

Abstract
Background
Wegener's granulomatosis (WG) is a necrotizing small‐vessel vasculitis that can affect any organ in the body but mainly affects the upper and lower respiratory tract, the kidneys, joints, skin and eyes. The current mainstay of remission induction therapy is systemic corticosteroids in combination with oral daily cyclophosphamide (CYC) which induces remission in 75% to 100% of cases. Although standard therapy is effective in inducing partial or complete remission, 50% of complete remissions are followed by at least one relapse. This is an update of a review first published in 2009.
Objectives
To determine if intravenous immunoglobulin (IVIg) adjuvant therapy provides a therapeutic advantage over and above treatment with systemic corticosteroids in combination with immunosuppressants for the treatment of WG.
Search methods
For this update the Cochrane Peripheral Vascular Diseases Group Trials Search Co‐ordinator (TSC) searched the Specialised Register (last searched November 2012) and CENTRAL (2012, Issue 11). Trial databases were searched by the TSC for details of ongoing and unpublished studies. No date or language restrictions were applied.
Selection criteria
Randomized controlled trials (RCTs), or quasi RCTs, or randomized cross‐over trials. Participants had to be adults with a confirmed diagnosis of WG.
Data collection and analysis
Two authors independently extracted data and assessed trial quality. Relative risk was used to analyze dichotomous variables, and mean difference (MD) was used to analyze continuous variables.
Main results
We included one RCT with 34 participants who were randomly assigned to receive IVIg or placebo once daily in addition to azathioprine and prednisolone for remission maintenance. There were no significant differences between adjuvant IVIg and adjuvant placebo in mortality, serious adverse events, time to relapse, open‐label rescue therapy, and infection rates. The fall in disease activity score, derived from patient‐reported symptoms, was slightly greater in the IVIg group than in the placebo group at one month (MD 2.30; 95% Confidence interval (CI) 1.12 to 3.48, P < 0.01) and three months (MD 1.80; 95% CI 0.35 to 3.25, P = 0.01). There was a significant increase in total adverse events in the IVIg group (relative risk (RR) 3.50; 95% CI 1.44 to 8.48, P < 0.01).
Authors' conclusions
There is insufficient evidence from one RCT that IVIg adjuvant therapy provides a therapeutic advantage compared with the combination of steroids and immunosuppressants for patients with WG. Given the high cost of IVIg (one dose at 2 g/kg for a 70 kg patient = $8,400), it should be limited to treat WG in the context of a well conducted RCT powered to detect patient‐relevant outcomes.
Keywords: Adult; Humans; Azathioprine; Azathioprine/therapeutic use; Chemotherapy, Adjuvant; Drug Therapy, Combination; Drug Therapy, Combination/methods; Glucocorticoids; Glucocorticoids/therapeutic use; Granulomatosis with Polyangiitis; Granulomatosis with Polyangiitis/therapy; Immunoglobulins, Intravenous; Immunoglobulins, Intravenous/therapeutic use; Immunologic Factors; Immunologic Factors/therapeutic use; Maintenance Chemotherapy; Maintenance Chemotherapy/methods; Prednisolone; Prednisolone/therapeutic use; Randomized Controlled Trials as Topic
Plain language summary
Intravenous immunoglobulin in addition to standard treatments for Wegener's granulomatosis
Wegener's granulomatosis is a rare disorder that causes inflammation of the blood vessels. This inflammation restricts blood flow to various organs which can eventually damage the organs. Organs most affected by Wegener's include the lungs, upper respiratory tract, kidneys, joints, skin and eyes. Wegener's granulomatosis also produces a granuloma (a mass or nodule of inflammatory tissue) which is found around the blood vessels and which can also damage surrounding tissue. The cause of Wegener's granulomatosis is unknown. Treatment is with corticosteroids and cytotoxic drugs which are often used for chemotherapy. Most patients get better with these drugs. However, the disorder returns in approximately half of patients. Intravenous immunoglobulin (IVIg) is an expensive and fairly rare blood product that has been used to treat Wegener's granulomatosis but its effects on the disorder are unknown. We asked if IVIg provided an advantage as an additive to standard treatments. We found one small randomized trial in which 34 participants were randomized to receive IVIg or placebo once daily in addition to azathioprine and prednisolone for remission maintenance. This trial did not provide enough evidence to determine if IVIg has an advantage over corticosteroids and immunosuppressants for the treatment of Wegener's granulomatosis.
Background
Description of the condition
Wegener's granulomatosis (WG) is a necrotizing small‐vessel vasculitis that can affect any organ in the body but mainly affects the upper and lower respiratory tract, the kidneys, joints, skin and eyes. It is characterized by chronic tissue inflammation and the formation of granuloma (a mass or nodule of chronically inflamed tissue with granulations that is usually associated with an infective process). The annual incidence is low and diagnostic criteria vary, but it is estimated to be about 5 to 10 per million. (Scott 2000).
Mild forms of the disease without renal involvement have been described and the course of the illness may vary from little activity to rapid progression. However, most patients with untreated generalized disease will experience a rapidly progressive fatal illness. Prior to the advent of immunosuppressive therapy, the five‐month survival rate was 18% and two‐year survival was 10% (Esper 1999).
Definitive diagnosis is established by biopsy of the involved organs and the use of special stains that exclude mycobacterial and fungal infection. Clinical criteria for the diagnosis of WG were developed by the American College of Rheumatology and must include at least two of the following criteria:
oral ulcers or nasal discharge;
abnormal findings on chest radiographs (nodules, cavities or fixed infiltrates);
abnormal urinary sediment (more than five red blood cells per high‐powered field);
granulomatous inflammation on biopsy;
sero‐positive for cytoplasmic anti‐neutrophil antibodies (C‐ANCA)
Anti‐neutrophil cytoplasmic antibodies (C‐ANCA) are present in approximately 90% of WG cases and can be used to assist diagnosis. There are limitations to C‐ANCA testing for the diagnosis of WG in that it is most useful in patients with known active disease. In a population where the prevalence of WG is low, a positive C‐ANCA test will be a false positive 37% of the time. This false positive rate drops to 16% in a population where the prevalence is high (Rao 1995). If clinical signs clearly indicate WG then the value of C‐ANCA for confirmation is limited as a tissue biopsy may be a more appropriate investigation. The value of serial C‐ANCA measurements to determine disease activity is still unknown as there are limited studies addressing the ability for C‐ANCA to be used to monitor disease activity. Also, the few studies that do address C‐ANCA in this regard have demonstrated conflicting results (Hoffman 1992). Rising C‐ANCA titres should therefore alert the clinician to potential progression of WG but should not be used as a signal to intensify immunosuppressive therapy (Reinhold‐Keller 2000).
There are two distinct phases of drug treatment for WG. The first is induction of remission and the second is the prevention of relapse. The current mainstay of remission induction therapy is systemic corticosteroids in combination with oral daily cyclophosphamide (CYC) which has been shown to induce remission in 75% to 100% of cases in cohort studies (Cohen 1980; De Groot 2001). Due to the serious morbidity (concomitant diseased states) associated with cumulative CYC, pulse doses can be used. A meta‐analysis of three randomized controlled trials of pulse CYC concluded that remission was more likely, CYC doses were lower, leucopenia (reduction in the number of circulating white blood cells) occurred less, and there were fewer adverse events compared with daily CYC therapy (Stegeman 1996). Trimethoprim‐sulfamethoxazole can be used for both remission induction and relapse prevention but only for localized WG (Hellmich 2006; Jayne 2003).
Methotrexate can be used to induce remission instead of CYC in early, less severe WG (De Groot 2005) and azathioprine can be used after remission (in place of CYC) to prevent relapse (Jayne 2003).
Mycophenolate, deoxyspergualin, ritixumab, and leflunomide are agents that have shown promise in maintenance of remission but more prospective randomized controlled trials involving these drugs are needed (Wung 2006). Infliximab has shown benefit in maintenance of remission (Booth 2004). However a similar agent etanercept did not show similar results (WGET Group 2005).
Description of the intervention
Intravenous immunoglobulin preparations are sterile solutions or lypholized concentrates of human immunoglobulin type G (IgG), obtained from pooled human plasma, that have been processed to remove polymers of immune globulin, thus enabling intravenous transfusion. Depending on the method of preparation, some products may also contain trace amounts of IgA and IgM.
How the intervention might work
Intravenous immunoglobulin is an expensive and fairly rare blood product that was originally used to treat immunodeficiencies and has been used to treat WG but its effects on the disorder is unknown. The mechanism of action of IVIg is complex and not well understood. A recent review (Chung 2009), hypothesizes that IVIg immunomodulatory effects may be due the clearance of anti‐idiotype antibodies, blockade of Fc receptors on phagocytic cells, downregulation of T‐cell and B‐cell function, and anticytokine effects.
Why it is important to do this review
Although standard therapy is effective in inducing partial or complete remission, 50% of complete remissions are followed by at least one relapse (Hoffman 1992). Furthermore, patients tend to develop increased morbidity from both the disease and treatment.
It is doubtful whether any single agent can trigger Wegener's granulomatosis, and until a better understanding of the pathophysiology of WG and the events that trigger relapse and perpetuate expression of the disease, broadly immunosuppressive therapies are currently the best known means of producing remission and minimizing disease morbidity. However, treatment related morbidity and toxicity from prolonged cyclophosphamide, methotrexate, azathioprine, and systemic corticosteroids therapy remains an ongoing concern, and leads to a search for safer and equally effective alternatives.
The effects of IVIg on WG is unknown. It is important to determine if IVIg used as adjuvant therapy has an advantage over corticosteroids and immunosuppressants for the treatment of WG.
Objectives
To determine from a systematic review of randomized and quasi‐randomized controlled trials if intravenous immunoglobulin adjuvant therapy provides a therapeutic advantage over and above treatment with systemic corticosteroids in combination with immunosuppressants for the treatment of Wegener's granulomatosis.
Methods
Criteria for considering studies for this review
Types of studies
All randomized controlled trials (RCTs), randomized cross‐over, and quasi‐RCTs (RCTs in which allocation to treatment was obtained by alternation, use of alternate medical records, date of birth, or other predictable methods).
Types of participants
Participants in studies were adults with a confirmed diagnosis of WG.
Any study that randomly assigned patients with a diagnosis of WG to IVIg. It was our intention to undertake sensitivity analyses of any studies that randomized patients with any of the following possible confounding characteristics:
patients on peritoneal dialysis;
patients with acute renal failure or undergoing continuous renal replacement;
patients treated with hemofiltration or hemodiafiltration;
patients who received IVIg therapy during the previous three months;
history of anaphylaxis to properly matched blood products;
selective IgA deficiency;
rapidly progressive glomerulonephritis (20% rise in serum creatinine within two weeks);
severe pulmonary hemorrhage.
If more trials are identified, we will undertake sensitivity analyses in future updates of this review.
Types of interventions
Intravenous immunoglobulin in addition to systemic corticosteroids and immunosuppressive therapy versus systemic corticosteroids and immunosuppressive therapy.
Types of outcome measures
Health outcome hierarchy (in descending order of importance):
all‐cause mortality;
non‐fatal serious adverse events;
time to remission;
length of remission;
organ damage: kidneys, respiratory tract, skin, etc.;
steroid/immunosuppressant sparing;
infection rates;
total adverse events;
withdrawals due to adverse drug reactions;
absolute fall in Birmingham Vasculitis Activity Score (BVAS) from baseline to end of follow up.
Note: Birmingham Vasculitis Activity score (BVAS) Appendix 1
The BVAS is a non‐linear rating scale used to assess disease activity and response to treatment for patients with WG. It consists of nine domains each representing a body system: systemic (i.e. fever, malaise), cutaneous, mucous membranes/eyes, ear, nose and throat, chest, cardiovascular, abdominal, renal and nervous system. Each domain has a different maximum number of points with a total possible maximum BVAS score of 63 (the greater the score the worse the disease). The score is only used to measure new or worsening changes occurring in the last month due to acute vasculitis. Within each domain each symptom is given a particular point value, which is entirely non‐linear and subjective. In addition, changes in BVAS scores in one system have very different clinical implications than do changes in another system. With this in mind individual patient data on details of BVAS changes would be needed (e.g. for each patient, what was the change in the score and where did the change occur specifically?) to interpret fully the effect of treatment (Luqmani 1994). In addition, we searched the biomedical literature and did not find any evidence of an identified minimal clinically relevant change or minimal clinically perceptible change in BVAS, making interpretation of these scores in clinical trials problematic.
Search methods for identification of studies
There was no restriction on language of publication.
Electronic searches
For this update the Cochrane Peripheral Vascular Diseases Group Trials Search Co‐ordinator (TSC) searched the Specialised Register (last searched November 2012) and the Cochrane Central Register of Controlled Trials (CENTRAL) 2012, Issue 11, part of The Cochrane Library, www.thecochranelibrary.com. See (Appendix 2) for details of the search strategy used to search CENTRAL. The Specialised Register is maintained by the TSC and is constructed from weekly electronic searches of MEDLINE, EMBASE, CINAHL, AMED, and through handsearching relevant journals. The full list of the databases, journals and conference proceedings which have been searched, as well as the search strategies used are described in the Specialised Register section of the Cochrane Peripheral Vascular Diseases Group module in The Cochrane Library (www.thecochranelibrary.com).
The following trial databases were searched by the TSC for details of ongoing and unpublished studies using the terms wegener in condition and immunoglobulin or IGG or IVIG in intervention.
World Health Organization International Clinical Trials Registry http://apps.who.int/trialsearch/
ClinicalTrials.gov http://clinicaltrials.gov/
For the 2008 version of the review the authors searched MEDLINE (1966 to Sept 2008) and EMBASE (1980 to Sept 2008) using the search strategies described in Appendix 3 and Appendix 4 .
Searching other resources
For the 2008 version of the review the authors searched conference proceedings, abstracts and poster presentations for potential trials. There was no restriction on language of publication.
Data collection and analysis
Selection of trials
The review was undertaken by all four authors. We used the search strategy described to obtain titles and abstracts of studies that were potentially relevant to the review. Two authors (PF and AT) independently screened titles and abstracts. The same two authors independently assessed retrieved abstracts and, if necessary the full text, of these studies to determine which studies satisfied the inclusion criteria. We planned to translate any studies reported in non‐English language journals before assessment and, where more than one publication of one trial existed, to include only the publication with the most complete data. We requested further information required from the original author by written correspondence and included in the review any relevant information obtained. Disagreements were resolved in consultation with KB and VM.
Data extraction
Data extraction was carried out independently by PF and AT using standard data extraction forms.
Study quality
The quality of studies was assessed independently by PF and AT without blinding to authorship or journal using the checklist developed for the Cochrane PVD Group. Discrepancies were resolved by discussion with KB and VM. The quality items assessed were method of randomization, allocation concealment, blinding (participants, investigators, outcome assessors and data analysis), intention‐to‐treat analysis and completeness of follow up.
In the event that more than one trial was included, and where these trials differed with respect to the identified trial quality indicators, we planned to perform sensitivity analyses to test the assumption that the presence or absence of trial quality indicators (e.g. whether or not allocation concealment was done) had a bearing on the intervention effect estimates (e.g. is the over‐all intervention effect estimate different than that in trials with adequate allocation concealment?).
Quality checklist
Allocation concealment
A. Adequate: randomization method described that would not allow investigator/participant to know or influence intervention group before eligible participant entered in the study. B. Unclear: randomization stated but no information on method used is available. C. Inadequate: method of randomization used such as alternate medical record numbers or unsealed envelopes; any information in the study that indicated that investigators or participants could influence intervention group.
Blinding
Blinding of investigators: yes/no/not stated. Blinding of participants: yes/no/not stated. Blinding of outcome assessor: yes/no/not stated. Blinding of data analysis: yes/no/not stated.
The above are considered not blinded if the treatment group can be identified in > 20% of participants because of the side effects of treatment.
Intention‐to‐treat analysis
Yes: specifically reported by authors that intention‐to‐treat analysis was undertaken and this was confirmed on study assessment. Yes: not stated but confirmed on study assessment. No: not reported and lack of intention‐to‐treat analysis confirmed on study assessment. (Patients who were randomized were not included in the analysis because they did not receive the study intervention, they withdrew from the study or were not included because of protocol violation.) No: stated but not confirmed upon study assessment. Not stated.
Completeness of follow up
Percentage of participants excluded or lost to follow up.
Statistical assessment
For dichotomous outcomes (death, non‐fatal serious adverse events, organ damage, steroid/immunosuppressant sparing, and total and withdrawal adverse drug reactions), we reported results as relative risk (RR) with 95% confidence intervals (CI). Where continuous scales of measurement were used to assess the effects of treatment (e.g. time to remission, length of remission, infection rates, and fall in BVAS score) the mean difference (MD) was used. If more than one trial had been included, we planned to undertake the following analyses:
1. Pooling of data using both the fixed‐effect model and the random‐effects model (the latter to ensure robustness of the model chosen and susceptibility to outliers).
2. Use of the standardized mean difference (SMD) where different continuous scales of measurement were used.
3. Testing of heterogeneity using a chi squared test on N‐1 degrees of freedom, with an alpha of 0.05 used for statistical significance and with the I2 test (Higgins 2003).
4. If necessary, use of subgroup analyses to explore possible sources of heterogeneity (e.g. participants, interventions and study quality). Heterogeneity among participants could be related to age and renal pathology. Heterogeneity in treatments could be related to prior agent(s) used and the agent, dose and duration of therapy.
5. Tabulation of adverse effects and assessment with descriptive techniques, as these are likely to be different for the various agents used. Where possible, we also would have calculated the risk difference with 95% confidence intervals for each adverse effect, either compared with no treatment or to another agent.
6. Use of funnel plots to assess for publication bias.
Results
Description of studies
No date or language restrictions were applied.
Results of the search
For this update 36 records were retrieved from CENTRAL but there were no new relevant studies. Similarly the search of the Specialised Register did not result in any new studies for consideration. There were no relevant ongoing studies.
Only one randomized controlled trial met our inclusion criteria (Jayne 2000). No ongoing clinical trials were identified and no RCTs were excluded see QUOROM flow chart (Figure 1).
1.
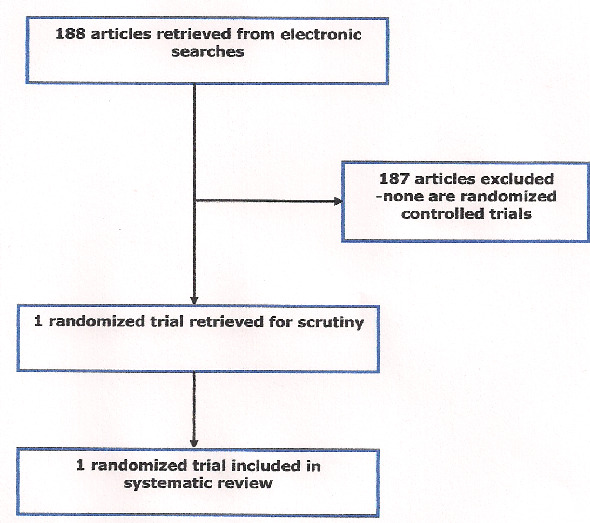
QUORUM (quality of reporting meta‐analyses) flow chart of study selection
Included studies
For details of the included study seeCharacteristics of included studies.
This study was a prospective, randomized, double‐blind, placebo‐controlled multicenter trial. Thirty four patients were randomly assigned to receive IVIg (total dose = 2 g/kg, n = 17) or placebo (n = 17) once daily in addition to azathioprine and prednisolone for remission maintenance. A prior diagnosis of Wegener’s granulomatosis or microscopic polyangiitis satisfying criteria of the Chapel Hill consensus conference was required for patients to enter the trial (including C‐ANCA positivity at diagnosis, active vasculitis with a requirement for further therapy, at least two months treatment with prednisolone and cyclophosphamide or azathioprine and ≥18 years. Another criterion for inclusion into the study was patients either relapsing on steroids alone (or steroids in combination with immunosuppressant therapy) or those that did not achieve full remission on steroids in combination with immunosuppressant therapy following initial presentation.
Patients were excluded from participating in the trial if they had any of the following characteristics: IVIg therapy during the previous three months, history of anaphylaxis to properly matched blood products, selective IgA deficiency, rapidly progressive glomerulonephritis (20% rise in serum creatinine within two weeks), severe pulmonary hemorrhage.
The primary outcome measure of the study was the number of patients achieving a reduction in BVAS of 50% after three months. Secondary outcome measures included a fall in BVAS, CRP (C‐Reactive Protein), and ANCA levels, relapse frequency between three and 12 months, reduction in immunosuppressive drug doses and adverse‐effect rates.
Excluded studies
There were no excluded studies.
Risk of bias in included studies
See Risk of Bias tables and Figure 2 and Figure 3.
2.
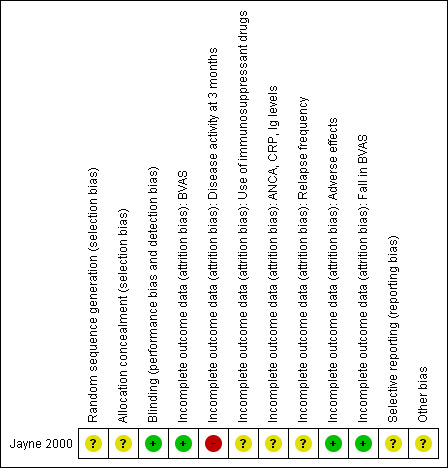
Methodological quality summary: review authors' judgements about each methodological quality item for the included study.
3.
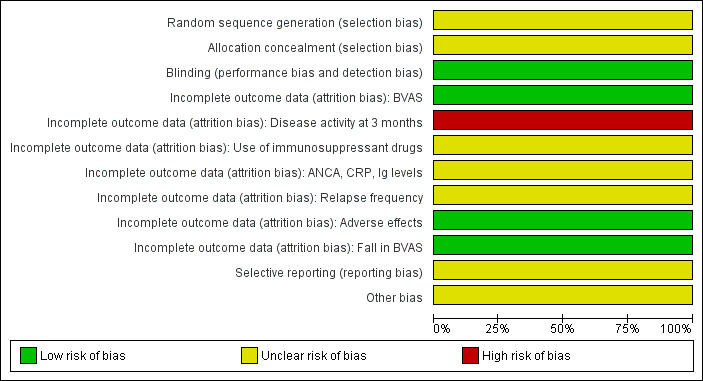
Methodological quality graph: review authors' judgements about each methodological quality item presented as percentages across the included study.
In general due to insufficient detail regarding the random sequence generation, unclear description of allocation concealment, and the possibility that the blinding procedures were compromised, the effects of IVIg seen in the trial could be overestimated. In addition, there is uncertainty as to whether an intention‐to‐treat analysis was conducted for all outcomes (i.e. five patients in each limb were not accounted for in the three‐month chest radiology or white cell scanning assessments). Again, the implication of this may be that the effects of IVIg have been overestimated, it may have been assumed that these patients did not experience a worsening of their condition, nor did they experience an adverse event.
Additional risks of bias include the following:
A. Purpose of the two‐week observation period prior to randomization.
The published trial report mentions a two‐week observation period for all enrolled patients that occurred prior to randomization. No details are given as to the purpose of this observation period however it was stated that 39 patients were enrolled. Subsequently four patients chose not to participate the assumption being this occurred during the observation period. The concern is the introduction of selection bias that may have excluded a particular type of patient prior to randomization limiting the external validity of subsequent findings. The lead author was contacted for clarification but has not provided a response to date.
B. The use of chlorpheniramine
Open‐label use of chlorpheniramine was permitted at the discretion of the treating physicians in the study. The reasons for use were not stated nor were details concerning the amount of chlorpheniramine used in each treatment group. This may be a concern as this drug may be used in response to treatment‐related side effects, and thus compromise blinding.The lead author has not provided the requested details to date.
C. Blinding effectiveness and infusion site reactions
In trials comparing active treatment IV infusions with placebo IV infusions, maintenance of blinding may be compromised. That being said it is important to know the rates of infusion site reactions for each group and whether the effectiveness of blinding was assessed in any way. If blinding was ineffective, this could lead to bias in the study and the results may not be reliable. The lead author has not provided these details to date.
Effects of interventions
Forest plots are not provided since only one trial met the criteria and results are presented as tables, see Table 1 for dichotomous outcomes and Table 2 for continuous outcomes. There were no significant differences between IVIg and placebo in mortality, serious adverse events, time to relapse, open‐label rescue therapy, and infection rates. The fall in BVAS score was significantly better in the IVIg group than in the placebo group at one month (mean difference (MD) 2.30; 95% Confidence Interval (CI) 1.12 to 3.48, P < 0. 01) and three months (MD 1.80; 95% CI 0.35 to 3.25, P = 0.01). The authors stated that there was no significant difference after three months. There was a significant increase in total adverse events in the IVIg group (relative risk (RR) 3.50; 95% CI 1.44 to 8.48, P < 0.01), (absolute risk increase 59; 95% CI 32 to 86), (number needed to harm (NNH) = two).
1. Dichotomous outcomes.
| Outcome |
IVIg n (%) |
Control n (%) |
RR (95% CI) |
ARR/ARI % |
NNT/H | P‐Value |
| All‐cause mortality | 0/17 (0) | 2/17 (12) | 0.20 (0.01 to 3.88) | n/a | n/a | P = 0.29 |
| Serious adverse events | 3/17 (18) | 2/17 (12) | 1.50 (0.29 to 7.87) | n/a | n/a | P = 0.63 |
| Time to remission | Not Reported | |||||
| Length of remission (reported as number of patients relapsed after 3 months) | 5/17 (29 ) | 4/17 (24 ) | 1.25 (0.40 to 3.87) | n/a | n/a | P = 0.70 |
| End‐organ damage | Not reported | |||||
| Steroid/immunosuppressant sparing open‐label: treatment for worsening vasculitis | ||||||
| Open‐label IVIg within 3 months | 0/17 (0) | 1/17 (6) | 0.33 (0.01 to 7.65) | n/a | n/a | P = 0.49 |
| Open‐label CYC within 3 months | 0/17 (12) | 1/17 (6) | 0.33 (0.01 to 7.65) | n/a | n/a | P = 0.49 |
| Open‐label IVIg after 3 months | 2/17 (12 ) | 1/17 (6) | 2.00 (0.20 to 20.04) | n/a | n/a | P = 0.56 |
| Open‐label CAMPATH‐1 after 3 months | 1/17 (6) | 1/17 (6) | 3.00 (0.13 to 68.84) | n/a | n/a | P = 0.49 |
| Infection rates (n = 1 aseptic meningitis in IVIg group, included in serious adverse events) | 1/17 (6) | 0/17 (0) | 3.0 (0.13 to 68.84) | n/a | n/a | P = 0.49 |
| Total adverse events | 14/17 (82) | 4/17 (24) | 3.50 (1.44 to 8.48) | 59 (95% CI 32 to 86) | 2 | P < 0.01 |
| Withdrawal due to adverse events | Not reported |
CYC: cyclophophamide IVIg: intravenous immunoglobulin NNT/H: number needed to harm RR: relative risk
2. Continuous outcomes: Birmingham Vasculitis Activity Score.
| IVIg | Control | MD (95%CI) | P‐Value | |
| At 1 month | 3.2 (SD 1.9) | 0.9 (SD1.6) | 2.30 (1.12 to 3.48) | P < 0.01 |
| At 3 months | 4.1 (SD 2.3) | 2.3 (SD 2.0) | 1.80 (0.35 to 3.25) | P = 0.01 |
| TOTAL 1 and 3 months | 2.10 (1.19 to 3.02) | P < 0.01 | ||
| After 3 months: stated “no significant difference” but no details reported |
IVIg: intravenous immunoglobulin MD: mean difference
Discussion
There are a number of issues relating to the internal/external validity, reliability, and clinical relevance of this trial:
A. Birmingham vasculitis activity score (BVAS)
Changes in BVAS scores in one system have very different clinical implications than do changes in another system. With this in mind individual patient data on details of BVAS changes would be needed (e.g. for each patient, what was the change in the score and where did the change occur specifically) to fully interpret the effect of treatment. This level of detail is not available in the published trial report and the author has not provided us with details to date.
It is difficult to determine what change in BVAS score would be considered a clinically relevant or minimally perceptible change due to drug therapy. In fact a search of the biomedical literature did not reveal any such study that attempted to correlate a particular change in BVAS to a minimally perceptible change from a patient’s perspective. In addition, as the score is only designed to measure changes within the last month it does not provide a good measure of disease activity in a more global sense. For example a change in BVAS of two points may indicate a particular symptom resolved in the past month of therapy but there is no guarantee that this symptom may not recur. Another scenario that may occur would be a new severe symptom appearing causing the score to increase by four points, but three minor symptoms were resolved (a three point reduction) indicating very little net change overall. This type of scenario is misleading as the presence of a new major symptom may hold more meaning clinically than the resolution of several minor symptoms.
The primary endpoint of the study was treatment response defined as: a > 50% reduction in BVAS from baseline to three months follow up. This endpoint would be useful if a 50% reduction from a high baseline BVAS was the same as from a low baseline BVAS (e.g. a reduction from BVAS of 20 points down to 10 points in a patient versus a reduction from BVAS of 6 points down to 3 points in another patient) but this is not the case. The more appropriate endpoint would be the absolute change from baseline in BVAS. This was reported as a secondary endpoint by the authors.
The results of fall in BVAS are as follows:
At one month, the fall in BVAS from baseline was greater in IVIg group (3.2, standard deviation (SD) 1.9) than in the placebo group (0.87, SD 1.6). This difference persisted at three months (4.1, SD 2.3) versus (2.3, SD 2.0). In other words at three months IVIg produced a greater reduction in BVAS by 1.8 points versus placebo. As stated earlier, the clinical relevance of a change of 1.8 points in BVAS is not known.
B. BVAS changes in different systems
In the published trial report details were provided on which systems had the greatest change in BVAS for the IVIg patients. This information would be necessary if there is incremental benefit in relation to system improvements with IVIg therapy compared to placebo in any particular systems. Details of system BVAS changes were not provided for the placebo treated patients. These details were requested from the author but have not been provided to date.
C. No difference in BVAS changes after three months
After three months the benefit in terms of additional BVAS reductions in the IVIg group were lost compared with the placebo group. This may imply that if there is a benefit from IVIg therapy that therapy would have to continue indefinitely or at least longer than one infusion. In addition, infusions would need to be spaced no more than three months apart. To interpret this correctly in the trial, doses of immunosuppressants and steroids for each group would need to be provided to see if they were actually reduced from baseline levels. Patients would then have to be followed to see if doses were then increased to baseline levels after IVIg effects wore off. The authors did not provide these details. In addition, it is difficult to interpret this correctly from the details reported in the trial as steroid and immunosuppressant doses were not allowed to change for the first three months. It is important to note that there were baseline differences between the groups with respect to doses of steroids and immunosuppressants (discussed in greater detail below). Doses of these agents could be adjusted after three months but details of changes if they did occur, were not provided. Another point to consider is that dose changes may have had an impact on patient outcomes. Hence patient outcomes may not be attributable solely to the differences in study intervention. This however is untested and needs to be confirmed in subsequent randomized controlled trials.
D. Total adverse events
The total number of patients experiencing at least one adverse event was greater in the IVIg patients compared with placebo patients (RR 3.50; 95% CI 1.44 to 8.48, P < 0.01). This is important to consider when interpreting the net effect of treatment with IVIg. However, as stated earlier, it is unclear what clinically relevant effect IVIg has on disease activity and on morbidity associated with WG (i.e. end‐organ dysfunction, infection rates). Therefore interpretation of the net effect is difficult with the existing evidence from this RCT.
E. All‐cause mortality and non‐fatal serious adverse events
The trial was inadequately powered and of too short a duration to detect differences in mortality and serious adverse events in the IVIg and placebo groups.
F. Open‐label use of IVIg and CYC during the study
In the study methods, no mention was made of explicit a‐priori criteria for the use of open‐label IVIg, open‐label CYC, or open‐label use of other agents for the management of worsening vasculitis. In the results section, open‐label use of these agents was reported for both groups. Within the first three months the placebo group received an escalation of therapy for worsening vasculitis, with one patient having cyclosphosphamide re‐introduced, and one patient receiving open‐label IVIg. After three months, five from the IVIg group and two from the placebo group had received escalation of therapy. Details of the exact reasons patients received open‐label agents, and the doses and duration of these agents would be useful to determine if this procedure may have led to bias either in favour of treatment or control, making the results less reliable. Also, the difference in measured effect between treatment groups may not be solely attributable to the study interventions.
Another potential problem of open‐label use of agents is the possibility that this may have led to the loss of blinding. Analyses were performed at the time of infusion, at two weeks, and three, six, nine and 12 months. It is not known if the effectiveness of the double‐blinding procedures were assessed at the points of analyses or at the end of the study making this difficult to interpret.
G. Baseline differences in patients
There were reported differences between the groups at baseline with respect to doses of steroids and immunosuppressants, for treatment of vasculitis. The mean steroid and AZA doses were higher in the IVIg group, however the mean CYC doses were lower in the IVIg group. The implication of an imbalance in baseline characteristics is that randomization may not have been successful due to the small sample size. This holds particular importance in this setting as differences in exposure to steroids or immunosuppressants between groups could have an impact on outcomes.
H. Mixed population of patients with Werner's granulomatosis and Microscopic polyangiitis
This trial enrolled patients with Wegeners granulomatosis and patients with Microscopic polyangiitis. Even though it has been suggested that WG and MP are pathologically indistinguishable, it is unknown whether patients with either type of vasculitis may respond differently. This study does not provide sufficient detail, nor have appropriate sensitivity analyses been done to identify if there is a different effect of IVIg depending on the type of vasculitis present. It is therefore impossible to determine the effect of IVIg specifically in WG patients in this study.
Summary
The net effect of IVIg for adjuvant therapy in WG is unknown. The length of remission induced by IVIg is also unknown. However, it appears that the time to relapse is no different with IVIg than with placebo. The effect of IVIg on end‐organ damage and the potential immunosuppressant or steroid sparing effect of IVIg is unclear as sufficient details were not reported in the trial. What was reported and presented here is the rescue therapy needed for relapsing patients, which showed no significant differences between IVIg and placebo. Withdrawals due to adverse events were not reported. However the total number of patients experiencing at least one adverse event was significantly greater in patients who received IVIg. Finally, the absolute fall in BVAS at three months was greater in patients who received IVIg but the clinical significance of the effect size seen in the trial is not known. This effect seen at three months was not present after six months where BVAS changes were similar for patients who received IVIg and placebo.
Limitations of the review
Improved quality of life and activities of daily living would be important clinical outcomes for patients with WG. We did not include these as outcomes in our hierarchy but, if possible they will be included in future updates. However, in a post‐hoc review of the one randomized trial included in this review, it is noted that the trial did not measure improvement in quality of life or activities of daily living.
Authors' conclusions
Implications for practice.
There is insufficient evidence from the one and only randomized controlled trial (Jayne 2000) that IVIg adjuvant therapy provides a therapeutic advantage compared with the combination of steroids and immunosuppressant therapy for patients with WG.
Given the high cost of IVIg (one dose at 2 g/kg for a 70 kg patient = $8,400), it should be limited to treat Wegener’s granulomatosis in the context of a well conducted randomized controlled trial powered to detect outcomes relevant to patients.
Implications for research.
Future randomized controlled trials should:
a) Use a triple‐blind, double dummy design, include a pilot assessment of blinding, and also assess the effectiveness of blinding at the completion of the study.
b) Individual patient data and details with regards to changes in BVAS should be provided. In addition an analysis to determine minimally perceptible changes to patients should be conducted. Absolute changes in BVAS should be reported.
c) The primary focus of the trial should be to determine the net effect of IVIg adjuvant therapy in patients with WG. Serious adverse events, all‐cause mortality, end‐organ dysfunction, quality of life, and infection rates should be measured, reported, and compared between treatment groups.
d) Be of a minimum length of one year and should include a group that receives only one 5‐day series of infusions of IVIg, and a group that receives one infusion of IVIg every three months as well as a placebo group.
What's new
| Date | Event | Description |
|---|---|---|
| 16 November 2012 | New search has been performed | Searches re‐run. No new included, excluded or ongoing studies identified. |
| 16 November 2012 | New citation required but conclusions have not changed | Searches re‐run, no new included, excluded or ongoing studies identified. Minor copy edits made. Conclusions not changed. |
History
Protocol first published: Issue 2, 2008 Review first published: Issue 3, 2009
| Date | Event | Description |
|---|---|---|
| 22 April 2008 | Amended | Converted to new review format. |
Acknowledgements
Dr. Jayne for responding to our e‐mails.
Members of the Therapeutics Initiative Drug Assessment Working Group for their contributions to critical appraisal.
Ms Sara Yaron from the Cochrane Consumer network for her peer referee comments.
Appendices
Appendix 1. Birmingham Vasculitis Activity Score
| Name: DOB: Sex: Unit No: Visit Date: Tick box only if abnormality is newly present or worsening within the previous 4 weeks and ascribable to vasculitis. |
|||||||
| WEIGHTED SCORE | |||||||
| 5. | CHEST | 6 (max total) | |||||
| None | [ ] | 0 | |||||
| Dyspnea or wheeze | [ ] | 2 | |||||
| Nodules or fibrosis | [ ] | 2 | |||||
| Pleural effusion / pleurisy | [ ] | 4 | |||||
| Infiltrate | [ ] | 4 | |||||
| Hemoptysis / hemorrhage | [ ] | 4 | |||||
| Massive hemoptysis | [ ] | 6 | |||||
| WEIGHTED SCORE | |||||||
| 1. | SYSTEMIC | 3 (max total) | 6. | CARDIOVASCULAR | 6 (max total) | ||
| None | [ ] | 0 | None | [ ] | 0 | ||
| Malaise | [ ] | 1 | Bruits | [ ] | 2 | ||
| Myalgia | [ ] | 1 | New loss of pulses | [ ] | 4 | ||
| Arthralgia/arthritis | [ ] | 1 | Aortic incompetence | [ ] | 4 | ||
| Fever (<38.5?C) | [ ] | 1 | Pericarditis | [ ] | 4 | ||
| Fever (>38.5 C) | [ ] | 2 | New myocardial infarct | [ ] | 6 | ||
| Wt loss (1‐2 kg) within past month | [ ] | 2 | CCF / cardiomyopathy | [ ] | 6 | ||
| Wt loss (> 2 kg) within past month | [ ] | 3 | |||||
| 2. | CUTANEOUS | 6 (max total) | 7. | ABDOMINAL | 9 (max total) | ||
| None | [ ] | 0 | None | [ ] | 0 | ||
| Infarct | [ ] | 2 | Abdominal pain | [ ] | 3 | ||
| Purpura | [ ] | 2 | Bloody diarrhea | [ ] | 6 | ||
| Other skin vasculitis | [ ] | 2 | Gallbladder perforation | [ ] | 9 | ||
| Ulcer | [ ] | 4 | Gut infarction | [ ] | 9 | ||
| Gangrene | [ ] | 6 | Pancreatitis | [ ] | 9 | ||
| Multiple digit gangrene | [ ] | 6 | |||||
| 3. | MUCOUS MEMBRANES / EYES | 6 (max total) | 8. | RENAL | 12 (max total) | ||
| None | [ ] | 0 | None | [ ] | 0 | ||
| Mouth ulcers | [ ] | 1 | Hypertension (diastolic >90) | [ ] | 4 | ||
| Genital ulcers | [ ] | 1 | Proteinuria (>1+ or >0.2g/24h) | [ ] | 4 | ||
| Conjunctivitis | [ ] | 1 | Hematuria (>1+ or >10rbc/mL) | [ ] | 8 | ||
| Epi / scleritis | [ ] | 2 | Creatinine 125‐249 µmol/L) | [ ] | 8 | ||
| Uveitis | [ ] | 6 | Creatinine 250‐499 µmol/L) | [ ] | 10 | ||
| Retinal exudates | [ ] | 6 | Creatinine >500 µmol/L | [ ] | 12 | ||
| Retinal hemorrhage | [ ] | 6 | Rise in creatinine >10% | [ ] | 12 | ||
| 4. | ENT | 6 (max total) | 9. | NERVOUS SYSTEM | 9 (max total) | ||
| Nil | [ ] | 0 | None | [ ] | 0 | ||
| Nasal discharge / obstruction | [ ] | 2 | Organic confusion / dementia | [ ] | 3 | ||
| Sinusitis | [ ] | 2 | Seizures (not hypertensive) | [ ] | 9 | ||
| Epistaxis | [ ] | 4 | Stroke | [ ] | 9 | ||
| Crusting | [ ] | 4 | Cord lesion | [ ] | 9 | ||
| Aural discharge | [ ] | 4 | Peripheral neuropathy | [ ] | 6 | ||
| Otitis media | [ ] | 4 | Motor mononeuritis multiplex | [ ] | 9 | ||
| New deafness | [ ] | 6 | |||||
| Hoarseness / laryngitis | [ ] | 2 | |||||
| Subglottic involvement | [ ] | 6 | |||||
| MAXIMUM SCORE | 63 | ||||||
Appendix 2. 2012 CENTRAL search strategy
#1 MeSH descriptor: [Wegener Granulomatosis] this term only 41 #2 wegen*:ti,ab,kw 84 #3 ANCA or C‐ANCA 122 #4 vasculitis 360 #5 granulomatosis 102 #6 polyangiitis 27 #7 #1 or #2 or #3 or #4 or #5 or #6 476 #8 MeSH descriptor: [Immunoglobulins, Intravenous] explode all trees 585 #9 *globulin* 10629 #10 Ig* in Trials 9859 #11 IVIG 529 #12 civacir or flebogamma or gamunex or carimune or gammagard or octagam or privigen 37 #13 #8 or #9 or #10 or #11 or #12 17334 #14 #7 and #13 in Trials 36
Appendix 3. 2008 MEDLINE (Ovid) search strategy
1. Wegener Granulomatosis/ 4941 2. Vasculitis/ 9305 3. (wegener$ adj granulomat$).ti,ab. 4430 4. (wegener$ adj5 vascul$).ti,ab. 452 5. (small adj5 vessel adj5 vascul$).ti,ab. 802 6. or/4‐5 14511 7.Immunoglobulins, Intravenous/ 7099 8. IVIG.mp. 2958 9. 7 or 8 7812 10. 6 and 9 139
Appendix 4. 2008 EMBASE search strategy
1. Wegener Granulomatosis/ 5295 2. (wegener$ adj granulomat$).ti,ab. 3722 3. (small adj5 vessel adj5 vascul$).ti,ab. 826 4. (wegener$ adj5 vascul$).ti,ab. 433 5.VASCULITIS/ 12812 6. or/1‐5 13057 7. Immunoglobulin G/ or Immunoglobulin/ 92643 8. intravenous immunoglobulin.mp. 4468 9. ivig.mp. 3010 10. 0r/7‐8 92986 11. 6 and 10 1065 12. Randomized Controlled Trial/ 168518 13.Randomization/ 26765 14 12 and 13 190258 15. 11 and 14 12
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Jayne 2000.
| Methods | Prospective, double‐blind, double dummy, multicenter, randomized trial. | |
| Participants | Inclusion criteria: Adults with a prior diagnosis of Wegener's granulomatosis or microscopic polyangiitis (based on Chapel Hill Consensus Conference criteria), ANCA positive at time of diagnosis, active disease requiring further therapy, and at least 2 months of therapy with prednisolone and cyclophosphamide or azathioprine. Exclusion criteria: IVIg therapy in the past 3 months, history of anaphylaxis to properly matched blood products, selective IgA deficiency, rapidly progressive glomerulonephritis (20% rise in serum creatinine within 2 weeks) or severe pulmonary hemorrhage. |
|
| Interventions | Background therapy: All patients received prednisolone and cyclophosphamide for remission induction and prednisolone and azathioprine for remission maintenance. Two‐week observation period then patients randomized to IVIg 0.4 mg/kg/day for 5 days (total dose 2 g/kg) or placebo. |
|
| Outcomes | Baseline, time of infusion, at 2 weeks, and monthly for 12 months: BVAS, weight, current immunosuppressive therapy, CRP, serum creatinine, liver function tests, hemoglobin, white cells, neutrophils, lymphocyte and platelet counts, hematuria, proteinuria, platelet counts, adverse effects of trial medication, Baseline and every 3 month (if vasculitis activity was detected): white cell scans and chest radiology |
|
| Notes | Contact with Dr. Jayne via email for clarification of certain aspects of the trial (i.e. clinical meaning of changes in the BVAS, and reason for initial observation period) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No description provided |
| Allocation concealment (selection bias) | Unclear risk | "Randomization and distribution of trial medication was centrally controlled by Novartis UK..." |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Double‐blind. "placebo identical in appearance to IVIg; patients and physicians were blinded to the treatment limb" |
| Incomplete outcome data (attrition bias) BVAS | Low risk | 2 patients died before the 3 month BVAS assessment however they did an ITT and per‐protocol analysis |
| Incomplete outcome data (attrition bias) Disease activity at 3 months | High risk | 5 patients in each limb (10 patients total) were not accounted for in the 3 month chest radiology or white cell scanning assessments |
| Incomplete outcome data (attrition bias) Use of immunosuppressant drugs | Unclear risk | No details given regarding the number of randomized patients assessed |
| Incomplete outcome data (attrition bias) ANCA, CRP, Ig levels | Unclear risk | No details given regarding the number of randomized patients assessed |
| Incomplete outcome data (attrition bias) Relapse frequency | Unclear risk | Two from the placebo group had escalation of therapy within the first 3 months for worsening vasculitis, with re‐introduction of cyclophosphamide in one and open IVIg in the other. After three months, five from the IVIg group had escalation of therapy with cyclophosphamide re‐introduced in two, two received open IVIg and one monoclonal anti‐T‐cell therapy with CAMPATH 1‐H. During the same period in the placebo group, one had cyclophosphamide re‐introduced, one open IVIg and one CAMPATH 1‐H. |
| Incomplete outcome data (attrition bias) Adverse effects | Low risk | Seventeen adverse effects were seen in 12 patients from the IVIg group as compared to six adverse effects in four patients in the placebo group |
| Incomplete outcome data (attrition bias) Fall in BVAS | Low risk | "BVAS at the time of infusion was 6.1 (SD 1.56) and 5.4 (SD 1.8) for the IVIg and placebo groups, respectively. Following infusion, the fall in BVAS was greater in the IVIg than the placebo group when compared at 1 month (fall in BVAS of 3.2, SD 1.9) versus 0.87, SD 1.6), P < 0.001), and 3 months (fall in BVAS of 4.1, SD 2.3 versus 2.3, SD 2.0, P < 0.01)" |
| Selective reporting (reporting bias) | Unclear risk | "The difference in clinical disease activity between the IVIg and placebo groups was present up to the analysis at 3 months. Subsequently, vasculitis activity, frequency of relapse and exposure to immunosuppression was the same in both limbs, indicating that the benefit of IVIg was not maintained beyond 3 months." See above |
| Other bias | Unclear risk | After entry into the trial and a 2‐week observation period, patients were randomly assigned to receive IVIg 0.4 g/kg/day for 5 days (total dose 2 g/kg) (Sandoglobulin, Novartis) or placebo |
ANCA: anti‐neutrophil cytoplasm antibody BVAS: Birmingham Vasculitis Activity Score CRP: C‐reactive protein IgA: immunoglobulin A ITT: intention to treat IVIg: intravenous immunoglobulin SD: standard deviation
Differences between protocol and review
The outcome adverse drug reactions was changed to total adverse events.
Contributions of authors
PF and AT independently selected studies, assessed study quality and extracted data.
KB and VM resolved any disagreements in the selection of trials and assessed study quality.
Sources of support
Internal sources
Department of Anesthesiology, Pharmacology and Therapeutics, University of British Columbia, Vancouver, Canada.
British Columbia Provincial Blood Coordinating Office, Canada.
Therapeutics Initiative, Drug Assessment Working Group, Canada.
External sources
-
Chief Scientist Office, Scottish Government Health Directorates, Scottish Government, UK.
The PVD Group editorial base is supported by the Chief Scientist Office.
Declarations of interest
None known
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Jayne 2000 {published data only}
- Jayne DRW, Chapel, H, Adu, D, Misbah, S, ODonoghue, D, Scott, D, Lockwood, CM. Intravenous immunoglobulin for ANCA‐associated systemic vasculitis with persistent disease activity. Quarterly Journal of Medicine 2000;93:433‐9. [DOI] [PubMed] [Google Scholar]
Additional references
Booth 2004
- Booth A, Harper L, Hammad T, Bacon P, Griffith M, Levy J, et al. Prospective study of TNFalpha blockade with infliximab in anti‐neutrophil cytoplasmic antibody‐associated systemic vasculitis. Journal of the American Society of Nephrology 2004;15(3):717‐21. [DOI] [PubMed] [Google Scholar]
Chung 2009
- Chung SA, Seo P. Advances in the use of biologic agents for the treatment of systemic vasculitis. Current Opinion in Rheumatology 2009;21:3‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cohen 1980
- Cohen RD, Conn DL, Ilstrup DM. Clinical features, prognosis, and response to treatment in polyarteritis. Mayo Clinic Proceedings 1980;55(3):146‐55. [PubMed] [Google Scholar]
De Groot 2001
- Groot K, Adu D, Savage CO, EUVAS (European vasculitis study group). The value of pulse cyclophosphamide in ANCA‐associated vasculitis: meta‐analysis and critical review. Nephrology Dialysis Transplantation 2001;16(10):2018‐27. [DOI] [PubMed] [Google Scholar]
De Groot 2005
- Groot K, Rasmussen N, Bacon PA, Tervaert JW, Feighery C, Gregorini G, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody‐associated vasculitis. Arthritis and Rheumatism 2005;52(8):2461‐9. [DOI] [PubMed] [Google Scholar]
Esper 1999
- Esper GJ, Johnson JS. Update on the treatment of Wegener's granulomatosis.. Bulletin on the Rheumatic Diseases. 1999;48(11):1‐4. [PubMed] [Google Scholar]
Hellmich 2006
- Hellmich B, Lamprecht P, Gross WL. Advances in the therapy of Wegener's granulomatosis. Current Opinion in Rheumatology 2006;18(1):25‐32. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hoffman 1992
- Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener's granulomatosis: an analysis of 158 patients. Annals of Internal Medicine 1992;116(6):488‐98. [DOI] [PubMed] [Google Scholar]
Jayne 2003
- Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW, Dadoniene J, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. New England Journal of Medicine 2003;349(1):36‐44. [DOI] [PubMed] [Google Scholar]
Luqmani 1994
- Luqmani RA, Bacon PA, Moots RJ, Janssen BA, Pall A, Emery P, et al. Birmingham Vasculitis Activity Score in systemic necrotizing vasculitis. QJM 1994;87(11):671‐8. [PubMed] [Google Scholar]
Rao 1995
- Rao JK, Weinberger M, Oddone EZ, Allen NB, Landsman P, Feussner JR. The role of antineutrophil cytoplasmic antibody (c‐ANCA) testing in the diagnosis of Wegener granulomatosis. A literature review and meta‐analysis. Annals of Internal Medicine 1995;123(12):925‐32. [DOI] [PubMed] [Google Scholar]
Reinhold‐Keller 2000
- Reinhold‐Keller E, Beuge N, Latza U, Groot K, Rudert H, Nolle B, et al. An interdisciplinary approach to the care of patients with Wegener's granulomatosis: long‐term outcome in 155 patients. Arthritis and Rheumatism 2000;43(5):1021‐32. [DOI] [PubMed] [Google Scholar]
Scott 2000
- Scott DG, Watts, RA. Systemic Vasculitis: epidemiology, classification, and environmental factors. Annals of the Rheumatic Diseases 2000;59(3):161‐3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Stegeman 1996
- Stegeman CA, Tervaert JW, Jong PE, Kallenberg CG. Trimethoprim‐sulfamethoxazole (co‐trimoxazole) for the prevention of relapses of Wegener's granulomatosis. Dutch Co‐Trimoxazole Wegener Study Group. New England Journal of Medicine 1996;335(1):16‐20. [DOI] [PubMed] [Google Scholar]
WGET Group 2005
- The Wegener's Granulomatosis Etanercept Trial (WGET) Research Group. Etanercept plus standard therapy for Wegener's granulomatosis. New England Journal of Medicine 2005;352(4):351‐61. [DOI] [PubMed] [Google Scholar]
Wung 2006
- Wung PK, Stone JH. Therapeutics of Wegener's granulomatosis. Nature Clinical Practice Rheumatology 2006;2(4):192‐200. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Fortin 2009
- Fortin PM, Tejani AM, Bassett K, Musini VM. Intravenous immunoglobulin as adjuvant therapy for Wegener's granulomatosis. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007057.pub2] [DOI] [PubMed] [Google Scholar]