

Abstract
Multiple myeloma is the second most common lymphoproliferative disorders, characterized by aberrant expansion of monoclonal plasma cells. In the last years, thanks to novel next generation sequencing technologies, multiple myeloma has emerged as one of the most complex hematological cancers, shaped over time by the activity of multiple mutational processes and by the acquisition of key driver events. In this review, we describe how whole genome sequencing is emerging as a key technology to decipher this complexity at every stage of myeloma development: precursors, diagnosis and relapsed/refractory. Defining the time windows when driver events are acquired improves our understanding of cancer etiology and paves the way for early diagnosis and ultimately prevention.
Keywords: Multiple Myeloma, Driver events, Timing, whole genome sequencing
INTRODUCTION
Multiple myeloma is the second most common lymphoproliferative disorders, characterized by aberrant expansion of monoclonal plasma cells. Historically, multiple myeloma has been thought to arise in the germinal center, where key initiating driver events are acquired.(1–3) The pre-malignant clone immortalized by these events then migrates to the bone marrow, where it expands and may be recognized clinically as monoclonal gammopathy of undetermined significance (MGUS) or smoldering multiple myeloma (SMM), which is detectable in virtually all patients, years to decades before the diagnosis of multiple myeloma.(4, 5) These multiple myeloma precursors can be detected using serum protein electrophoresis in up to 3% of the normal population above 60 years old. On average, the risk of progression from these precursors to MM ranges from 1% to 10% per year.(6, 7) The trajectory towards MM can be understood as an evolutionary process, where driver events accumulate over time conferring a competitive advantage to individual subclones, progressively shaping the genomic landscape and clinical phenotype (Figure 1).(8–11) Determining when and where these events are acquired is critical to design strategies for prevention of multiple myeloma, which, despite considerable improvements in treatment, remains incurable in the majority of patients.
Figure 1.
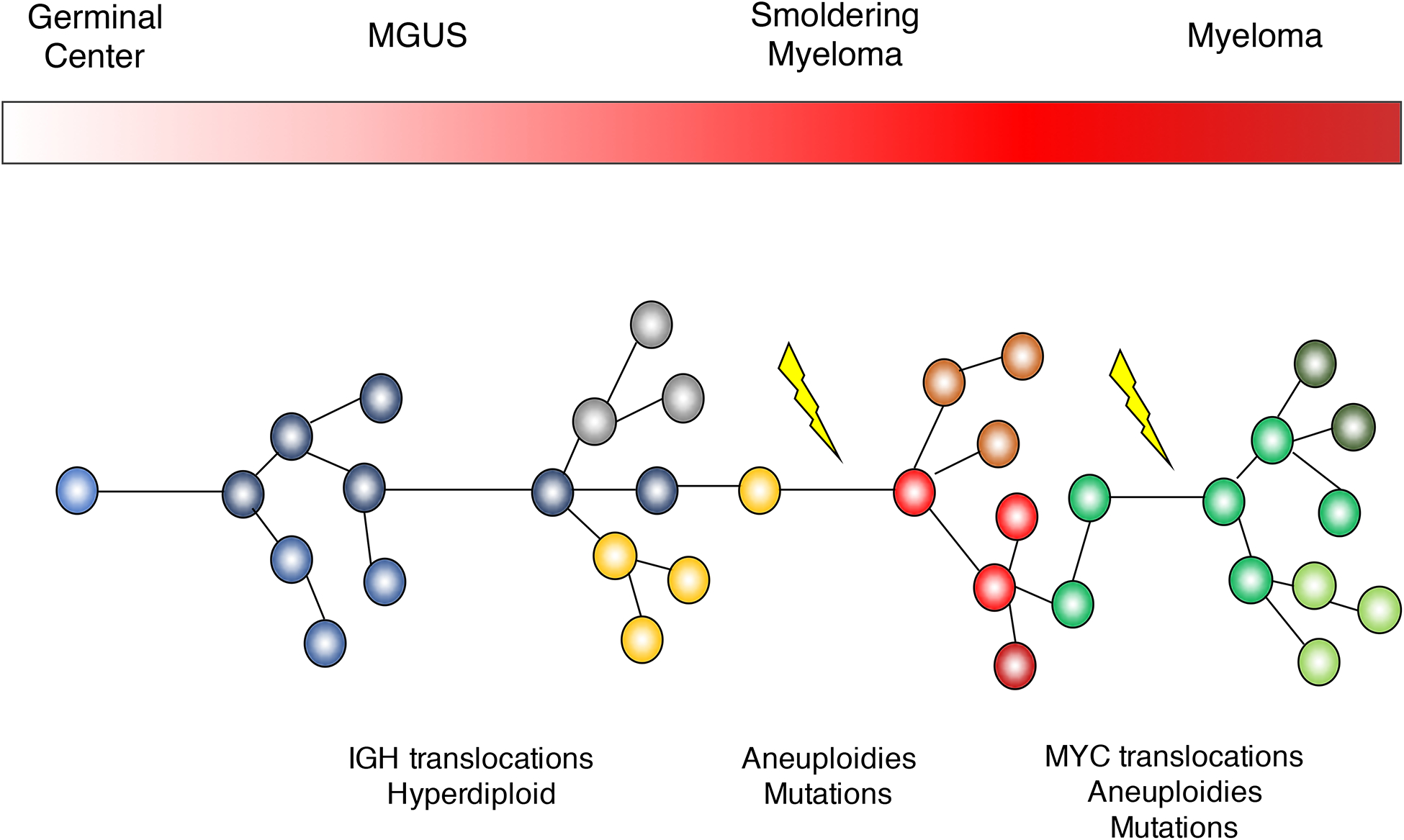
Multiple myeloma pathogenetic model.
In this review, we summarize the essential role of next generation sequencing in defining how the multiple myeloma genome is shaped over time.
BEFORE NEXT GENERATION SEQUENCING
For many years the understanding of multiple myeloma pathogenesis relied upon cytogenetic approaches such as metaphase cytogenetics of cell lines or interphase Fuorescent In-Situ Hybridization (FISH) and gene expression microarrays of patient samples.(1–3) These approaches allowed the identification of recurrent translocations involving the IGH locus and key oncogenes which were overexpressed as a consequence, including CCND1, CCND2, CCND3, MMSET, MAF, MAFB and MAFA.(12–16) These structural events are present in all cells, during all phases of cancer development (i.e., precursor disease stages, newly-diagnosed MM and relapsed MM), and are considered initiating events (Figure 1).(17) The acquisition of these translocations are believed to be linked to aberrant AID activity in the germinal center, introducing DNA double strand breaks (DBS) in the IGH locus, which are then joined to another DBS occurring elsewhere in the genome.
Genome-wide SNP arrays used to map recurrent copy number abnormalities highlighted the complex landscape of recurrent aneuploidies (i.e., copy number abnormalities, CNA) in multiple myeloma. Of note, more than 60% of multiple myelomas have a karyotype characterized by multiple trisomies involving the odd numbered chromosomes.(1–3) This “hyperdiploid” profile is observed in similar proportions between MM and precursors, and has a mutually exclusive pattern with the main IGH translocations (though exceptions do occur), suggesting an early etiologic role.
Several cytogenetic and structural variants events have been historically linked to poor outcome [e.g. t(4;14)(MMSET;IGH) and del17p13 (TP53)] and have been integrated in clinical/biological prognostic scoring systems.(18) Many of these features are still associated with poor outcomes despite the introduction of novel drug combinations.(19, 20)
THE NEXT GENERATION SEQUENCING REVOLUTION
In the last 8 years, our understanding of the clinical and biological landscape of multiple myeloma has rapidly changed. The introduction of novel agents dramatically improved survival, (19) raising two important questions: “what is the best front-line combination therapy” and “how can we develop a patient-specific precision medicine”. In parallel to these clinical improvements, the introduction of next generation sequencing has significantly expanded the landscape of recognized “driver events” involved in multiple myeloma pathogenesis. In addition to the IGH translocations and recurrent aneuploidies, a large catalogue of novel drivers has been described, including recurrent translocations (i.e. MYC) and single nucleotide variants (SNVs).(21–25) Some of these new drivers have been associated with prognosis, but their real impact in the therapeutic decision process and in predicting disease progression remains unclear.(8, 11, 22–24, 26–30)
These biological advances have been mostly based on targeted and whole exome sequencing (WXS) approaches, intrinsically limited by their focus on the coding part of the genome and unable to fully capture important features such as structural variants (SVs) and copy number abnormalities (CNAs). In contrast, whole genome sequencing (WGS) can interrogate the full repertoire of SNVs, CNVs, and SVs distributed throughout the entire genome.(31) Using such WGS to investigate the genomic profile of a longitudinal series of 30 MM patients, we recently found that the MM genome is characterized by a large number of SVs not detectable using exome sequencing.(22, 29) Furthermore, we identified several distinct patterns of complex structural events, including chromoplexy, templated insertions and chromothripsis (Figure 2).(32) Chromothripsis also has an important prognostic role, being associated with poor outcomes.(33) A recent study investigating the landscape of translocations in multiple myeloma within 795 newly-diagnosed patients with available low coverage long insert WGSs, revealed 20 recurrent hotspots, including novel driver genes.(21) Interestingly, patients with translocations involving IGL were characterized by poor outcome. These recent investigations suggest that a comprehensive characterization of SVs using WGS data can expand the catalogue of drivers and improve the risk stratification of myeloma patients.
Figure 2.
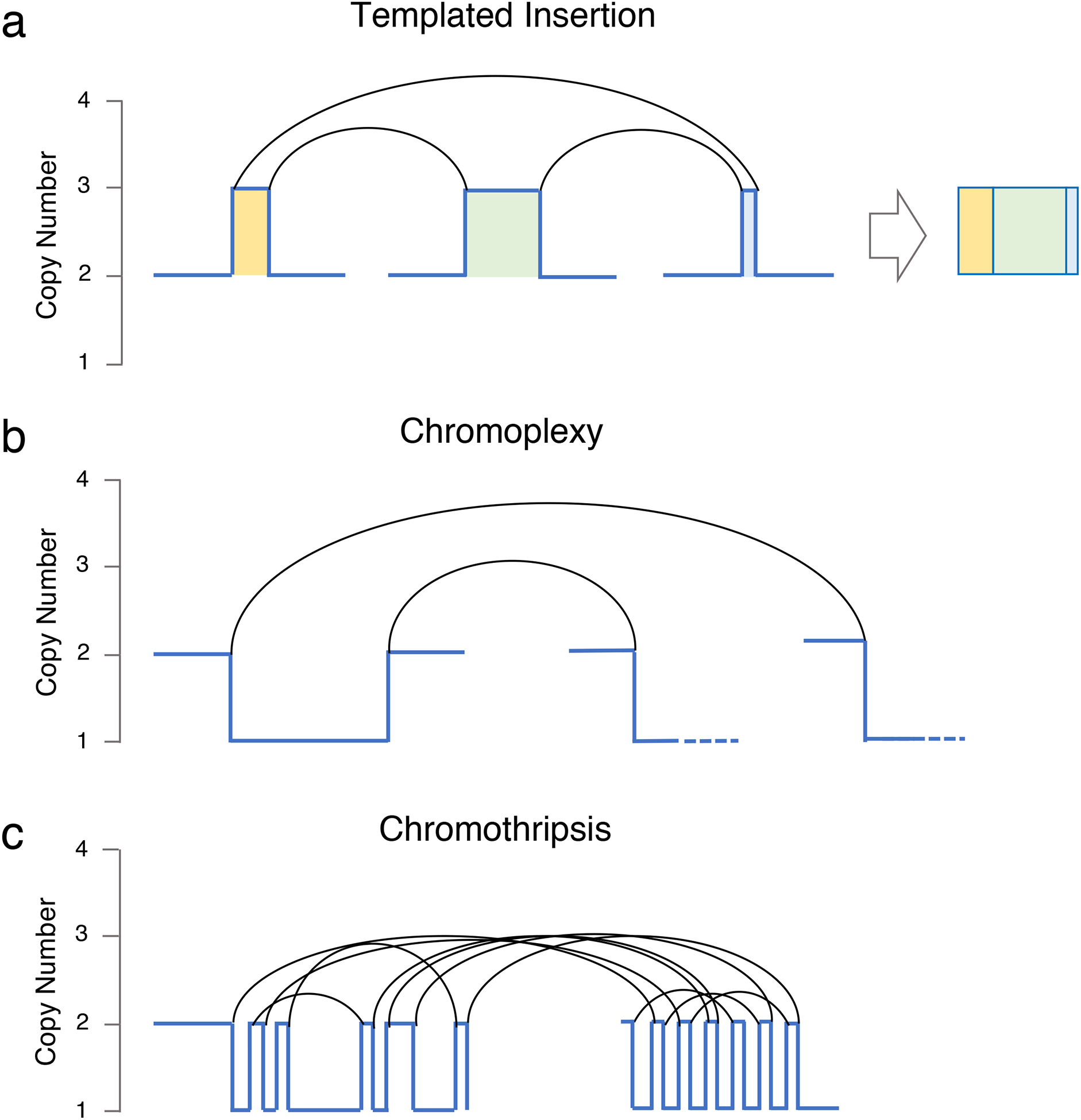
Three classed of complex events observed in multiple myeloma.
While, in a standard cytogenetic/SNP array/exomes investigation, each aneuploidy is counted as single event, using SVs we can link different chromosomal aberration to the same structural event, highlighting how multiple known drivers can be deregulated at the same time. This provides a strong oncogenic drive in a single molecular event and can potentially lead to rapid changes in clinical behavior and punctuated evolution.
TIMELINE OF DRIVER EVENTS IN MULTIPLE MYELOMA
Next generation sequencing has created an unprecedented catalogue of multiple myeloma driver events, but it still unclear when each of these events exert their role in the natural history of myeloma progression. The selective advantage provided by these events could be limited either to the phase of cancer development (e.g. initiation, pre-clinical expansion or treatment-resistance), or may be important during the entire cancer life history. This critical question can be answered by reconstructing the chronological order of acquisition of driver events.
Novel computational tools allow to reconstruct the natural history of cancer evolution. These tools are based on the idea that cancers are shaped by the accumulation of drivers and the activity of different mutational processes over time that determine the ultimate behavior of the disease. To estimate the timeline in which driver events are acquired, we rely on the cancer cell fraction (i.e. number of cells carrying a distinct alteration; CCF) of different alterations across large cohorts of patients.(22, 24, 34) This has revealed that trisomies of the odd numbered chromosomes and gain of chromosome 1q have high CCF and are usually clonal, suggesting their acquisition during the early phases of cancer development. In contrast, recurrent deletions (e.g. del13q, del1p, del14q and del16q) and non-synonymous mutations in driver genes have a heterogenous landscape, with a significant proportion only being present in a minor sub-clonal fraction, suggesting a late role.(22, 24, 34)
In multiple myeloma, more than 50% of all mutations are clonal (i.e. trunk of the phylogenetic tree).(35) These events are usually considered as a common group of early events; however, they may reflect the sum of multiple selection events over time (Figure 1). CCF is no longer helpful in reconstructing the timeline of these events, because they are all detectable in 100% of the tumor cells. Recently, a new computational approach to define the relative order of driver events among clonal somatic variants has been presented. This relies on the fact that when an allele is duplicated, all the genomic events acquired until that point will also be duplicated (Figure 3). Thus, while their cancer cell fraction will be always 100%, the variant allele frequency (VAF) of the duplicated mutations will change from 50% to 66%, now being present on two out of the three alleles. Conversely, all the other clonal mutations acquired after the gain, or before the gain on the minor allele, will have a VAF of 33%. Applying this concept on multiple myeloma WGS data, we have shown that it is possible to estimate the relative timing of driver events in multiple myeloma. Interestingly, in a significant fraction of hyperdiploid patients, the final karyotype is the sum of multiple independent events over time. We also found a subset of patient with whole genome duplication, usually associated with relapse and poor outcome, consisting of a complete duplication of most of the alleles.(22, 36–38) Combing the molecular time and CCF-based approaches, we can divide multiple myeloma driver events for each patient in early (clonal pre-gain), intermediate (clonal post-gain) and late (subclonal) (Figure 3). Applying this workflow to a large WGS cohort of MM patients will reveal in which time window each driver event was acquired, potentially revealing the main evolutionary trajectories of disease.
Figure 3.
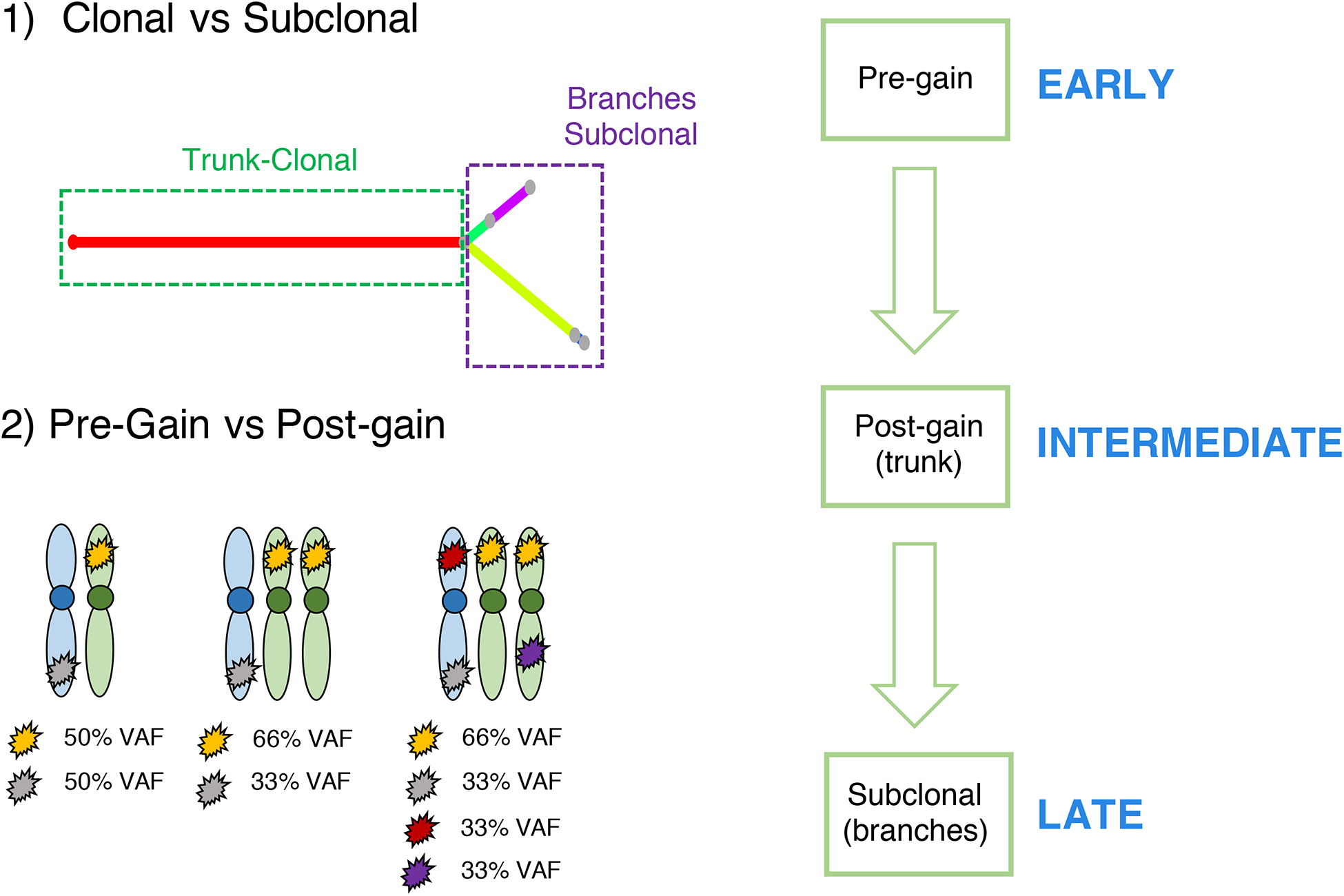
The methodological workflow to reconstruct the chronological order of driver events in multiple myeloma.
MUTATIONAL PROCESSES IN MULTIPLE MYELOMA
The SNV catalogue of each cancer represents the sum of the activity of multiple different mutational processes over time. Each of these processes leaves a characteristic mutational signature in the cancer genome, which is defined by the DNA damage and repair processes involved.(39–41) Considering the bases 5’ and 3’ of each mutated nucleotide, we can generate a classification system based on 96 classes (i.e. 6 possible SNVs * 16 possible trinucleotide contexts). Applying mathematical approaches such as non-negative matrix factorization, we can extract the main components, or signatures, which could potentially reflect a distinct mutational process.(39, 40, 42, 43) More than 50 mutational signatures have been reported and linked to the specific intrinsic and extrinsic exposures .(39) In multiple myeloma, different whole exome studies showed the importance of an APOBEC signature (SBS2 and SBS13), present in a significant fraction of patients.(8, 26, 28) APOBEC is a deaminase whose physiological function is largely unknown. Interestingly, high APOBEC mutational burden is associated with MAF/s translocation and poor outcome.(26, 28) Two additional signatures have been identified in whole exome data using de novo extraction approaches: SBS1 and SBS5 (Figure 4a). These two processes were defined as being “clock-like” due to their association with cell ageing and their constant activity over time in both normal and tumor cells.(42)
Figure 4.
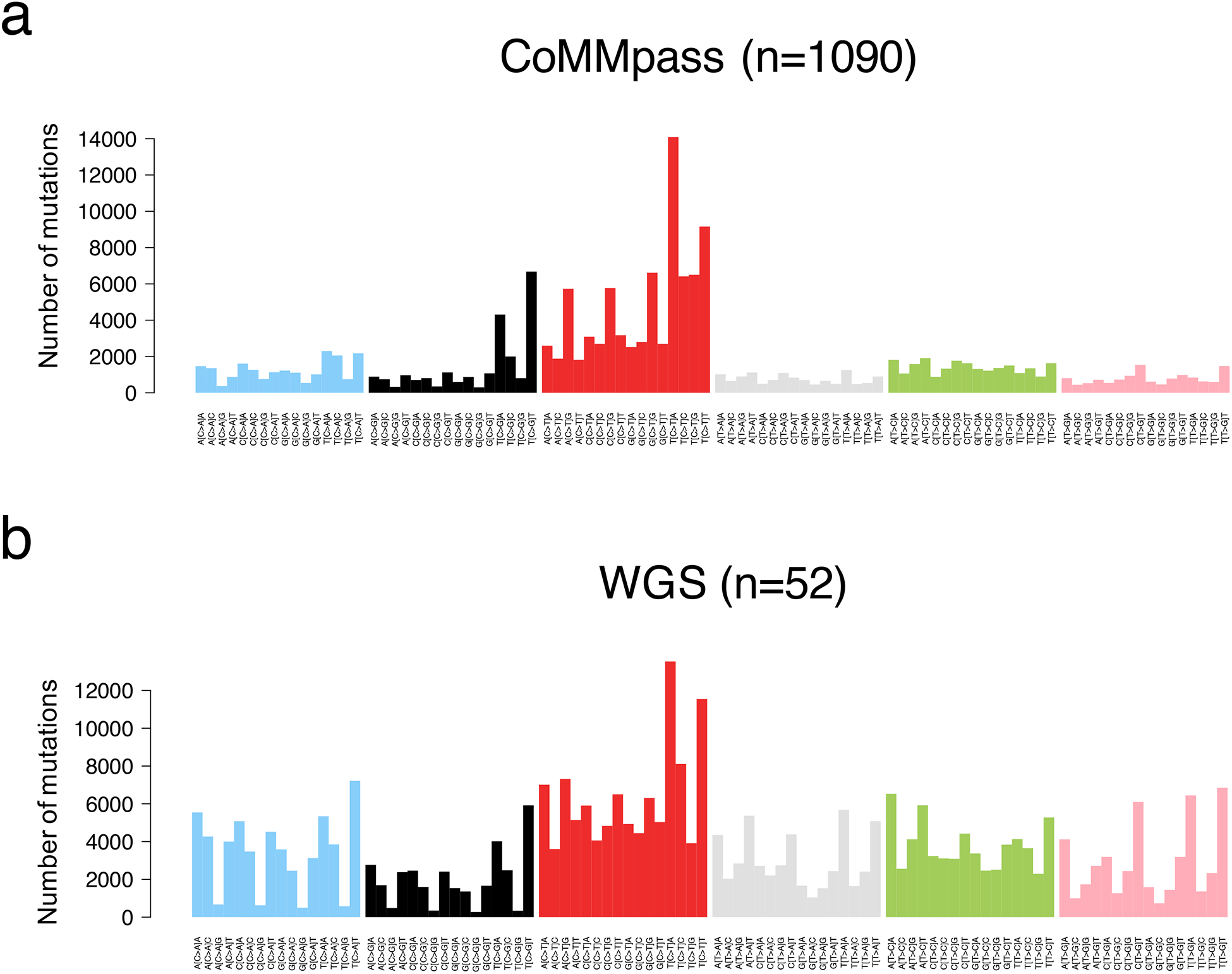
Multiple myeloma mutational signature landscape. The 96-classes mutational profile from exome (a, CoMMpass) and genome data (b).
Differently from WXS, WGS has the potential to define the entire mutational catalogue acquired by the major cancer clone (Figure 4b). Thanks to this comprehensive approach, four additional mutational processes have now been identified in multiple myeloma: SBS8, SBS9, SBS18 and SBS-MM1.(29, 41, 44). These signatures are mostly active during late replication time, heterochromatin and non-coding regions.(29, 41, 44) SBS8 has been described in solid and hematological cancers, but its etiology is largely unknown. SBS18 has been linked to oxidative stress(39) and has been identified in only a small fraction of MM patients. SBS-MM1 has been recently reported to be a consequence of exposure to the mutagenic activity melphalan; one of the most commonly used therapies in MM.(44) Despite the strong mutagenic activity of melphalan, its impact on tumor evolution remains unknown. SBS9 is a characteristic mutational signature of post germinal center B-cell cancers, which has been linked to AID exposure in the germinal center and somatic hypermutation of the immunoglobulin loci.(39–41, 45–47)
Reconstructing the timeline of mutational signature activity in multiple myeloma in several recent studies, we have developed a two-step model for multiple myeloma development.(29, 44) The early phase is characterized by high AID activity, persisting for some time after disease initiation, indicating prolonged GC exposure. Drivers that are linked to the GC-phase are IGH-translocations, multiple independent trisomies and chromosomal gains, the first 1q gain, and mutations in driver genes involved by the off-target AID activity. The late evolutionary phase is characterized by low or absent AID activity, indicating that the malignant clone has become GC independent. SBS8, SBS18 and APOBEC, having been largely absent in the GC-phase, become increasingly active in the late phase. In particular, APOBEC has been shown to play a critical role in subclonal diversification after the emergence of the most common recent ancestor and is associated with non-synonymous mutations. There is however one group of patients where this model does not fit, defined by MAF/MAFB translocations and high APOBEC mutational burden.(26, 28) Our data suggests that APOBEC has been highly active from the earliest evolutionary phase in these patients, while the AID mutational signature is barely detectable, indicating a different evolutionary trajectory where the tumor becomes GC independent right after the initiation. This history is in line with the known aggressive clinical course of these patients and with the low prevalence of MAF/MAFB translocations and high APOBEC among MM precursors.(48)
CONCLUSION
Multiple myeloma and its precursors represent a unique setting where we can explore the chronological order of drivers. The understanding of these evolutionary trajectories will have a major impact to determine when and how patients should be treated. MM precursors can be detected up to 6% of the healthy population older than 65 years.(49) The clinical management of these entities is still based on disease burden-based prognostic scores where high-risk groups are defined by high disease burden. From a genomic perspective, in most of these patients, transformation to multiple myeloma has already occurred, and the time to progression reflects the time that the dominant clone needs to expand and become symptomatic. Understanding the key drivers involved in progression of multiple myeloma precursors has the potential to identify high-risk patients before the clonal expansion, enabling strategies to prevent disease development. Integration of comprehensive genomic profiles with established clinical biomarkers will allow the generation of multi-stage prediction models to define the optimal therapy for each patient.
PRACTICE POINTS.
Whole genome sequencing can identify all known genomic drivers in multiple myeloma
The multiple myeloma life-history is shaped over time by the acquisition of drivers and the activity of different mutational processes.
APOBEC mutational burden and complex structural variants are emerging as new potential markers for high-risk multiple myeloma
RESEARCH AGENDA.
Large cohorts of whole genome and transcriptome data at different stage of multiple myeloma evolution will reveal the key evolutionary trajectories.
Defining these trajectories will open new avenues of research into cancer etiology and prevention.
ACKNOWLEDGMENTS
This work is supported by the Memorial Sloan Kettering Cancer Center NCI Core Grant (P30 CA 008748).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE OF CONFLICTS OF INTEREST
No conflict of interests to declare.
REFERENCES
- 1.Corre J, Munshi N, Avet-Loiseau H. Genetics of multiple myeloma: another heterogeneity level? Blood. 2015;125(12):1870–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Manier S, Salem KZ, Park J, Landau DA, Getz G, Ghobrial IM. Genomic complexity of multiple myeloma and its clinical implications. Nat Rev Clin Oncol. 2017;14(2):100–13. [DOI] [PubMed] [Google Scholar]
- 3.Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012;12(5):335–48. [DOI] [PubMed] [Google Scholar]
- 4.Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smoldering multiple myeloma. Hematol Oncol Clin North Am. 2007;21(6):1093–113, ix. [DOI] [PubMed] [Google Scholar]
- 5.Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009;113(22):5412–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kyle RA, Larson DR, Therneau TM, Dispenzieri A, Kumar S, Cerhan JR, et al. Long-Term Follow-up of Monoclonal Gammopathy of Undetermined Significance. N Engl J Med. 2018;378(3):241–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kyle RA, Remstein ED, Therneau TM, Dispenzieri A, Kurtin PJ, Hodnefield JM, et al. Clinical course and prognosis of smoldering (asymptomatic) multiple myeloma. N Engl J Med. 2007;356(25):2582–90. [DOI] [PubMed] [Google Scholar]
- 8.Bolli N, Avet-Loiseau H, Wedge DC, Van Loo P, Alexandrov LB, Martincorena I, et al. Heterogeneity of genomic evolution and mutational profiles in multiple myeloma. Nat Commun. 2014;5:2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bolli N, Maura F, Minvielle S, Gloznik D, Szalat R, Fullam A, et al. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun. 2018;9(1):3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, et al. Clonal competition with alternating dominance in multiple myeloma. Blood. 2012;120(5):1067–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lohr JG, Stojanov P, Carter SL, Cruz-Gordillo P, Lawrence MS, Auclair D, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25(1):91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol. 2005;23(26):6333–8. [DOI] [PubMed] [Google Scholar]
- 13.Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J Jr. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood. 2005;106(1):296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agnelli L, Bicciato S, Mattioli M, Fabris S, Intini D, Verdelli D, et al. Molecular classification of multiple myeloma: a distinct transcriptional profile characterizes patients expressing CCND1 and negative for 14q32 translocations. J Clin Oncol. 2005;23(29):7296–306. [DOI] [PubMed] [Google Scholar]
- 15.Chesi M, Bergsagel PL, Brents LA, Smith CM, Gerhard DS, Kuehl WM. Dysregulation of cyclin D1 by translocation into an IgH gamma switch region in two multiple myeloma cell lines. Blood. 1996;88(2):674–81. [PubMed] [Google Scholar]
- 16.Chesi M, Nardini E, Brents LA, Schrock E, Ried T, Kuehl WM, et al. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Nat Genet. 1997;16(3):260–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barwick BG, Gupta VA, Vertino PM, Boise LH. Cell of Origin and Genetic Alterations in the Pathogenesis of Multiple Myeloma. Front Immunol. 2019;10:1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palumbo A, Avet-Loiseau H, Oliva S, Lokhorst HM, Goldschmidt H, Rosinol L, et al. Revised International Staging System for Multiple Myeloma: A Report From International Myeloma Working Group. J Clin Oncol. 2015;33(26):2863–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thorsteinsdottir S, Dickman PW, Landgren O, Blimark C, Hultcrantz M, Turesson I, et al. Dramatically improved survival in multiple myeloma patients in the recent decade: results from a Swedish population-based study. Haematologica. 2018;103(9):e412–e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Avet-Loiseau H, Leleu X, Roussel M, Moreau P, Guerin-Charbonnel C, Caillot D, et al. Bortezomib plus dexamethasone induction improves outcome of patients with t(4;14) myeloma but not outcome of patients with del(17p). J Clin Oncol. 2010;28(30):4630–4. [DOI] [PubMed] [Google Scholar]
- 21.Barwick BG, Neri P, Bahlis NJ, Nooka AK, Dhodapkar MV, Jaye DL, et al. Multiple myeloma immunoglobulin lambda translocations portend poor prognosis. Nat Commun. 2019;10(1):1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maura F, Bolli N, Angelopoulos N, Dawson KJ, Leongamornlert D, Martincorena I, et al. Genomic landscape and chronological reconstruction of driver events in multiple myeloma. Nat Commun. 2019;10(1):3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies F, et al. A high-risk, Double-Hit, group of newly diagnosed myeloma identified by genomic analysis. Leukemia. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walker BA, Mavrommatis K, Wardell CP, Ashby TC, Bauer M, Davies FE, et al. Identification of novel mutational drivers reveals oncogene dependencies in multiple myeloma. Blood. 2018;132(6):587–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker BA, Wardell CP, Brioli A, Boyle E, Kaiser MF, Begum DB, et al. Translocations at 8q24 juxtapose MYC with genes that harbor superenhancers resulting in overexpression and poor prognosis in myeloma patients. Blood Cancer J. 2014;4:e191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maura F, Petljak M, Lionetti M, Cifola I, Liang W, Pinatel E, et al. Biological and prognostic impact of APOBEC-induced mutations in the spectrum of plasma cell dyscrasias and multiple myeloma cell lines. Leukemia. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walker BA, Boyle EM, Wardell CP, Murison A, Begum DB, Dahir NM, et al. Mutational Spectrum, Copy Number Changes, and Outcome: Results of a Sequencing Study of Patients With Newly Diagnosed Myeloma. J Clin Oncol. 2015;33(33):3911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker BA, Wardell CP, Murison A, Boyle EM, Begum DB, Dahir NM, et al. APOBEC family mutational signatures are associated with poor prognosis translocations in multiple myeloma. Nat Commun. 2015;6:6997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bolli F, Maura M, Minvielle M, Gloznik D, Szalat R, Fullam A, et al. Genomic patterns of progression in smoldering multiple myeloma. Nat Commun. 2018;9(1)(3363). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bolli N, Biancon G, Moarii M, Gimondi S, Li Y, de Philippis C, et al. Analysis of the genomic landscape of multiple myeloma highlights novel prognostic markers and disease subgroups. Leukemia. 2018;32(12):2604–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458(7239):719–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li YRN, Weischenfeldt j, Wala JA, Shapira O, Schumacher SE, Khurana W,, Korbel JIM, Beroukhim R, Campbell PJ on behalf of the PCAWG-Structural Variation Working Group ^ and the PCAWG Network. Patterns of structural variation in human cancer. bioRxiv. 2017. [Google Scholar]
- 33.Magrangeas F, Avet-Loiseau H, Munshi NC, Minvielle S. Chromothripsis identifies a rare and aggressive entity among newly diagnosed multiple myeloma patients. Blood. 2011;118(3):675–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aktas Samur A, Minvielle S, Shammas M, Fulciniti M, Magrangeas F, Richardson PG, et al. Deciphering the chronology of copy number alterations in Multiple Myeloma. Blood Cancer J. 2019;9(4):39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maura F, Rustad EH, Yellapantula V, Luksza M, Hoyos D, Maclachlan KH, et al. Role of AID in the temporal pattern of acquisition of driver mutations in multiple myeloma. Leukemia. 2019. [DOI] [PubMed] [Google Scholar]
- 36.Ashby C, Tytarenko RG, Wang Y, Weinhold N, Johnson SK, Bauer M, et al. Poor overall survival in hyperhaploid multiple myeloma is defined by double-hit bi-allelic inactivation of TP53. Oncotarget. 2019;10(7):732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Peterson JF, Rowsey RA, Marcou CA, Pearce KE, Williamson CM, Frederick LA, et al. Hyperhaploid plasma cell myeloma characterized by poor outcome and monosomy 17 with frequently co-occurring TP53 mutations. Blood Cancer J. 2019;9(3):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sawyer JR, Morgan GJ. Hyperhaploid karyotypes in multiple myeloma. Oncotarget. 2017;8(45):78259–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexandrov L, Kim J, Haradhvala N, Huang M, Ng A, Boot A, et al. The Repertoire of Mutational Signatures in Human Cancer. bioRxiv. 2018. [Google Scholar]
- 40.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maura F, Degasperi A, Nadeu F, Leongamornlert D, Davies H, Moore L, et al. A practical guide for mutational signature analysis in hematological malignancies. Nat Commun. 2019;10(1):2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, et al. Clock-like mutational processes in human somatic cells. Nat Genet. 2015;47(12):1402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alexandrov LB, Nik-Zainal S, Wedge DC, Campbell PJ, Stratton MR. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3(1):246–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rustad H, Yellapantula V, Bolli N, Leongamornlert D, Nadeu F, N. A, et al. Timing the Initiation of Multiple Myeloma. Sneak Peek. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kasar S, Kim J, Improgo R, Tiao G, Polak P, Haradhvala N, et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat Commun. 2015;6:8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Puente XS, Bea S, Valdes-Mas R, Villamor N, Gutierrez-Abril J, Martin-Subero JI, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526(7574):519–24. [DOI] [PubMed] [Google Scholar]
- 47.Hoang PH, Dobbins SE, Cornish AJ, Chubb D, Law PJ, Kaiser M, et al. Whole-genome sequencing of multiple myeloma reveals oncogenic pathways are targeted somatically through multiple mechanisms. Leukemia. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maura F, Bolli N, Rustad EH, Hultcrantz M, Munshi N, Landgren O. Moving From Cancer Burden to Cancer Genomics for Smoldering Myeloma: A Review. JAMA Oncol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajkumar SV, Landgren O, Mateos MV. Smoldering multiple myeloma. Blood. 2015;125(20):3069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]