.cl |
| Notes | Recruitment status: recruiting Prospective completion date: December 2020 Sponsor/funding: Pontificia Universidad Catolica de Chile, Fundacion Arturo Lopez Perez, Principal Investigator: Maria Elvira Balcells, MD ebalcells@uc.cl |
NCT04376034.
| Study name | Convalescent plasma collection from individuals that recovered from COVID19 and treatment of critically ill individuals with donor convalescent plasma |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 16 April 2020 |
| Contact information | Brian Peppers, DO, PhD 304‐594‐2483 brian.peppers@hsc.wvu.edu Lisa Giblin Sutton, Pharm D 304‐293‐0928 giblinl@wvumedicine.org |
| Notes | Recruitment status: not yet recruiting Prospective completion date: 30 March 2021 Sponsor/funding: West Virginia University |
NCT04376788.
| Study name | Exchange transfusion versus plasma from convalescent patients with methylene blue in patients with COVID‐19 |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 5 May 2020 |
| Contact information | Mohamed M Moussa +201001553744 drmohamed_metwali1@med.asu.edu.eg Essam A Hassan, MD +201001839394 essam.abdelwahed@yahoo.com |
| Notes | Recruitment status: not yet recruiting Prospective completion date: 1 June 2020 Sponsor/funding: Ain Shams University Investigators: Principal Investigator: Mohamed M Moussa, Ain Shams University |
NCT04377568.
| Study name | CONCOR‐KIDS: a randomized, multicentered, open‐label phase 2 clinical trial of the safety and efficacy of human coronavirus‐immune convalescent plasma for the treatment of COVID‐19 disease in hospitalized children |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 May 2020 |
| Contact information | Contact: Julia Upton 416 813 7654 ext 208634 julia.upton@sickkids.ca Contact: Christoph Licht christoph.licht@sickkids.ca |
| Notes | Recruitment status: not yet recruiting Prospective completion date: 1 May 2022 Sponsor/funding: The Hospital for Sick Children, C17 Council (regulatory sponsor) |
NCT04377672.
| Study name | Human convalescent plasma for high risk children exposed or infected with SARS‐CoV‐2 (COVID‐19) |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 28 May 2020 |
| Contact information | Contact: Oren Gordon, MD 4106141211 ogordon3@jhmi.edu Contact: Mary Katherine Brosnan 410‐955‐8264 mbrosna1@jhmi.edu |
| Notes | Estimated primary completion date 28 May 2021 Institution ‐ John Hopkins University |
NCT04380935.
| Study name | Effectiveness and safety of convalescent plasma therapy on COVID‐19 patients with acute respiratory distress syndrome |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 11 May 2020 Estimated completion date 31 August 2020 |
| Contact information | Contact: Robert Sinto, MD +628158835432 rsinto@yahoo.com |
| Notes | Recruitment status: recruiting Prospective completion date: 31 August 2020 Sponsor/funding: Indonesia University/NR |
NCT04381858.
| Study name | Convalescent plasma vs human immunoglobulin to treat COVID‐19 pneumonia |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 6 May 2020 Completion 30 September 2020 |
| Contact information | Jose Manuel Arreola, MD, PhD 4494632049 dr.jmag@gmail.com |
| Notes | Recruitment status: recruiting Prospective completion date: 30 September 2020 Sponsor/funding: Centenario Hospital Miguel Hidalgo |
NCT04381936.
| Study name | Randomised evaluation of COVID‐19 therapy (RECOVERY) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 19 March 2020 |
| Contact information | Richard Haynes +44 (0)1865 743743 recoverytrial@ndph.ox.ac.uk |
| Notes | Recruitment status: recruiting Prospective completion date: June 2021 Sponsor/funding: University of Oxford |
NCT04383535.
| Study name | Convalescent plasma and placebo for the treatment of COVID‐19 severe pneumonia |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 15 May 2020 |
| Contact information | Contact: Waldo H Belloso, PhD +541149590200 waldo.belloso@hiba.org.ar Contact: Ventura Simonovich, MD +541149590200 ventura.simonovich@hiba.org.ar |
| Notes | Recruitment status: recruiting Prospective completion date: August 2020 Sponsor/funding: Hospital Italiano de Buenos Aires/NR |
NCT04383548.
| Study name | Clinical study for efficacy of anti‐corona VS2 immunoglobulins prepared from COVID19 convalescent plasma prepared by VIPS mini‐pool IVIG medical devices in prevention of SARS‐CoV‐2 infection in high risk groups as well as treatment of early cases of COVID19 patients |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 June 2020 |
| Contact information | Contact: Alshaimaa M Selim, specialist 01003580480 shaimaamokhtargood@yahoo.com Contact: Maha A Mohamed, Professor 01000004572 atwa_maha@yahoo.com |
| Notes | Recruitment status: not yet recruiting Prospective completion date: 1 January 2021 Sponsor/funding: Assiut University |
NCT04384497.
| Study name | Convalescent plasma for treatment of COVID‐19: an exploratory dose identifying study |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 7 May 2020 |
| Contact information | Contact: Joakim Dillner, MD, PhD +46 (0) 72‐468 24 60 joakim.dillner@ki.se Contact: Johan Ursing, MD, PhD +46 (0) 70‐475 15 30 johan.ursing@sll.se |
| Notes | Recruitment status: recruiting Prospective completion date: December 2020 Sponsor/funding: Karolinska University Hospital/NR |
NCT04384588.
| Study name | COVID19‐convalescent plasma for treating patients with active symptomatic COVID 19 infection (FALP‐COVID) (FALP‐COVID) |
| Methods |
|
| Participants | Inclusion criteria for all patients:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 7 April 2020 |
| Contact information | Contact: Christian Caglevic, MD56981369487 christian.caglevic@falp.org |
| Notes | Recruitment status: recruiting Prospective completion date: 6 April 20201 Sponsor/funding: Fundacion Arturo Lopez PerezConfederación de la Producción y del Comercio (CPC)Bolsa de Santiago |
NCT04385043.
| Study name | Phase 2b/3 trial to evaluate the safety and efficacy of plasma transfusion from convalescent patients with SARS‐CoV‐2 infection on severity and mortality of COVID‐19 in hospitalized patients |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 May 2020 |
| Contact information | Luca Gallelli, University of Catanzaro |
| Notes | Recruitment status: recruiting Prospective completion date: 15 October 2020 (primary), 15 May 2021 (study) Sponsor/funding: University of Catanzaro; Azienda Ospedaliera Policlinico "Mater Domini", Azienda Sanitaria Provinciale Di Catanzaro, Annunziata Hospital, Cosenza, Italy, Azienda Ospedaliera Bianchi‐Melacrino‐Morelli |
NCT04385186.
| Study name | Inactivated convalescent plasma as a therapeutic alternative in hospitalized patients COVID‐19 |
| Methods |
|
| Participants | Inclusion criteria:
Exclusion criteria:
History of immunosuppression |
| Interventions |
|
| Outcomes |
|
| Starting date | 20 June 2020 |
| Contact information | Andrés F Zuluaga, MD, MSc, MeH 3014020291 andres.zuluaga@udea.edu.co Ana L Muñoz, MSc, PhD ana.munoz@hemolifeamerica.org |
| Notes |
|
NCT04385199.
| Study name | The use of convalescent plasma for patients hospitalized with COVID‐19 disease |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 4 May 2020 |
| Contact information | Geneva Tatem, MD313‐587‐6775, gtatem1@hfhs.org |
| Notes |
|
NCT04388410.
| Study name | Phase 2b/3 trial to evaluate the safety and efficacy of plasma transfusion from convalescent patients with SARS‐CoV‐2 infection on severity and mortality of COVID‐19 in hospitalized patients. |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 June 2020 |
| Contact information | Not provided |
| Notes |
|
NCT04388527.
| Study name | An open‐label, single arm, phase 1, safety and exploratory efficacy study of convalescent plasma for severely ill mechanically ventilated participants with COVID‐19 caused by SARS‐CoV‐2 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 30 April 2020 |
| Contact information |
|
| Notes |
|
NCT04389710.
| Study name | Convalescent plasma for the treatment of patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 15 April 2020 |
| Contact information |
|
| Notes |
|
NCT04389944.
| Study name | Amotosalen‐ultraviolet a pathogen‐inactivated convalescent plasma in addition to best supportive care and antiviral therapy on clinical deterioration in adults presenting with moderate to severe coronavirus disease 2019 infectious disease (COVID‐19) |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 31 March 2020 |
| Contact information |
|
| Notes |
|
NCT04390178.
| Study name | Plasma from individuals who have recovered from severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2) infection as treatment for acute COVID‐19 disease |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 10 April 2020 |
| Contact information | Principal Investigator: Johan Ursing, MD, PhD, Danderyd Hospital |
| Notes |
|
NCT04390503.
| Study name | A phase 2 randomized, double‐blinded trial to evaluate the efficacy and safety of human anti‐ SARS‐CoV‐2 plasma in close contacts of COVID‐19 cases |
| Methods |
|
| Participants |
Group B: SARS‐CoV‐2 PCR‐positive but asymptomatic or mild symptoms at screening
Group C: SARS‐CoV‐2 PCR‐negative (uninfected) at time of screening but asymptomatic or mildly symptomatic at screening
*Close contact is defined by CDC as being within approximately 6 feet (2 meters) of a COVID‐19 case for a prolonged period of time (without PPE); close contact can occur while caring for, living with, visiting, or sharing a healthcare waiting area or room with a COVID‐19 case +Mild symptoms are rated by participant as mild and not interfering with normal daily activities
Group B: SARS‐CoV‐2 PCR‐positive but asymptomatic or mild symptoms at screening
Group C: SARS‐CoV‐2 PCR‐negative (uninfected) at time of screening
|
| Interventions |
|
| Outcomes |
|
| Starting date | May 2020 |
| Contact information |
|
| Notes |
|
NCT04391101.
| Study name | Efficacy of convalescent plasma for the treatment of severe SARS‐CoV‐2 infection: a randomized, open label clinical trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | June 2020 |
| Contact information |
|
| Notes |
|
NCT04392232.
| Study name | A phase 2 study of COVID 19 convalescent plasma in high risk patients with COVID 19 infection |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 5 May 2020 |
| Contact information |
|
| Notes |
|
NCT04392414.
| Study name | Randomized, open label, prospective study of the safety and efficacy of hyperimmune convalescent plasma in moderate and severe COVID‐19 disease |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 May 2020 |
| Contact information |
|
| Notes |
|
NCT04393727.
| Study name | Transfusion of convalescent plasma for the early treatment of pneumonIa due to SARSCoV2: a multicenter open label randomized control trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 May 2020 |
| Contact information |
|
| Notes |
|
NCT04395170.
| Study name | A multicenter randomized clinical trial to evaluate the efficacy and safety of the use of convalescent plasma (PC) compared to anti‐COVID‐19 human immunoglobulin and standard treatment in hospitalized patients |
| Methods |
|
| Participants |
|
| Interventions |
CP:
hyperimmune immunoglobulin:
|
| Outcomes |
|
| Starting date | June 2020 |
| Contact information |
|
| Notes |
|
NCT04397523.
| Study name | Use of COVID‐19 convalescent plasma in the patients infected with COVID‐19 (SARS‐CoV‐2) ‐ efficacy and safety |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 30 April 2020 |
| Contact information | Rada Grubovic Rastvorceva, MD MSci PhD +38923226923 ext 126 drgrubovic@gmail.com |
| Notes |
|
NCT04397757.
| Study name | An an open‐label, controlled, phase 1, safety and exploratory efficacy study of convalescent plasma for severely ill, hospitalized participants with COVID‐19 pneumonia caused by SARS‐CoV‐2 |
| Methods |
|
| Participants |
Note ‐ An exception must be requested to the Sponsor if ≥ 72 h since positive test
|
| Interventions |
|
| Outcomes |
|
| Starting date | 13 March 2020 |
| Contact information |
|
| Notes |
|
NCT04403477.
| Study name | Convalescent plasma transfusion therapy in severe COVID‐19 patients‐ a tolerability, efficacy and dose‐response phase II RCT |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 May 2020 |
| Contact information | Mohammad S Rahman, M Phil, FCPS +88 01971840757 srkhasru@gmail.com Fazle R Chowdhury, FCPS; PhD +88 01916578699 mastershakil@hotmail.com |
| Notes |
|
NCT04404634.
| Study name | Convalescent plasma to limit coronavirus associated complications: a randomized blinded phase 2 study comparing the efficacy and safety of anti‐SARS‐CoV‐2 plasma to placebo in COVID‐19 hospitalized patients |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | May 2020 |
| Contact information | Mahalia Desruisseaux, MD203‐737‐4057 mahalia.desruisseaux@yale.edu Alessandro Santin, MD203‐737‐4450 alessandro.santin@yale.edu |
| Notes |
|
NCT04405310.
| Study name | Plasma from convalescent donors with COVID‐19 for the management of patients with SARS‐COV‐2 phase II and III, a double center randomized double blind trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 20 May 2020 |
| Contact information | Angela Perez‐Calatayud, MD +525542389377 gmemiinv@gmail.com Yanet Ventura, MD +52554848965 yanereb@gmail.com |
| Notes |
Recruitment status: recruiting Prospective completion date: 20 June 2020 Sponsor/funding: Grupo Mexicano para el Estudio de la Medicina Intensiva Hospital General Naval de Alta Especialidad ‐ Escuela Medico Naval National Institute of Pediatrics, Mexico Instituto Nacional de Enfermedades Respiratorias |
NCT04407208.
| Study name | Convalescent plasma therapy in patients with COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 May 2020 |
| Contact information | Marliana Sri Rejeki, Sp.FK +6281323756199 marlianasr@gmail.com Familia Bela, Sp. PA +6285228878818 |
| Notes |
Recruitment status: recruiting Prospective completion date: 1 August 2020 Sponsor/funding: Biofarma Rumah Sakit Pusat Angkatan Darat Gatot Soebroto Eijkman Institute for Molecular Biology |
NCT04408040.
| Study name | Use of convalescent plasma collected from donors recovered from COVID‐19 virus disease for transfusion, as an empirical and preemptive treatment during viral pandemic outbreak |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | June 2020 |
| Contact information | Stacey Brown 404‐780‐7965 stacey.brown@northside.com |
| Notes |
|
NCT04408209.
| Study name | Convalescent plasma for the treatment of patients with severe COVID‐19 infection ‐ a multicenter phase II trial |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 23 April 2020 |
| Contact information | Aikaterini Niarchou +30 6949124743 aniarchou@med.uoa.gr Ioanna Charitaki +30 6976156403 j.charitaki@gmail.com |
| Notes |
Recruitment status: recruiting Prospective completion date: 30 June 2020 Sponsor/funding: National and Kapodistrian University of Athens Hellenic Society of Hematology |
NCT04412486.
| Study name | An open label trial of transfusion of COVID‐19 convalescent plasma (CCP) to patients with moderate to severe COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 1 June 2020 |
| Contact information | Gailen D Marshall, Jr., MD, PhD 601‐815‐5527 gmarshall@umc.edu |
| Notes |
Recruitment status: recruiting Prospective completion date: 31 May 2022 Sponsor/funding: Gailen D. Marshall Jr., MD PhD University of Mississippi Medical Center |
U1111‐1251‐9286.
| Study name | Use of convalescent plasma submitted to pathogen inactivation for the treatment of patients with severe COVID‐19 |
| Methods |
|
| Participants |
|
| Interventions |
|
| Outcomes |
|
| Starting date | 19 April 2020 |
| Contact information | Pedro Kurtz Address: Rua do Resende 156 City: Ro de Janeiro / Brazil Zip Code: 20231092 Telephone: 2122779352 E‐mail: kurtzpedro@mac.com |
| Notes |
|
AE: adverse event; ALT: alanine transaminase; ARDS: acute respiratory distress syndrome; AST: aspartate transaminase; BAL: bronchoalveolar lavage; BAT: best available therapy; BMI: body mass index; CDC: Centers for Disease Control and Prevention; COI: conflict of interest; COPD: chronic obstructive pulmonary disease; CP: convalescent plasma; CPAP: continuous positive airway pressure; CPK: creatine phosphokinase; CRP: C‐reactive protein; CT: computed tomography; DFPP: double‐filtration plasmapheresis; DVT: deep vein thrombosis; ECMO: extracorporeal membrane oxygenation; ED: emergency department; FDA: US Food and Drug Administration; FiO2: fractional inspired oxygen; GFR: glomerular filtration rate; HBV/HCV: hepatitis B/C; HCPOA: healthcare power of attorney; HLA: human leukocyte antigen; ICU: intensive care unit; IgA (B/G/M): immunoglobulin A (B/G/M); IL‐6: interleukin‐6; IV: intravenous; IVIG: intravenous immunoglobulin; LAR: legal authorised representative; LDH: lactate dehydrogenase; NR: not reported; NYHA: New York Heart Association; PaO2: arterial blood oxygen partial pressure; PCR: polymerase chain reaction; PE: pulmonary embolism; QoL: quality of life; RCT: randomised controlled trial; RNA: ribonucleic acid; RT‐PCR: reverse transcription polymerase chain reaction; SAE: serious adverse event; SARS: severe acute respiratory syndrome; SC: subcutaneous; SOFA: Sequential Organ Failure Assessment; SpO2: peripheral capillary oxygen saturation; TACO: transfusion‐associated circulatory overload; TAD: transfusion‐associated dyspnoea; TB: tuberculosis; TRALI: transfusion‐related acute lung injury; TTP: thrombotic thrombocytopenic purpura; UIP: usual interstitial pneumonia; ULN: upper limit of normal; WBC: white blood count; WHO: World Health Organization
Differences between protocol and review
Types of studies
As the evidence we found from the RCT was at unclear or high risk of bias and at serious risk of bias for the controlled NRSIs, and as none of these studies reported safety data for the control arm, we also included safety data from prospective and retrospective non‐comparative study designs (e.g. case series) and followed the methodology as specified in the protocol (Piechotta 2020). Because of the missing comparator, efficacy data of non‐controlled studies cannot be placed in context and therefore do not provide any useful evidence. In contrast to the protocol, we therefore decided to only include safety data of non‐controlled studies.
Types of interventions
We added standard immunoglobulin as an eligible control treatment.
Types of outcome measures
We revised the secondary outcome 'Improvement of clinical symptoms, assessed through need for respiratory support at up to 7 days; 8 to 15 days; 16 to 30 days' and added to the fourth bullet point: 'plus high‐flow oxygen', to differentiate from the third bullet point. It now reads:
Improvement of clinical symptoms, assessed by need for respiratory support at up to 7 days; 8 to 15 days; 16 to 30 days:
oxygen by mask or nasal prongs
oxygen by NIV (non‐invasive ventilation) or high flow
intubation and mechanical ventilation
mechanical ventilation plus high‐flow oxygen
extracorporeal membrane oxygenation (ECMO)
We added the outcome, 'quality of life' after discussion with a patient representative.
Electronic searches
As publication bias might influence all subsequent analyses and conclusions, we searched all potential relevant trials registries in detail to detect ongoing as well as completed studies, but not yet published studies. Nowadays, it is mandatory to provide results at least in the trials registry. In case results were not published elsewhere, we had planned to extract and analyse these data. However, no outcome data had yet been added to the trials registries. We decided to exclude study registries in the search strategy, because they are already included in the Cochrane COVID‐19 Study Register, which is updated Monday to Friday and exclude the WHO COVID‐19 Global Research Database. The WHO COVID‐19 Global Research Database and LitCov are included in the collection of Center for Disease Control and Prevention COVID‐19 Research Article Database. The search part for COVID‐19 was updated for the search strategies from IM and CD peer reviewed it.
Data extraction and management
We had planned to extract data using a standardised data extraction form developed in Covidence. However, we could not adapt the standardised form to our needs. Therefore we generated a customised data extraction form in Microsoft Excel (Microsoft Corporation 2018).
Assessment of risk of bias in included studies
Randomised controlled trials
We had planned to use the Risk of Bias 2.0 (RoB 2) tool to analyse the risk of bias in the underlying study results (Sterne 2019). However, RoB 2 is not yet available in RevMan Web (Review Manager Web), and the Cochrane Editorial and Methods Department recommended for us to use the former 'Risk of bias' tool for this version of the review (Higgins 2011), instead. Please refer to Appendix 6 for further information on the planned bias assessment with RoB 2.
Summary of findings and assessment of the certainty of the evidence
At protocol stage we had planned to assess the certainty of the evidence for our primary outcomes (all‐cause mortality at hospital discharge and time to death), only. However, for the first (rapid) version of this review, we decided to assess the certainty of the evidence also for prioritised secondary outcomes (clinical improvement, grade 3 and 4 adverse events, and serious adverse events) to increase the informative value on effectiveness and safety of convalescent plasma therapy. For the living systematic review we also prioritised patient quality of life as an important patient outcome and added this outcome to the 'Summary of findings' table. We specified in the methods how we graded the certainty of the evidence, especially for non‐randomised controlled trials using ROBINS‐I for 'Risk of bias' assessment, for calculation of absolute effects for time‐to‐event outcomes and for writing informative statements for the findings and certainty of the evidence.
Some passages in this protocol, especially in the methods section, are from the standard template of Cochrane Haematology.
Contributions of authors
VP: methodological expertise, and conception and writing of the review
KLC: clinical expertise, and conception and writing of the review
SJV: clinical expertise, and conception and writing of the review
CD: development of the search strategy
IM: development of the search strategy
EMW: clinical expertise and advice
AL: clinical expertise and advice
CK: clinical expertise and advice
ZM: clinical expertise and advice
CS‐O: clinical expertise and advice
LJE: clinical and methodological expertise, and conception and writing of the review
NS: methodological expertise and advice, and conception and writing of the review
Sources of support
Internal sources
-
Sanquin Blood Supply, Netherlands
Center for Clinical Transfusion Research
-
University Hospital of Cologne, Germany
Cochrane Cancer, Department I of Internal Medicine
-
Monash University, Australia
Transfusion Research Unit, Department of Epidemiology and Preventive Medicine
-
NHS Blood and Transplant, UK
NHS Blood and Transplant
External sources
No sources of support supplied
Declarations of interest
VP: none known
KLC: HSANZ Leukaemia Foundation PhD scholarship to support studies at Monash University. This is not related to the work in this review.
SJV: none known
CD: none known
IM: none known
EMW: I have sought funding support from Australian Medical Research Future Fund for a trial of convalescent plasma. I will not be involved in bias assessment, data extraction or interpretation, but will serve as a content expert.
AL: none known
CK: none known
ZM: I have sought funding support from Australian Medical Research Future Fund for a trial of convalescent plasma. I will not be involved in bias assessment, data extraction or interpretation, but will serve as a content expert.
CS‐O: is a member of the BEST Collaborative Clinical Study Group. I will not be involved in bias assessment, data extraction or interpretation, but will serve as a content expert.
LJE: co‐lead of the COVID‐19 immunoglobulin domain of the REMAP‐CAP trial. I will not be involved in bias assessment, data extraction or interpretation, but will serve as a content expert.
NS: none known
contributed equally
contributed equally
contributed equally
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Ahn 2020 {published data only}
- Ahn JY, Sohn Y, Lee SH, Cho Y, Hyun JH, Baek YJ, et al. Use of convalescent plasma therapy in two COVID-19 patients with acute respiratory distress syndrome in Korea. Journal of Korean Medical Science 2020;35(14):e149. [DOI] [PMC free article] [PubMed] [Google Scholar]
Anderson 2020 {published data only}
- Anderson J, Schauer J, Bryant S, Graves CR. The use of convalescent plasma therapy and remdesivir in the successful management of a critically ill obstetric patient with novel coronavirus 2019 infection: a case report. Case Reports in Women's Health 2020;27:e00221. [DOI: 10.1016/j.crwh.2020.e00221] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bao 2020b {published data only}
- Bao Y, Lin SY, Cheng ZH, Xia J, Sun YP, Zhao Q, et al. Clinical features of COVID-19 in a young man with massive cerebral hemorrhage—case report. SN Comprehensive Clinical Medicine 2020 May 23 [Epub ahead of print]. [DOI: 10.1007/s42399-020-00315-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
Çınar 2020 {published data only}
- Çınar OE, Sayınalp B, Karakulak EA, Karataş AA, Velet M, İnkaya AÇ, et al. Convalescent (immune) plasma treatment in a myelodysplastic COVID-19 patient with disseminated tuberculosis. Transfusion and Apheresis Science 2020 May 29 [Epub ahead of print]. [DOI: 10.1016/j.transci.2020.102821] [DOI] [PMC free article] [PubMed] [Google Scholar]
Duan 2020 {published data only}
- Duan K, Liu B, Li C, Zhang H, Yu T, Qu J, et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proceedings of the National Academy of Sciences of the United States of America 2020;202004168. [DOI: 10.1073/pnas.2004168117] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan K, Liu B, Li C, Zhang H, Yu T, Qu J, et al. The feasibility of convalescent plasma therapy in severe COVID-19 patients: a pilot study. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Jin 2020 {published data only}
- Jin C, Gu J, Yuan Y, Long Q, Zhang Q, Zhou H, et al. Treatment of 6 COVID-19 patients with convalescent plasma. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.05.21.20109512] [DOI] [Google Scholar]
Joyner 2020 {published data only}
- Joyner M, Wright RS, Fairweather DL, Senefeld J, Bruno K, Klassen S, et al. Early safety indicators of COVID-19 convalescent plasma in 5,000 patients. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.05.12.20099879] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT04338360. Expanded access to convalescent plasma for the treatment of patients with COVID-19. clinicaltrials.gov/show/NCT04338360 (first received 8 April 2020).
Kong 2020 {published data only}
- Kong Y, Cai C, Ling L, Zeng L, Wu M, Wu Y, et al. Successful treatment of a centenarian with coronavirus disease 2019 (COVID-19) using convalescent plasma. Transfusion and Apheresis Science 2020 May 21 [Epub ahead of print]. [DOI: 10.1016/j.transci.2020.102820] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2020 {published data only}
- ChiCTR2000029757. Convalescent plasma for the treatment of severe and critical novel coronavirus pneumonia (COVID-19): a prospective randomized controlled trial. www.chictr.org.cn/showproj.aspx?proj=49081 (first received 12 February 2020).
- Li L, Zhang W, Hu Y, Tong X, Zheng S, Yang J, et al. Effect of convalescent plasma therapy on time to clinical improvement in patients with severe and life-threatening COVID-19: a randomized clinical trial. JAMA 2020 Jun 3 [Epub ahead of print]. [DOI: 10.1001/jama.2020.10044] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2020 {published data only}
- Liu ST, Lin HM, Baine I, Wajnberg A, Gumprecht JP, Rahman F, et al. Convalescent plasma treatment of severe COVID-19: a matched control study. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.05.20.20102236] [DOI] [PubMed] [Google Scholar]
Pei 2020 {published data only}
- Pei S, Yuan X, Zhimin ZZ, Run YR, Xie Y, Minxue SM, et al. Convalescent plasma to treat COVID-19: Chinese strategy and experiences. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.04.07.20056440] [DOI] [Google Scholar]
Perotti 2020 {published data only}
- NCT04321421. Plasma from donors recovered from new coronavirus 2019 as therapy for critical patients with COVID-19. clinicaltrials.gov/show/NCT04321421 (first received 25 March 2020).
- Perotti C, Baldanti F, Bruno R, Delfante C, Seminari E, Casari S, et al. Mortality reduction in 46 severe COVID-19 patients treated with hyperimmune plasma. A proof of concept single arm multicenter interventional trial. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.05.26.20113373] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perotti C, Del Fante C, Baldanti F, Franchini M, Percivalle E, Vecchio Nepita E, et al. Plasma from donors recovered from the new coronavirus 2019 as therapy for critical patients with COVID-19 (COVID-19 plasma study): a multicentre study protocol. Internal and Emergency Medicine 2020 May 28 [Epub ahead of print]. [DOI: 10.1007/s11739-020-02384-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Salazar 2020 {published data only}
- Salazar E, Perez KK, Ashraf M, Chen J, Castillo B, Christensen PA, et al. Treatment of COVID-19 patients with convalescent plasma. American Journal of Pathology 2020 May 27 [Epub ahead of print]. [DOI: 10.1016/j.ajpath.2020.05.014] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar E, Perez KK, Ashraf M, Chen J, Castillo B, Christensen PA, et al. Treatment of COVID-19 patients with convalescent plasma in Houston, Texas. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.05.08.20095471] [DOI] [PMC free article] [PubMed] [Google Scholar]
Shen 2020 {published data only}
- Shen C, Wang Z, Zhao F, Yang Y, Li J, Yuan J, et al. Treatment of 5 critically ill patients with COVID-19 with convalescent plasma. JAMA 2020;323(16):1582-9. [DOI: 10.1001/jama.2020.4783] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tan 2020 {published data only}
- Tan L, Kang X, Zhang B, Zheng S, Liu B, Yu T, et al. A special case of COVID-19 with long duration of viral shedding for 49 days. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.03.22.20040071] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yang 2020 {published data only}
- Yang X, Sui Y, Liu F, Kang Z, Wu S, Zhao J, et al. Clinical characteristics and convalescent plasma therapy in severe and critically ill COVID-19 patients. Social Science Research Network 2020.
Ye 2020 {published data only}
- Ye M, Fu D, Ren Y, Wang F, Wang D, Zhang F, et al. Treatment with convalescent plasma for COVID-19 patients in Wuhan, China. Journal of Medical Virology 2020. [DOI: 10.1002/jmv.25882] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zeng 2020 {published data only}
- Zeng QL, Yu ZJ, Gou JJ, Li GM, Ma SH, Zhang GF, et al. Effect of convalescent plasma therapy on viral shedding and survival in COVID-19 patients. Journal of Infectious Diseases 2020 Jun 16 [Epub ahead of print]. [DOI: 10.1093/infdis/jiaa228] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020a {published data only}
- Zhang B, Liu S, Tan T, Huang W, Dong Y, Chen L, et al. Treatment with convalescent plasma for critically ill patients with SARS-CoV-2 infection. Chest 2020 Mar 31 [Epub ahead of print]. [DOI: 10.1016/j.chest.2020.03.039] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhang 2020b {published data only}
- Zhang L, Pang R, Xue X, Bao J, Ye S, Dai Y, et al. Anti-SARS-CoV-2 virus antibody levels in convalescent plasma of six donors who have recovered from COVID-19. Aging 2020 Apr 22 [Epub ahead of print];12. [DOI: 10.18632/aging.103102] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Alzoughool 2020 {published data only}
- Alzoughool F, Alanagreh L. Coronavirus drugs: using plasma from recovered patients as a treatment for COVID-19. International Journal of Risk & Safety in Medicine 2020;31(2):47-51. [DOI: 10.3233/JRS-201017] [DOI] [PMC free article] [PubMed] [Google Scholar]
Barone 2020 {published data only}
- Barone P, DeSimone RA. Convalescent plasma to treat coronavirus disease 2019 (COVID-19): considerations for clinical trial design. Transfusion 2020;60(6):1123-7. [DOI: 10.1111/trf.15843] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bloch 2020 {published data only}
- Bloch EM, Shoham S, Casadevall A, Sachais BS, Shaz B, Winters JL, et al. Deployment of convalescent plasma for the prevention and treatment of COVID-19. Journal of Clinical Investigation 2020. [DOI: 10.1172/JCI138745] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brasil Ministerio 2020 {published data only}
- Brasil Ministério da Saúde, Secretaria de Ciência. Tratamento farmacológico para casos internados com SARS-COV-2, do Hospital das Clínicas da Faculdade de Medicina de Ribeirão Preto da Universidade de São Paulo. portalarquivos.saude.gov.br/images/pdf/2020/May/11/TratamentoFarmacologico-SARS-COV-2-HC.RP.pdfhttp://fi-admin.bvsalud.org/document/view/27t7v (accessed prior to 25 June 2020).
Budhai 2020 {published data only}
- Budhai A, Wu AA, Hall L, Strauss D, Paradiso S, Alberigo J, et al. How did we rapidly implement a convalescent plasma program? Transfusion 2020 May 25 [Epub ahead of print]. [DOI: 10.1111/trf.15910] [DOI] [PMC free article] [PubMed] [Google Scholar]
Cao 2020a {published data only}
- Cao W, Liu X, Bai T, Fan H, Hong K, Song H, et al. High-dose intravenous immunoglobulin as a therapeutic option for deteriorating patients with coronavirus disease 2019. Open Forum Infectious Diseases 2020;7(3):ofaa102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cao 2020b {published data only}
- Cao H, Shi Y. Convalescent plasma: possible therapy for novel coronavirus disease 2019. Transfusion 2020;60(5):1078-83. [DOI: 10.1111/trf.15797] [DOI] [PMC free article] [PubMed] [Google Scholar]
Casadevall 2020a {published data only}
- Casadevall A, Pirofski LA. The convalescent sera option for containing COVID-19. Journal of Clinical Investigation 2020;130(4):1545-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Casadevall 2020b {published data only}
- Casadevall A, Joyner MJ, Pirofski LA. A randomized trial of convalescent plasma for COVID-19 - potentially hopeful signals. JAMA 2020 Jun 3 [Epub ahead of print]. [DOI: 10.1001/jama.2020.10218] [DOI] [PubMed] [Google Scholar]
Chen 2020a {published data only}
- Chen L, Xiong J, Bao L, Shi Y. Convalescent plasma as a potential therapy for COVID-19. Lancet Infectious Diseases 2020;27:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020b {published data only}
- Chen X, Li Y, Wang J, Cai H, Cao H, Sheng J. Pregnant women complicated with COVID-19: a clinical analysis of 3 cases. Zhejiang da Xue Xue Bao. Yi Xue Ban = Journal of Zhejiang University. Medical Sciences 2020;49(2):240-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chen 2020c {published data only}
- Chen Q, Quan B, Li X, Gao G, Zheng W, Zhang J, et al. A report of clinical diagnosis and treatment of nine cases of coronavirus disease 2019. Journal of Medical Virology 2020;92(6):683-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
ChiCTR2000030312 {published data only}
- ChiCTR2000030312. Cancelled, due to modify the protocol A single-center, open-label and single arm trial to evaluate the efficacy and safety of anti-SARS-CoV-2 inactivated convalescent plasma in the treatment& [A single-center, open-label and single arm trial to evaluate the efficacy and safety of anti-SARS-CoV-2 inactivated convalescent plasma in the treatment of novel coronavirus pneumonia (COVID-19) patient]. www.chictr.org.cn/showproj.aspx?proj=50258 (first received 23 April 2020).
ChiCTR2000030381 {published data only}
- ChiCTR2000030381. Cancelled by investigator A randomized, open-label, controlled and single-center trial to evaluate the efficacy and safety of anti-SARS-CoV-2 inactivated convalescent plasma in the treatment of novel coronavirus pneumonia (COVID-19) patient [A randomized, open-label, controlled and single-center trial to evaluate the efficacy and safety of anti-SARS-CoV-2 inactivated convalescent plasma in the treatment of novel coronavirus pneumonia (COVID-19) patient]. www.chictr.org.cn/showproj.aspx?proj=50290 (first received 23 April 2020).
ChiCTR2000030442 {published data only}
- ChiCTR2000030442. Combination of tocilizumab, IVIG and CRRT in severe patients with novel coronavirus pneumonia (COVID-19). www.chictr.org.cn/showproj.aspx?proj=50380 (first received 23 April 2020).
Datta 2020 {published data only}
- Datta SS, Basu S. Randomization amid a pandemic - a critical appraisal regarding convalescent plasma therapy clinical trials for COVID-19 patients. ISBT Science Series 2020. [DOI: 10.1111/voxs.12564] [DOI] [Google Scholar]
de Assis 2020 {published data only}
- Assis RR, Jain A, Nakajima R, Jasinskas A, Felgner J, Obiero JM, et al. Analysis of SARS-CoV-2 antibodies in COVID-19 convalescent plasma using a coronavirus antigen microarray. bioRxiv [Preprint] 2020. [DOI: 10.1101/2020.04.15.043364] [DOI] [PMC free article] [PubMed] [Google Scholar]
Díez 2020 {published data only}
- Díez JM, Romero C, Gajardo R. Currently available intravenous immunoglobulin (Gamunex-C and Flebogamma© DIF) contains antibodies reacting against SARS-CoV-2 antigens. bioRxiv [Preprint] 2020. [DOI: 10.1101/2020.04.07.029017] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dzik 2020 {published data only}
- Dzik S. COVID-19 convalescent plasma: now is the time for better science. Transfusion Medicine Reviews 2020 Apr 23 [Epub ahead of print]. [DOI: 10.1016/j.tmrv.2020.04.002] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fleming 2020 {published data only}
- Fleming AB, Raabe V. Current studies of convalescent plasma therapy for COVID-19 may underestimate risk of antibody-dependent enhancement. Journal of Clinical Virology 2020;127:104388. [DOI] [PMC free article] [PubMed] [Google Scholar]
Franchini 2020 {published data only}
- Franchini M, Marano G, Velati C, Pati I, Pupella S, Liumbruno GM. Operational protocol for donation of anti-COVID-19 convalescent plasma in Italy. Vox Sanguinis 2020. [DOI: 10.1111/vox.12940] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hammarström 2020 {published data only}
- Hammarström L, Abolhassani H, Baldanti F, Marcotte H, Pan-Hammarström Q. Development of passive immunity against SARS-CoV-2 for management of immunodeficient patients - a perspective. Journal of Allergy and Clinical Immunology 2020. [DOI: 10.1016/j.jaci.2020.04.043] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hu 2020 {published data only}
- Hu H, Ma F, Wei X, Fang Y. Coronavirus fulminant myocarditis saved with glucocorticoid and human immunoglobulin. European Heart Journal 2020;16:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
ISRCTN86534580 {published data only}
- ISRCTN86534580. A trial evaluating treatments for suspected coronavirus infection in people aged 50 years and above with pre-existing conditions and those aged 65 years and above. www.isrctn.com/ISRCTN86534580 (first received 20 March 2020).
Jawhara 2020 {published data only}
- Jawhara S. Could intravenous immunoglobulin collected from recovered coronavirus patients protect against COVID-19 and strengthen the immune system of new patients? International Journal of Molecular Sciences 2020;21(7):2272. [DOI: 10.3390/ijms21072272] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jiang 2020 {published data only}
- Jiang Y, He S, Zhang C, Wang X, Chen X, Jin Y, et al. Clinical characteristics of 60 discharged cases of 2019 novel coronavirus-infected pneumonia in Taizhou, China. Annals of Translational Medicine 2020;8(8):547. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kesici 2020 {published data only}
- Kesici S, Yavuz S, Bayrakci B. Get rid of the bad first: therapeutic plasma exchange with convalescent plasma for severe COVID-19. Proceedings of the National Academy of Sciences of the United States of America 2020;117(23):12526-7. [DOI: 10.1073/pnas.2006691117] [DOI] [PMC free article] [PubMed] [Google Scholar]
Khanna 2020 {published data only}
- Khanna SS, Qayyum MA, Patley RB, Patley A, Rathod D, Shah R, et al. Convalescent plasma therapy for coronavirus in critically ill patients. Journal of Advanced Medical and Dental Sciences Research 2020;8(4):57-60. [Google Scholar]
Knudson 2020 {published data only}
- Knudson CM, Jackson JB. COVID-19 convalescent plasma: phase 2. Transfusion 2020;60(6):1332-3. [DOI: 10.1111/trf.15842] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kominers 2020 {published data only}
- Kominers SD, Pathak PA, Sonmez T, Ünver MU. Paying it backward and forward: expanding access to convalescent plasma therapy through market design. SSRN 2020. [DOI: 10.2139/ssrn.3594465] [DOI] [Google Scholar]
Kumar 2020 {published data only}
- Kumar S, Sharma V, Priya K. Battle against COVID-19: efficacy of convalescent plasma as an emergency therapy. American Journal of Emergeny Medicine 2020;S0735-6757(20):30465-4. [DOI: 10.1016/j.ajem.2020.05.101] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lancet Haematology 2020 {published data only}
- Lancet Haematology. The resurgence of convalescent plasma therapy. Lancet Haematology 2020;7(5):e353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lanza 2020 {published data only}
- Lanza F, Seghatchian J. Reflection on passive immunotherapy in those who need most: some novel strategic arguments for obtaining safer therapeutic plasma or autologous antibodies from recovered COVID -19 infected patients. British Journal of Haematology 2020 May 14 [Epub ahead of print]. [DOI: 10.1111/bjh.16814] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lin 2020 {published data only}
- Lin JH, Chen YC, Lu CL, Hsu YN, Wang WJ. Application of plasma exchange in association with higher dose CVVH in cytokine storm complicating COVID-19. Journal of the Formosan Medical Association 2020;119(6):1116-8. [DOI: 10.1016/j.jfma.2020.04.023] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ministerio de Salud 2020 {published data only}
- Ministerio de salud - Instituto Nacional de Salud. Lineamientos técnicos para uso de plasma convaleciente en pacientes con COVID-19. fi-admin.bvsalud.org/document/view/nruba 2020;1:20.
NCT04261426 {published data only}
- NCT04261426. The efficacy of intravenous immunoglobulin therapy for severe 2019-nCoV infected pneumonia. clinicaltrials.gov/ct2/show/NCT04261426 (first received 23 April 2020).
NCT04323800 {published data only}
- NCT04323800. Convalescent plasma to stem coronavirus: a randomized, blinded phase 2 study comparing the efficacy and safety human coronavirus immune plasma (HCIP) vs. control (SARS-CoV-2 non-immune plasma) among adults exposed to COVID-19. clinicaltrials.gov/show/NCT04323800 (first received 23 April 2020).
NCT04325672 {published data only}
- NCT04325672. Convalescent plasma to limit coronavirus associated complications: an open label, phase 2A study of high-titer anti-SARS-CoV-2 plasma in hospitalized patients with COVID-19. clinicaltrials.gov/show/NCT04325672 (first received 23 April 2020).
NCT04344015 {published data only}
- NCT04344015. COVID-19 plasma collection. clinicaltrials.gov/show/NCT04344015 (first received 23 April 2020).
NCT04344379 {published data only}
- NCT04344379. Prevention of SARS-CoV-2 in hospital workers exposed to the virus. clinicaltrials.gov/show/NCT04344379 (first received 14 April 2020).
NCT04344977 {published data only}
- NCT04344977. COVID-19 plasma collection. clinicaltrials.gov/ct2/show/NCT04344977 (first received 14 April 2020).
NCT04350580 {published data only}
- NCT04350580. Polyvalent immunoglobulin in COVID-19 related ARDS. ClinicalTrials.gov/show/NCT04350580 (first received 17 April 2020).
NCT04360278 {published data only}
- NCT04360278. Plasma collection from convalescent and/or immunized donors for the treatment of COVID-19. clinicaltrials.gov/show/NCT04360278 (first received 24 April 2020).
NCT04368013 {published data only}
- NCT04368013. Host-pathogen interactions, immune response, and clinical prognosis at COVID-19 - the CoVUm trial. clinicaltrials.gov/show/NCT04368013 (first received 20 April 2020).
Pawar 2020 {published data only}
- Pawar AY, Hiray AP, Sonawane DD, Bhambar RS, Derle DV, Ahire YS. Convalescent plasma: a possible treatment protocol for COVID- 19 patients suffering from diabetes or underlying liver diseases. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 2020;14(4):665-9. [DOI: 10.1016/j.dsx.2020.05.023] [DOI] [PMC free article] [PubMed] [Google Scholar]
Qiu 2020 {published data only}
- Qiu T, Wang J, Zhou J, Zou J, Chen Z, Ma X, et al. The report of two cases infection with novel coronavirus (2019-NCcoV) after kidney transplantation and the association literature analyzation. Chinese Journal of Organ Transplantation 2020;41(0):E004. [Google Scholar]
Roback 2020 {published data only}
- Roback JD, Guarner J. Convalescent plasma to treat COVID-19: possibilities and challenges. JAMA 2020. [DOI: 10.1001/jama.2020.4940] [DOI] [PubMed] [Google Scholar]
Robbiani 2020 {published data only}
- Robbiani DF, Gaebler C, Muecksch F, Cetrulo LJ, Wang Z, Cho A, et al. Convergent antibody responses to SARS-CoV-2 infection in convalescent individuals. bioRxiv [Preprint] 2020. [DOI: 10.1101/2020.05.13.092619] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rubin 2020 {published data only}
- Rubin R. Testing an old therapy against a new disease: convalescent plasma for COVID-19. JAMA 2020 Apr 30 [Epub ahead of print]. [DOI: 10.1001/jama.2020.7456] [DOI] [PubMed] [Google Scholar]
Seghatchian 2020 {published data only}
- Seghatchian J, Lanza F. Convalescent plasma, an apheresis research project targeting and motivating the fully recovered COVID 19 patients: a rousing message of clinical benefit to both donors and recipients alike. Transfusion and Apheresis Science 2020 Apr 22 [Epub ahead of print]:102794. [DOI: 10.1016/j.transci.2020.102792] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sheridan 2020 {published data only}
- Sheridan C. Convalescent serum lines up as first-choice treatment for coronavirus. Nature Biotechnology 2020;38(6):655-8. [DOI: 10.1038/d41587-020-00011-1] [DOI] [PubMed] [Google Scholar]
Shi 2020 {published data only}
- Shi H, Zhou C, He P, Huang S, Duan Y, Wang X, et al. Successful treatment of plasma exchange followed by intravenous immunoglobulin in a critically ill patient with 2019 novel coronavirus infection. International Journal of Antimicrobial Agents 2020:105974. [DOI: 10.1016/j.ijantimicag.2020.105974] [DOI] [PMC free article] [PubMed] [Google Scholar]
Syal 2020 {published data only}
- Syal K. COVID-19: herd immunity and convalescent plasma transfer therapy. Journal of Medical Virology 2020;13:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tanne 2020 {published data only}
- Tanne JH. Covid-19: FDA approves use of convalescent plasma to treat critically ill patients. BMJ 2020;368:m1256. [DOI] [PubMed] [Google Scholar]
Tiberghien 2020 {published data only}
- Tiberghien P, Lambalerie X, Morel P, Gallian P, Lacombe K, Yazdanpanah Y. Collecting and evaluating convalescent plasma for COVID-19 treatment: why and how. Vox Sanguinis 2020. [DOI: 10.1111/vox.12926] [DOI] [PubMed] [Google Scholar]
Tu 2020 {published data only}
- Tu Y, Wu X, Liu F, Wang J, Luo Y, Cai Z, et al. Two clinical cases of novel coronavirus pneumonia (NCP) in renal transplant recipients. Chinese Journal of Organ Transplantation 2020;41(0):E005. [Google Scholar]
Wong 2020 {published data only}
- Wong HK, Lee CK. Pivotal role of convalescent plasma in managing emerging infectious diseases. Vox Sanguinis 2020. [DOI: 10.1111/vox.12927] [DOI] [PMC free article] [PubMed] [Google Scholar]
Xie 2020 {published data only}
- Xie Y, Cao S, Li Q, Chen E, Dong H, Zhang W, et al. Effect of regular intravenous immunoglobulin therapy on prognosis of severe pneumonia in patients with COVID-19. Journal of Infection 2020:S0163-4453(20)30172-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yoo 2020 {published data only}
- Yoo JH. Convalescent plasma therapy for corona virus disease 2019: a long way to go but worth trying. Journal of Korean Medical Science 2020;35(14):e150. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zeng 2020a {published data only}
- Zeng F, Chen X, Deng G. Convalescent plasma for patients with COVID-19. Proceedings of the National Academy of Sciences of the United States of America 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhao 2020b {published data only}
- Zhao Q, He Y. Challenges of convalescent plasma therapy on COVID-19. Journal of Clinical Virology 2020;127:104358. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhu 2020 {published data only}
- Zhu M, Kaiming H, Zhu Z. Use of convalescent plasma in COVID-19 patients in China. Transfusion Clinical Biology 2020;16:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
ChiCTR2000029850 {published data only}
- ChiCTR2000029850. Efficacy and safety of convalescent plasma treatment for severe patients with novel coronavirus pneumonia (COVID-19): a prospective cohort study. www.chictr.org.cn/showproj.aspx?proj=49533 (first received 15 February 2020).
ChiCTR2000030010 {published data only}
- ChiCTR2000030010. A randomized, double-blind, parallel-controlled, trial to evaluate the efficacy and safety of anti-SARS-CoV-2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia patients (COVID-19). www.chictr.org.cn/showproj.aspx?proj=49777 (first received 19 February 2020).
ChiCTR2000030039 {published data only}
- ChiCTR2000030039. Clinical study for infusing convalescent plasma to treat patients with new coronavirus pneumonia (COVID-19). www.chictr.org.cn/showproj.aspx?proj=49544 (first received 21 February 2020).
ChiCTR2000030179 {published data only}
- ChiCTR2000030179. Experimental study of novel coronavirus pneumonia rehabilitation plasma therapy severe novel coronavirus pneumonia (COVID-19). www.chictr.org.cn/showproj.aspx?proj=50059 (first received 24 February 2020).
ChiCTR2000030627 {published data only}
- ChiCTR2000030627. Study on the application of convalescent plasma therapy in severe COVID-19. www.chictr.org.cn/showproj.aspx?proj=50727 (first received 8 March 2020).
ChiCTR2000030702 {published data only}
- ChiCTR2000030702. Convalescent plasma for the treatment of common COVID-19: a prospective randomized controlled trial. www.chictr.org.cn/showproj.aspx?proj=50537 (first received 10 March 2020).
ChiCTR2000030929 {published data only}
- ChiCTR2000030929. A randomized, double-blind, parallel-controlled trial to evaluate the efficacy and safety of anti-SARS-CoV-2 virus inactivated plasma in the treatment of severe novel coronavirus pneumonia (COVID-19). www.chictr.org.cn/showproj.aspx?proj=50696 (first received 17 March 2020).
ChiCTR2000031501 {published data only}
- ChiCTR2000031501. The efficacy of convalescent plasma in patients with critical novel coronavirus pneumonia (COVID-19): a pragmatic, prospective cohort study. www.chictr.org.cn/showproj.aspx?proj=50254 (first received 2 April 2020).
EUCTR2020‐001310‐38 {published data only}
- EUCTR2020-001310-38. A randomized, prospective, open label clinical trial on the use of convalescent plasma compared to best supportive care in patients with severe COVID-19. www.clinicaltrialsregister.eu/ctr-search/search?query=eudract_number:2020-001310-38 (first received 23 April 2020).
IRCT20151228025732N53 {published data only}
- IRCT20151228025732N53. Therapeutic effects of plasma of recovered people from COVID-19 on hospitalized patients with this disease. en.irct.ir/trial/46931 (first received 10 April 2020).
IRCT20200310046736N1 {published data only}
- IRCT20200310046736N1. Comparison of the therapeutic effect of convalescent plasma and plasma-derived immunoglobulin-enriched solution on COVID-19 patients. en.irct.ir/trial/46424 (first received 1 April 2020).
IRCT20200325046860N1 {published data only}
- IRCT20200325046860N1. Convalescent plasma therapy for COVID-19 patients. en.irct.ir/trial/46759 (first received 30 March 2020).
IRCT20200404046948N1 {published data only}
- IRCT20200404046948N1. Efficacy and safety of convalescent plasma in the treatment of COVID-19. en.irct.ir/trial/46973 (first received 15 April 2020).
IRCT20200409047007N1 {published data only}
- IRCT20200409047007N1. Effect of COVID 19 survivors plasma in COVID 19 patients with ARDS. en.irct.ir/trial/47058 (first received 12 April 2020).
IRCT20200413047056N1 {published data only}
- IRCT20200413047056N1. Comparison between the efficacy of intravenous immunoglobulin and convalescent plasma in COVID-19. en.irct.ir/trial/47212 (first received 17 April 2020).
NCT04264858 {published data only}
- ChiCTR2000030841. Treatment of acute severe COVID-19 with immunoglobulin from cured COVID-19 patients. www.chictr.org.cn/showproj.aspx?proj=51072 (first received 15 March 2020).
- NCT04264858. An exploratory clinical study on the treatment of acute severe 2019-nCoV pneumonia with immunoglobulin from cured 2019-nCoV pneumonia patients. clinicaltrials.gov/show/NCT04264858 (first received 11 February 2020).
NCT04292340 {published data only}
- NCT04292340. The efficacy and safety of anti-SARS-CoV-2 inactivated convalescent plasma in the treatment of novel coronavirus pneumonia patient (COVID-19): an observational study. clinicaltrials.gov/show/NCT04292340 (first received 3 March 2020).
NCT04327349 {published data only}
- NCT04327349. Investigating effect of convalescent plasma on COVID-19 patients outcome: a clinical trial. clinicaltrials.gov/show/NCT04327349 (first received 31 March 2020).
NCT04332380 {published data only}
- NCT04332380. Convalescent plasma for patients with COVID-19: a pilot study. clinicaltrials.gov/show/NCT04332380 (first received 2 April 2020).
NCT04332835 {published data only}
- NCT04332835. Convalescent plasma for patients with COVID-19: a randomized, open label, parallel, controlled clinical study. clinicaltrials.gov/show/NCT04332835 (first received 3 April 2020).
NCT04333251 {published data only}
- NCT04333251. Evaluating convalescent plasma to decrease coronavirus associated complications. A phase I study comparing the efficacy and safety of high-titer anti-Sars-CoV-2 plasma vs best supportive care in hospitalized patients with interstitial pneumonia due to COVID-19. clinicaltrials.gov/show/NCT04333251 (first received 3 April 2020).
NCT04333355 {published data only}
- NCT04333355. Phase 1 study to evaluate the safety of convalescent plasma as an adjuvant therapy in patients with SARS-CoV-2 infection. clinicaltrials.gov/show/NCT04333355 (first received 3 April 2020).
NCT04338360 {published data only}
- NCT04338360. Expanded access to convalescent plasma for the treatment of patients with COVID-19. clinicaltrials.gov/show/NCT04338360 (first received 8 April 2020).
NCT04340050 {published data only}
- NCT04340050. COVID-19 convalescent plasma. clinicaltrials.gov/show/NCT04340050 (first received 9 April 2020).
NCT04342182 {published data only}
- NCT04342182. Convalescent plasma as therapy for COVID-19 severe SARS-CoV-2 disease (CONCOVID Study) (ConCoVid-19). cinicaltrials.gov/show/NCT04342182 (first received 10 April 2020).
NCT04343261 {published data only}
- NCT04343261. Convalescent plasma in the treatment of COVID 19. clinicaltrials.gov/show/NCT04343261 (first received 13 April 2020).
NCT04343755 {published data only}
- NCT04343755. Convalescent plasma as treatment for hospitalized subjects with COVID-19 infection. clinicaltrials.gov/show/NCT04343755 (first received 13 April 2020).
NCT04344535 {published data only}
- NCT04344535. Convalescent plasma vs. standard plasma for COVID-19. clinicaltrials.gov/show/NCT04344535 (first received 14 April 2020).
NCT04345289 {published data only}
- EUCTR2020-001367-88-DK. Efficacy and safety of novel treatment options for adults with COVID-19 pneumonia. apps.who.int/trialsearch/Trial2.aspx?TrialID=EUCTR2020-001367-88-DK (first received 14 April 2020).
- NCT04345289. Efficacy and safety of novel treatment options for adults with COVID-19 pneumonia (CCAP). clinicaltrials.gov/show/NCT04345289 (first received 14 April 2020).
NCT04345523 {published data only}
- NCT04345523. Convalescent plasma therapy vs. SOC for the treatment of COVID19 in hospitalized patients (ConPlas-19). clinicaltrials.gov/show/NCT04345523 (first received 14 April 2020).
NCT04345679 {published data only}
- NCT04345679. Anti COVID-19 convalescent plasma therapy. clinicaltrials.gov/show/NCT04345679 (first received 14 April 2020).
NCT04345991 {published data only}
- NCT04345991. Efficacy of convalescent plasma to treat COVID-19 patients, a nested trial in the CORIMUNO-19 cohort. clinicaltrials.gov/show/NCT04345991 (first received 15 April 2020).
NCT04346446 {published data only}
- NCT04346446. Efficacy of convalescent plasma therapy in severely sick COVID-19 patients. clinicaltrials.gov/show/NCT04346446 (first received 15 April 2020).
NCT04346589 {published data only}
- NCT04346589. Convalescent antibodies infusion in critically ill COVID 19 patients. clinicaltrials.gov/ct2/show/NCT04346589 (first received 15 April 2020).
NCT04347681 {published data only}
- NCT04347681. Potential efficacy of convalescent plasma to treat severe COVID-19 and patients at high risk of developing severe COVID-19. clinicaltrials.gov/show/NCT04347681 (first received 15 April 2020).
NCT04348656 {published data only}
- NCT04348656. Convalescent plasma for hospitalized adults with COVID-19 respiratory illness (CONCOR-1). clinicaltrials.gov/show/NCT04348656 (first received 16 April 2020).
NCT04348877 {published data only}
- NCT04348877. Plasma rich antibodies from recovered patients from COVID19. clinicaltrials.gov/show/NCT04348877 (first received 16 April 2020).
NCT04352751 {published data only}
- NCT04352751. Experimental use of convalescent plasma for passive immunization in current COVID-19 pandemic in Pakistan in 2020. clinicaltrials.gov/show/NCT04352751 (first received 20 April 2020).
NCT04353206 {published data only}
- NCT04353206. Convalescent plasma in ICU patients with COVID-19-induced respiratory failure. clinicaltrials.gov/show/NCT04353206 (first received 20 April 2020).
NCT04354831 {published data only}
- NCT04354831. A study evaluating the efficacy and safety of high-titer anti-SARS-CoV-2 plasma in hospitalized patients with COVID-19 infection. clinicaltrials.gov/ct2/show/NCT04354831 (first received 21 April 2020).
NCT04355767 {published data only}
- NCT04355767. Convalescent plasma vs. placebo in emergency room patients with COVID-19. clinicaltrials.gov/ct2/show/NCT04355767 (first received 21 April 2020).
NCT04355897 {published data only}
- NCT04355897. CoVID-19 plasma in treatment of COVID-19 patients. clinicaltrials.gov/ct2/show/NCT04355897 (first received 21 April 2020).
NCT04356482 {published data only}
- NCT04356482. Convalescent plasma for ill patients by COVID-19. clinicaltrials.gov/show/NCT04356482 (first received 22 April 2020).
NCT04356534 {published data only}
- NCT04356534. Convalescent plasma trial in COVID -19 patients. clinicaltrials.gov/show/NCT04356534 (first received 22 April 2020).
NCT04357106 {published data only}
- NCT04357106. COPLA study: treatment of severe forms of coronavirus infection with convalescent plasma. clinicaltrials.gov/show/NCT04357106 (first received 22 April 2020).
NCT04358211 {published data only}
- NCT04358211. Expanded access to convalescent plasma to treat and prevent pulmonary complications associated with COVID-19. clinicaltrials.gov/show/NCT04358211 (first received 24 April 2020).
NCT04358783 {published data only}
- NCT04358783. Convalescent plasma compared to the best available therapy for the treatment of SARS-CoV-2 pneumonia. clinicaltrials.gov/show/NCT04358783 (first received 24 April 2020).
NCT04359810 {published data only}
- NCT04359810. Plasma therapy of COVID-19 in critically ill patients. clinicaltrials.gov/show/NCT04359810 (first received 24 April 2020).
NCT04360486 {published data only}
- NCT04360486. Treatment of COVID-19 with anti-SARS-CoV-2 convalescent plasma (ASCoV2CP). clinicaltrials.gov/show/NCT04360486 (first received 24 April 2020).
NCT04361253 {published data only}
- NCT04361253. Evaluation of SARS-CoV-2 (COVID-19) antibody-containing plasma therapy. clinicaltrials.gov/show/NCT04361253 (first received 24 April 2020).
NCT04362176 {published data only}
- NCT04362176. Passive immunity trial of Nashville II. clinicaltrials.gov/show/NCT04362176 (first received 24 April 2020).
NCT04363034 {published data only}
- NCT04363034. Arkansas expanded access COVID-19 convalescent plasma treatment program. clinicaltrials.gov/ct2/show/NCT04363034 (first received 27 April 2020).
NCT04364737 {published data only}
- NCT04364737. Convalescent plasma to limit COVID-19 complications in hospitalized patients. clinicaltrials.gov/show/NCT04364737 (first received 28 April 2020).
NCT04365439 {published data only}
- NCT04365439. Convalescent plasma for COVID-19. clinicaltrials.gov/show/NCT04365439 (first received 28 April 2020).
NCT04366245 {published data only}
- NCT04366245. Clinical trial to evaluate the efficacy of treatment with hyperimmune plasma obtained from convalescent antibodies of COVID-19 infection. clinicaltrials.gov/show/NCT04366245 (first received 28 April 2020).
NCT04372368 {published data only}
- NCT04372368. Convalescent plasma for the treatment of patients with COVID-19. clinicaltrials.gov/show/NCT04372368 (first received 04 May 2020).
NCT04372979 {published data only}
- NCT04372979. Efficacy of convalescent plasma therapy in the early care of COVID-19 patients. clinicaltrials.gov/show/NCT04372979 (first received 04 May 2020).
NCT04373460 {published data only}
- NCT04373460. Convalescent plasma to limit SARS-CoV-2 associated complications. clinicaltrials.gov/show/NCT04373460 (first received 04 May 2020).
NCT04374370 {published data only}
- NCT04374370. SARSCoV2 (COVID-19) convalescent plasma (CP) expanded access protocol (EAP). clinicaltrials.gov/show/NCT04374370 (first received 5 May 2020).
NCT04374487 {published data only}
- NCT04374487. A phase II, open label, randomized controlled trial to assess the safety and efficacy of convalescent plasma to limit COVID-19 associated complications. clinicaltrials.gov/show/NCT04374487 (first received 5 May 2020).
NCT04374526 {published data only}
- NCT04374526. Early transfusIon of convalescent plasma in elderly COVID-19 patients to prevent disease progression. clinicaltrials.gov/show/NCT04374526 (first received 5 May 2020).
NCT04374565 {published data only}
- NCT04374565. Convalescent plasma for treatment of COVID-19 patients with pneumonia. clinicaltrials.gov/show/NCT04374565 (first received 5 May 2020).
NCT04375098 {published data only}
- NCT04375098. Efficacy and safety of early COVID-19 convalescent plasma in patients admitted for COVID-19 infection. clinicaltrials.gov/show/NCT04375098 (first received 5 May 2020).
NCT04376034 {published data only}
- NCT04376034. Convalescent plasma collection and treatment in pediatrics and adults. clinicaltrials.gov/show/NCT04376034 (first received 6 May 2020).
NCT04376788 {published data only}
- NCT04376788. Exchange transfusion versus plasma from convalescent patients with methylene blue in patients with COVID-19. clinicaltrials.gov/show/NCT04376788 (first received 6 May 2020).
NCT04377568 {published data only}
- NCT04377568. Efficacy of human coronavirus-immune convalescent plasma for the treatment of COVID-19 disease in hospitalized children. clinicaltrials.gov/show/NCT04377568 (first received 6 May 2020).
NCT04377672 {published data only}
- NCT04377672. Human convalescent plasma for high risk children exposed or infected with SARS-CoV-2. clinicaltrials.gov/show/NCT04377672 (first received 6 May 2020).
NCT04380935 {published data only}
- NCT04380935. Effectiveness and safety of convalescent plasma therapy on COVID-19 patients with acute respiratory distress syndrome. clinicaltrials.gov/show/NCT04380935 (first received 6 May 2020).
NCT04381858 {published data only}
- NCT04381858. Convalescent plasma vs human immunoglobulin to treat COVID-19 pneumonia. clinicaltrials.gov/show/NCT04381858 (first received 11 May 2020).
NCT04381936 {published data only}
- NCT04381936. Randomised evaluation of COVID-19 therapy (RECOVERY). clinicaltrials.gov/ct2/show/NCT04381936 (amended to include convalescent plasma 27 May 2020).
NCT04383535 {published data only}
- NCT04383535. Convalescent plasma and placebo for the treatment of COVID-19 severe pneumonia. clinicaltrials.gov/show/NCT04383535 (first received 12 May 2020).
NCT04383548 {published data only}
- NCT04383548. Clinical study for efficacy of anti-corona VS2 immunoglobulins prepared from COVID19 convalescent plasma prepared by VIPS mini-pool IVIG medical devices in prevention of SARS-CoV-2 infection in high risk groups as well as treatment of early cases of COVID. clinicaltrials.gov/show/NCT04383548 (first received 12 May 2020).
NCT04384497 {published data only}
- NCT04384497. Convalescent plasma for treatment of COVID-19: an exploratory dose identifying study. clinicaltrials.gov/show/NCT04384497 (first received 12 May 2020).
NCT04384588 {published data only}
- NCT04384588. COVID19-convalescent plasma for treating patients with active symptomatic COVID 19 infection (FALP-COVID). clinicaltrials.gov/show/NCT04384588 (first received 12 May 2020).
NCT04385043 {published data only}
- NCT04385043. Hyperimmune plasma in patients with COVID-19 severe infection. clinicaltrials.gov/show/NCT04385043 (first received 12 May 2020).
NCT04385186 {published data only}
- NCT04385186. Inactivated convalescent plasma as a therapeutic alternative in patients CoViD-19. clinicaltrials.gov/show/NCT04385186 (first received 12 May 2020).
NCT04385199 {published data only}
- NCT04385199. Convalescent plasma for patients with COVID-19. clinicaltrials.gov/show/NCT04385199 (first received 12 May 2020).
NCT04388410 {published data only}
- NCT04388410. Safety and efficacy of convalescent plasma transfusion for patients with SARS-CoV-2 infection. clinicaltrials.gov/show/NCT04388410 (first received 14 May 2020).
NCT04388527 {published data only}
- NCT04388527. COVID-19 convalescent plasma for mechanically ventilated population. clinicaltrials.gov/show/NCT04388527 (first received 14 May 2020).
NCT04389710 {published data only}
- NCT04389710. Convalescent plasma for the treatment of COVID-19. clinicaltrials.gov/show/NCT04389710 (first received 15 May 2020).
NCT04389944 {published data only}
- NCT04389944. Amotosalen-ultraviolet a pathogen-inactivated convalescent plasma in addition to best supportive care and antiviral therapy on clinical deterioration in adults presenting with moderate to severe COVID-19. clinicaltrials.gov/show/NCT04389944 (first received 15 May 2020).
NCT04390178 {published data only}
- NCT04390178. Convalescent plasma as treatment for acute coronavirus disease (COVID-19). clinicaltrials.gov/show/NCT04390178 (first received 15 May 2020).
NCT04390503 {published data only}
- NCT04390503. Convalescent plasma for COVID-19 close contacts. clinicaltrials.gov/ct2/show/NCT04390503 (first received 15 May 2020).
NCT04391101 {published data only}
- NCT04391101. Convalescent plasma for the treatment of severe SARS-CoV-2 (COVID-19). clinicaltrials.gov/show/NCT04391101 (first received 18 May 2020).
NCT04392232 {published data only}
- NCT04392232. A phase 2 study of COVID 19 convalescent plasma in high risk patients with COVID 19 infection. clinicaltrials.gov/show/NCT04392232 (first received 18 May 2020).
NCT04392414 {published data only}
- NCT04392414. Hyperimmune convalescent plasma in moderate and severe COVID-19 disease. clinicaltrials.gov/show/NCT04392414 (first received 18 May 2020).
NCT04393727 {published data only}
- NCT04393727. Transfusion of convalescent plasma for the early treatment of pneumonIa due to SARSCoV2. clinicaltrials.gov/show/NCT04393727 (first received 19 May 2020).
NCT04395170 {published data only}
- NCT04395170. Convalescent plasma compared to anti-COVID-19 human immunoglobulin and standard treatment (TE) in hospitalized patients. clinicaltrials.gov/show/NCT04395170 (first received 20 May 2020).
NCT04397523 {published data only}
- NCT04397523. Efficacy and safety of COVID-19 convalescent plasma. clinicaltrials.gov/show/NCT04397523 (first received 21 May 2020).
NCT04397757 {published data only}
- NCT04397757. COVID-19 convalescent plasma for the treatment of hospitalized patients with pneumonia caused by SARS-CoV-2. clinicaltrials.gov/show/NCT04397757 (first received 21 May 2020).
NCT04403477 {published data only}
- NCT04403477. Convalescent plasma therapy in severe COVID-19 infection. clinicaltrials.gov/show/NCT04403477 (first received 27 May 2020). [Google Scholar]
NCT04404634 {published data only}
- NCT04404634. Convalescent plasma to limit coronavirus associated complications. clinicaltrials.gov/show/NCT04404634 (first received 28 May 2020).
NCT04405310 {published data only}
- NCT04405310. Convalescent plasma of COVID-19 to treat SARS-COV-2 a randomized double blind 2 center trial (CPC-SARS). clinicaltrials.gov/show/NCT04405310 (first received 28 May 2020).
NCT04407208 {published data only}
- NCT04407208. Convalescent plasma therapy in patients with COVID-19. clinicaltrials.gov/show/NCT04407208 (first received 29 May 2020).
NCT04408040 {published data only}
- NCT04408040. Use of convalescent plasma for COVID-19. clinicaltrials.gov/show/NCT04408040 (first received 29 May 2020).
NCT04408209 {published data only}
- NCT04408209. Convalescent plasma for the treatment of patients with severe COVID-19 infection. clinicaltrials.gov/show/NCT04408209 (first received 29 May 2020).
NCT04412486 {published data only}
- NCT04412486. COVID-19 convalescent plasma (CCP) transfusion. clinicaltrials.gov/show/NCT04412486 (first received 02 June 2020).
U1111‐1251‐9286 {published data only}
- U1111-1251-9286. Effect of convalescent plasma in patients with severe COVID-19. www.ensaiosclinicos.gov.br/rg/RBR-4vm3yy/ (first received 11 May 2020).
Additional references
Balshem 2011
- Balshem H, Helfand M, Schünemann HJ, Oxman AD, Kunz R, Brozek J, et al. GRADE guidelines: 3. Rating the quality of evidence. Journal of Clinical Epidemiology 2011;64(4):401-6. [DOI] [PubMed] [Google Scholar]
Bao 2020a
- Bao L, Deng W, Gao H, Xiao C, Liu J, Xue J, et al. Reinfection could not occur in SARS-CoV-2 infected rhesus macaques. bioRxiv [Preprint] 2020. [DOI: 10.1101/2020.03.13.990226] [DOI] [Google Scholar]
Baudel 2020
- Baudel JL, Vigneron C, Pras-Landre V, Joffre J, Marjot F, Ait-Oufella H, et al. Transfusion-related acute lung injury (TRALI) after intravenous immunoglobulins: French multicentre study and literature review. Clinical Rheumatology 2020;39(2):541-6. [DOI] [PubMed] [Google Scholar]
Beigel 2017
- Beigel JH, Tebas P, Elie-Turenne MC, Bajwa E, Bell TE, Cairns B, et al. Immune plasma for the treatment of severe influenza: an open-label, multicentre, phase 2 randomised study. Lancet Respiratory Medicine 2017;5(6):500-11. [DOI: 10.1016/S2213-2600(17)30174-1] [DOI] [PMC free article] [PubMed] [Google Scholar]
Beigel 2019
- Beigel JH, Aga E, Elie-Turenne M-C, Cho J, Tebas P, Clark CL, et al. Anti-influenza immune plasma for the treatment of patients with severe influenza A: a randomised, double-blind, phase 3 trial. Lancet Respiratory Medicine 2019;7(11):941-50. [DOI: 10.1016/S2213-2600(19)30199-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
Beigel 2020
- Beigel JH, Tomashek KM, Dodd LE, Mehta AK, Zingman BS, Kalil AC, et al. Remdesivir for the treatment of COVID-19 — preliminary report. New England Journal of Medicine 2020. [DOI: 10.1056/NEJMoa2007764] [DOI] [PubMed] [Google Scholar]
Brennan 2003
- Brennan VM, Salomé-Bentley NJ, Chapel HM, Immunology Nurses Study. Prospective audit of adverse reactions occurring in 459 primary antibody-deficient patients receiving intravenous immunoglobulin. Clinical and Experimental Immunology 2003;133(2):247-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
CDC 2020a
- Centers for Disease Control and Prevention (CDC). Coronavirus Disease 2019 (COVID-19). Available at www.cdc.gov/coronavirus/2019-ncov/covid-data/covidview/index.html (accessed 13 April 2020).
CDC 2020b
- Centers for Disease Control and Prevention (CDC). Interim clinical guidance for management of patients with confirmed coronavirus disease (COVID-19). Available at www.cdc.gov/coronavirus/2019-ncov/hcp/clinical-guidance-management-patients.html (last accessed 26 April 2020).
Chun 2016
- Chun S, Chung CR, Ha YE, Han TH, Ki CS, Kang ES, et al. Possible transfusion-related acute lung injury following convalescent plasma transfusion in a patient with Middle East respiratory syndrome. Annals of Laboratory Medicine 2016;36(4):393-5. [DOI: 10.3343/alm.2016.36.4.393] [DOI] [PMC free article] [PubMed] [Google Scholar]
COMET 2020
- Core outcome set developers’ response to COVID-19 (2nd April 2020). Available at www.comet-initiative.org/Studies/Details/1538 (accessed 9 April 2020).
Covidence [Computer program]
- Veritas Health Innovation Covidence. Version accessed 20 April 2020. Melbourne, Australia: Veritas Health Innovation.Available at covidence.org.
Davey 2019
- Davey RT Jr, Fernández-Cruz E, Markowitz N, Pett S, Babiker AG, Wentworth D, et al. Anti-influenza hyperimmune intravenous immunoglobulin for adults with influenza A or B infection (FLU-IVIG): a double-blind, randomised, placebo-controlled trial. Lancet Respiratory Medicine 2019;7(11):951-63. [DOI: 10.1016/S2213-2600(19)30253-X] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2019
- Deeks JJ, Higgins JP, Altman DG, editor(s). Chapter 10: Analysing data and undertaking meta-analyses. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Driggin 2020
- Driggin E, Madhavan MV, Bikdeli B, Chuich T, Laracy J, Biondi-Zoccai G, et al. Cardiovascular considerations for patients, health care workers, and health systems during the COVID-19 pandemic. Journal of the American College of Cardiology 2020;75(18):2352-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Eibl 2008
- Eibl MM. History of immunoglobulin replacement. Immunology and Allergy Clinics of North America 2008;28(4):737-64. [DOI: 10.1016/j.iac.2008.06.004] [DOI] [PubMed] [Google Scholar]
EPOC 2017
- Cochrane Effective Practice and Organisation of Care (EPOC). What study designs can be considered for inclusion in an EPOC review and what should they be called? EPOC Resources for review authors. Available from epoc.cochrane.org/resources/epoc-resources-review-authors (accessed 23 April 2019).
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Version accessed 20 April 2020. Hamilton (ON): McMaster University (developed by Evidence Prime).Available at gradepro.org.
Higgins 2011
- Higgins JP, Altman DG, Sterne JA, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2019a
- Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook. [DOI] [PMC free article] [PubMed]
Higgins 2019b
- Higgins JP, Deeks JJ, editor(s). Chapter 6: Choosing effect measures and computing estimates of effect. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Ho 2005
- Ho MS, Chen WJ, Chen HY, Lin SF, Wang MC, Di J, et al. Neutralizing antibody response and SARS severity. Emerging Infectious Diseases 2005;11(11):1730-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Horby 2020
- Horby P, Lim WS, Emberson J, Mafham M, Bell J, Linsell L, et al. Effect of dexamethasone in hospitalized patients with COVID-19: preliminary report. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Huang 2020
- Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020;395(10223):497-506. [DOI: 10.1016/S0140-6736(20)30183-5] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hung 2013
- Hung IF, To KK, Lee CK, Lee KL, Yan WW, Chan K, et al. Hyperimmune IV immunoglobulin treatment: a multicenter double-blind randomized controlled trial for patients with severe 2009 influenza A(H1N1) infection. Chest 2013;144(2):464-73. [DOI: 10.1378/chest.12-2907] [DOI] [PubMed] [Google Scholar]
Kim 2020
- Kim D-H, Choe YJ, Jeong J-Y. Understanding and interpretation of case fatality rate of coronavirus disease 2019. Journal of Korean Medical Science 2020;35(12):e137. [DOI: 10.3346/jkms.2020.35.e137] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kreijtz 2011
- Kreijtz JH, Fouchier RA, Rimmelzwaan GF. Immune responses to influenza virus infection. Virus Research 2011;162(1-2):19-30. [DOI: 10.1016/j.virusres.2011.09.022] [DOI] [PubMed] [Google Scholar]
Lauer 2020
- Lauer SA, Grantz KH, Bi Q, Jones FK, Zheng Q, Meredith HR, et al. The incubation period of coronavirus disease 2019 (COVID-19) from publicly reported confirmed cases: estimation and application. Annals of Internal Medicine 2020:M20-0504. [DOI: 10.7326/M20-0504] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lefebvre 2019
- Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf MI, et al. Chapter 4: Searching for and selecting studies. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Li 2019
- Li T, Higgins JP, Deeks JJ, editor(s). Chapter 5: Collecting data. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Liang 2020
- Liang W, Guan W, Chen R, Wang W, Li J, Xu K, et al. Cancer patients in SARS-CoV-2 infection: a nationwide analysis in China. Lancet Oncology 2020;21(3):335-7. [DOI: 10.1016/S1470-2045(20)30096-6] [DOI] [PMC free article] [PubMed] [Google Scholar]
Luke 2006
- Luke TC, Kilbane EM, Jackson JL, Hoffman SL. Meta-analysis: convalescent blood products for Spanish influenza pneumonia: a future H5N1 treatment? Annals of Internal Medicine 2006;145(8):599-609. [DOI] [PubMed] [Google Scholar]
Mair‐Jenkins 2015
- Mair-Jenkins J, Saavedra-Campos M, Baillie JK, Cleary P, Khaw FM, Lim WS, et al. The effectiveness of convalescent plasma and hyperimmune immunoglobulin for the treatment of severe acute respiratory infections of viral etiology: a systematic review and exploratory meta-analysis. Journal of Infectious Diseases 2015;211(1):80-90. [DOI: 10.1093/infdis/jiu396] [DOI] [PMC free article] [PubMed] [Google Scholar]
McGuinness 2020
- McGuinness LA, Higgins JP. Risk-of-bias VISualization (robvis): An R package and Shiny web app for visualizing risk-of-bias assessments. Research Synthesis Methods 2020:1-7. [DOI: 10.1002/jrsm.1411] [DOI] [PubMed] [Google Scholar]
McKenzie 2019
- McKenzie JE, Brennan SE, Ryan RE, Thomson HJ, Johnston RV, Thomas J. Chapter 3: Defining the criteria for including studies and how they will be grouped for the synthesis. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al (editors). Cochrane Handbook for Systematic Reviews of Interventions version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Microsoft Corporation 2018 [Computer program]
- Microsoft Corporation, available at: office.microsoft.com/excel Mircosoft Excel. Microsoft Corporation. Microsoft Corporation, available at: office.microsoft.com/excel, 2018.
Mo 2006
- Mo H, Zeng G, Ren X, Li H, Ke C, Tan Y, et al. Longitudinal profile of antibodies against SARS-coronavirus in SARS patients and their clinical significance. Respirology 2006;11(1):49-53. [DOI: 10.1111/j.1440-1843.2006.00783.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Journal of Clinical Epidemiology 2009;62(10):1006-12. [DOI] [PubMed] [Google Scholar]
Morens 1994
- Morens DM. Antibody-dependent enhancement of infection and the pathogenesis of viral disease. Clinical Infectious Diseases 1994;19(3):500-12. [DOI] [PubMed] [Google Scholar]
Mulder 2019
- Mulder RL, Bresters D, Van den Hof M, Koot BG, Castellino SM, Loke YK, et al. Hepatic late adverse effects after antineoplastic treatment for childhood cancer. Cochrane Database of Systematic Reviews 2019, Issue 4. [DOI: 10.1002/14651858.CD008205.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Otrock 2017
- Otrock ZK, Liu C, Grossman BJ. Transfusion‐related acute lung injury risk mitigation: an update. Vox Sanguinis 2017;112(8):694-703. [DOI] [PubMed] [Google Scholar]
Pandey 2012
- Pandey S, Vyas GN. Adverse effects of plasma transfusion. Transfusion 2012;52 Suppl 1:65S-79S. [DOI] [PMC free article] [PubMed] [Google Scholar]
Parmar 1998
- Parmar MK, Torri V, Stewart L. Extracting summary statistics to perform meta-analyses of the published literature for survival endpoints. Statistics in Medicine 1998;17(24):2815-34. [DOI] [PubMed] [Google Scholar]
Payne 2016
- Payne DC, Iblan I, Rha B, Algasrawi S, Hin A, Al Nsour M, et al. Persistence of antibodies against Middle East respiratory syndrome coronavirus. Emerging Infectious Disease Journal 2016;22(10):1824-6. [DOI: 10.3201/eid2210.160706] [DOI] [PMC free article] [PubMed] [Google Scholar]
Reeves 2019
- Reeves BC, Deeks JJ, Higgins JP, Shea B, Tugwell P, Wells GA. Chapter 24: Including non-randomized studies on intervention effects. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Review Manager Web [Computer program]
- The Cochrane Collaboration Review Manager Web (RevMan Web). The Cochrane Collaboration, 2019.Available from revman.cochrane.org.
Ricke 2020
- Ricke D, Malone R. Medical countermeasures analysis of 2019-nCoV and vaccine risks for antibody-dependent enhancement (ADE). Preprints [Preprint] 2020. [DOI: 10.20944/preprints202003.0138.v1] [DOI] [Google Scholar]
Robbins 1995
- Robbins JB, Schneerson R, Szu SC. Perspective: hypothesis: serum IgG antibody is sufficient to confer protection against infectious diseases by inactivating the inoculum. Journal of Infectious Diseases 1995;171(6):1387-98. [DOI] [PubMed] [Google Scholar]
Rock 2011
- Rock G. A comparison of methods of pathogen inactivation of FFP. Vox Sanguinis 2011;100(2):169-78. [DOI] [PubMed] [Google Scholar]
Santesso 2020
- Santesso N, Glenton C, Dahm P, Garner P, Akl A, Alper B, et al. GRADE guidelines 26: informative statements to communicate the findings of systematic reviews of interventions. Journal of Clinical Epidemiology 2020;119:126-35. [DOI] [PubMed] [Google Scholar]
Schünemann 2019a
- Schünemann HJ, Vist GE, Higgins JP, Santesso N, Deeks JJ, Glasziou P, et al. Chapter 15: Interpreting results and drawing conclusions. In: Higgins JP, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, et al, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 6.0 (updated July 2019). Cochrane, 2019. Available from www.training.cochrane.org/handbook.
Schünemann 2019b
- Schünemann HJ, Cuello C, Akl EA, Mustafa RA, Meerpohl JJ, Thayer K, et al. GRADE guidelines: 18. How ROBINS-I and other tools to assess risk of bias in nonrandomized studies should be used to rate the certainty of a body of evidence. Journal of Clinical Epidemiology 2019;111:105-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sekul 1994
- Sekul EA, Cupler EJ, Dalakas MC. Aseptic meningitis associated with high-dose intravenous immunoglobulin therapy: frequency and risk factors. Annals of Internal Medicine 1994;121(4):259-62. [DOI] [PubMed] [Google Scholar]
Skoetz 2020
- Skoetz N, Goldkuhle M, Van Dalen EC, Akl EA, Trivella M, Mustafa RA, et al. GRADE guidelines 27: how to calculate absolute effects for time-to-event outcomes in summary of findings tables and Evidence Profiles. Journal of Clinical Epidemiology 2020;118:124-31. [DOI] [PubMed] [Google Scholar]
Sterne 2016
- Sterne JA, Hernán MA, Reeves BC, Savović J, Berkman ND, Viswanathan M, et al. ROBINS-I: a tool for assessing risk of bias in non-randomised studies of interventions. BMJ 2016;355:i4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2019
- Sterne JA, Savovic J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ 2019;366:l4898. [DOI] [PubMed] [Google Scholar]
Stiehm 2013
- Stiehm ER. Adverse effects of human immunoglobulin therapy. Transfusion Medicine Reviews 2013;27(3):171-8. [DOI] [PubMed] [Google Scholar]
Team 2020
- Team NCPERE . Vital surveillances: the epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) – China. China CDC Weekly 2020;2(8):113-22. [DOI: 10.3760/cma.j.issn.0254-6450.2020.02.003] [DOI] [PMC free article] [PubMed] [Google Scholar]
Tierney 2007
- Tierney JF, Stewart LA, Ghersi D, Burdett S, Sydes MR. Practical methods for incorporating summary time-to-event data into meta-analysis. Trials 2007;8:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tolouian 2020
- Tolouian R, Vahed SZ, Ghiyasvand S, Tolouian A, Ardalan M. COVID-19 interactions with angiotensin-converting enzyme 2 (ACE2) and the kinin system; looking at a potential treatment. Journal of Renal Injury Prevention 2020;9(2):e19. [DOI: 10.34172/jrip.2020.19] [DOI] [Google Scholar]
US Covid Plasma 2020
- US Covid Plasma. COVID-19 expanded access program. Available from www.uscovidplasma.org (accessed 9 July 2020).
Van de Veerdonk 2020
- Van de Veerdonk F, Netea MG, Van Deuren M, Van der Meer JW, De Mast Q, Bruggemann RJ, et al. Kinins and cytokines in COVID-19: a comprehensive pathophysiological approach. Preprints [Preprint] 2020. [DOI: 10.20944/preprints202004.0023.v1] [DOI] [Google Scholar]
Wan 2020
- Wan Y, Shang J, Sun S, Tai W, Chen J, Geng Q, et al. Molecular mechanism for antibody-dependent enhancement of coronavirus entry. Journal of Virology 2020;94(5):e02015-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 2014
- Wang S-F, Tseng S-P, Yen C-H, Yang J-Y, Tsao C-H, Shen C-W, et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochemical and Biophysical Research Communications 2014;451(2):208-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2007
- World Health Organization (WHO). Cumulative number of reported probable cases of SARS. www.who.int/csr/sars/country/2003_07_11/en/ (accessed 13 April 2020).
WHO 2019
- World Health Organization (WHO). Middle East respiratory syndrome coronavirus (MERS-CoV). www.who.int/emergencies/mers-cov/en/ (accessed 13 April 2020).
WHO 2020a
- World Health Organization (WHO). Report of the WHO‐China Joint Mission on coronavirus disease 2019 (COVID‐19); February 2020. www.who.int/docs/default-source/coronaviruse/who-china-joint-mission-on-covid-19-final-report.
WHO 2020b
- World Health Organization (WHO). Rolling updates on coronavirus diseases (COVID‐19). www.who.int/emergencies/diseases/novel-coronavirus-2019/events-as-they-happen (accessed 8 July 2020).
WHO 2020c
- World Health Organization (WHO). Coronavirus disease (COVID-19) situation report-169; 7 July 2020. www.who.int/docs/default-source/coronaviruse/situation-reports/20200707-covid-19-sitrep-169.pdf.
WHO 2020d
- World Health Organization (WHO). Clinical management of severe acute respiratory infection (SARI) when COVID19 disease is suspected: interim guidance. World Health Organization 2020;WHO/2019-nCoV/clinical/2020.4.
Wu 2020a
- Wu C, Chen X, Cai Y, Xia J, Zhou X, Xu S, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Internal Medicine 2020. [DOI: 10.1001/jamainternmed.2020.0994] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wu 2020b
- Wu F, Wang A, Liu M, Wang Q, Chen J, Xia S, et al. Neutralizing antibody responses to SARS-CoV-2 in a COVID-19 recovered patient cohort and their implications. medRxiv [Preprint] 2020. [DOI: ] [Google Scholar]
Xu 2020
- Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respiratory Medicine 2020;8(4):420-2. [DOI: 10.1016/S2213-2600(20)30076-X] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zhao 2020a
- Zhao J, Yuan Q, Wang H, Liu W, Liao X, Su Y, et al. Antibody responses to SARS-CoV-2 in patients of novel coronavirus disease 2019. medRxiv [Preprint] 2020. [DOI: 10.1101/2020.03.02.20030189] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Piechotta 2020
- Piechotta V, Valk SJ, Chai KL, Wood EM, Lamikanra A, Kimber C, et al. Safety and effectiveness of convalescent plasma or hyperimmune globulin for people with COVID-19: a rapid review. available at: doi.org/10.17605/OSF.IO/DWF53 . [DOI: 10.17605/OSF.IO/DWF53] [DOI]
Valk 2020
- Valk SJ, Piechotta V, Chai KL, Doree C, Monsef I, Wood EM, et al. Convalescent plasma or hyperimmune immunoglobulin for people with COVID-19: a rapid review. Cochrane Database of Systematic Reviews 2020, Issue 5. [DOI: 10.1002/14651858.CD013600] [DOI] [PMC free article] [PubMed] [Google Scholar]