

Abstract
X-连锁鱼鳞病(XLI)是一种类固醇硫酸酯酶缺乏的代谢性疾病,常于出生时或生后不久发病,编码类固醇硫酸酯酶的基因(STS)位于X染色体短臂上,STS基因发生缺失或突变时可导致此病的发生。本研究收集一个家系的临床表型资料,其中先证者,男,足月顺产,11岁,全身皮肤干燥、粗糙、呈黑褐色鳞片状,主要累及腹部和肢体伸侧。采集家系中各成员的外周血提取DNA,采用多重连接依赖式探针扩增(MLPA)技术对家系各成员的X染色体上的STS基因拷贝数进行检测,用全基因组芯片进一步明确X染色体微缺失片段的大小,随后采用MLPA技术对先证者母亲再生育进行产前诊断。结果发现家系中先证者及2个患者均为STS缺失的男性半合子,基因芯片鉴定出Xp22.31存在缺失,缺失大小为1.6Mb(chrX:6,516,735-8,131,442),另鉴定出2个女性家庭成员为携带者。先证者母亲再生育产前诊断结果证实胎儿为携带者。本研究表明该XLI家系存在STS基因缺失,该缺失引发出XLI特有的皮肤病变,MLPA是XLI分子诊断与产前诊断的便捷可靠技术。
Keywords: X-连锁鱼鳞病, STS基因, 基因突变, 儿童
Abstract
X-linked ichthyosis (XLI) is a metabolic disease with steroid sulfatase deficiency and often occurs at birth or shortly after birth. The encoding gene of steroid sulfatase, STS, is located on the short arm of the X chromosome, and STS deletion or mutation can lead to the development of this disease. This study collected the data on the clinical phenotype from a family, and the proband, a boy aged 11 years with full-term vaginal delivery, had dry and rough skin and black-brown scaly patches, mainly in the abdomen and extensor aspect of extremities. Peripheral blood samples were collected from each family member and DNA was extracted. Multiplex ligation-dependent probe amplification (MLPA) was used to measure the copy number of STS on the X chromosome. Whole-genome microarray was used to determine the size of the segment with microdeletion in the X chromosome. MLPA was then used for prenatal diagnosis for the mother of the proband. The results revealed that the proband and another two male patients had hemizygotes in STS deletion. Gene microarray identified a rare deletion with a size of 1.6 Mb at Xp22.31 (chrX:6, 516, 735-8, 131, 442). Two female family members were found to be carriers. Prenatal diagnosis showed that the fetus carried by the proband's mother was a carrier of this microdeletion. This study showed STS gene deletion in this family of XLI, which causes the unique skin lesions of XLI. MLPA is a convenient and reliable technique for the molecular and prenatal diagnosis of XLI.
Keywords: X-linked ichthyosis, STS gene, Gene mutation, Child
遗传性鱼鳞病多表现为全身性皮肤干燥、粗糙、过多角化、鱼鳞状鳞屑。非综合征鱼鳞病主要分为六型,即寻常性鱼鳞病、板层状鱼鳞病、表皮松解性角化过度鱼鳞病、限局性线状鱼鳞病、获得性鱼鳞病、胶样婴儿,X-连锁鱼鳞病(X-linked ichthyosis, XLI)属于寻常性鱼鳞病[1]。该病常发生在男性,女性多为携带者。患者于出生时或生后不久即发病,表现为全身皮肤干燥、粗糙,覆着黑褐色鳞片,主要累及肢体伸侧,也可累及屈侧,颈部及耳前区受累为该病的特征[2]。1978年,Webster等[3]发现XLI患者皮肤成纤维细胞缺乏一种酶,即类固醇硫酸酯酶(steroid sulfatase, STS),并首次证实了该酶与XLI有关。本文分析了广西地区一个XLI家系STS基因的缺失情况。
1. 资料与方法
1.1. 研究对象
本研究中的XLI家系先证者为广西柳州市妇幼保健院2015年3月的门诊患者。先证者男,足月顺产,11岁,全身皮肤干燥、粗糙、呈黑褐色鳞片状,主要累及腹部和肢体伸侧(图 1)。家系中有患同种疾病的患者3人,详细收集该家系成员共14人的临床表现和遗传资料,先证者父母为非近亲婚配,其母孕3产2,第1胎患有先天性心脏病,先证者为第2胎,第3胎现孕17周。
1.
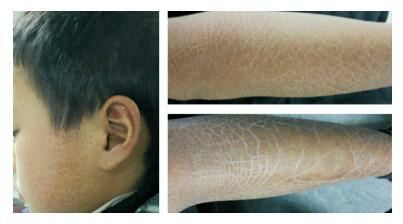
XLI家系先证者皮肤表型
1.2. DNA提取
抽取7个家庭成员外周血2 mL,2%乙二胺四乙酸二钾抗凝,包括先证者父母(Ⅱ1及Ⅱ2),先证者姐姐(Ⅲ1),先证者(Ⅲ2)、先证者舅舅(Ⅱ3)、先证者表妹(Ⅲ4)和先证者表弟(Ⅲ5)。抽取先证者母亲羊水10 mL。用标准酚氯仿提取外周血和羊水DNA,利用ND-1000-UV-VIS波长紫外/可见光扫描分光光度计(NanoDrop,美国)对样本的基因组DNA的提取质量和浓度进行检测。DNA样本的OD260 nm/OD280 nm比值应在1.6~2.0之间,OD260 nm/OD230 nm比值需≥2.0,浓度需≥50 ng/μL。-70℃冷冻保存。
1.3. 多重连接依赖式探针扩增分析
本研究采用荷兰MRC公司研发的多重连接依赖式探针扩增(multiplex ligation-dependent probe amplification, MLPA)P160-STS试剂盒,MLPA探针覆盖了STS基因全部10个外显子其周围1 Mb区域。
1.4. 单核苷酸多态性微阵列分析
芯片检测样本按试剂盒操作手册。单核苷酸多态性微阵列分析(SNP array-based comparative genomic hybridization, SNP-array)采用Human CytoSNP-12芯片试剂盒(Illumina公司)。芯片经过洗涤和离心甩干处理后,扫描和分析结果。芯片结果经数据库查询、比对后根据其临床意义分为致病性、良性、临床意义不明确3类[4]。本实验室所采用的数据库为实验室内部数据库,DGV数据库(Database of Genomic Variant),DECIPHER数据库(Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources),OMIM数据库(Online Mendelian Inheritance in Man),ISCA数据库(International Standard for Cytogenomic Arrays Consortium)等。
2. 结果
2.1. MLPA检测结果
MLPA对7个家庭成员的基因组DNA进行检测,发现家系中包括先证者在内的3名患者(Ⅱ3、Ⅲ2、Ⅲ5)均为STS基因缺失半合子,3名患者均为出生后不久发病,全身皮肤干燥、粗糙、呈黑褐色鳞片状;2名女性(Ⅱ1、Ⅲ4)均为STS基因缺失杂合子,杂合子未见明显临床表型。产前诊断表明先证者母亲孕第三胎(Ⅲ3)为STS缺失杂合子,见表 1,图 2。
1.
家系成员临床表型与基因检测结果
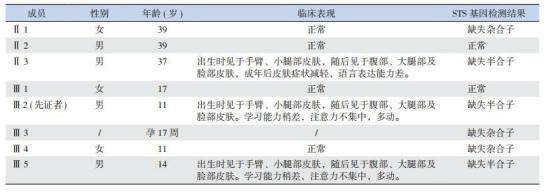
|
2.
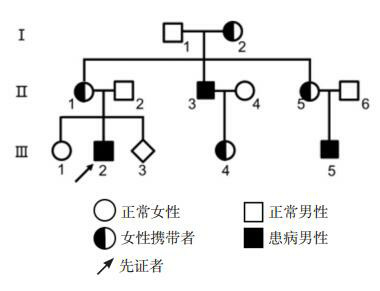
家系图
2.2. SNP-array检测结果
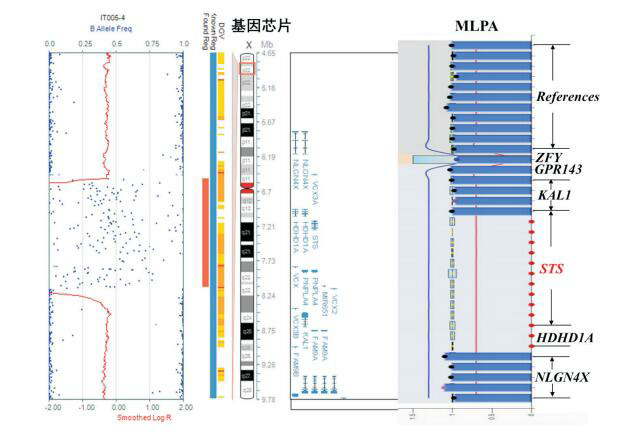
基因芯片检测先证者外周血全基因组DNA拷贝数变异,发现先证者X染色体短臂p22.31-pter存在缺失,缺失大小为1.6 Mb,缺失范围为chrX:6, 516, 735~8, 131, 442,丢失的基因有HDHD1A、STS、VCX、PNPLA4和MIR651(图 3)。该微缺失与一个已报道的微缺失致病区域在位置和大小上匹配,并且缺失内包含XLI的致病基因STS,XLI是由STS基因点突变或缺失导致的,其中STS基因缺失占主要部分,根据美国医学遗传学会关于染色体基因组拷贝数变异的分类标准[4],可将此微缺失定义为致病性拷贝数变异。
3.
先证者SNP-array及MLPA检测结果
左图为基因芯片(SNP-array)检测结果,蓝色散点为Xp22区带SNP探针信号;红色条柱为1.6 Mb的微缺失;X染色体上红色方框内为Xp22区带,丢失的基因包括HDHD1A、STS、VCX、PNPLA4和MIR651。右图为MLPA检测结果:NLGN4X、HDHD1A、STS、KAL1、GPR143、ZFY为被检测基因,References为对照,探针检测信号在0.7~1.3之间为正常,小于0.7为缺失,大于1.3为拷贝数增加,STS基因区探针信号为0说明存在缺失。
3. 讨论
XLI为X连锁的隐性遗传病,属寻常型鱼鳞病的一种,发病率为1/4 000,男性发病率远高于女性,患者大多在出生时或者出生后不久发病,鳞屑大而显著,褐色或黑色,遍布全身,无毛囊角化,掌趾皮肤正常,少部分患者并发皮肤外的症状,如角膜浑浊、支气管哮喘等过敏反应[5],本研究中3名患者尚未见皮肤外的并发症。XLI患者皮肤成纤维细胞缺乏类固醇硫酸酯酶是导致本病的病因。健康人中STS主要存在于角质层细胞的细胞膜区,活性也最高,此酶的主要功能是将角质层中的胆固醇硫酸盐分解为胆固醇和硫酸盐,这一过程对表皮细胞的粘合及正常脱落起重要作用[6]。当STS缺乏时,此分解过程被阻断,角质层的胆固醇硫酸盐堆积,激发典型的鳞屑表现和渗透屏障功能障碍。另一方面胆固醇硫酸盐分解产生胆固醇对皮肤起到屏障作用,当STS缺乏时,胆固醇硫酸盐分解过程被阻断,皮肤的屏障作用受到干扰,pH值下降,中性pH环境下活性最佳的丝氨酸蛋白酶活性被抑制,角质层黏附性增加,进一步使角质层细胞滞留,促进鳞屑堆积[7]。
STS基因位于X染色体短臂远端Xp22.32-peter,基因全长164 kb,含10个外显子,STS基因被包括在具有重复结构的侧翼序列内,容易发生非等位基因同源重组导致基因缺失,因此STS基因突变类型85%~90%为缺失,包括完全缺失、部分缺失,点突变较罕见[8-9]。X短臂微缺失也是STS基因丢失的原因之一[10]。在本研究的家系中,STS基因的缺失即为Xp22.32微缺失导致,微缺失大小为1.6 Mb,缺失范围内包括5个基因,分别为HDHD1A、STS、VCX、PNPLA4和MIR651,该微缺失尚未见文献报道。STS的缺失不仅导致胆固醇硫酸酯酶的表达减少,间接控制表皮颗粒层的类固醇合成,从而导致角质层细胞持久性粘着,干扰了角质层正常脱屑过程;另一方面,STS还可导致表皮屏障功能的损害,并作为转录因子影响转谷氨酞胺酶1基因的表达和活性,引起表皮的角化过度改变[11]。有研究报道VCX基因缺失与精神发育迟缓有关[12],这一家系中3例患者表现出轻度精神发育迟缓,学习能力稍差、注意力不集中、多动,可能与VCX缺失相关。目前在3例患者中尚未观察到与HDHD1A、PNPLA4和MIR651基因缺失相关的表现。
本研究中的家系成员共13人,3例患者,他们的母亲均为突变携带者。家系分析符合X连锁隐性遗传病的特点:(1)本病由女性携带者向后代传递;(2)男性患者只能将致病基因传递给女儿,而不会出现“父-子”传递方式;(3)女性携带者(杂合子)可以将致病基因传递给男性和女性后代;(4)女性携带者的男性后代有50%的概率发病,而女性后代由于受X染色体失活的影响,其表型有较大的差异,由于STS基因位于X染色体短臂远端Xp22.3,在遗传选择的影响下,女性含短臂缺失的X染色体通常处于失活状态,因而多数没有表现出临床症状[13]。男性只有一条X染色体,为半合子,因此X染色体发生畸变通常导致较为严重的临床表现型[14]。本研究中先证者及其余2例患者均表现出严重的临床表型。本研究采用MLPA技术对STS基因全部外显子及其上下游1 Mb进行拷贝数分析,有效的发现了导致该家系疾病的病因为STS基因缺失,并通过基因芯片技术证明STS基因的缺失是X染色体微缺失所致。另外根据基因检测的结果,我们可以知道家系成员的再发风险,为该家庭的遗传咨询和产前诊断提供了分子基础和科学依据。
Biography
黄际卫, 男, 博士, 医师, 实验师
Funding Statement
柳州市出生缺陷预防与控制重点实验室项目(2014G020404)
References
- 1.Takeichi T, Akiyama M. Inherited ichthyosis:non-syndromic forms. J Dermatol. 2016;43(3):242–251. doi: 10.1111/jde.2016.43.issue-3. [DOI] [PubMed] [Google Scholar]
- 2.唐 志平, 赵 恬, 张 芳, et al. 先天性鱼鳞病基因遗传学研究进展. http://www.cnki.com.cn/Article/CJFDTOTAL-LPFZ201306031.htm 皮肤性病诊疗学杂志. 2013;20(6):447–451. [Google Scholar]
- 3.Webster D, France JT, Shapiro LJ, et al. X-linked ichthyosis due to steroid-sulphatase deficiency. https://www.researchgate.net/publication/23059240_X-linked_ichthyosis_due_to_steroid-sulphatase_deficiency. Lancet. 1978;1(8055):70–72. doi: 10.1016/s0140-6736(78)90005-3. [DOI] [PubMed] [Google Scholar]
- 4.Kearney HM, Thorland EC, Brown KK, et al. American College of Medical Genetics standards and guidelines for interpretation and reporting of postnatal constitutional copy number variants. Genet Med. 2011;13(7):680–685. doi: 10.1097/GIM.0b013e3182217a3a. [DOI] [PubMed] [Google Scholar]
- 5.Traupe H, Fischer J, Oji V. Nonsyndromic types of ichthyosesan update. J Dtsch Dermatol Ges. 2014;12(2):109–121. doi: 10.1111/ddg.12229. [DOI] [PubMed] [Google Scholar]
- 6.Reed MJ, Purohit A, Woo LW, et al. Steroid sulfatase:molecular biology, regulation, and inhibition. Endocr Rev. 2005;26(2):171–202. doi: 10.1210/er.2004-0003. [DOI] [PubMed] [Google Scholar]
- 7.Elias PM, Williams ML, Choi EH, et al. Role of cholesterol sulfate in epidermal structure and function:lessons from X-linked ichthyosis. Biochim Biophys Acta. 2014;1841(3):353–361. doi: 10.1016/j.bbalip.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akiyama M, Shimizu H. An update on molecular aspects of the non-syndromic ichthyoses. Exp Dermatol. 2008;17(5):373–382. doi: 10.1111/exd.2008.17.issue-5. [DOI] [PubMed] [Google Scholar]
- 9.Alperin ES, Shapiro LJ. Characterization of point mutations in patients with X-linked ichthyosis.Effects on the structure and function of the steroid sulfatase protein. J Biol Chem. 1997;272(33):20756–20763. doi: 10.1074/jbc.272.33.20756. [DOI] [PubMed] [Google Scholar]
- 10.Hand JL, Runke CK, Hodge JC. The phenotype spectrum of X-linked ichthyosis identified by chromosomal microarray. J Am Acad Dermatol. 2015;72(4):617–627. doi: 10.1016/j.jaad.2014.12.020. [DOI] [PubMed] [Google Scholar]
- 11.何 玉清, 曾 抗, 张 锡宝, et al. 四种遗传性鱼鳞病基因型与临床表型的相关性分析. 中国皮肤性病学杂志. 2009;23(5):267–270. [Google Scholar]
- 12.Ben Khelifa H, Soyah N, Ben-Abdallah-Bouhjar I, et al. Xp22.3 interstitial deletion:a recognizable chromosomal abnormality encompassing VCX3A and STS genes in a patient with X-linked ichthyosis and mental retardation. Gene. 2013;527(2):578–583. doi: 10.1016/j.gene.2013.06.018. [DOI] [PubMed] [Google Scholar]
- 13.Brown CJ, Willard HF. Localization of a gene that escapes inactivation to the X chromosome proximal short arm:implications for X inactivation. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1684990/ Am J Hum Genet. 1990;46(2):273–279. [PMC free article] [PubMed] [Google Scholar]
- 14.Ellegren H. Characteristics, causes and evolutionary consequences of male-biased mutation. Proc Biol Sci. 2007;274(1606):1–10. doi: 10.1098/rspb.2006.3720. [DOI] [PMC free article] [PubMed] [Google Scholar]