

Abstract
患儿,男,2岁,因咳嗽、发热第6次入院。患儿2月龄时发现腋下淋巴结结核,多次因发热、咳嗽住院,并发现嗜酸性粒细胞增多,以及多次免疫全套提示IgG、IgA、IgM降低。入院后予以抗结核、抗细菌、抗真菌治疗,仍反复发热、肝脾进行性增大,入院第35天出现果酱样大便、B超提示左侧腹局部肠管横断面呈“同心圆征”,于全麻下行剖腹探查、肠套叠复位+回盲部成形术以及肠壁结节、肠系膜淋巴结活检、肝活检术,术后患儿出现肝功能衰竭、DIC、电解质紊乱,救治无效死亡。入院后血培养及肝活检均发现马尔尼菲青霉菌;并进一步对患儿、患儿父母及弟弟进行免疫缺陷病相关基因检测,提示患儿存在CD40L基因半合突变(IVS1-3T→G),确诊为高IgM综合征。患儿母亲为此拼接点错误基因的携带者、父亲正常,患儿弟弟与患儿有相同的CD40L基因突变。
Keywords: 嗜酸性粒细胞增多, 马尔尼菲青霉菌, 高IgM综合征, 幼儿
Abstract
A 2-year-old boy was admitted into the hospital because of cough and fever. Lymph node tuberculosis was noted when he was 2 months old and he was subsequently hospitalized several times because of cough and fever. After hospitalization the laboratory examination showed an increased eosinophia level in blood. The immune function tests shows decreased levels of IgG, IgA, and IgM. The patient had no response to anti-tuberculosis, anti-bacterial, and anti-fungal treatment, resulting in recurrent fever and progressive enlargement of the liver and spleen. Jam-like stools were noted 35 days after admission. B ultrasonography showed suspected intussusception. Laparotomy, reduction of intussusception and ileocecum angioplasty, biopsies of intestinal wall nodules and lymphoglandulae mesentericae, and hepatic biopsy were then performed under general anesthesia. The patient eventually died because of postoperative severe liver damage, disseminated intravascular coagulation and electrolyte disorder. Both the blood culture and hepatic biopsy tests showed Penicillium marneffei infecton. Immunodeficiency gene test was performed on the patient, his bother and their parents. T→G base substitution mutation (IVS1-3 T→G) in the CD40L gene was found in the patient. X-linked hyper-IgM syndrome was thus diagnosed in the patient. His mother was a carrier of the mutated CD40L gene, but his father was normal in the gene test. Hemizygous mutation in the CD40L gene was found in both the patient and his bother.
Keywords: Eosinophilia, Penicillium marneffei, Hyper-IgM syndrome, Young child
1. 病例介绍
患儿,男,2岁,因咳嗽10+ d、发热5 d第6次入院。患儿10余天前因受凉后出现阵发性连声咳、早晚为多,有痰,无气促、呕泻等,外院予以头孢硫脒、头孢地嗪、炎琥宁等治疗5 d,咳嗽无明显好转,近5 d出现发热、最高体温39℃。转诊我院,以“发热查因”收入院。起病以来,精神、睡眠可,食纳稍差,大小便正常,体重无减轻。
既往史:患儿2个月时发现腋下淋巴结肿大(出生时接种卡介苗),淋巴结活检见增生的淋巴细胞背景中散在类上皮样细胞、考虑“腋下淋巴结结核”,予以“异烟肼”治疗3个月。患儿5个月时曾因咳嗽、气促住外院,诊断为弥漫性肺间质病变、重症肺炎、败血症、真菌感染、腋下淋巴结结核、呼吸衰竭(I型),予以呼吸机治疗55 d。9个月时因喘息、发热于我院第一次住院,诊断为重症肺炎、闭塞性细支气管炎、喉梗阻(声门下腔狭窄)。以后患儿因声门下腔狭窄激光介入治疗于我院住院3次。第4次我院住院过程中,血常规:WBC 17.79×109/L,N 25%、L49%、E 27%,余项正常;肺泡灌洗液细胞成分分析:N 24%,L 10%,E 64%,上皮细胞2%;骨髓细胞学检查:骨髓增生活跃,嗜酸性粒细胞比值52%,巨核细胞少见,血小板散在分布。患儿外周血、骨髓涂片、肺泡灌洗液均发现嗜酸性粒细胞增高,考虑嗜酸性粒细胞增多综合征。患儿1岁6个月时因发热、咳嗽、流涕第5次住我院,免疫全套示IgA 0.24 g/L(正常值:0.31~0.67 g/L),IgG 2.57 g/L(正常值:5.09~10.09 g/L),IgM 0.60 g/L(正常值:0.98~1.78 g/L);T、B淋巴细胞计数正常。
个人史及家族史:患儿系第1胎第1产,足月平产出生,出生体重2.55 kg,无产伤窒息史。生长发育无特殊。患儿父母体健,非近亲结婚,家族中无类似病史可询。
入院体查:T 36.6℃,P 130次/min,R 32次/min,体重11 kg。发育正常,营养欠佳,无特殊面容,自动体位,查体不合作,全身皮肤未见黄染、皮疹及出血点,全身浅表淋巴结未触及肿大。唇无紫绀,口腔黏膜无疱疹和溃疡,扁桃体Ⅱ°肿大,无脓性分泌物。颈软、无抵抗,气管居中。胸廓无畸形,双侧呼吸运动度对称,双肺呼吸音粗,未闻及明显干湿性啰音和胸膜摩擦音。心率130次/min,律齐,各瓣膜听诊区未闻及病理性杂音,心音有力,无心包摩擦音。腹部平软,未触及腹部包块,肝脏肋下1.5 cm、质软,脾脏肋下未触及。肠鸣音6~7次/min。肛门、外生殖器正常。双下肢无浮肿,四肢肌力、肌张力正常。巴氏征、克氏征、布氏征阴性。
辅助检查:血常规示白细胞增高、嗜酸性粒细胞比例增高(WBC 19.20×109/L、E 12.7%),余正常。C反应蛋白101 mg/L(正常值:0~10 mg/L)。肝功能:谷丙转氨酶183 U/L(正常值9~50 U/L)、谷草转氨酶105 U/L(正常值15~40 U/L),余正常。免疫全套:IgA 0.26 g/L(正常值:0.31~0.67 g/L),IgG 1.35 g/L(正常值:5.09~10.09 g/L),IgM 0.73 g/L(参考值:0.98~1.78 g/L)。中性粒细胞活化率99.9%(正常: > 90%),T、B细胞亚群无异常,1, 3-β-葡聚糖正常、半乳糖甘露醇聚糖:0.8 μg/L(正常 < 0.5 μg/L)。狼疮全套、ANCA(-)。寄生虫全套(-)。过敏原、IgE、维生素B12均无异常。骨髓细胞学检查无异常,骨髓FIP1L1/PDGFRα融合基因(-),TCR、Ig基因重排检测(-)。输血前常规(-),结核斑点试验220 pg/mL(正常值0~14 pg/mL)。呼吸道病毒7项(包括呼吸道合胞病毒,腺病毒,流感病毒A、B型,副流感病毒1、2、3型)、EB病毒-Ab、MP-Ab、CP-Ab均阴性,EB病毒、呼吸道腺病毒、肺炎支原体DNA无异常。痰培养、细菌培养:无致病菌生长。脑脊液常规、生化均大致正常,脑脊液墨汁染色、G染色阴性,脑脊液真菌涂片阴性。胸部CT提示闭塞性支气管炎、双肺肺炎、隆突下淋巴结明显肿大。入院第2天行纤维支气管镜检查,可见左主支气管后外侧壁向内隆起,中段近膜部可见黄白色坏死物,考虑:支气管内膜炎(真菌感染?结核感染?)、声门下腔狭窄。入院第4天行第2次纤维支气管镜检查,见左主支气管后外侧壁向内隆起较前好转,下段开口处近膜部可见少许黄白坏死物。并行支气管粘膜活检,考虑肉芽肿性炎症,结核不能完全排除。入院第13天行第3次纤维支气管镜,镜下较第2次无明显变化。腹部B超(入院第3天)提示肝脾肿大,肝肋下40 mm,脾肋下21 mm,腹腔局部积液;复查腹部B超(入院第14天):肝下界于脐下7.2 mm,脾肋下33 mm,肝内外胆管壁稍增粗,腹腔局部积液;入院第31天腹部B超提示肝下界于脐下25 mm,肝实质回声欠均匀,脾肋下20 mm,肝内外胆管壁稍增粗,腹腔局部积液,腹腔内多个低回声结节、考虑淋巴结肿大,腹腔内于左右髂窝位置不规则回声团、疑来自肠管病变。
2. 诊断思维
2岁男性幼儿,出生后反复感染:2个月时发现淋巴结结核,多次因发热、咳嗽住院:其中5个月时因“肺弥漫性病变”呼吸机治疗55 d,肺间质疾病相关基因检测未见异常;因气管插管后的声门下腔狭窄行介入治疗时多次发现外周血、骨髓涂片、肺泡灌洗液中嗜酸性粒细胞增高,未找到血液系统恶性肿瘤依据,也排除了可致嗜酸性粒细胞增多的常见因素,如寄生虫和病毒感染、过敏性疾病、药物或化学物质引起、结缔组织病和内分泌疾病等,考虑特发性嗜酸性粒细胞增多症。患儿1岁6个月起发现IgA、IgG、IgM均降低;本次住院过程中患儿反复发热40 d,经强有力抗结核、抗细菌、抗真菌治疗效果不佳,病情持续进展,最终死亡。患儿接种卡介苗后出现腋下淋巴结结核、反复感染、免疫球蛋白降低,要考虑存在免疫缺陷基础疾病:男性患儿需要注意X-连锁无丙种球蛋白血症(X-linked agammaglobulinemia, XLA),但该患儿有扁桃体及淋巴结大,且CD19不低,不支持XLA;同时也需注意与普通变异性免疫缺陷病(common variable immunodeficiency, CVID)、X-连锁淋巴细胞异常增生症(X-linked lymphoproliferative disease, XLP)等疾病相鉴别,患儿无EB病毒感染的证据,XLP可能性不大;CVID也表现为免疫球蛋白低,T、B淋巴细胞计数可大致正常,但T细胞功能异常是致病的关键,但本院不能行T细胞功能检测,在诊断不明情况下,进一步完善免疫缺陷病相关基因检测以协助诊断。
3. 进一步检查
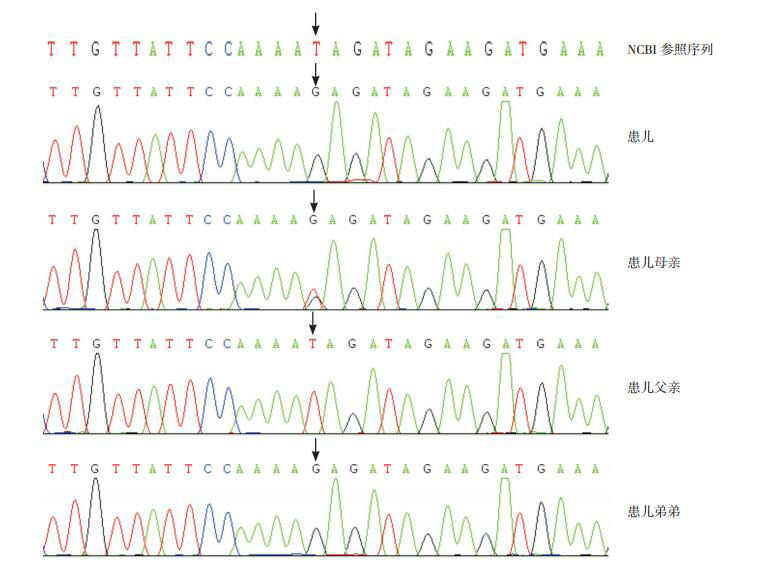
抽取患儿静脉血2 mL(EDTA抗凝血),进行4 000种单基因遗传病基因突变筛查,发现患儿CD40L基因的内含子碱基T突变为G(IVS1-3 T→G)。进一步采集患儿父母及弟弟的外周血进行CD40L基因检测,发现患儿母亲为此拼接点错误基因的携带者,其父亲正常,并且患儿弟弟有相同的突变,见图 1,提示高IgM综合征。患儿入院后38 d血培养发现马尔尼菲青霉菌典型帚状枝及孢子链,菌落表面丝绒状,形成放射状皱褶。
1.
患儿家系CD40L基因突变位点测序图
患儿及弟弟均为CD40L基因半合子突变(IVS1-3 T → G),母亲为杂合突变,父亲为IVS1-3 T野生型。箭头所指为突变位点。
4. 临床经过
患儿本次以“咳嗽、发热”入院,因患儿既往有声门下腔狭窄病史,故于入院后第2天和第4天两次行纤维支气管镜检查,镜下可见隆突下淋巴结肿大,中段近膜部可见黄白坏死物,支气管粘膜活检病理考虑肉芽肿性炎症。结合患儿有结核接触史(反复追问病史,患儿邻居有肺结核),既往有腋下淋巴结结核病史,以及因特发性嗜酸性粒细胞增多症口服过激素治疗,同时患儿结核斑点实验阳性,考虑结核可能性大,遂予利福平、异烟肼、乙胺丁醇、丁胺卡那联合抗结核,并予抗感染及间断输注丙种球蛋白免疫支持、甲强龙抗炎及保护脏器等对症处理,患儿仍反复发热,肝脾进行性增大,伴肝功能损害。患儿GM试验阳性,而且有服用激素等宿主因素,抗结核、抗细菌治疗仍反复发热,不排除真菌感染可能,先后加用伏立康唑、卡泊芬净抗真菌治疗,仍持续发热,精神差,伴烦躁不安,肝脏进行性增大至脐下25 mm,入院第35天出现果酱样大便,床旁B超提示左侧腹局部肠管横断面呈“同心圆征”,考虑肠套叠可能。于全麻下行急诊剖腹探查、肠套叠复位+回盲部成形术,以及肠壁结节、肠系膜淋巴结活检、肝活检术。术后患儿病情持续进展,出现肝功能衰竭、DIC难以纠正,住院第38天血培养及肝活检均提示马尔尼菲青霉菌感染,停卡泊芬净,改为两性霉素B抗真菌治疗,住院第40天出现难以纠正的高血钾、严重代谢性酸中毒、低血糖、血压不稳、心律失常,死亡。
5. 确诊依据
(1)高IgM综合征(hyper-immunoglobulin M syndromes, HIGM):患儿,男,2岁,接种卡介苗后出现腋下淋巴结结核,自幼反复感染、伴嗜酸性粒细胞增多,多次查免疫全套示IgG、IgA、IgM均低,基因诊断提示CD40L基因半合突变(IVS1-3 T→G),其母亲为此拼接点错误基因的携带者,父亲正常,患儿弟弟与患儿有相同的点突变。(2)马尔尼菲青霉菌败血症:患儿发热、肝脾肿大,血培养及肝活检均找到马尔尼菲青霉菌。
6. 讨论
高IgM综合征是一种较罕见的原发性免疫缺陷病。20世纪60年代由Asselain和Rosen等首次报道,其发病机制主要为免疫球蛋白类别转换障碍,导致患者IgM不能正常转化为IgG和IgA,血清IgM正常或升高,IgG和IgA降低或缺乏,伴或不伴体细胞高频突变缺陷[1]。据文献报道,约有6.4%HIGM患者的血清IgM水平低下[2]。本例IgM水平也降低。因此,有学者认为以HIGM命名可能造成临床医生对该病的错误认识,近年来主张直接以突变基因命名各类HIGM[3]。根据基因缺陷不同,HIGM分为7个亚型,1型(HIGM 1)最常见,约占65%~70%,为X连锁隐性遗传[4],是由CD40L基因(位于Xq26.3-27.1)突变引起。CD40L基因包括5个外显子,第1号外显子编码胞质区、跨膜区和胞外区的6个氨基酸,第2号与第3号外显子编码胞外茎区,第4号、第5号外显子编码C末端的147个氨基酸。CD40L是TNF超家族成员,主要表达于活化的CD4+T淋巴细胞表面,可作用于CD40分子,使B淋巴细胞分泌的免疫球蛋白发生转化类别转换。CD40L基因一旦发生突变,可使T淋巴细胞表面的CD40L表达降低,或导致CD40L不能与CD40结合,或影响CD40分子三聚体的形成。因此CD40L缺陷可损伤T淋巴细胞和B淋巴细胞相互作用,破坏生发中心形成,并影响免疫球蛋白类别转换[5]。基因检测是确诊HIGM 1的金标准[6]。
HIGM 1发病率约为1/1 030 000,所有HIGM 1患者均有明显的IgG缺乏,绝大多数有IgA缺乏,但只有一半的患者IgM水平升高,有的患者IgM水平甚至降低。本例患者的IgG明显降低发现于第5次我院住院期间,伴IgA、IgM均降低,早期一直未发现免疫球蛋白降低,可能与患儿间断输注丙种球蛋白有关。
超过一半的HIGM 1患者在1岁以内、90%的在4岁以内出现临床症状,主要表现为反复感染,多以卡氏肺孢子菌和隐孢子虫感染多见,弓形虫、隐球菌和组织胞浆菌感染亦见报道[7]。HIGM 1感染症状重,死亡率高,早期严重感染和随后并发的肝脏疾病为主要致死原因。该患儿本次住院过程中血培养及肝活检均发现马尔尼菲青霉菌,伴马尔尼菲青霉菌败血症。马尔尼菲青霉菌为条件致病菌,免疫功能低下者易感,在艾滋病病人中马尔尼菲青霉菌感染有增多趋势[8],主要是单核巨噬细胞受累,可引起皮肤、淋巴结和内脏的致命性感染。
中性粒细胞缺乏是HIGM 1最常见的特征之一,但本患儿中性粒细胞一直正常,多次血常规、肺泡灌洗液、骨髓涂片均提示嗜酸性粒细胞增高,而患儿寄生虫、过敏原和IgE检查均无异常,骨髓细胞学及嗜酸增值性疾病基因筛查均未见异常;而且仅本次住院有马尔尼菲青霉菌感染依据,不能解释反复嗜酸性粒细胞增多的全过程。因此,嗜酸性粒细胞增多原因不明。HIGM 1合并嗜酸性粒细胞增多临床少见,其具体机制有待进一步研究。有文献报道伴嗜酸性粒细胞增多的HIGM 1采用丙种球蛋白替代治疗联合小剂量糖皮质激素效果较好[9]。
HIGM 1是一种严重的免疫缺陷病,常常发生多种严重和难治性感染,病死率高,需免疫球蛋白(每次500 mg/kg,每月1次)规律替代治疗以预防机会性感染,根治则需造血干细胞移植,基因治疗尚在试验阶段。国外报道干细胞移植治疗HIGM 1的成功率为72%[10],2006年我国首例HIGM 1患者骨髓移植成功[11]。
7. 结语
高IgM综合征是一种较罕见的原发性免疫缺陷病。本患儿卡介苗接种后出现腋下淋巴结结核,而且反复感染,免疫球蛋白降低,考虑存在免疫缺陷基础疾病,基因诊断提示CD40L基因半合突变(IVS1-3 T→G),确诊HIGM 1。患儿弟弟在家系基因测序中得以早期诊断,每月定期输注丙种球蛋白,口服复方新诺明预防卡氏肺囊虫感染,无反复感染发生,生长发育正常。因此,该病如果能够早期诊断、规范管理,反复严重感染是可能避免的,死亡率也可以大大降低。临床医生应提高对卡介苗接种异常反应,以及反复发热、肝脾大、嗜酸性粒细胞增多的警惕性,不能仅仅满足于针对某次感染的诊断,应注意免疫缺陷基础疾病的可能。
Biography
刘丹, 女, 硕士, 主治医师
References
- 1.唐 文静, 赵 晓东. 高IgM综合征发病机制研究进展. http://mall.cnki.net/magazine/article/gwsx200403010.htm 国际儿科杂志. 2013;40(1):10–13. [Google Scholar]
- 2.Heinoid A, Hanebeck B, Daniel V, et al. Pitfalls of hyper-IgM syndrome:a new CD40 ligand mutation in the presence of low IgM levels.A case report and a critical review of the literature. Infection. 2010;38(6):491–496. doi: 10.1007/s15010-010-0061-9. [DOI] [PubMed] [Google Scholar]
- 3.Hirbod-Mobarakeh A, Aghamohammadi A, Rezaei N. Immunoglobulin class switch recombination deficiency type 1 or CD40 ligand deficiency:from bedside to bench and back again. Expert Rev Clin Immunol. 2014;10(1):91–105. doi: 10.1586/1744666X.2014.864554. [DOI] [PubMed] [Google Scholar]
- 4.贺 建新, 王 荃, 刘 秀云, et al. 以卡氏肺孢子菌肺炎起病的X连锁高IgM综合征婴儿1例. http://www.cnki.com.cn/Article/CJFDTOTAL-ZSEK201308025.htm 中国实用儿科杂志. 2013;28(8):635–637. [Google Scholar]
- 5.陈 同辛, 金 莹莹. 高IgM综合征研究进展. http://www.cnki.com.cn/Article/CJFDTOTAL-SYQK201221003.htm 实用儿科临床杂志. 2012;27(21):1624–1628. [Google Scholar]
- 6.Davies EG, Thrasher AJ. Update on the hyper immunoglobulin M syndromes. Br J Haematol. 2010;149(2):167–180. doi: 10.1111/bjh.2010.149.issue-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winkelstein JA, Marino MC, Ochs H, et al. The X-linked hyper-IgM syndrome:clinical and immunologic features of 79 patients. Medicine (Baltimore) 2003;82(6):373–384. doi: 10.1097/01.md.0000100046.06009.b0. [DOI] [PubMed] [Google Scholar]
- 8.覃 川, 温 小凤, 蒋 忠胜. 艾滋病合并马尔尼菲青霉菌病20例临床分析. http://www.cnki.com.cn/Article/CJFDTOTAL-YJYX201301043.htm 右江医学. 2013;41(1):88–89. [Google Scholar]
- 9.Guo Li, Chen Bo, Xu B, et al. X-linked hyper-IgM syndrome with eosinophilia in a male child:A case report. https://www.researchgate.net/publication/272388434_X-linked_hyper-IgM_syndrome_with_eosinophilia_in_a_male_child_A_case_report. Exp Ther Med. 2015;9(4):1328–1330. doi: 10.3892/etm.2015.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tomizawa D, Imai K, Ito S, et al. Allogeneic hematopoietic stem cell transplantationfor seven children with X-linked hyper-IgM syndrome:a single center experience. Am J Hematol. 2004;76(1):33–39. doi: 10.1002/(ISSN)1096-8652. [DOI] [PubMed] [Google Scholar]
- 11.王 坚敏, 陈 同辛, 陈 静, et al. 造血干细胞移植治疗X-连锁重症联合免疫缺陷病临床观察. http://med.wanfangdata.com.cn/Paper/Detail?id=PeriodicalPaper_nkllysj201001010 内科临床与实践. 2010;5(1):48–52. [Google Scholar]