

Abstract
目的
探讨葡萄糖转运子1缺陷综合征(GLUT1-DS)的临床特征和诊疗方法,分析运动障碍的诊断意义。
方法
收集4例GLUT1-DS患儿的临床资料,分析其临床特点和治疗随访情况。
结果
4例中男2例、女2例,起病年龄2~15个月。表现为运动障碍、癫癎发作和发育迟缓,均以癫癎发作为首诊原因。4例均有持续性共济失调、肌张力异常和构音障碍,2例有持续性震颤,发作性肢体瘫痪和眼球运动障碍各2例,劳累易诱发发作性症状。4例患儿的脑脊液葡萄糖及其与血糖的比值均降低。4例均检测到SLC2A1基因突变,均接受生酮饮食治疗,生酮比3:1~2:1,发作性症状5周内完全缓解。
结论
对于合并多样化运动障碍的智力运动发育迟缓的癫癎患儿需考虑GLUT1-DS,生酮饮食的生酮比维持在3:1~2:1可起效。
Keywords: 葡萄糖转运子1缺陷综合征, 运动障碍, 发育迟缓, 癫癎, SLC2A1基因, 儿童
Abstract
Objective
To investigate the clinical features, diagnosis and treatment of glucose transporter 1 deficiency syndrome (GLUT1-DS), as well as the diagnostic value of movement disorders.
Methods
The clinical data of four children with GLUT1-DS were collected, and their clinical features, treatment, and follow-up results were analyzed.
Results
There were two boys and two girls, with an age of onset of 2-15 months. Clinical manifestations included movement disorders, seizures, and developmental retardation. Seizures were the cause of the first consultation in all cases. The four children all had persistent ataxia, dystonia, and dysarthria; two had persistent tremor, two had paroxysmal limb paralysis, and two had eye movement disorders. Paroxysmal symptoms tended to occur in fatigue state. All four children had reductions in the level of cerebrospinal fluid glucose and its ratio to blood glucose, as well as SLC2A1 gene mutations. The four children were given a ketogenic diet, at a ketogenic ratio of 2:1 to 3:1, and achieved complete remission of paroxysmal symptoms within 5 weeks.
Conclusions
GLUT1-DS should be considered for epileptic children with mental retardation and motor developmental delay complicated by various types of movement disorders. The ketogenic diet is effective at a ketogenic ratio of 2:1 to 3:1 for the treatment of GLUT1-DS.
Keywords: Glucose transporter 1 deficiency syndrome, Movement disorder, Developmental retardation, Epilepsy, SLC2A1 gene, Child
葡萄糖转运子1缺陷综合征(glucose transporter 1 deficiency syndrome, GLUT1-DS, OMIM606777)是一种罕见的常染色体显性遗传病, 发病率为1/90 000[1]。GLUT1-DS的致病基因为SLC2A1基因, 该基因突变导致GLUT1功能障碍, 葡萄糖不能有效通过血脑屏障, 脑组织能量供应不足, 进而引起一系列神经系统症状, 表现为多样化的运动障碍、药物难治性癫癎、发育落后。生酮饮食为特效治疗, 早期治疗者认知预后较好。然而, 癫癎发作和发育落后为神经系统疾病常见症状, 对于早期诊断缺乏特异性, 因此多样化的运动障碍对GLUT1-DS诊断至关重要。本文对4例GLUT1-DS患儿临床资料进行总结, 重点分析多样化运动障碍的特点, 旨在提高对本病的认识, 发现有助于早期诊断的临床线索。
1. 资料与方法
1.1. 研究对象
以2016年3月至2017年3月于首都儿科研究所神经内科经遗传学确诊的4例GLUT1-DS患儿为研究对象。收集患儿临床资料, 分析运动障碍等临床特征、辅助检查和治疗随访情况。辅助检查包括血生化、尿气相质谱有机酸分析、血串联质谱酰基肉碱谱分析、头颅MRI检查、脑电图和脑脊液检查。每例患儿分别于生酮饮食治疗前和治疗3个月后行脑电图检测。腰穿前患儿禁食4 h, 测快速血糖后行腰椎穿刺术, 并测定脑脊液葡萄糖、计算脑脊液葡萄糖/血糖比值。饮食中脂肪与碳水化合物和蛋白质的质量比(生酮比)从2:1起始, 逐渐引入生酮饮食, 每月至少电话随访1次:记录饮食情况, 询问所有与治疗目的无关的反应, 监测血糖、血β-羟丁酸, 观察疗效和不良反应。
本研究获得患儿监护人知情同意, 以及医院临床研究伦理委员会批准(批准文号:SHERLL 2016033)。
1.2. 基因检测
采集患儿及其父母静脉血5 mL, 采用全遗传病基因靶向捕获二代测序技术进行基因检测(信诺佰世检验所检测)。错义突变采用Mutation Taster、SIFT和PolyPhen2软件预测其对蛋白功能的影响。
2. 结果
2.1. 临床特点
4例中男2例、女2例, 起病年龄2~15个月, 确诊年龄3岁4个月~11岁1个月。4例均有持续性运动障碍, 表现为共济失调、肌张力异常和构音障碍, 例1、例3还伴有震颤。共济失调轻者仅表现行走欠稳, 四肢协调性欠佳, 重者表现为持物不稳, 不能独走, 剪刀步态呈“Z”形前进; 肌张力异常表现为双下肢肌张力增高, 上肢肌张力尚可; 构音障碍表现为语言含糊不清, 说话费力, 不自然的中断。3例有发作性运动障碍, 例1表现为发作性肢体瘫痪, 例3为发作性眼球运动障碍, 例2既有发作性肢体瘫痪也有发作性眼球运动障碍。发作性运动障碍均于清醒状态发生, 玩耍或晨起空腹时多见, 抗癫癎治疗无效。见表 1。
1.
4例GLUT1-DS患儿的临床表现和SLC2A1基因突变分析
| 病例 | 性别 | 起病年龄(月) | 确诊年龄 | 肌张力异常 | 共济失调 | 构音障碍 | 震颤 | 发作性眼球运动障碍 | 发作性肢体瘫痪 | 癫癎发作 | 发育落后 | 踝阵挛 | EEG | 头颅MRI | SLC2A1突变 | 突变来源 |
| 注:[GLUT1-DS]葡萄糖转运子1缺陷综合征。 | ||||||||||||||||
| 1 | 女 | 11 | 4岁2个月 | + | + | + | + | - | + | 强直, 失张力 | + | + | 全导阵发性高幅慢波, 夹杂癫癎放电 | 正常 | c.787_791del TTCCG | 新生 |
| 2 | 男 | 15 | 11岁1个月 | + | + | + | - | + | + | 强直阵挛, 失张力, 复杂部分性 | + | + | 右侧半球持续性慢波, 右侧半球、左侧额区癫癎放电 | 双侧额叶皮层下异常信号 | c.376G > A | 新生 |
| 3 | 男 | 2 | 3岁4个月 | + | + | + | + | + | - | 强直阵挛 | + | + | 全导多量癫癎放电, 额区、枕区著 | 双侧额颞叶脑沟增宽 | c.377G > A | 新生 |
| 4 | 女 | 6 | 6岁 | + | + | + | - | - | - | 强直阵挛 | + | + | 双侧额、中央、额中线区偶见不典型尖波 | 正常 | c.688_689 insT | 新生 |
4例患儿首诊原因均为癫癎发作, 例1表现为强直发作和失张力发作, 例2为强直阵挛发作、失张力发作、复杂部分性发作, 例3、例4仅为强直阵挛发作。例1应用托吡酯, 例2应用拉莫三嗪和丙戊酸钠, 例3应用左乙拉西坦和奥卡西平, 例4联用丙戊酸钠、左乙拉西坦和奥卡西平治疗。
4例患儿均有不同程度发育落后, 理解力、计算力差, 语言含糊不清, 运动协调性差。例1身高、体重均低于同年龄、同性别儿童身高、体重的2个标准差, 其余3例体格发育大致正常。4例患儿头围均正常。
4例患儿起病年龄平均8个月, 确诊年龄平均为5岁5个月。其中例1、例3、例4曾经诊断为癫癎, 未进一步行病因诊断; 例2曾因发作性四肢瘫就诊, 因脑脊液葡萄糖2.09 mmol/L, 诊断为中枢神经系统感染。
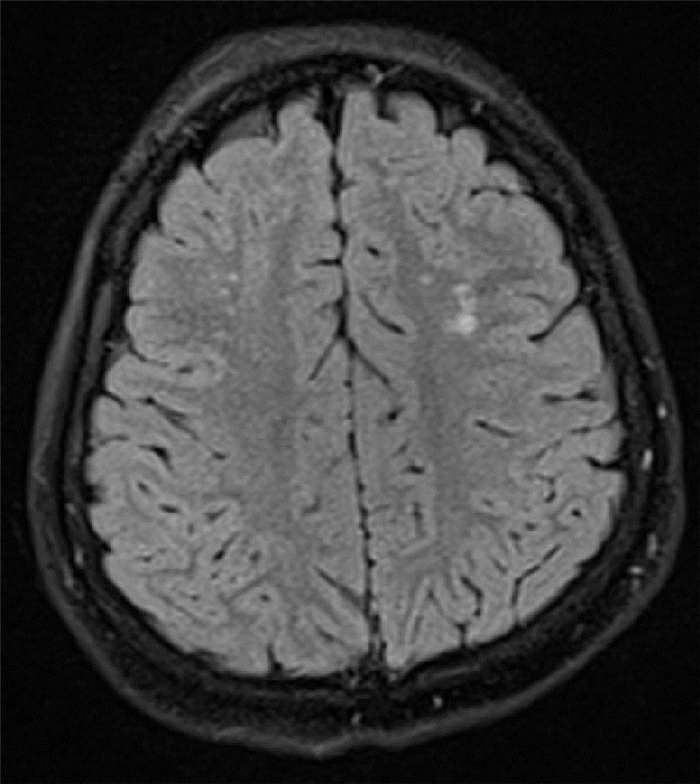
4例患儿肝、肾功能均正常, 血糖、血气分析、血氨、乳酸、同型半胱氨酸正常; 血、尿代谢筛查均未发现有机酸、氨基酸和脂肪酸代谢障碍。头颅MRI异常者2例, 例2表现为双侧额叶皮层下白质T2 FLAIR高信号(图 1), 例3为双侧额颞叶脑沟增宽。例2发作间期脑电图示右侧半球持续性慢波和大量癫癎样放电; 例1、例3脑电图示全导联癫癎样放电; 例4脑电图仅记录到少量不典型尖波。见表 1。4例患儿脑脊液葡萄糖以及脑脊液葡萄糖/血糖比值均明显降低。
1.
病例2确诊时头颅MRI
双侧额叶皮层下白质T2 FLAIR高信号(如箭头所示)。
2.2. 基因检测结果
4例患儿均检测到SLC2A1基因突变, 均为新生突变。例1第6外显子c.787_791delTTCCG(p.F263Lfs)突变(图 2); 例2第4外显子c.376G > A(p.R126C)突变; 例3第6外显子c.377G > A(p.R126H)突变; 例4第6外显子c.688_689insT突变(图 3)。例2、例3为错义突变, 经Mutation Taster、SIFT和PolyPhen2软件预测, 为致病性突变。例1、例4为移码突变, 均为致病性突变。
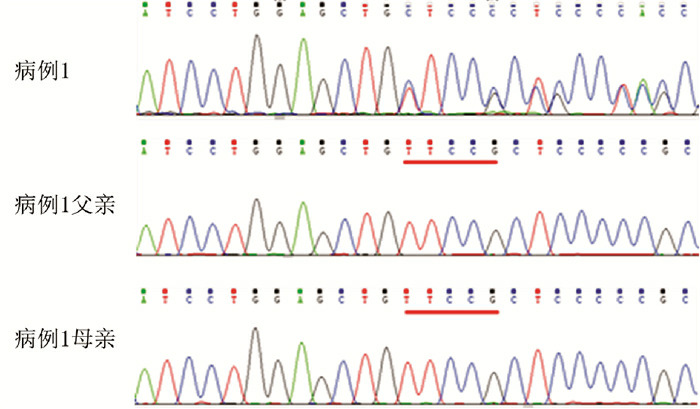
2.
病例1的SLC2A1基因突变及其父母的一代测序图
病例1存在SLC2A1基因c.787_791delTTCCG杂合突变(缺失的碱基如红线所示), 其父母该位点正常。
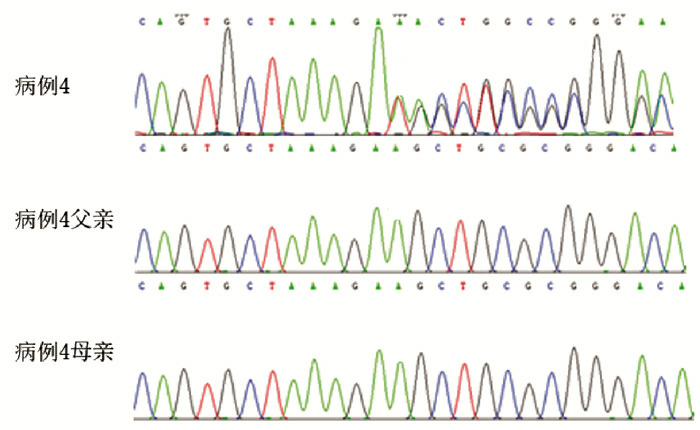
3.
病例4的SLC2A1基因突变及其父母的一代测序图
病例4存在SLC2A1基因c.688_689insT杂合突变(突变位点如红箭头所示), 其父母该位点正常。
2.3. 治疗及随访
4例患儿确诊后均加用生酮饮食治疗, 耐受性和依从性好, 随访4个月至1年5个月。例1、例2、例3的生酮比为2:1, 例4为3:1, 血β-羟丁酸维持在1.80~3.04 mmol/L, 平均2.38 mmol/L。例1、例4于生酮饮食2周内, 例2于4周内, 例3于5周内, 癫癎发作以及发作性运动障碍均完全缓解。治疗3个月后复查长程视频脑电图均正常。生酮饮食治疗3个月内, 1例患儿有过低血糖发生; 4例患儿均有消化道症状:表现为呕吐、恶心、腹胀、便秘, 3个月后明显减轻。生酮饮食治疗6个月后, 例1、例3、例4抗癫癎药物开始减量, 例1已减停, 例3、例4仍在减量中。
3. 讨论
GLUT1-DS是常染色体显性遗传病, SLC2A1基因为致病基因, 定位于1p34.2, 编码的钠依赖葡萄糖转运子1蛋白具有492个氨基酸。神经系统的GLUT1分布在血管内皮细胞膜和神经胶质细胞膜上。神经胶质细胞通过细胞膜上的GLUT1摄取葡萄糖, 葡萄糖经糖酵解途径转化成乳酸, 乳酸通过单羧酸转运体进入神经元细胞, 参与线粒体氧化代谢, 为脑组织提供能量[2]。当GLUT1表达异常, 葡萄糖不能有效通过血脑屏障, 导致脑组织持续缺乏能量供应, 进而引起一系列神经系统症状。
经典型GLUT1-DS以癫癎发作, 运动障碍和发育落后为主征[3]。本研究患儿均有多种运动障碍、癫癎发作和发育迟缓, 故为经典型GLUT1-DS。Akman等[4]认为, GLUT1-DS早期诊断困难, 多样化的运动障碍是特征性表现, 识别各种运动障碍有助于诊断。GLUT1-DS典型的运动障碍包括步态异常(89%)、肌张力障碍(86%)、意向性震颤(70%), 不典型的运动异常包括眼球运动障碍、偏瘫、单瘫、四肢瘫等, 运动障碍可为持续性或发作性[5]。本组患儿持续性的运动障碍表现为共济失调、肌张力异常、构音障碍、震颤; 3例患儿有包括发作性眼球运动障碍在内的发作性运动障碍, 均于清醒状态下出现, 空腹或玩耍时多发, 抗癫癎药物治疗无效, 视频脑电图监测也未记录到发作性肢体无力。但国外文献[6]报道, GLUT1-DS患儿发作性偏瘫的同期脑电图示偏瘫对侧的背景活动变慢, 故发作性症状为非癫癎事件。国外文献[7]报道, 约32%的GLUT1-DS患儿有发作性眼球运动障碍, 多于病程早期出现, 表现为眼阵挛、斜视、眼球旋转, 眼球水平或旋转性震颤, 眼球异常运动有较固定的间隔时间:约200~800 ms, 而且发作性眼球运动障碍同期的脑电图无癫癎样放电。眼球运动障碍病因尚不清楚, 推测与大脑发育不成熟或能量耗竭相关[8]。例2于10岁10个月首次出现眼球运动障碍, 而非病程早期, 故推测能量耗竭可能性更大。文献报道GLUT1-DS患儿的头颅MRI大多表现为脑外间隙增宽或髓鞘化延迟[3], 而例2头颅MRI可见额叶白质T2Flaire高信号, 提示脑白质损伤, 推测与治疗前病史较长有关。文献[9]报道, GLUT1-DS的癫癎发作常表现为全面强直-阵挛发作(53%)、失神发作(49%)、复杂部分性发作(37%)、肌阵挛发作(27%)、失张力发作(26%)、强直发作(12%)和痉挛发作(3%)。而4岁以内起病的失神癫癎患儿中GLUT1-DS占12%[10]。本研究4例患儿无失神发作, 可能与观察病例尚少且失神发作不易被发现相关。国外文献报道在GLUT1-DS经典型患儿中小头畸形为突出表现[11]。但刘燕燕等[12]报道的6例患儿中仅1例有小头畸形。本研究4例患儿均无小头畸形。提示头围正常不能除外本病。
GLUT1-DS的诊断需排除中枢神经系统感染, 而且脑脊液葡萄糖浓度 < 2.2 mmol/L、葡萄糖/血糖的比值< 0.45即可临床诊断GLUT1-DS[13]。基因检测发现SLC2A1基因突变可确诊[14]。对于基因检测阴性的患儿, 外周血红细胞3-氧甲基-D-葡萄糖摄取率 < 60%可诊断本病。文献[3]报道SLC2A1基因新生突变占90%。本研究4例患儿的脑脊液葡萄糖小于2.2 mmol/L, 脑脊液葡萄糖/血糖比值小于0.45;患儿均携带SLC2A1基因突变, 均为新生突变, 其中2例移码突变为国内外尚未报道的新突变。
生酮饮食为本病特效治疗, 早期治疗可明显改善预后[15]。约8%的患儿可通过抗癫癎药物控制癫癎发作, 但应避免苯巴比妥、水合氯醛、丙戊酸钠等药物[8]。生酮饮食近期不良反应为消化道症状、代谢性酸中毒、低血糖, 远期不良反应包括肾结石、高脂血症、生长发育迟缓等。本研究患儿均应用生酮饮食, 以较低的生酮比(3:1~2:1)维持, 耐受性和依从性好, 发作性症状在治疗5周内完全控制, 脑电图亦明显改善。因此GLUT1-DS患儿的生酮饮食可以较低的生酮比维持[16]。
综上, GLUT1-DS为可治疗的遗传性疾病, 早期诊断至关重要。在发育落后的癫癎患儿, 共济失调、肌张力障碍、构音障碍、震颤、发作性肢体瘫痪和发作性眼球运动异常等多种运动障碍可为诊断提供线索。脑脊液葡萄糖/血糖比值降低和SLC2A1基因突变可明确诊断。生酮饮食以较低的生酮比维持即可控制症状。
Biographies
姬辛娜, 女, 硕士, 主治医师
Chen Q,Email:chenqianxhl@163.com
References
- 1.Coman DJ, Sinclair KG, Burke CJ, et al. Seizures, ataxia, developmental delay and the general paediatrician:glucose transporter 1 deficiency syndrome. J Paediatr Child Health. 2006;42(5):263–267. doi: 10.1111/jpc.2006.42.issue-5. [DOI] [PubMed] [Google Scholar]
- 2.Benarroch EE. Brain glucose transporters:implications for neurologic disease. Neurology. 2014;82(15):1374–1379. doi: 10.1212/WNL.0000000000000328. [DOI] [PubMed] [Google Scholar]
- 3.Pearson TS, Akman C, Hinton VJ, et al. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome. Curr Neurol Neurosci Rep. 2013;13(4):342. doi: 10.1007/s11910-013-0342-7. [DOI] [PubMed] [Google Scholar]
- 4.Akman CI, Yu J, Alter A, et al. Diagnosing glucose transporter 1 deficiency at initial presentation facilitates early treatment. J Pediatr. 2016;171:220–226. doi: 10.1016/j.jpeds.2015.12.030. [DOI] [PubMed] [Google Scholar]
- 5.Pons R, Collins A, Rotstein M, et al. The spectrum of movement disorders in Glut-1 deficiency. Mov Disord. 2010;25(3):275–281. doi: 10.1002/mds.22808. [DOI] [PubMed] [Google Scholar]
- 6.Pellegrin S, Cantalupo G, Opri R, et al. EEG findings during "paroxysmal hemiplegia" in a patient with GLUT1-deficiency. Eur J Paediatr Neurol. 2017;21(3):580–582. doi: 10.1016/j.ejpn.2017.01.002. [DOI] [PubMed] [Google Scholar]
- 7.Pearson TS, Pons R, Engelstad K, et al. Paroxysmal eye-head movements in GLUT1 deficiency syndrome. Neurology. 2017;88(17):1666–1673. doi: 10.1212/WNL.0000000000003867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Giorgis V, Varesio C, Baldassari C, et al. Atypical manifestations in Glut1 deficiency syndrome. J Child Neurol. 2016;31(9):1174–1180. doi: 10.1177/0883073816650033. [DOI] [PubMed] [Google Scholar]
- 9.Pong AW, Geary BR, Engelstad KM, et al. Glucose transporter type Ⅰ deficiency syndrome:epilepsy phenotypes and outcomes. Epilepsia. 2012;53(9):1503–1510. doi: 10.1111/epi.2012.53.issue-9. [DOI] [PubMed] [Google Scholar]
- 10.Suls A, Mullen SA, Weber YG, et al. Early-onset absence epilepsy caused by mutations in the glucose transporter GLUT1. Ann Neurol. 2009;66(3):415–419. doi: 10.1002/ana.v66:3. [DOI] [PubMed] [Google Scholar]
- 11.Brockmann K. The expanding phenotype of GLUT1-deficiency syndrome. Brain Dev. 2009;31(7):545–552. doi: 10.1016/j.braindev.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 12.刘 燕燕, 包 新华, 王 爽, et al. 葡萄糖转运子1缺乏综合征的临床特点与基因突变分析. http://www.doc88.com/p-8028028736694.html. 中华儿科杂志. 2013;51(6):443–447. [PubMed] [Google Scholar]
- 13.Klepper J. GLUT1 deficiency syndrome in clinical practice. Epilepsy Res. 2012;100(3):272–277. doi: 10.1016/j.eplepsyres.2011.02.007. [DOI] [PubMed] [Google Scholar]
- 14.Fajardo M, Cirillo ML. Understanding the spectrum of SLC2A1-associated disorders. Pediatr Neurol Briefs. 2017;31(2):4. doi: 10.15844/pedneurbriefs-31-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujii T, Ito Y, Takahashi S, et al. Outcome of ketogenic diets in GLUT1 deficiency syndrome in Japan:A nationwide survey. Brain Dev. 2016;38(7):628–637. doi: 10.1016/j.braindev.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 16.Kass HR, Winesett SP, Bessone SK, et al. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure. 2016;35:83–87. doi: 10.1016/j.seizure.2016.01.011. [DOI] [PubMed] [Google Scholar]