

SUMMARY
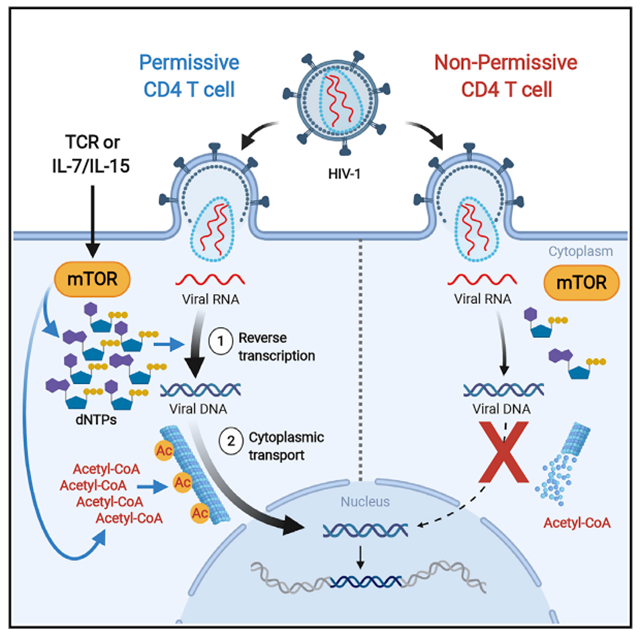
Cellular metabolism governs the susceptibility of CD4 T cells to HIV-1 infection. Multiple early post-fusion steps of HIV-1 replication are restricted in resting peripheral blood CD4 T cells; however, molecular mechanisms that underlie metabolic control of these steps remain undefined. Here, we show that mTOR activity following T cell stimulatory signals overcomes metabolic restrictions in these cells by enabling the expansion of dNTPs to fuel HIV-1 reverse transcription (RT), as well as increasing acetyl-CoA to stabilize microtubules that transport RT products. We find that catalytic mTOR inhibition diminishes the expansion of pools of both of these metabolites by limiting glucose and glutamine utilization in several pathways, thereby suppressing HIV-1 infection. We demonstrate how mTOR-coordinated biosyntheses enable the early steps of HIV-1 replication, add metabolic mechanisms by which mTOR inhibitors block HIV-1, and identify some metabolic modules downstream of mTOR as druggable targets for HIV-1 inhibition.
Grphaical Abstract
In Brief
Taylor et al. show that mTOR links cellular metabolism to the susceptibility of CD4 T cells to HIV-1 infection by expanding the pools of metabolites that facilitate the synthesis of reverse transcription products and their cytoplasmic transport toward the nucleus. They also characterize targets for HIV-1 interventions downstream of mTOR signaling.
INTRODUCTION
Resting peripheral blood CD4 T cells are non-permissive at multiple post-entry steps of HIV-1 infection, in contrast to their tissue-resident counterparts (Eckstein et al., 2001; Grivel and Margolis, 2009; Kinter et al., 2003). After HIV-1 enters these metabolically quiescent blood cells, reverse transcription (RT) is minimal (Korin and Zack, 1999), and RT products are not found in the nucleus (Sun et al., 1997). Ex vivo exposure of blood resting CD4 T cells to a multitude of extracellular signals present in tissue microenvironments enables their infection (Bolduc et al., 2017; Cameron et al., 2010; Sun et al., 1997; Unutmaz et al., 1999).
There is increasing evidence that cellular metabolism is a major determinant of HIV-1 susceptibility; for example, limiting glucose transport and glycolysis impairs both HIV-1 replication and expansion of its latent reservoirs (Loisel-Meyer et al., 2012; Palmer et al., 2017; Valle-Casuso et al., 2019). However, the cellular mechanisms by which such metabolic perturbations inhibit HIV-1 replication in susceptible CD4 T cells remain undefined. We studied how intracellular metabolic effects downstream of T cell activation caused by extracellular signals present in tissue microenvironments may counter restrictions to HIV-1 RT, and transit of RT products, in resting blood CD4 T cells.
It is well established that the activation of CD4 T cells via the T cell receptor (TCR) or common gamma chain (γc)-cytokine stimulation triggers the mechanistic target of rapamycin (mTOR) (Delgoffe et al., 2011; Marçais et al., 2014), a “master regulator” of metabolism as the catalytic kinase subunit of 2 distinct signaling complexes, mTOR complex 1 (mTORC1) and mTORC2 (Delgoffe et al., 2011; Li et al., 2011; Marçais et al., 2014). Therefore, we focused our study on the downstream metabolic effects of these mTOR-activating stimuli on HIV-1 replication. We reported previously that the inhibition of an upstream pathway that activates mTOR can block the synthesis of deoxyribonucleotide triphosphates (dNTPs) essential for HIV-1 RT in TCR-stimulated cells (Taylor et al., 2015). HIV-1 infection of cell lines and myeloid cells induces the acetylation of α-tubulin to stabilize a subset of microtubules (MTs) that facilitate the transport of HIV-1 RT products through the cytoplasm to the nuclear membrane (Sabo et al., 2013). It has not yet been reported whether this also occurs in activated blood CD4 T cells, or whether its absence contributes to the inefficient nuclear entry of the limited HIV-1 RT products made in resting blood CD4 T cells as documented by the lack of nuclear-produced viral 2-long terminal repeat (2-LTR circle) DNA (Sun et al., 1997). Here, we show how direct catalytic inhibition of both mTOR complexes (using catalytic inhibitors called mTORi here) or allosteric inhibition of mTORC1 by rapamycin reverses intracellular metabolic effects to enable these early steps in HIV-1 replication after TCR or γc-cytokine stimulation. Here, we identify which biosynthetic pathways downstream of T cell activation-associated mTOR activity enable multiple post-entry steps in HIV-1 replication. In addition to advancing the understanding of how cellular metabolism governs HIV-1 susceptibility, this report identifies potential targets for anti-HIV-1 strategies.
RESULTS
mTOR Activity Facilitates Multiple Intracellular Steps of Early HIV-1 Replication
To evaluate the effects of mTORi on the post-entry steps of HIV-1 replication, we performed experiments with X4-tropic HIV-1 that could enter CD4 T cells independently of the C-C chemokine receptor type 5 (CCR5) co-receptor, whose expression has been shown to be sensitive to both rapamycin and mTORi (Heredia et al., 2003, 2015). Resting CD4 T cells were pretreated with 3 chemically distinct catalytic mTOR inhibitors, rapamycin, or DMSO for 24 h before α-CD3/CD28 stimulation. After 48 h of stimulation, they were each infected with HIV-1 NL4-3, which uses C-X-C chemokine receptor type 4 (CXCR4) as a co-receptor. We have previously demonstrated that phospholipase D1 (PLD1), a known upstream activator of mTORC1 (Xu et al., 2011; Yoon et al., 2011), drives both dNTP pool expansion and HIV-1 replication in activated CD4 T cells (Taylor et al., 2015). Here, as a control, cells were also exposed to a specific PLD1 inhibitor (PLDi). Consistent with our earlier report, PLDi inhibited HIV-1 infection by 6-fold (Figures 1A and S1). Rapamycin caused a degree (4.5-fold) of inhibition similar to that of PLDi. Each catalytic mTORi potently suppressed HIV infection in α-CD3/CD28-stimulated cells in a concentration-dependent manner, to a magnitude exceeding that observed with rapamycin or PLDi (Figures 1A and S1). Since AZD2014 (mTORi #1) was extensively optimized for selectivity to reduce potential off-target effects (Pike et al., 2013), we selected mTORi #1 for all subsequent experiments.
Figure 1. mTOR Activity Is Essential for Early Replication of HIV-1 in CD4 T Cells after TCR and γ Chain (γc)-Cytokine Stimulation.
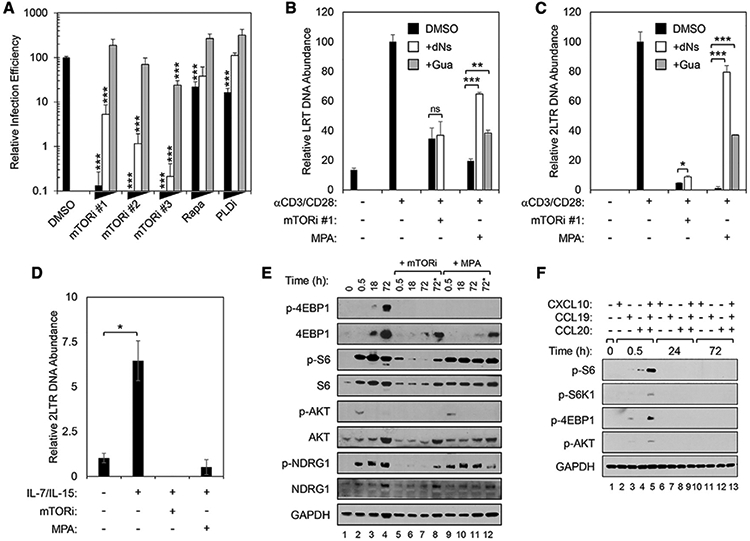
(A) mTORi inhibits HIV-1 replication. Resting primary CD4 T cells were pretreated and cultured with titrations of rapamycin, PLDi, or catalytic mTOR inhibitors mTORi #1, mTORi #2, or mTORi #3 for 24 h, and then stimulated for 48 h with α-CD3/CD28 beads before infection with an X4-envelope-pseudotyped NL4-3-derived GFP reporter virus. Three days post-infection, cells were analyzed for GFP expression by fluorescence-activated cell sorting (FACS). Means and SDs of triplicate samples are shown. Statistical significance was determined by 2-tailed Student’s t test, comparing DMSO to each condition. *p < 0.05, **p < 0.01, and ***p < 0.005.
(B) mTOR inhibition decreases late reverse transcripts (LRT) after TCR stimulation. (C) mTOR inhibition decreases 2-LTR circle DNA after TCR stimulation. (B and C) Resting CD4 T cells were pretreated with either mTORi #1 (B) or mycophenolic acid (MPA) (C) and then stimulated with α-CD3/CD28 beads as in (A).
(D) mTOR inhibition decreases 2-LTR circle DNA after gc-cytokine stimulation. Resting CD4 T cells were pretreated with mTORi or MPA and then stimulated with IL-7/IL-15 for 5 days before infection with HIV-1 NL4-3 (50 ng p24 per million cells).
In (B)–(D), total DNA was harvested 16 and 72 h after infection for qPCR analysis of accumulation of LRT (B) and 2-LTR circle (C and D) DNA, respectively. As indicated, cells were treated with 50 μM guanosine (+Gua) or each of all 4 deoxyribonucleosides (+dNs). Means and SDs of triplicates are shown. Data are from 3 independent donors. Statistical significance was determined by 2-tailed Student’s t test. *p < 0.05, **p < 0.01, and ***p < 0.005.
(E) mTOR signaling with and with inhibitor pre-treatment after TCR stimulation. (F) mTOR signaling with and with inhibitor pre-treatment after stimulation with selected chemokines. (E and F) Immunoblots are of indicated targets in whole-cell lysates (WCLs) of CD4 T cells pretreated with mTORi, as in (A), or MPA, as in (B), followed by α-CD3/CD28 (E) or chemokine (F) stimulation. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was a loading control. Data are from 3 donors. In (E), the 2 lanes labeled 72* had mTORi or MPA and were added 18 h after stimulation with α-CD3/CD28 beads and analyses were done at 72 h after stimulation.
Because different metabolites are required for the early steps of HIV-1 replication (Bukrinsky et al., 1992; Fassati et al., 2003; Gao et al., 1993), we assessed the anabolism-coordinating role of mTOR in the accumulation of viral late reverse transcripts (LRTs) and 2-LTR circles in α-CD3/CD28-stimulated CD4 T cells using qPCR (Figures 1B and 1C). LRT detection indicates the near completion of RT, and the detection of 2-LTR circles indicates cytoplasmic trafficking and nuclear import of the HIV-1 RT products. Treatment with mTORi suppressed the accumulation of both LRT and, more markedly, 2-LTR circles after α-CD3/CD28-stimulation, consistent with effects on both RT and at least one of the subsequent steps (Figures 1B and 1C). An inhibitor of a rate-limiting step in purine syntheses catalyzed by inosine monophosphate dehydrogenase (IMPDH), mycophenolic acid (MPA), was used as a positive control here as it is known to decrease HIV-1 replication (Chapuis et al., 2000; Margolis et al., 1999). We demonstrate that the addition of exogenous deoxyribonucleosides (dNs) or guanosine (G) to bypass the IMPDH1-dependent step in purine biosynthesis rescued the accumulation of both LRT and 2-LTR circle DNA after MPA pretreatment of α-CD3/CD28-stimulated cells (Figures 1B and 1C). However, we observed no significant rescue of LRT DNA, and only marginal recovery of 2-LTR circle DNA, in mTORi-treated cells (Figures 1B and 1C). Stimulation by γc-cytokines (a combination of interleukin-7 [IL-7] and IL-15 [IL-7/IL-15]) also increased 2-LTR circle formation; pretreatment with either mTORi or MPA blocked this effect (Figure 1D).
mTORC1 and mTORC2 Are Activated in CD4 T Cells by Extracellular Signals
Next, we confirmed that α-CD3/CD28 stimulation increased the activity of mTOR complexes in resting CD4 T cells. Here, α-CD3/CD28 beads increased the phosphorylation of mTORC1 downstream targets eukaryotic translation initiation factor 4E (eIF4E)-binding protein 1 (4E-BP1) and ribosomal protein S6 (S6) (Figure 1E) (Delgoffe et al., 2011; Li et al., 2011; Marçais et al., 2014). α-CD3/CD28 stimulation induced robust mTORC1 activity within 30 min that was sustained over 72 h, as determined by both the expression and the level of phosphorylated ribosomal protein S6 (p-S6) (Figure 1E). Pretreatment with either mTORi or MPA abrogated the phosphorylation of both 4E-BP1 and S6, thus confirming the efficacy of both inhibitors in suppressing mTORC1-dependent phosphorylation events in stimulated CD4 T cells (Figure 1E). α-CD3/CD28 stimulation also induced the mTORC2-specific phosphorylation of AKT (Ser473, p-AKT) and NDRG1 (Thr346, p-NDRG1) (Figure 1E) (Delgoffe et al., 2011; García-Martínez and Alessi, 2008). These mTORC2-specific phosphorylation events were inhibited by mTORi, but not by MPA. These data provide evidence that mTORi suppresses the catalytic activity of both mTORC1 and mTORC2 complexes in stimulated CD4 T cells and confirm that MPA selectively inhibits mTORC1 (Emmanuel et al., 2017; He et al., 2011).
Next, we examined resting CD4 T cell treatment with the γc-cytokines and chemokines known to facilitate HIV-1 replication. Simulation with IL-7, IL-15, or IL-7/IL-15 increased abundance of p-S6 that was suppressed by mTORi treatment (Figure S2). We observed that CXCL10, CCL19, and CCL20, chemokines that increase the susceptibility of resting CD4 T cells to HIV-1 infection (Anderson et al., 2020; Cameron et al., 2010), also activate mTOR (Figure 1F). When tested individually, only CCL20 increased transient S6 phosphorylation (Figure 1F). CCL19 induced detectable levels of p-4E-BP1. Notably, a combination of CXCL10, CCL19, and CCL20 more markedly activated the phosphorylation of both of these mTORC1 targets than any of the individually tested chemokines (Figure 1F). Phosphorylated AKT was also found to be transiently and minimally increased after exposure to a combination of all 3 chemokines (Figure 1F). Thus, diverse stimuli that increase the susceptibility of resting CD4 T cells to HIV-1 infection each enhance mTORC1 and/or mTORC2 activity, albeit with different kinetics, magnitude, and profile. Given greater mTOR activation with α-CD3/CD28 and IL-7/IL-15 stimulation, we focused on defining mTOR-related mechanisms that affect HIV replication after those stimuli thereafter.
mTOR Activates Multiple Pathways to Expand RT-Enabling dNTP Pools
We evaluated the impact of mTOR on dNTP pool expansion because LRT accumulation is known to require it. α-CD3/CD28 stimulation elevated the pools of all dNTPs as determined by mass spectrometry (liquid chromatography-tandem mass spectrometry [LC-MS/MS]) (Bushman et al., 2011) (Figure 2A). Pretreatment with mTORi significantly reduced dNTP levels by 25%, 91%, 78%, and 88%, for deoxycytidine triphosphate (dCTP), deoxyguanosine triphosphate (dGTP), thymidine triphosphate (TTP), and deoxyadenosine triphosphate (dATP), respectively. Rapamycin had more modest effects on dNTP pool expansion in treated cells.
Figure 2. mTOR Activity Is Essential for Deoxyribonucleotide Triphosphates (dNTPs) Pool Expansion via Coordination of Programs for Nutrient Uptake, Pentose Phosphate Pathway (PPP), and Other Nucleotide Biosynthesis Pathways.

(A) Levels of dNTPs in CD4 T cells treated as indicated were determined by LC-MS/MS. Experiments were repeated 3 times using cells from independent donors. Means and SDs of triplicate samples from a single representative donor are shown. Statistical significance was determined by 2-tailed Student’sttests. *p < 0.05, **p < 0.01, and ***p < 0.005.
(B and C). Immunoblots are shown of phosphorylated CAD (p-CAD) and total CAD protein (CAD) in WCLs of CD4 T cells pretreated with mTORi or MPA as in Figure 1, followed by stimulation with α-CD3/CD28 beads for 3 days or γc-cytokines (IL-7 or IL-15) for 5 days (B), or a time course following stimulation with α-CD3/CD28 beads (C). GAPDH is a loading control in both panels. In the lanes labeled 72* in (C), mTORi or MPA (as indicated) were added only at 18 h after stimulation with α-CD3/CD28 beads and analyses done at 72 h after stimulation. (C) includes the relative densitometry values of CAD and p-CAD.
(D) Immunoblots of CD4 T cell WCLs for indicated targets following α-CD3/CD28 or γc-cytokine stimulation of cells following pretreatment with mTORi or MPA, as in (B). The rightmost 5 lanes show decreases in mTOR-dependent phosphorylation of 4E-BP1 (4E-BP1/p-4E-BP1) and expression of ASCT2, Glut1, and RRM1 with MPA pretreatment that were rescued by the positive feedback of guanine nucleotides. Exogenous nucleosides (deoxyribonucleosides [dNs], deoxycytidine [dC], deoxyguanosine [dG], or guanosine [G]) were added at the time of α-CD3/CD28 stimulation, as indicated. GAPDH was a loading control.
(E and F) Immunoblot analyses of the indicated proteins in WCLs of CD4 T cells pretreated with mTORi, as above, followed by stimulation for the indicated time with α-CD3/CD28 beads. In the lanes labeled 72* in (E) and (F), mTORi was added only at 18 h after stimulation with α-CD3/CD28 beads and analyses done at 72 h after stimulation. Below each blot, relative densitometry values are indicated for each target, but for SREBP-1 in (F), the p:m densitometry ratios are presented. Data are representative of 3 independent donors.
(G) Schematic of the PPP and metabolic profiling of pathway intermediates in resting CD4 T cell (U), stimulated for 48 h with α-CD3/CD28 beads in the absence (S) or presence of mTORi pretreatment (S + I). Means and SDs are shown. Data are from 3 independent donors.
We next studied carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase (CAD), the rate-limiting enzyme for de novo synthesis of all pyrimidine nucleotides, including the dNTPs, dCTP, and TTP. CAD expression is regulated by c-Myc (Eberhardy and Farnham, 2001; Wang et al., 2011), but its function is stimulated after phosphorylation (Ben-Sahra et al., 2016; Robitaille et al., 2013). Stimulation of human CD4 T cells with α-CD3/CD28 beads, as well as either IL-7 or IL-15, increased the level of phosphorylation at the activating residue S1859 of CAD (p-CAD), and mTORi exposure reduced this phosphorylation (Figure 2B). Increased p-CAD was evident at 18 h after stimulation and persisted for at least 72 h in the absence of mTORi (Figure 2C). Any detection of p-CAD after stimulation was abrogated by treatment with mTORi before α-CD3/CD28 stimulation (Figures 2C and S3A). The decrease in p-CAD at 72 h after stimulation was also seen when mTORi exposure started 18 h after stimulation (Figure 2C, p-CAD lane 72*, +mTORi). The latter result is consistent with the dependence of p-CAD on mTOR activity across this entire time course, including after mTOR-dependent upregulation of c-Myc at 18 h after stimulation (Figure 2E). In contrast, mTORi exposure before α-CD3/CD28 stimulation, or starting at 18 h after stimulation, did not completely block increases in total CAD expression (Figures 2C and S3A). The significant reduction in the ratio of p-CAD:CAD by mTORi exposure after 18 h of stimulation confirms the preferential reduction of p-CAD when compared to the total CAD levels (Figures 2C and S3A). Furthermore, metabolic profiling experiments revealed that mTORi decreased orotidine (Figure S3B), a pyrimidine intermediate downstream of CAD action, thereby confirming that the lack of activated p-CAD impaired pyrimidine biosynthetic activity (Figures 2C and S3A) in the presence of mTORi.
We next analyzed processes upstream and downstream of ribonucleotide syntheses to further evaluate the mechanisms of the effect of mTORi on purine dNTPs, since the elevation of CAD activity cannot explain the mTOR-dependent increases in purine nucleotides following CD4 T cell stimulation (Figures 2A and S3C). The cell metabolic steps needed for the de novo synthesis of nucleotide precursors of dNTPs begin with the coordinated uptake of glucose and glutamine via the activation-induced expression of glucose transporter 1 (Glut1/SLC2A1) and alanine, serine, cysteine-preferring transporter 2 (ASCT2/SLC1A5), respectively, in CD4 T cells (Buck et al., 2015). Notably, Glut1 expression and the level of glucose uptake by activated CD4 T cells were both previously reported to correlate with HIV-1 infection (Valle-Casuso et al., 2019). α-CD3/CD28 and IL-7/IL-15 stimulation each induced Glut1 and ASCT2 expression in parallel with mTORC1 target p-4E-BP1, and the effects of both stimuli on each transporter were suppressed by mTORi (Figure 2D).
We also found that the expression of IMPDH1, the key enzyme that regulates the synthesis of guanine-containing purines, was dependent on mTOR activity after stimulation via the TCR or IL-7/IL-15 (Figure 2D). Since ribonucleotide reductase is required to convert both purine and pyrimidine ribonucleotides to deoxyribonucleotides, and its inhibition by hydroxyurea (HU) inhibition blocks HIV-1 RT in activated T cells (Korin and Zack, 1999), we also assessed the effects of catalytic mTOR inhibition on ribonucleotide reductase catalytic subunit M1 (RRM1). TCR or IL-7/IL-15 stimulation induced the expression of RRM1, which was also mTOR dependent after either stimulus (Figure 2D).
Both IMPDH1 and RRM1 are well characterized as targets of c-Myc (Liu et al., 2008; Mannava et al., 2012). Therefore, these results are consistent with T cell activation’s mediating the induction of the c-Myc-dependent transcriptional program via mTOR signaling (Taylor et al., 2015). We assessed the effects of mTORi treatment on the expression of two additional c-Myc targets known to catalyze key steps in purine and pyrimidine synthesis, respectively: methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) and thymidylate synthase (TYMS) (Liu et al., 2019; Pikman et al., 2016) (Figure 2E). These 2 mitochondrial 1-carbon (1C) metabolism enzymes are enhanced by mitochondrial proteome remodeling triggered by α-CD3/CD28 stimulation (Ron-Harel et al., 2016). The expression of both c-Myc targets was suppressed in CD4 T cells pretreated with mTORi before this stimulus, along with decreased c-Myc expression (Figure 2E). Consistent with these mTOR-dependent effects being mediated via c-Myc, the robust suppression of c-Myc targets was not observed when mTORi exposure started after the known early post-stimulation peak of c-Myc expression (18 h post-stimulation) (Figure 2E, lane labeled 72*) (Wang et al., 2011).
The pentose phosphate pathway (PPP) was also studied because glucose is converted to ribose phosphate precursors of ribonucleotides by enzymes in the PPP (Wang et al., 2011). In fact, increasing the flux of glucose-derived carbons through the PPP by inhibiting glycolysis increases reverse transcription and HIV-1 infection (Clerc et al., 2019). We determined that the PPP enzymes ribulose-5-phosphate 3-epimerase (RPE) and phosphogluconate dehydrogenase (PGD) accumulated in CD4 T cells after α-CD3/CD28 stimulation (Figure 2F). Each was reduced by mTORi treatment (Figure 2F), which is consistent with the dependence on mTORC1 shown in mouse embryonic fibroblasts (Düvel et al., 2010). The conversion of sterol regulatory element-binding protein 1 (SREBP-1), a transcription factor critical for PPP enzyme expression, from its inactive membrane-bound precursor (p) to a mature (m) transcriptionally active form by α-CD3/CD28 stimulation was reduced by mTORi in CD4 T cells, as evidenced by an increased p:m ratio after 30 min of stimulation (Figure 2F). To confirm the modulation of the PPP pathway, we performed metabolic profiling of CD4 T cells stimulated with α-CD3/CD28 in the absence and presence of mTORi (Figure 2G). Here, we observed a significant accumulation of the PPP products sedoheptulose-7-phosphate (S7P) and 5-phosphoribosyl diphosphate (PRPP) in stimulated CD4 T cells. This induction (S7P and PRPP) and the maintenance of the levels of PPP metabolites (for 6-phosphogluconate [6PG]) were each blocked by mTORi (Figure 2G). In sum, mTORi affected several metabolic pathways to abrogate the expansion of all dNTPs (Figure 2A).
Guanine Nucleotides Mediate an mTORC1-Dependent Positive Feedback Loop
Supplementation of MPA-treated cell cultures with exogenous G, a mixture of all dNs, or deoxyguanosine (dG) but not deoxycytidine (dC) alone rescued the inhibitory effects of MPA on p-4E-BP1, confirming the dependence on the guanine nucleotide pool depletion of inhibition of mTORC1 activity by MPA (Figure 2D). In addition, the MPA-dependent inhibition of Glut1, ASCT2, and RRM1 expression were each rescued by exogenous G, dNs, or dG, but not dC (Figure 2D). This indicates that IMPDH activity can promote sustained mTORC1 activity in activated CD4 T cells and that IMPDH inhibitor-mediated decreases in mTOR activity can be bypassed by providing G or dG (Figures 1B and 1C). In contrast, an inhibitor that specifically and directly impairs mTOR catalysis is not subject to this positive feedback loop such that decreases in p-4E-BP1, Glut1, ASCT2, RRM1, and HIV-1 LRTs and 2-LTR circles are not reversed by exogenous dNs (Figures 1B, 1C, and 2D).
mTOR Regulates Expansion of Acetyl-Coenzyme A (Ac-CoA) Pools That Enable α-Tubulin Lys40 Acetylation
The cumulative effects of mTORi on the sequential metabolic processes leading to dNTP pool expansion can explain the decreased HIV-1 reverse transcription (LRT) (Figure 1B), but not decreases in nuclear import (2-LTR circles) observed in mTORi-treated, activated CD4 T cells (Figure 1C). We hypothesized that mTORi diminished the nuclear import of HIV-1 RT products by decreasing the Ac-CoA pools needed for the acetylation of α-tubulin at Lys40 (Ac-Tub). This acetylation stabilizes MTs and has been reported to enhance the cytoplasmic transport of HIV-1 RT products toward the nucleus (Sabo et al., 2013). Synthesis of the cytoplasmic pool of Ac-CoA has been shown to be dependent on both AKT and ATP citrate lyase (ACLY) (Lee et al., 2014). We also postulated that mTOR-dependent increases in glucose-derived pyruvate and glutamine-derived α-ketoglutarate (α-KG) would each enhance the tricarboxylic acid (TCA) cycle-derived citrate that fuels Ac-CoA production, adding to the mTOR activation of both AKT and ACLY.
CD4 T cells rapidly increase coordinated glucose uptake and glycolysis after activation (Macintyre et al., 2014). We determined that both the Glut1 transporter and the glycolytic pathway enzyme lactate dehydrogenase A (LDHA) increased in synchrony after either α-CD3/CD28 or IL-7/IL-15 stimulation; mTORi diminished both Glut1 and LDHA expression (Figure 3A, top 2 panels). LDHA induction affected the CD4 T cells since increases were observed in ATP after either α-CD3/CD28 or IL-7/IL-15 stimulation and blocked by mTORi (Figure S4), consistent with the results in other cell types (Broecker-Preuss et al., 2017; Wang et al., 2011).
Figure 3. Stimulation of CD4 T Cells Cause mTOR-Dependent Coordination of Glycolytic Pathway and Microtubule (MT) Stabilization.

(A) Immunoblot analyses of the indicated proteins in WCLs of CD4 T cells pretreated with mTORi as in Figure 2, followed by stimulation with α-CD3/CD28 beads, γc-cytokines (IL-7/IL-15), or TLR-2 ligand pam3CSK4. GAPDH was a loading control. Data are representative of 3 independent donors.
(B) Quantification of glycolytic metabolites in CD4 T cells (n = 3) stimulated with α-CD3/CD28 beads for 48 h or IL-7/IL-15 for 72 h in the absence or presence of pretreatment with inhibitors, as indicated by global metabolomic analysis. Significant increases in p < 0.05 are depicted in red and significant decreases of p < 0.05 are depicted in blue.
(C) Schematic representation of glycolysis and tricarboxylic acid (TCA) cycle supporting cellular acetyl-coenzyme A (Ac-CoA) synthesis in the presence of metabolic inhibitors 2-DG, UK5099, or DON.
(D) Quantification of levels of α-ketoglutarate (α-KG) in resting CD4 T cells (U) stimulated with α-CD3/CD28 beads for 48 h in the absence (S) or presence of inhibitor pretreatment (S + I), as in (B). Means and SDs of data from 3 independent donors are shown. Significant differences between U and S or S and S + I were determined by 2-tailed Student’s t test. *p < 0.05.
(E) Flow cytometric analysis of CD4 T cells stimulated (S) with α-CD3/CD28 beads as in (A) in the absence or presence of mTORi, 2-DG, DON, or UK5099 (UK) pretreatment as indicated. Means and SDs of samples from 3 independent donors are shown. Statistical significance of differences between stimulated and mock or inhibitor-treated CD4 T cells were determined by 2-tailed Student’s t test.
(F) Correlation analysis between Ac-Tub abundance and mTOR activity using data presented in (E).
We confirmed the impact of mTORi on CD4 T cell glucose metabolism via the metabolic profiling of glycolytic intermediates. α-CD3/CD28 stimulation increased the lactate levels, a product of aerobic glycolysis, and both mTORi and rapamycin suppressed this (Figure 3B); this is consistent with the effect of mTORi on LDHA expression (Figure 3A). Notably, IL-7/IL-15-stimulated CD4 T cells produced much less lactate compared to cells stimulated via the TCR (Figure 3B). This is evidence of more activation of aerobic glycolysis in cells stimulated by α-CD3/CD28 than IL-7/IL-15, which is consistent with a similar differential in the increased expression of Glut1 and LDHA (Figure 3A). Glut1 facilitates glycolysis by increasing the uptake of glucose, and LDHA restores the pools of nicotinamide adenine dinucleotide (NAD+) from NADH. Moreover, there were significant increases in pyruvate that enters the TCA cycle after either α-CD3/CD28 or IL-7/IL-15 stimulation; these increases were abrogated by mTORi and, to a lesser extent, by rapamycin (Figure 3B).
The production of citrate from glutamine-derived α-KG and pyruvate also fuels the Ac-CoA syntheses necessary for acetylation events (DeBerardinis et al., 2007) (Figure 3C). α-KG has also been identified as a TCA metabolite that is a critical determinant of HIV-1 infection (Clerc et al., 2019). Since the expression of ASCT2, a glutamine transporter necessary for the maintenance of mitochondrial α-KG levels, is suppressed by mTOR inhibition (Figure 2D), we assessed the levels of the glutaminolysis product α-KG (Figure 3D). TCR stimulation of resting CD4 T cells increased α-KG levels nearly 4-fold (p = 0.0476), and mTORi treatment blocked this increase (p = 0.0275).
These results suggest that the inhibition of mTOR activity results in the depletion of both glucose- and glutamine-derived carbon sources, pyruvate and α-KG, respectively, that support the biosynthesis of citrate in the TCA cycle via anaplerosis to enable Ac-CoA pool expansion. This raised questions about the relative contributions of these metabolites in generating the Ac-CoA necessary for the acetylation of α-tubulin. To address this, we treated CD4 T cells with the glutamine antagonist 6-diazo-5-oxo-l-norleucine (DON), which blocks glutamine utilization via the TCA cycle, or with 2-deoxy glucose (2-DG), a competitive inhibitor of glycolysis (Figure 3C). Both DON and 2-DG significantly reduced the accumulation of Ac-Tub in α-CD3/CD28-stimulated CD4 T cells (Figure 3E, left panel). Similarly, the treatment of cells with UK5099, a compound that inhibits the utilization and import of pyruvate into mitochondria, also significantly suppressed Ac-Tub levels after stimulation (Figure 3E, left panel). Furthermore, 2-DG, UK5099, and DON each also reduced p-S6, a marker of mTOR activity (Figure 3E, right panel). The decreases in Ac-Tub were strongly correlated with the magnitude of mTOR inhibition, based on p-S6 levels, achieved by each of the inhibitors (Figure 3F); this is consistent with another report involving only 2-DG (Dennis et al., 2001). This suggests that de novo Ac-Tub production after the α-CD3/CD28 stimulation of CD4 T cells is dependent on mTOR activity-related changes in both glucose and glutamine utilization.
The dependence of Ac-Tub on the synthesis of Ac-CoA was confirmed in a separate experiment. Increases in Ac-Tub after α-CD3/CD28 stimulation were prevented by a combination of direct inhibitors of Ac-CoA syntheses via either salvage (via ACLY) or de novo (via Ac-CoA synthetase short-chain family member 2 [ACSS2]) pathways, as well as by the mTORi (Figure S5) (Zhao et al., 2016).
Citrate is converted to Ac-CoA by ACLY. ACLY expression is governed by mTORC1-dependent SREBP-1 activation (Assmann et al., 2017), similar to the PPP enzymes RPE and PGD noted here (Figure 2F). Moreover, the activation of ACLY has been reported to be mediated via phosphorylation by the mTORC2 downstream target AKT (Lee et al., 2014; Martinez Calejman et al., 2020). In agreement with these previous reports, we found that mTORi, which blocks the activity of both mTORC1 (p-S6) and mTORC2 (p-AKT), suppressed the levels of both total ACLY and p-ACLY after α-CD3/CD28 stimulation (Figure 3A). However, these treatments had no detectable effects on the enzyme upstream of ACLY, citrate synthase (CS). Increased Ac-Tub and increased total ACLY and p-ACLY, which can elevate the Ac-CoA synthetic capacity, were present at 72 h after α-CD3/CD28 stimulation and were attenuated by mTORi (Figure 3A).
mTOR Regulates HIV-Induced MT Acetylation-Mediated Stabilization in Activated CD4 T Cells
We next used microscopy to study MT-stabilizing Ac-Tub increases enabled by the enhancement of Ac-CoA pools by mTOR in activated CD4 T cells. This MT-stabilizing acetylation was previously described both to be induced by HIV-1 infection via the HIV-1 matrix protein and to play an important role in the long-range intracellular transport of HIV-1 replication intermediates to the nucleus in cell lines and myeloid cells (Sabo et al., 2013). Here, we compared the effects of α-CD3/CD28 stimulation alone, HIV-1 infection alone, and the combination of both on the acetylation of MTs in primary CD4 T cells. The infection of α-CD3/CD28-stimulated cells with vesicular stomatitis virus (VSV)-G-pseudotyped HIV-1 induced significantly more Ac-Tub-stabilized MTs than did α-CD3/CD28 stimulation alone (Figures 4A and 4B). Most important, mTORi pretreatment blocked this increase (Figures 4A–4C), adding the mTORi inhibition of MT acetylation to decreased RT as a mechanism underlying the greater decrease in 2-LTR circles than LRTs seen with mTORi (Figures 1B and 1C). Neither α-CD3/CD28 stimulation in the absence of HIV-1 infection nor HIV-1 infection of resting cells significantly increased Ac-Tub in CD4 T cells from 3 donors (Figure 4C).
Figure 4. HIV-1 Infection Induces mTOR-Dependent Acetyl-Lys (40) α-Tubulin Formation in Activated CD4 T Cells.

(A and B) Resting CD4 T cells or CD4 T cells stimulated with α-CD3/CD28 for 3 days in the absence or presence of mTORi pretreatment were either mock infected (A) or infected with HIV-1-VSV (B) for 4 h. Then, cells were fixed and stained with α-pan-tubulin and α-acetyl-Lys (40) α-tubulin antibodies.
(C) Quantitation of fluorescence signal intensity from images in (A) and (B). Data derived from independent experiments using CD4 T cells from 3 different donors. Means and SDs of data are shown. The significance of differences was determined by 2-tailed Student’s t tests. *p < 0.05, **p < 0.01, and ***p < 0.005.
IL-7/IL-15 stimulation also increased Ac-Tub, as assessed differently by the flow cytometry of resting CD4 T cells sorted from peripheral blood mononuclear cells (PBMCs) from 3 different blood donors (Figure 5, black line). IL-15 and other γc-cytokines have been reported to enhance the susceptibility of CD4 T cells to HIV-1 infection via Janus kinase 1 (JAK1); tofacitinib and ruxolitinib, 2 US Food and Drug Administration (FDA)-approved JAK inhibitors, can block HIV-1 replication in vitro (Gavegnano et al., 2017; Manganaro et al., 2018). Among other effects, JAK1 stimulates phosphatidylinositol 3-kinase (PI3K) (Lai et al., 2013), which is an upstream activator of mTOR. Therefore, we tested whether tofacitinib (JAKi) could ablate mTOR-dependent increases in Ac-Tub after IL-7/IL-15. Resting CD4 T cells were pretreated with JAKi (blue line) or mTORi (red line) and then stimulated with IL-7/IL-15, as indicated. Both JAKi and mTORi decreased Ac-Tub, p-STAT5, as well as the markers of cell-cycle entry (Ki-67), cell growth (FSC-A), and the mTOR downstream target, CD71 (Zheng et al., 2007) Figure 5). Furthermore, both this JAKi (Figure 5, blue line) and a second JAKi (ruxolitinib, not shown) also decreased B cell lymphoma 2 (Bcl-2) after IL-7/IL-15 stimulation; mTORi did not have this effect (Figure 5).
Figure 5. mTOR Activity Is Essential for the Induction of Ac-MT after Common γc-Cytokine Stimulation.

(A) Sorted resting CD4 T cell peripheral blood mononuclear cells (PBMCs) were left untreated (gray-filled histogram) or stimulated with IL-7/IL-15 for 5 days in the absence (black line) or presence of pretreatment with mTORi (red line) or tofacitinib (JAKi) (blue line). Cells were treated with an inhibitor 24 h before stimulation and the inhibitor continued for the duration of stimulation. The effects of each inhibitor on post-stimulation accumulation of stabilized microtubules (Ac-Tub), cell size (FSC-A), proliferation (Ki-67), STAT5 activity (p-STAT5), Bcl-2, and CD71 expression were determined by flow cytometry. Representative histograms are presented (A) and data from 3 independent donors for all of the parameters are plotted with the means and SDs of the data shown.
(B) Significance of differences was determined by 2-tailed Student’s t tests. *p < 0.05, **p < 0.01, and ***p < 0.005. Comparisons are between mock (gray-filled bars) versus +IL-7/IL-15 (in black outline), between +IL-7/IL-15 versus +IL-7/IL-15/mTORi (in red outline), and between +IL-7/IL-15 versus +IL-7/IL-15/Jaki (in blue outline).
Elevated mTOR Activity Occurs in Blood CD4 T Cells from Viremic HIV-Infected Subjects
Next, we sought evidence of the physiological relevance of mTOR activation for HIV-1 replication in vivo. Since mTOR-triggering IL-7 and IL-15 are elevated in plasma during chronic HIV infection (Napolitano et al., 2001; Swaminathan et al., 2016), we assessed mTOR activity in CD4 T cells present in PBMCs from HIV-infected, antiretroviral therapy (ART)-naive research cohort participants. These cells were not stimulated ex vivo. We observed significantly higher (p = 0.002) levels of intracellular p-S6 in unstimulated CD4 T cells from viremic, HIV-infected subjects (n = 4) when compared to cells from uninfected controls (n = 4) (Figure 6A). These results are consistent with the increased mTOR signaling in blood CD4 T cells of untreated HIV-1-infected individuals in vivo. To test the hypothesis that mTOR inhibitors may have therapeutic value by ameliorating the potentially deleterious effects of mTOR activation with and without ART suppression of viremia, we studied PBMCs from 13 viremic and 14 ART-suppressed, HIV-infected study participants matched for age, BMI, and race. We exposed participant PBMCs to mTORi for 8 h and found a significant reduction in mTOR activity (p-S6) in treated cells from both viremic (p = 0.0059) and ART-suppressed (p = 0.0001) participants (Figures 6B and 6C).
Figure 6. Blood CD4 T Cells from Research Participants with HIV-1 Viremia Show Evidence of Active mTOR Signaling In Vivo.

(A) Cells from HIV-1+ participants have increased mTOR signaling, compared to cells from healthy controls. PBMCs from viremic HIV-1+ research participants and healthy controls (HC) were surface phenotyped to identify CD4 T cells and analyzed without any ex vivo stimulation for the expression of the canonical phosphorylated target downstream of mTOR, phosphorylated ribosomal protein S6 (p-S6), by multiparameter flow cytometry. Each group had cells from 4 independent donors. Means and SDs of data are shown.
(B) mTOR inhibition decreased p-S6 in cells from HIV-1+ participants. (C) mTOR inhibition decreased p-S6 in cells from both viremic and ART-treated HIV-1+ participants. (B and C) PBMCs from viremic HIV-infected (n = 13) (B) and ART-suppressed HIV-infected (n = 14) (C) study participants were treated with either mTORi or DMSO vehicle for 8 h ex vivo and analyzed for mTOR activity (p-S6) by flow cytometry. A representative histogram is presented for PBMCs obtained from a HIV-infected donor cultured in the absence and presence of mTORi. Data from PBMCs for all of the participants are plotted in (C). The means and SDs of data are shown. Significant differences were determined by 2-tailed Student’s t tests: *p < 0.05, **p < 0.01, and ***p < 0.005.
DISCUSSION
We identified that catalytic mTOR inhibition affects cellular metabolism to impair the early post-entry steps of HIV-1 replication. The identification here of mTOR-dependent metabolic mechanisms enabling the early steps of RT and the cytoplasmic transport of RT products describes how inhibiting T cell glucose transport and glycolysis impairs HIV-1 replication (Loisel-Meyer et al., 2012; Palmer et al., 2017; Valle-Casuso et al., 2019) (Figure 7).
Figure 7. Summary of Mechanisms by Which the mTOR Pathway Determines Permissivity to HIV-1 by Regulating Cellular Metabolite Pools.

Activation of the mTORC1 signaling cascade coordinates increases in glucose uptake (via Glut1 induction) and the PPP and c-Myc activities that are each needed for nucleotide biosyntheses. These processes and the c-Myc-dependent increases in ribonucleotide reductase expand the pools of all 4 dNTPs to facilitate HIV-1 reverse transcription (step 1). mTORC1 also increases the transport of glutamine into the cell via the induction of ASCT2 expression, thereby elevating the level of α-KG that enters the TCA cycle for citrate production. Glut1-mediated glucose transport also enhances the entry of another citrate precursor, pyruvate, into the TCA cycle. ATP citrate lyase (ACLY) is also activated by actions of both mTORC1 and mTORC2 and facilitates conversion of the expanded pool of citrate to Ac-CoA. Increased Ac-CoA fuels the acetylation of the α-tubulin that stabilizes microtubules resulting from α-tubulin polymerization, which are used for cytoplasmic trafficking of HIV-1 toward the nucleus (step 2).
The mTOR-dependent processes required for HIV-1 RT include increases in glucose and glutamine transporter expression (Glut1 and ASCT2, respectively). These transporters were previously shown to be associated with HIV-1 infection levels in CD4 T cells subsets displaying differential susceptibility to HIV-1 infection (Valle-Casuso et al., 2019). Notably, the increased uptake of glucose and glutamine supply key nucleotide precursors (Tong et al., 2009) used by multiple mTOR-dependent rate-limiting nucleotide biosynthetic enzymes (e.g., CAD, IMPDH1, MTHFD2, TYMS, RRM1) that facilitate the expansion of all dNTPs necessary for RT.
Here, we determine that the stabilization of MTs by acetylation, which has previously been documented to enable the cytoplasmic transport of RT products (Sabo et al., 2013), also requires an mTOR-mediated metabolic cascade. Both cellular glutamine and glycolytic products, which are each enhanced by mTOR activity, contributed to MT acetylation via the TCA cycle. TCA cycle-derived citrate serves as substrate for the ACLY-catalyzed expansion of Ac-CoA pools. ACLY expression and activation by phosphorylation were also both mTOR dependent after CD4 T cell stimulation. Increased Ac-CoA pools enhanced the acetylation of tubulin that stabilizes the MTs important for the cytoplasmic transport of HIV RT products (Sabo et al., 2013). HIV-1, via its matrix protein, was shown by Sabo et al. (2013) to induce the stable subset of MTs that facilitate the transport of HIV-1 replication intermediates in cell lines and myeloid cells (. In recent studies, HIV-1 envelope-mediated signaling was shown to also trigger Ac-Tub (Cabrera-Rodríguez et al., 2019; Casado et al., 2018). Results here now show that increased Ac-Tub also occurs in α-CD3/CD28-stimulated CD4 T lymphocytes after HIV-1 infection, but not in resting CD4 T cells with limited metabolic activity to fuel microtubule stabilization. We posit that this mTOR-dependent acetylation explains why 2-LTR circles decreased with catalytic mTOR inhibition and were not rescued by exogenous deoxyribonucleosides (Figures 1B and 1C); blocking this additional mTOR-enabled cascade added to the impact of decreasing RT via the mTORi impairment of dNTP pool expansion. Thus, the mTORi treatment of stimulated CD4 T cells recapitulates the non-permissive phenotype of resting CD4 T cells, including impairment in both RT and the nuclear entry of HIV DNA (Korin and Zack, 1999; Sun et al., 1997; Zack et al., 1990).
This work also characterized targets downstream of mTOR that have previously been studied as targets for anti-HIV interventions. Inhibitors of 2 of these targets (the IMPDH inhibitor MPA and the RRM1 inhibitorHU) were not successfully translated into clinical use (Hawley et al., 2013; Lori et al., 2005). Furthermore, we show that MPA impairs HIV-1 nuclear entry as well as RT in vitro. Results also indicate that G or dG repletion rescued the effects of the MPA inhibition of IMPDH1 on both mTORC1 activity (Figure 2C) and these separate steps in early HIV-1 replication (Figures 1B and 1C). Thus, MPA-caused decreases in guanine nucleotides downmodulated the mTOR activity needed for both RT and the cytoplasmic transport of RT products. In contrast, G or dG repletion did not rescue mTORi interruption of this circuit at the biochemical or the virological level. This suggests that the mTORi suppression of HIV-1 replication may be less influenced by various dietary or microbial sources of guanine nucleotides than would MPA. JAK inhibitors have also been shown to block HIV-1 replication in vitro (Gavegnano et al., 2017; Manganaro et al., 2018). Results here document that the activation of mTOR downstream of JAKs may account for effects that are countered by these JAK inhibitors. Moreover, JAK inhibitors also impaired Bcl-2 expression, a CD4 T cell survival mechanism, while mTORi did not; this suggests that inhibiting mTOR may have an advantage over inhibiting JAKs. Finally, the identification here that the expansion of Ac-CoA pools via the lipogenic SREBP-ACLY pathway enables the Ac-Tub stabilization of MT needed for the cytoplasmic transport of RT products adds to evidence suggesting that specific inhibition of that axis may impair HIV-1 replication (Taylor et al., 2011). An ACLY inhibitor is now in advanced clinical trials to reduce low-density lipoprotein (LDL) cholesterol (Ray et al., 2019). Targeting a factor downstream of mTOR, such as ACLY, could be better tolerated than the catalytic mTOR inhibitors now in clinical development for cancer (Basu et al., 2015; Burris et al., 2017; Naing et al., 2012; Powles et al., 2016).
The physiological relevance of these mTOR-enabled metabolic pathways for antiviral defense is supported by the fact that antiviral interferon-stimulated genes (ISGs) such as SAMHD1 target metabolic pathways downstream of mTOR (Raniga and Liang, 2018). The effects of mTORi on the steps of HIV-1 replication that block provirus establishment suggest the relevance of future research that may lead to virus resistance-proof HIV-1 prevention. The study of the potential impact of adjunctive mTORi on HIV reservoirs that may be established when ART initiates is also pertinent (Abrahams et al., 2019). Since recent reports noted that mTOR is required for the reversal of HIV-1 latency (Besnard et al., 2016) and that glycolysis inhibition blocked latency reversal (Valle-Casuso et al., 2019), the inhibition of mTOR and/or processes downstream of it also offers promise for research on sustaining HIV-1 remission after stopping antiretrovirals. Further study is also warranted to explore whether the inhibition of the in vivo mTOR activation observed here can delay the early onset of aging-related diseases among well-treated individuals living with HIV (Yang et al., 2019), given the links between aging-related diseases and mTOR activity (Stallone et al., 2019).
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Harry E. Taylor (taylorha@upstate.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
PBMCs and resting CD4 T cells isolated from healthy donors’ PBMC (Lifesource, Rosemont, IL) were maintained in RPMI 1640 culture. 293T cells were grown in DMEM medium. All media contained 10% fetal bovine serum (FBS) (Hyclone), 2 mM L-glutamine, 100 μg/mL streptomycin, and 100 U/mL penicillin. Cryopreserved PBMC were also obtained from HIV-infected, viremic persons who had signed IRB-approved consent to participate in an observational cohort of young men who have sex with men in Chicago (U01 DA 036939) (Table S1).
METHOD DETAILS
Analysis of HIV Replication and mTOR activity in CD4 T cells
Isolation of resting CD4 T cells from uninfected subjects’ PBMC (Lifesource, Rosemont, IL), culture, preparation of virus stocks, virus infection, qPCR for HIV-1 DNA were performed as described elsewhere (Taylor et al., 2015). Single-round CXCR4-tropic envelope-pseudo-typed GFP-expressing HIV-1 (Zhou et al., 2005) and HIV-1 NL4-3, a CXCR4-tropic virus, were used specifically to obviate any possible effect of mTORi on cell CCR5 expression (Heredia et al., 2003). Flow cytometric analysis of single-round infections allowed a quantitative analysis of viral replication over a 4-log dynamic range (Zhang et al., 2004). Where indicated, cells were treated with cytokines or chemokines (Peprotech) at 100 ng/ml, or pretreated with VU0359595 (PLDi) (10, 2, or 0.4 μM, Cayman Chemical), rapamycin (1, 0.2, or 0.04 μM, Cayman Chemical), AZD2014 (mTORi #1) (5, 1, 0.2 μM, Selleckchem), AZD8055 (mTORi #2) (5, 1, 0.2 μM, Selleckchem), TAK-228 (mTORi #3) (5, 1, 0.2 μM, Cayman Chemical), mycophenolic acid (10 μM, Cayman Chemical), or treated with indicated nucleoside (Sigma) at 50 μM at the time of stimulation.
Quantification of dNTP pools
Analysis of dNTP pools was performed as previously described (Bushman et al., 2011). Briefly, for each condition 5 × 106 cells were pelleted and lysed in 70:30 methanol: water. 1.5 × 106 cell equivalents were applied to Waters QMA Solid Phase Extraction (SPE) cartridges for separation of monophosphate (MP), diphosphate (DP), and triphosphate (TP) fractions using potassium chloride concentration gradient washes. The triphosphate fraction was dephosphorylated using an excess of alkaline phosphatase. Following dephosphorylation, stable labeled internal standards were added to the samples, which were then de-salted and concentrated using Phenomenex Strata-X-CW SPE cartridges. A validated ultra-performance liquid chromatography tandem mass spectrometry (UPLC-MS/MS) method was used for quantitation of each dN (molar equivalent to dNTP), with a range between 50 and 2500 fmol/sample for each dNTP.
Immunoblot (IB)
IB analysis was conducted as described previously (Taylor et al., 2015). WCL were prepared using RIPA buffer [50mM Tris-HCl pH 8.0, 150mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, and 1mM EDTA] with Protease Inhibitor Cocktail (Roche). Clarified WCL were resolved by SDS-PAGE on 4%–12% gradient Bis-Tris or 3%–8% Tris-acetate polyacrylamide gel and transferred to a nitrocellulose membrane, then blocked with SuperBlock Blocking Buffer (Thermo Scientific) and incubated with indicated antibodies overnight at 4°C. After sequential washes, the immunoreactive protein bands were visualized by Enhanced Chemiluminescence (SuperSignal West Dura Chemiluminescent Substrate, Thermo Scientific) according to the manufacturer’s instructions. Densitometry was performed using ImageJ software (Schneider et al., 2012). Protein levels were normalized to the respective loading control. The following antibodies were used: CAD (Cell Signaling Technology, #11933), p-CAD (Cell Signaling Technology, #12662), RRM1 (Cell Signaling Technology, #8637), thymidylate synthase (Cell Signaling Technology, #9045), IMPDH1 (Novus, #NBP-52933), 4E-BP1 (Cell Signaling Technology, #9452), p-4E-BP1 (Cell Signaling Technology, #2855), S6 (Cell Signaling Technology, #2217), p-S6 (Cell Signaling Technology, #5364), p-S6K1 (Cell Signaling Technology, #9234), AKT (Cell Signaling Technology, #9272), p-AKT (S473, Cell Signaling Technology, #4060), c-Myc (Cell Signaling Technology, #5605), ASCT2 (Cell Signaling Technology, #8057), GLUT1 (Millipore, #07-1401), LDHA (Cell Signaling Technology, #3582), acetyl-α-tubulin (Cell Signaling Technology, #5335), α-tubulin (Cell Signaling Technology, #2125), SREBP-1 (Santa Cruz, #sc-367), citrate synthase (Santa Cruz, #sc-390693), PGD (Proteintech, #14718-1-AP), MTHFD2 (Proteintech, #12270-1-AP), RPE (Proteintech, #12168-2-AP), and GAPDH (Sigma, #G9545).
Flow cytometry
Cryopreserved PBMCs were thawed, washed, stained with LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Thermofisher) to exclude nonviable cells and surface phenotyped by staining with flurochrome-conjugated antibodies at 4°C for 30 min. For intracellular staining, cells were subsequently fixed and permeabilized using Cytofix/Cytoperm solution (BD Biosciences). Flow cytometric data was obtained on a LSRFortessa (Becton Dickinson) and analyzed with FlowJo software (TreeStar). The following antibodies were used: CD3-BV510 (BD Biosciences, #563109), CD4-PE-Cy7 (Biolegend, #344612), CD71-AF700 (BD Biosciences, #563769), Bcl-2-PE (BD Biosciences, # 340651), pStat5-PE-CF594 (pY694) (BD Biosciences, #562501), and pS6-AF488 (Cell Signaling Technology, #5018).
Metabolic profiling Sample preparation
Cells from three different donors were harvested at the times indicated and washed once in cold PBS, and pellets were snap-frozen in a dry ice-ethanol bath. Samples were stored at −80°C until shipment to Metabolon (Durham, NC).
Ultrahigh Performance Liquid Chromatography-Tandem Mass Spectroscopy (UPLC-MS/MS)
All methods utilized a Waters ACQUITY ultra-performance liquid chromatography (UPLC) and a Thermo Scientific Q-Exactive high resolution/accurate mass spectrometer interfaced with a heated electrospray ionization (HESI-II) source and Orbitrap mass analyzer operated at 35,000 mass resolution. The sample extract was dried then reconstituted in solvents compatible to each of the four methods. Each reconstitution solvent contained a series of standards at fixed concentrations to ensure injection and chromatographic consistency. One aliquot was analyzed using acidic positive ion conditions, chromatographically optimized for more hydrophilic compounds. In this method, the extract was gradient eluted from a C18 column (Waters UPLC BEH C18-2.1x100 mm, 1.7 μm) using water and methanol, containing 0.05% perfluoropentanoic acid (PFPA) and 0.1% formic acid (FA). Another aliquot was also analyzed using acidic positive ion conditions; however, it was chromatographically optimized for more hydrophobic compounds. In this method, the extract was gradient eluted from the same afore mentioned C18 column using methanol, acetonitrile, water, 0.05% PFPA and 0.01% FA and was operated at an overall higher organic content. Another aliquot was analyzed using basic negative ion optimized conditions using a separate dedicated C18 column. The basic extracts were gradient eluted from the column using methanol and water, however with 6.5mM Ammonium Bicarbonate at pH 8. The fourth aliquot was analyzed via negative ionization following elution from a HILIC column (Waters UPLC BEH Amide 2.1x150 mm, 1.7 μm) using a gradient consisting of water and acetonitrile with 10mM Ammonium Formate, pH 10.8. The MS analysis alternated between MS and data-dependent MSn scans using dynamic exclusion. The scan range varied slighted between methods but covered 70-1000 m/z. Raw data files are archived and extracted as described below.
Data Analysis:
The informatics system consisted of four major components, the Laboratory Information Management System (LIMS), the data extraction and peak-identification software, data processing tools for QC and compound identification, and a collection of information interpretation and visualization tools for use by data analysts. The hardware and software foundations for these informatics components were the LAN backbone, and a database server running Oracle 10.2.0.1 Enterprise Edition.
LIMS:
The purpose of the Metabolon LIMS system was to enable fully auditable laboratory automation through a secure, easy to use, and highly specialized system. The scope of the Metabolon LIMS system encompasses sample accessioning, sample preparation and instrumental analysis and reporting and advanced data analysis. All of the subsequent software systems are grounded in the LIMS data structures. It has been modified to leverage and interface with the in-house information extraction and data visualization systems, as well as third party instrumentation and data analysis software.
Data Extraction and Compound Identification:
Raw data was extracted, peak-identified and QC processed using Metabolon’s hardware and software. These systems are built on a web-service platform utilizing Microsoft’s .NET technologies, which run on high-performance application servers and fiber-channel storage arrays in clusters to provide active failover and loadbalancing. Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities. Metabolon maintains a library based on authenticated standards that contains the retention time/index (RI), mass to charge ratio (m/z), and chromatographic data (including MS/MS spectral data) on all molecules present in the library. Furthermore, biochemical identifications are based on three criteria: retention index within a narrow RI window of the proposed identification, accurate mass match to the library ± 10 ppm, and the MS/MS forward and reverse scores between the experimental data and authentic standards. The MS/MS scores are based on a comparison of the ions present in the experimental spectrum to the ions present in the library spectrum. While there may be similarities between these molecules based on one of these factors, the use of all three data points can be utilized to distinguish and differentiate biochemicals. More than 3300 commercially available purified standard compounds have been acquired and registered into LIMS for analysis on all platforms for determination of their analytical characteristics. Additional mass spectral entries have been created for structurally unnamed biochemicals, which have been identified by virtue of their recurrent nature (both chromatographic and mass spectral). These compounds have the potential to be identified by future acquisition of a matching purified standard or by classical structural analysis.
Curation:
A variety of curation procedures were carried out to ensure that a high-quality dataset was made available for statistical analysis and data interpretation. The QC and curation processes were designed to ensure accurate and consistent identification of true chemical entities, and to remove those representing system artifacts, mis-assignments, and background noise. Metabolon data analysts use proprietary visualization and interpretation software to confirm the consistency of peak identification among the various samples. Library matches for each compound were checked for each sample and corrected if necessary.
Metabolite Quantification and Data Normalization:
Peaks were quantified using area-under-the-curve. For studies spanning multiple days, a data normalization step was performed to correct variation resulting from instrument inter-day tuning differences. Essentially, each compound was corrected in run-day blocks by registering the medians to equal one (1.00) and normalizing each data point proportionately (termed the “block correction”; Figure 2). For studies that did not require more than one day of analysis, no normalization is necessary, other than for purposes of data visualization. In certain instances, biochemical data may have been normalized to an additional factor (e.g., cell counts, total protein as determined by Bradford assay, osmolality, etc.) to account for differences in metabolite levels due to differences in the amount of material present in each sample.
Measurement of Intracellular ATP
ATP levels were quantified using the ENLITEN ATP Assay System (Promega) according to the manufacturer’s instructions.
Indirect immunofluorescence and Microscopy
HIV-1 virus to infect primary T cells were generated by transfecting 293T cells on a 15cm dish with 18 μg of R7 ΔEnv mcherry, derived from R7ΔEnvGFP by replacing the GFP cassette with mcherry and 7 μg of pCMV-VSVg (Dharan et al., 2016). For microscopy experiments, primary T cells were added to Poly-L-Lysine treated coverslips and synchronized infection was performed as described before (Campbell et al., 2008). In brief, cells were spinoculated at 13°C for 2 hours at 1200xg, after which virus containing media was replaced with 37°C media. Following a two-hour incubation, coverslips containing cells were fixed and permeabilized using solutions containing microtubule stabilizing buffer, PFA, glutaraldehyde and Triton X-100. Prior to antibody labeling, coverslips were washed in MgPBS containing NaBH4 to reduce any residual free aldehyde groups. Coverslips were incubated with a primary labeling mix containing Rat mAb for tubulin (Abcam ab6161) and Rabbit mAb for acetylated-α-tubulin (Cell Signaling 5335) for one hour at room temperature. Following primary antibody incubation coverslips were washed with PBS. Secondary antibody labeling mix was added to the coverslips and allowed to incubate for 40 minutes at room temperature. Lastly, coverslips were washed with PBS, mounted, and stored at 4°C. Z stack images were collected with a DeltaVision wide-field fluorescent microscope (Applied Precision, GE) equipped with a digital camera (CoolSNAP HQ; Photometrics) using a 1.4-numerical aperture 100x objective lens. Excitation light was generated using a solid-state illumination module (Applied Precision, GE), and deconvolved using SoftWoRx deconvolution software. All images underwent identical acquisition parameters. Following acquisition, images were quantified using Imaris software (Bitplane). An algorithm was generated using a surface function within Imaris in order to calculate sum fluorescence intensities of Ac-Tub. Datasets were assembled using Prism, version 6.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using Prism 7 (GraphPad), and the statistical analysis methods used are indicated in Figure Legends. p < 0.05 was considered significant. All image analysis was performed with ImageJ (NIH).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CAD | Cell Signaling Technology | Cat# 11933 |
| p-CAD | Cell Signaling Technology | Cat# 12662 |
| RRM1 | Cell Signaling Technology | Cat# 8637 |
| thymidylate synthase | Cell Signaling Technology | Cat# 9045 |
| 4E-BP1 | Cell Signaling Technology | Cat# 9452 |
| P-4E-BP1 | Cell Signaling Technology | Cat# 2855 |
| S6 | Cell Signaling Technology | Cat# 2217 |
| p-S6 | Cell Signaling Technology | Cat# 5364 |
| NDRG1 | Cell Signaling Technology | Cat# 5196 |
| P-NDRG1 | Cell Signaling Technology | Cat# 5482 |
| P-S6K1 | Cell Signaling Technology | Cat# 9234 |
| AKT | Cell Signaling Technology | Cat# 9272 |
| p-AKT | Cell Signaling Technology | Cat# 4060 |
| c-Myc | Cell Signaling Technology | Cat# 5605 |
| ASCT2 | Cell Signaling Technology | Cat# 8057 |
| LDHA | Cell Signaling Technology | Cat# 3582 |
| acetyl-α-tubulin | Cell Signaling Technology | Cat# 5335 |
| α-tubulin | Cell Signaling Technology | Cat# 2125 |
| ACLY | Cell Signaling Technology | Cat# 4332 |
| p-ACLY | Cell Signaling Technology | Cat# 4331 |
| IMPDH1 | Novus | Cat# NBP-52933 |
| GLUT1 | Millipore | Cat# 07-1401 |
| SREBP-1 | Santa Cruz | Cat# sc-367 |
| citrate synthase | Santa Cruz | Cat# sc-390693 |
| PGD | Proteintech | Cat# 14718-1-AP |
| RPE | Proteintech | Cat# 12168-2-AP |
| MTHFD2 | Proteintech | Cat# 12270-1-AP |
| GAPDH | Sigma | Cat# G9545 |
| CD3-BV510 | BD Biosciences | Cat# 563109 |
| CD71-AF700 | BD Biosciences | Cat# 563769 |
| Bcl-2-PE | BD Biosciences | Cat# 340651 |
| pStat5-PE-CF594 (pY694) | BD Biosciences | Cat# 562501 |
| pS6-AF488 | Cell Signaling Technology | Cat# 5018 |
| Ki-67-BV421 | Biolegend | Cat# 350505 |
| CD4-PE-Cy7 | Biolegend | Cat# 344612 |
| Anti-Tubulin antibody | Abcam | Cat# ab6161 |
| Biological Samples | ||
| Leukopaks (Buffy Coats) | Lifesouce, Rosemont, IL | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| VU0359595 | Cayman Chemical | Cat# 10955 |
| Rapamycin | Cayman Chemical | Cat# 13346 |
| TAK-228/INK128 | Cayman Chemical | Cat# 11811 |
| Mycophenolic Acid | Cayman Chemical | Cat# 21716 |
| AZD8055 | Selleckchem | Cat# 16978 |
| AZD2014 | Selleckchem | Cat#.S2783 |
| 2′-Deoxyadenosine monohydrate | Sigma | Cat# D8668 |
| 2′-Deoxycytidine hydrochloride | Sigma | Cat# D0776 |
| 2′-Deoxyguanosine monohydrate | Sigma | Cat# D0901 |
| Thymidine | Sigma | Cat# T1895 |
| Guanosine | Sigma | Cat# G6264 |
| 6-Diazo-5-oxo-L-norleucine (DON) | Sigma | Cat# D2141 |
| 2-deoxy-glucose | Sigma | Cat# D3179 |
| UK5099 | Sigma | Cat# PZ0160 |
| CXCL-10 | Peprotech | Cat# 300-12 |
| CCL19 | Peprotech | Cat# 300-29B |
| CCL20 | Peprotech | Cat# 300-29A |
| IL-7 | Peprotech | Cat# 200-07 |
| IL-15 | Peprotech | Cat# 200-15 |
| Critical Commercial Assays | ||
| ENLITEN ATP Assay System | Promega | Cat# FF2000 |
| Experimental Models: Cell Lines | ||
| 293T | ATCC | CRL-3216 |
| Oligonucleotides | ||
| LRT F: TGT GTG CCC GTC TGT TGT GTG A | Taylor et al., 2015 | N/A |
| LRT R: GAG TCC TGC GTC GAG AGA TCT | Taylor et al., 2015 | N/A |
| 2LTR F: AAC TAG GGA ACC CAC TGC TTA AG | Taylor et al., 2015 | N/A |
| 2LTR R: TCC ACA GAT CAA GGA TCT CTT GTC | Taylor et al., 2015 | N/A |
| GAPDH F: GAA GGT GAA GGT CGG AGT | Taylor et al., 2015 | N/A |
| GAPDH R: GAA GAT GGT GAT GGG ATT TC | Taylor et al., 2015 | N/A |
| Recombinant DNA | ||
| HIV-1 NL4-3 ΔEnv EGFP reporter vector | Zhou et al., 2005 | N/A |
| X4 HIV-1 envelope expression vector | Zhou et al., 2005 | N/A |
| pCMV-VSVg | Dharan et al., 2016 | N/A |
| R7ΔEnvGFP | Dharan et al., 2016 | N/A |
| Software and Algorithms | ||
| GraphPad Prism (GraphPad) | Motulsky and Brown, 2006 | https://www.graphpad.com |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| FACSDiva software version 8.0.1 (BD Biosciences) | Becton, Dickinson and Company | https://www.bdbiosciences.com/en-us/instruments/research-instruments/research-software/flow-cytometry-acquisition/facsdiva-software |
| FlowJo software version 10 | FlowJo LLC | https://www.flowjo.com/ |
Highlights.
mTOR activity controls the susceptibility of CD4 T cells to HIV-1 infection
mTOR signaling coordinates anabolic activities for dNTP synthesis that fuel HIV RT
mTOR induces stabilization of microtubules co-opted for cytoplasmic transport of HIV-1
Catalytic mTOR inhibitors potently suppress HIV-1 infection
ACKNOWLEDGMENTS
This work was supported by NIH P01 AI 131346, a Northwestern Medicine Catalyst Award to R.T.D., and a Developmental Core Pilot Project award to H.E.T. from the Third Coast Center for AIDS Research (CFAR), an NIH-funded center (P30 AI117943). H.E.T. was also supported by start-up funds from the State of New York and the Research Foundation for SUNY. N.C. was supported by NIH F30 AI 131937. Flow cytometry was conducted at the Northwestern UniversityFlow Cytometry Core Facility, supported by Cancer Center Support Grant NCI CA060553. Also appreciated are Third Coast CFAR Viral Pathogenesis Core services (P30 AI117943); cryopreserved cells from the biorepository of U01 DA 036939; the expert dNTP quantitation and consultation from Peter L. Anderson, PharmD, at the Skaggs School of Pharmacy and Pharmaceutical Sciences of the University of Colorado Anschutz Medical Campus; advice from Leonidas C. Platanias, MD, PhD; and critical review of the manuscript by members of the Northwestern HIV Translational Research Center (Chisu Song and Gael Scholtes).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107810.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abrahams MR, Joseph SB, Garrett N, Tyers L, Moeser M, Archin N, Council OD, Matten D, Zhou S, Doolabh D, et al. (2019). The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med 11, eaaw5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JL, Khoury G, Fromentin R, Solomon A, Chomont N, Sinclair E, Milush JM, Hartogensis W, Bacchetti P, Roche M, et al. (2020). Human Immunodeficiency Virus (HIV)-Infected CCR6+ Rectal CD4+ T Cells and HIV Persistence On Antiretroviral Therapy. J. Infect. Dis 221, 744–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann N, O’Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, Heinrich P, Oefner PJ, Lynch L, Gardiner CM, et al. (2017). Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat. Immunol 18, 1197–1206. [DOI] [PubMed] [Google Scholar]
- Basu B, Dean E, Puglisi M, Greystoke A, Ong M, Burke W, Cavallin M, Bigley G,Womack C, Harrington EA, et al. (2015). First-in-Human Pharmacokinetic and Pharmacodynamic Study of the Dual m-TORC 1/2 Inhibitor AZD2014. Clin. Cancer Res 21, 3412–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Sahra I, Hoxhaj G, Ricoult SJH, Asara JM, and Manning BD (2016). mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351, 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A, Bassik MC, Verschueren E, Battivelli E, Chan J, et al. (2016). The mTOR Complex Controls HIV Latency. Cell Host Microbe 20, 785–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolduc JF, Ouellet M, Hany L, andTremblay MJ (2017). Toll-Like Receptor 2 Ligation Enhances HIV-1 Replication in Activated CCR6+ CD4+ T Cells by Increasing Virus Entry and Establishing a More Permissive Environment to Infection. J. Virol 91, e01402–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broecker-Preuss M, Becher-Boveleth N, Bockisch A, Dührsen U, and Müller S (2017). Regulation of glucose uptake in lymphoma cell lines by c-MYC- and PI3K-dependent signaling pathways and impact of glycolytic pathways on cell viability. J. Transl. Med 15, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, and Pearce EL (2015). T cell metabolism drives immunity. J. Exp. Med 212, 1345–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukrinsky MI, Sharova N, Dempsey MP, Stanwick TL, Bukrinskaya AG, Haggerty S, and Stevenson M (1992). Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. USA 89, 6580–6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burris HA 3rd, Kurkjian CD, Hart L, Pant S, Murphy PB, Jones SF, Neuwirth R, Patel CG, Zohren F, and Infante JR (2017). TAK-228 (formerly MLN0128), an investigational dual TORC1/2 inhibitor plus paclitaxel, with/without trastuzumab, in patients with advanced solid malignancies. Cancer Chemother. Pharmacol 80, 261–273. [DOI] [PubMed] [Google Scholar]
- Bushman LR, Kiser JJ, Rower JE, Klein B, Zheng JH, Ray ML, and Anderson PL (2011). Determination of nucleoside analog mono-, di-, and triphosphates in cellular matrix by solid phase extraction and ultra-sensitive LC-MS/MS detection. J. Pharm. Biomed. Anal 56, 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera-Rodríguez R, Hebmann V, Marfil S, Pernas M, Marrero-Hernández S, Cabrera C, Urrea V, Casado C, Olivares I, Márquez-Arce D, et al. (2019). HIV-1 envelope glycoproteins isolated from Viremic Non-Progressor individuals are fully functional and cytopathic. Sci. Rep 9, 5544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron PU, Saleh S, Sallmann G, Solomon A, Wightman F, Evans VA, Boucher G, Haddad EK, Sekaly RP, Harman AN, et al. (2010). Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc. Natl. Acad. Sci. USA 107, 16934–16939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell EM, Perez O, Anderson JL, and Hope TJ (2008). Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5alpha. J. Cell Biol 180, 549–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casado C, Marrero-Hernández S, Márquez-Arce D, Pernas M, Marfil S, Borràs-Grañana F, Olivares I, Cabrera-Rodríguez R, Valera MS, de Armas-Rillo L, et al. (2018). Viral Characteristics Associated with the Clinical Nonprogressor Phenotype Are Inherited by Viruses from a Cluster of HIV-1 Elite Controllers. MBio 9, e02338–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapuis AG, Paolo Rizzardi G, D’Agostino C, Attinger A, Knabenhans C, Fleury S, Acha-Orbea H, and Pantaleo G (2000). Effects of mycophenolic acid on human immunodeficiency virus infection in vitro and in vivo. Nat. Med 6, 762–768. [DOI] [PubMed] [Google Scholar]
- Clerc I, Moussa DA, Vahlas Z,Tardito S, Oburoglu L, Hope TJ, Sitbon M, Dardalhon V, Mongellaz C, and Taylor N (2019). Entry of glucose- and glutamine-derived carbons into the citric acid cycle supports early steps of HIV-1 infection in CD4T cells. Nat. Metab 1, 717–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, and Thompson CB (2007). Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. USA 104, 19345–19350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, and Powell JD (2011). The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol 12, 295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis PB, Jaeschke A, Saitoh M, Fowler B, Kozma SC, and Thomas G (2001). Mammalian TOR: a homeostatic ATP sensor. Science 294, 1102–1105. [DOI] [PubMed] [Google Scholar]
- Dharan A, Talley S, Tripathi A, Mamede JI, Majetschak M, Hope TJ, and Campbell EM (2016). KIF5B and Nup358 Cooperatively Mediate the Nuclear Import of HIV-1 during Infection. PLoS Pathog. 12, e1005700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardy SR, and Farnham PJ (2001). c-Myc mediates activation of the cad promoter via a post-RNA polymerase II recruitment mechanism. J. Biol. Chem 276, 48562–8571. [DOI] [PubMed] [Google Scholar]
- Eckstein DA, Penn ML, Korin YD, Scripture-Adams DD, Zack JA, Kreisberg JF, Roederer M, Sherman MP, Chin PS, and Goldsmith MA (2001). HIV-1 actively replicates in naive CD4(+) T cells residing within human lymphoid tissues. Immunity 15, 671–682. [DOI] [PubMed] [Google Scholar]
- Emmanuel N, Ragunathan S, Shan Q, Wang F, Giannakou A, Huser N, Jin G, Myers J, Abraham RT, and Unsal-Kacmaz K (2017). Purine Nucleotide Availability Regulates mTORC1 Activity through the Rheb GTPase. Cell Rep. 19,2665–2680. [DOI] [PubMed] [Google Scholar]
- Fassati A, Görlich D, Harrison I, Zaytseva L, and Mingot JM (2003). Nuclear import of HIV-1 intracellular reverse transcription complexes is mediated by importin 7. EMBO J. 22, 3675–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao WY, Cara A, Gallo RC, and Lori F (1993). Low levels of deoxynucleotides in peripheral blood lymphocytes: a strategy to inhibit human immunodeficiency virus type 1 replication. Proc. Natl. Acad. Sci. USA 90, 8925–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Martínez JM, and Alessi DR (2008). mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J 416, 375–385. [DOI] [PubMed] [Google Scholar]
- Gavegnano C, Brehm JH, Dupuy FP, Talla A, Ribeiro SP, Kulpa DA, Cameron C, Santos S, Hurwitz SJ, Marconi VC, et al. (2017). Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLoS Pathog. 13, e1006740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivel JC, and Margolis L (2009). Use of human tissue explants to study human infectious agents. Nat. Protoc. 4, 256–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley T, Spear M, Guo J, and Wu Y (2013). Inhibition of HIV replication in vitro by clinical immunosuppressants and chemotherapeutic agents. Cell Biosci. 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Smeets RL, Koenen HJ, Vink PM, Wagenaars J, Boots AM, and Joosten I (2011). Mycophenolic acid-mediated suppression of human CD4+ T cells: more than mere guanine nucleotide deprivation. Am. J. Transplant 11, 439–49. [DOI] [PubMed] [Google Scholar]
- Heredia A, Amoroso A, Davis C, Le N, Reardon E, Dominique JK, Klin-gebiel E, Gallo RC, and Redfield RR (2003). Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV beta-chemokines: an approach to suppress R5 strains of HIV-1. Proc. Natl. Acad. Sci. USA 100, 10411–10416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heredia A, Le N, Gartenhaus RB, Sausville E, Medina-Moreno S, Zapata JC, Davis C, Gallo RC, and Redfield RR (2015). Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 112, 9412–9417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinter A, Moorthy A, Jackson R, and Fauci AS (2003). Productive HIV infection of resting CD4+ T cells: role of lymphoid tissue microenvironment and effect of immunomodulating agents. AIDS Res. Hum. Retroviruses 19, 847–856. [DOI] [PubMed] [Google Scholar]
- Korin YD, and Zack JA (1999). Nonproductive human immunodeficiency virus type 1 infection in nucleoside-treated G0 lymphocytes. J. Virol 73, 6526–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YG, Hou MS, Lo A, Huang ST, Huang YW, Yang-Yen HF, and Liao NS (2013). IL-15 modulates the balance between Bcl-2 and Bim via a Jak3/1-PI3K-Akt-ERK pathway to promote CD8αα+ intestinal intraepithelial lymphocyte survival. Eur. J. Immunol 43, 2305–2316. [DOI] [PubMed] [Google Scholar]
- Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, et al. (2014). Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20, 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Rao RR, Araki K, Pollizzi K, Odunsi K, Powell JD, and Shrikant PA (2011). A central role for mTOR kinase in homeostatic proliferation induced CD8+ T cell memory and tumor immunity. Immunity 34, 541–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Li F, Handler J, Huang CR, Xiang Y, Neretti N, Sedivy JM, Zeller KI, and Dang CV (2008). Global regulation of nucleotide biosynthetic genes by c-Myc. PLOS ONE 3, e2722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Han Y, Yu C,Ji Y, Wang C, Chen X, Wang X, Shen J, Zhang Y, and Lang JY (2019). MYC predetermines the sensitivity of gastrointestinal cancer to antifolate drugs through regulating TYMS transcription. EBioMedicine 48, 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loisel-Meyer S, Swainson L, Craveiro M, Oburoglu L, Mongellaz C, Costa C, Martinez M, Cosset FL, Battini JL, Herzenberg LA, et al. (2012). Glut1-mediated glucose transport regulates HIV infection. Proc. Natl. Acad. Sci. USA 109, 2549–2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lori F, Foli A, Groff A, Lova L, Whitman L, Bakare N, Pollard RB, and Lisziewicz J (2005). Optimal suppression of HIV replication by low-dose hydroxyurea through the combination of antiviral and cytostatic (‘virostatic’) mechanisms. AIDS 19, 1173–1181. [DOI] [PubMed] [Google Scholar]
- Macintyre AN, Gerriets VA, Nichols AG, Michalek RD, Rudolph MC, Deoliveira D, Anderson SM, Abel ED, Chen BJ, Hale LP, and Rathmell JC (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganaro L, Hong P, Hernandez MM, Argyle D, Mulder LCF, Potla U, Diaz-Griffero F, Lee B, Fernandez-Sesma A, and Simon V (2018). IL-15 regulates susceptibility of CD4+ T cells to HIV infection. Proc. Natl. Acad. Sci. USA 115, E9659–E9667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannava S, Moparthy KC, Wheeler LJ, Leonova KI, Wawrzyniak JA, Bianchi-Smiraglia A, Berman AE, Flanagan S, Shewach DS, Zeitouni NC, et al. (2012). Ribonucleotide reductase and thymidylate synthase or exogenous deoxyribonucleosides reduce DNA damage and senescence caused by C-MYC depletion. Aging (Albany N.Y.) 4, 917–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marçais A, Cherfils-Vicini J, Viant C, Degouve S, Viel S, Fenis A, Rabil-loud J, Mayol K, Tavares A, Bienvenu J, et al. (2014). The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol 15, 749–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis D, Heredia A, Gaywee J, Oldach D, Drusano G, and Redfield R (1999). Abacavir and mycophenolic acid, an inhibitor of inosine monophosphate dehydrogenase, have profound and synergistic anti-HIV activity. J. Acquir. Immune Defic. Syndr 21, 362–370. [PubMed] [Google Scholar]
- Martinez Calejman C, Trefely S, Entwisle SW, Luciano A, Jung SM, Hsiao W, Torres A, Hung CM, Li H, Snyder NW, et al. (2020). mTORC2-AKT signaling to ATP-citrate lyase drives brown adipogenesis and de novo lipogenesis. Nat. Commun. 11, 575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky HM, and Brown RE (2006). Detecting outliers when fitting data with nonlinear regression – a new method based on robust nonlinear regression and the false discovery rate. BMC Bioinformatics 7, 123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naing A, Aghajanian C, Raymond E, Olmos D, Schwartz G, Oelmann E, Grinsted L, Burke W, Taylor R, Kaye S, et al. (2012). Safety, tolerability, pharmacokinetics and pharmacodynamics of AZD8055 in advanced solid tumours and lymphoma. Br. J. Cancer 107, 1093–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano LA, Grant RM, Deeks SG, Schmidt D, De Rosa SC, Herzenberg LA, Herndier BG, Andersson J, and McCune JM (2001). Increased production of IL-7 accompanies HIV-1-mediated T-cell depletion: implications for T-cell homeostasis. Nat. Med 7, 73–79. [DOI] [PubMed] [Google Scholar]
- Palmer CS, Duette GA, Wagner MCE, Henstridge DC, Saleh S, Pereira C, Zhou J, Simar D, Lewin SR, Ostrowski M, et al. (2017). Metabolically active CD4+ T cells expressing Glut1 and OX40 preferentially harbor HIV during in vitro infection. FEBS Lett. 591, 3319–3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike KG, Malagu K, Hummersone MG, Menear KA, Duggan HM, Gomez S, Martin NM, Ruston L, Pass SL, and Pass M (2013). Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: the discovery of AZD8055 and AZD2014. Bioorg. Med. Chem. Lett 23, 1212–1216. [DOI] [PubMed] [Google Scholar]
- Pikman Y, Puissant A, Alexe G, Furman A, Chen LM, Frumm SM, Ross L, Fenouille N, Bassil CF, Lewis CA, et al. (2016). Targeting MTHFD2 in acute myeloid leukemia. J. Exp. Med 213, 1285–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powles T, Wheater M, Din O, Geldart T, Boleti E, Stockdale A, Sundar S, Robinson A, Ahmed I, Wimalasingham A, et al. (2016). A Randomised Phase 2 Study of AZD2014 Versus Everolimus in Patients with VEGF-Refractory Metastatic Clear Cell Renal Cancer. Eur. Urol. 69, 450–456. [DOI] [PubMed] [Google Scholar]
- Raniga K, and Liang C (2018). Interferons: Reprogramming the Metabolic Network against Viral Infection. Viruses 10, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray KK, Bays HE, Catapano AL, Lalwani ND, Bloedon LT, Sterling LR, Robinson PL, and Ballantyne CM; CLEAR Harmony Trial (2019). Safety and Efficacy of Bempedoic Acid to Reduce LDL Cholesterol. N. Engl. J. Med 380, 1022–1032. [DOI] [PubMed] [Google Scholar]
- Robitaille AM, Christen S, Shimobayashi M, Cornu M, Fava LL, Moes S, Prescianotto-Baschong C, Sauer U, Jenoe P, and Hall MN (2013). Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339, 1320–1323. [DOI] [PubMed] [Google Scholar]
- Ron-Harel N, Santos D, Ghergurovich JM, Sage PT, Reddy A, Lovitch SB, Dephoure N, Satterstrom FK, Sheffer M, Spinelli JB, et al. (2016). Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab. 24, 104–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabo Y, Walsh D, Barry DS, Tinaztepe S, de Los Santos K, Goff SP, Gundersen GG, and Naghavi MH (2013). HIV-1 induces the formation of stable microtubules to enhance early infection. Cell Host Microbe 14, 535–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallone G, Infante B, Prisciandaro C, and Grandaliano G (2019). mTOR and Aging: An Old Fashioned Dress. Int. J. Mol. Sci 20, 2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Pinchuk LM, Agy MB, and Clark EA (1997). Nuclear import of HIV-1 DNA in resting CD4+ T cells requires a cyclosporin A-sensitive pathway. J. Immunol 158, 512–517. [PubMed] [Google Scholar]
- Swaminathan S, Qiu J, Rupert AW, Hu Z, Higgins J, Dewar RL, Stevens R, Rehm CA, Metcalf JA, Sherman BT, et al. (2016). Interleukin-15 (IL-15) Strongly Correlates with Increasing HIV-1 Viremia and Markers of Inflammation. PLOS ONE 11, e0167091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor HE, Linde ME, Khatua AK, Popik W, and Hildreth JE (2011). Sterol regulatory element-binding protein 2 couples HIV-1 transcription to cholesterol homeostasis and T cell activation. J. Virol 85, 7699–7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor HE, Simmons GE Jr., Mathews TP, Khatua AK, Popik W, Lindsley CW, D’Aquila RT, and Brown HA (2015). Phospholipase D1 Couples CD4+ T Cell Activation to c-Myc-Dependent Deoxyribonucleotide Pool Expansion and HIV-1 Replication. PLoS Pathog. 11, e1004864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X, Zhao F, and Thompson CB (2009). The molecular determinants of de novo nucleotide biosynthesis in cancer cells. Curr. Opin. Genet. Dev 19, 32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unutmaz D, KewalRamani VN, Marmon S, and Littman DR (1999). Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J. Exp. Med 189, 1735–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle-Casuso JC, Angin M, Volant S, Passaes C, Monceaux V, Mikhailova A, Bourdic K, Avettand-Fenoel V, Boufassa F, Sitbon M, et al. (2019). Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4(+)T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 29, 611–626.e5. [DOI] [PubMed] [Google Scholar]
- Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger J, and Green DR (2011). The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35, 871–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Salloum D, Medlin PS, Saqcena M, Yellen P, Perrella B, and Foster DA (2011). Phospholipase D mediates nutrient inputto mammalian target of rapamycin complex 1 (mTORC1). J. Biol. Chem. 286, 25477–25486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang HY, Beymer MR, and Suen SC (2019). Chronic Disease Onset Among People Living with HIV and AIDS in a Large Private Insurance Claims Dataset. Sci. Rep 9, 18514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon MS, Du G, Backer JM, Frohman MA, and Chen J (2011). Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J. Cell Biol 795, 435–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack JA, Arrigo SJ, Weitsman SR, Go AS, Haislip A, and Chen IS (1990). HIV-1 entry into quiescent primary lymphocytes: molecularanalysis reveals a labile, latent viral structure. Cell 61, 213–222. [DOI] [PubMed] [Google Scholar]
- Zhang H, Zhou Y, Alcock C, Kiefer T, Monie D, Siliciano J, Li Q, Pham P, Cofrancesco J, Persaud D, and Siliciano RF (2004). Novel single-cell-level phenotypic assay for residual drug susceptibility and reduced replication capacity of drug-resistant human immunodeficiency virus type 1. J. Virol 78, 1718–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo YM, et al. (2016). ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep. 17, 1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Collins SL, Lutz MA, Allen AN, Kole TP, Zarek PE, and Powell JD (2007). A role for mammalian target of rapamycin in regulating T cell activation versus anergy. J. Immunol 178, 2163–2170. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhang H, Siliciano JD, and Siliciano RF (2005). Kinetics of human immunodeficiency virus type 1 decay following entry into resting CD4+ T cells. J. Virol 79, 2199–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.