

Abstract
We have previously shown a more potent impact of knockout of Cu,Zn-superoxide dismutase (SOD1) than that of Se-dependent glutathione peroxidase-1 (GPX1) on murine hepatotoxicity induced by an intraperitoneal (ip) injection of a high dose of acetaminophen (APAP, 600 mg/kg). The objective of this experiment was to compare the temporal impacts of knockouts of GPX1 and SOD1 alone or together on mouse susceptibility to an injection of a low dose of APAP (300 mg/kg). The APAP-mediated rises in plasma alanine aminotransferase activity and nitrate/nitrite concentrations, hepatic GSH depletion, and hepatic protein nitration at 5 and (or) 24 h were nearly abolished (P < 0.05) in SOD1−/− or GPX1 and SOD1 double-knockout (DKO) mice, while GPX1−/− mice exerted only moderate or no change compared with the WT. Despite an increased (P < 0.05) APAP-N-acetylcysteine and decreased APAP-glucuronide (P < 0.05) relative to the total APAP metabolites in urine collected for 24 h after the APAP injection, the SOD1−/− mice displayed no shift in urinary APAP-cysteine compared with the WT mice. Knockout of SOD1 prevented the APAP-induced hepatic GPX inactivation (P < 0.05), whereas knockout of GPX1 aggravated the APAP-induced hepatic SOD activity loss (P < 0.05). However, the APAP-mediated activity changes of these enzymes in liver accompanied no protein alterations. In conclusion, knockout of GPX1 or SOD1 exerted differential impact on mouse susceptibility to this low dose of APAP, but neither shifted urinary APAP-cysteine formation.
Keywords: acetaminophen; Cu,Zn-superoxide dismutase; glutathione peroxidase; oxidative stress; protein nitration; toxicity
Introduction
Acetaminophen (APAP) is one of the most widely used analgesic-antipyretic drugs and causes centrilobular liver necrosis, acute liver failure, and death at high doses (1). As probably the best supported mechanism of APAP toxicity, hepatic glutathione (GSH) depletion results from the oxidation of excess APAP into reactive N-acetyl p-benzoquinoneimine (NAPQI) (2–4). This reaction is catalyzed by cytochrome P450 enzyme (CYP2E1), and is accelerated when glucuronidation and sulfation of APAP are saturated (5, 6). While low levels of NAPQI are inactivated by conjugation with GSH in a reaction catalyzed by glutathione S-transferases (GST) (7), high levels of NAPQI deplete cellular GSH, binding to cysteine residues on proteins as 3-(cystein-S-yl)-APAP adducts and inactivating the modified proteins (3, 8).
Reactive oxygen species (ROS), including superoxide anion (O2•) and hydrogen peroxide (H2O2), are produced upon APAP exposure (9). Likewise, APAP has been shown to induce vascular production of peroxynitrite (PN, OONO-), a strong reactive nitrogen species (RNS) formed by the spontaneous reaction of nitric oxide (NO) and O2• (10, 11). Furthermore, Knight et al. (11) have suggested PN formation and the subsequent protein nitration as a mediator of APAP-mediated hepatotoxicity. This notion is supported by the fact that reducing NO and O2• formation protects against the APAP-overdose (12).
However, roles of antioxidant enzymes in APAP overdose remain controversial. Following an intraperitoneal (ip) injection of a lethal dose of APAP (450 mg/kg), mice overexpressing Cu,Zn-superoxide dismutase (EC 1.15.1.1, SOD1) were more resistant, but mice overexpressing Se-dependent cellular glutathione peroxidase-1 (EC 1.11.19, GPX1) were more susceptible than wild-type (WT) mice to the APAP-induced lethality and liver injuries (13). Because knockouts of GPX1 and SOD1 have demonstrated similar impacts on mouse susceptibility to oxidative stress induced by ROS generators such as paraquat and diquat (14–16), the opposite effects of overexpression of these two enzymes on APAP toxicity (13) were intriguing. As SOD1 or GPX1 overexpression does not necessarily represent a physiological status, we have used GPX1 knockout (GPX1−/−), SOD1 knockout (SOD1−/−), and GPX1 and SOD1 double knockout (DKO) mice to determine the impacts of these two enzymes on mouse lethality induced by an ip injection of 600 mg of APAP/kg body weight (17).
Our results indicate that knockout of SOD1 alone or with GPX1 protected mice against the APAP-mediated mouse mortality. While 75% of WT and GPX1−/− mice died within 20 h, all SOD1−/− and DKO mice survived for at least 2 weeks. However, the rather high dose of APAP might preclude a possible protection of GPX1−/− against or a potentially different impact of DKO from SOD1−/− on the hepatotoxicity induced by a lower or a more clinically relevant dose of APAP. Furthermore, mechanistic characterizations of the APAP toxicity in our previous study were limited to comparisons between only the WT and SOD1−/− mice. Body status of NO production was not determined to relate to the induced hepatic protein nitration, whereas hepatic GPX1, SOD1, and GST protein levels were not assayed to compare with their respective activity changes. Therefore, this consecutive experiment was conducted with the four genotypes of mice given an ip injection of 300 mg of APAP/kg to address those unanswered questions.
Materials and Methods
Chemicals and Antibodies.
All chemicals were purchased from Sigma (St. Louis, MO) unless indicated otherwise. Monoclonal antibodies against mouse nitrotyrosine were purchased from Upstate Biotechnology (Lake Placid, NY). Antibodies against mouse IgG were purchased from Pierce (Rockford, IL). Anti-human GPX1 antibody was purchased from Lab Frontier (Seoul, Korea). Antibovine SOD1 antibody was from Chemicon (Temecula, CA), and anti-rat GST-π antibody was from Alpha Diagnostic (San Antonio, TX). Anti-rabbit IgG was purchased from Bio-Rad Laboratories (Hercules, CA).
Mice.
Wild-type, GPX1−/− (18), and SOD1−/− (14) mice with the same genetic background (129/SVJ x C57BL/6), were initially provided by Dr. Y. S. Ho, Wayne State University (Detroit, MI) and were bred in our facility. Mice lacking both GPX1 and SOD1 (DKO) were generated in our laboratory by crossing the individual knockouts. Genotypes were confirmed by PCR and enzyme activity assays. All mice were fed a Torula yeast-based diet (18), given free access to feed and distilled water, and housed in shoebox cages in a constant (22°C) animal room with a 12-h light:dark cycle. All experiments were approved by the Institutional Animal Care and Use Committee at Cornell University and conducted in accordance with NIH guidelines for animal care.
APAP Treatment and Sample Collection.
Male mice (8–12 week old) were given ip injections of phosphate-buffered saline (PBS) or 300 mg of APAP/kg of body weight (prepared in warm PBS) following an overnight (8 h) fast. Mice were euthanized (16) at various time-points (0 to 24 h, n = 5 to 10 per genotype at each time-point) post the injection to collect plasma and tissue samples. To compare the APAP metabolite profiles in the urine of the four genotypes by mass spectrometry (see below), mice (n = 5/genotype) were housed individually in metabolic cages for 24 h after the injection of APAP to collect urine. All samples were frozen in liquid nitrogen and stored at −80°C prior to analysis.
Biochemical Analyses.
Plasma ALT (EC 2.6.1.2) activity was determined following the method outlined by Sigma. Hepatic GSH was measured spectrophotometrically (19). Cellular GPX1 activity was measured using the coupled assay of NADPH oxidation and H2O2 as a substrate (18). Total SOD activity was determined using a water-soluble formazan dye kit (Dojindo Molecular Technologies Inc., Gaithersburg, MD). Hepatic GST activity was measured using 1-chloro-2,4-dinitrobenzene as a substrate (20) Plasma nitrate and nitrite was determined to monitor the APAP-induced NO production (21). Protein was determined as described by Lowry et al. (22). To compare the APAP metabolites profiles in the urine collected for 24 h after the APAP injection among the four genotypes, a 5 μL aliquot of urine sample was injected into a Waters UPLC-QTOFMS system (Mildford, MA) and analyzed as described by Chen et al. (23).
Western Blot Analyses.
Liver samples were homogenized in 50 mM potassium phosphate buffer, pH 7.8, containing 0.1% Triton X-100, 1.34 mM diethylenetri-aminepentaacetic acid, 1 mM PMSF, 10 μg peptstain A/ml, 10 μg leupeptin/ml, and 10 μg aprotinin/ml. The homogenates were centrifuged at 14,000 g for 20 min at 4°C. The supernatant (100, 10, 10, and 20 μg protein per lane for the assay of nitrotyrosine, GPX1, SOD1, and GST-π, respectively) was loaded onto SDS-PAGE (12% gel). The relative amount of the protein was determined using an IS-1000 Digital Imaging System (Alpha Innotech Co., San Leandro, CA).
Statistical Analyses.
Data were analyzed as one way-ANOVA using the GLM procedure of SAS (release 6.11, SAS Institute, Cary, NC). The Bonferroni t test was used for mean comparisons. The significance level was set at P < 0.05 unless indicated otherwise.
Results
The APAP-Induced Plasma ALT Activity Rise Was Abolished in SOD1−/− and DKO Mice.
Compared with the PBS-treated controls, plasma ALT activity was increased by 117-fold (P < 0.05) and 183-fold (P < 0.05) in the APAP-treated WT mice at 5 and 24 h, respectively (Fig. 1A). The increases in the APAP-treated GPX1−/− mice (67- and 91-fold, respectively) were relatively less than those in the WT mice, but the differences between the two genotypes were not statistically significant. In contrast, little rise in plasma ALT was caused by the APAP treatment at either 5 or 24 h in the SOD1−/− or DKO mice.
Figure 1.
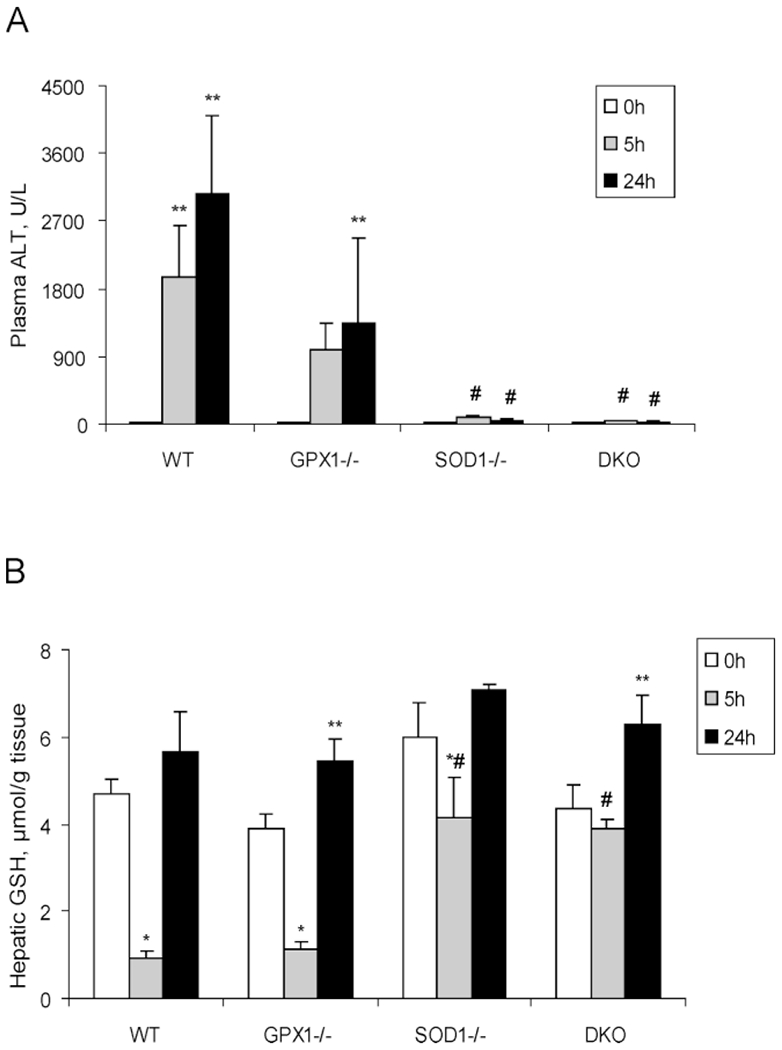
Plasma ALT activities (A) and hepatic GSH concentrations (B) of different genotypes of mice treated with PBS or APAP (300 mg/kg) at various time points. Mice were injected (ip) with PBS and killed immediately after the injection (0 h) or with APAP and euthanized at 5 and 24 h after the injection. In both panels, values are means ± SE (n=5 to 10). Asterisk (*) indicates the APAP-induced decreases (P < 0.05) and ** indicates the APAP-induced increases (P < 0.05) as compared to the PBS-treated controls (0 h) within genotypes, whereas # indicates the genotype effect (P < 0.05) within the same treatment/time-point compared with the WT mice.
The APAP-Induced Hepatic GSH Depletion Was Attenuated in SOD1−/− and DKO Mice.
At 5 h following the APAP injection, hepatic GSH in the WT and GPX1−/− mice was reduced (P < 0.05) by 81% and 72% compared to their initial values, respectively (Fig. 1B). In contrast, the reduction was only 30% in the SOD1−/− mice and negligible in the DKO mice. Thus, hepatic GSH concentrations in the SOD1−/− and DKO mice were 71% to 78% higher (P < 0.05) than in the WT and GPX1−/− mice at 5 h following the APAP injection. In all genotypes, hepatic GSH was replenished to or exceeded the initial levels at 24 h following the APAP injection.
Urine APAP Metabolites Were Different in SOD1−/− Mice.
No significant differences were detected between WT and GPX1−/− or DKO (Table 1) in the relative concentrations of APAP and four major metabolites in the urine collected for 24 h after the APAP administration. However, the percentage of APAP-N-acetylcysteine was higher while the percentage of APAP-glucuronide was lower (P < 0.05) in SOD1−/− urine compared with that in WT.
Table 1.
Effects of Genotype on Relative Peak Area of APAP Metabolites in Urine Collected for 24 h After APAP (300 mg/kg) Injectiona
| WT | GPX1−/− | SOD1−/− | DKO | |
|---|---|---|---|---|
| APAP | 11.3 ± 1.4 | 13.9 ± 3.3 | 9.1 ± 2.6 | 10.8 ± 1.7 |
| APAP-cysteine | 48.9 ± 1.5 | 49.2 ± 2.2 | 46.2 ± 4.9 | 51.4 ± 2.7 |
| APAP-N-acetylcysteine | 2.7 ± 0.8 | 4.8 ± 0.6 | 18.9 ± 7.0* | 4.8 ± 1.5 |
| APAP-glucuronide | 37.0 ± 2.0 | 32.1 ± 3.2 | 25.7 ± 3.3* | 32.9 ± 1.8 |
Values (means ± SE, n = 5) represent the relative percentage of the peak area (total = 100 for each genotype).
indicates P < 0.05 as compared to WT.
The APAP-Induced Hepatic Protein Nitration Was Blocked in SOD1−/− and DKO Mice.
Hepatic protein nitration was induced in the WT and GPX1−/− mice 5 h after the APAP injection, but not in the SOD1−/− or DKO mice (Fig. 2A). At 24 h following the APAP injection, hepatic protein nitration was detectable in only the WT mice (Fig. 2B). In the WT and GPX1−/− mice, plasma nitrate/nitrite concentrations were increased (P < 0.05) over the baseline (0 h) by the APAP treatment at 5 h, followed by decreases at 24 h (Fig. 3). In contrast, plasma nitrate/nitrite in the SOD1−/− or DKO mice showed little response to the APAP treatment. The WT had higher (P < 0.05) plasma nitrate/nitrite concentration at 5 h than that of the other three genotypes of mice.
Figure 2.
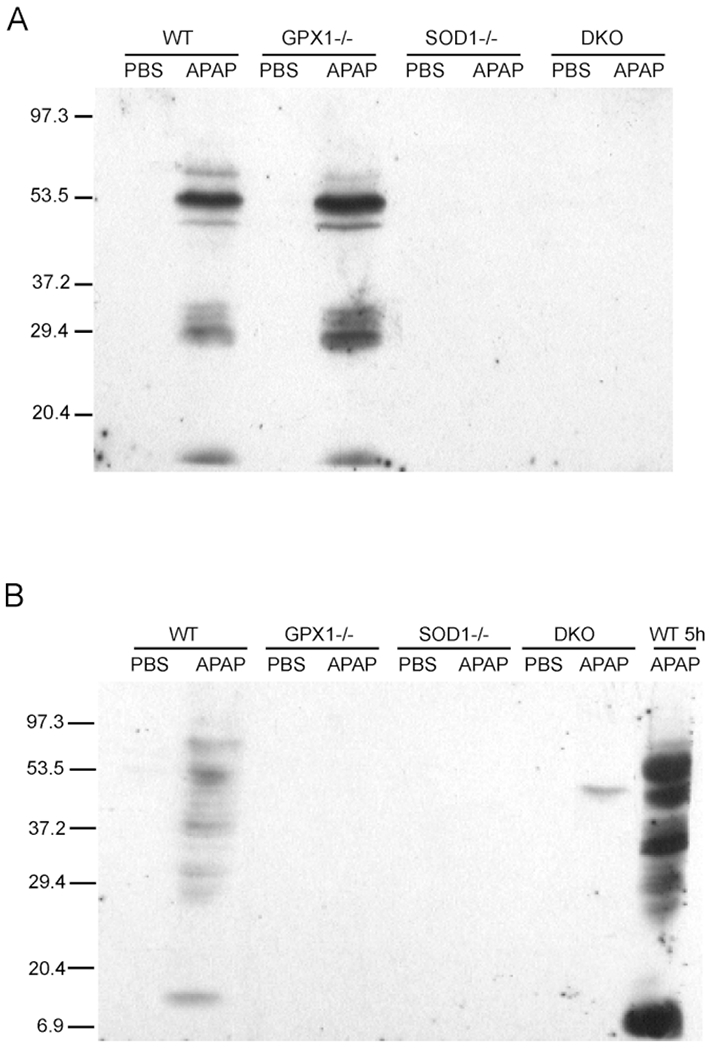
Hepatic protein nitration in different genotypes of mice at 5 h (A) and 24 h (B) following an ip injection of PBS or 300 mg of APAP/kg. Mice were killed at the designated time points, and the Western blot analysis was conducted as described in Materials and Methods. The blot image is a representation of three independent experiments.
Figure 3.
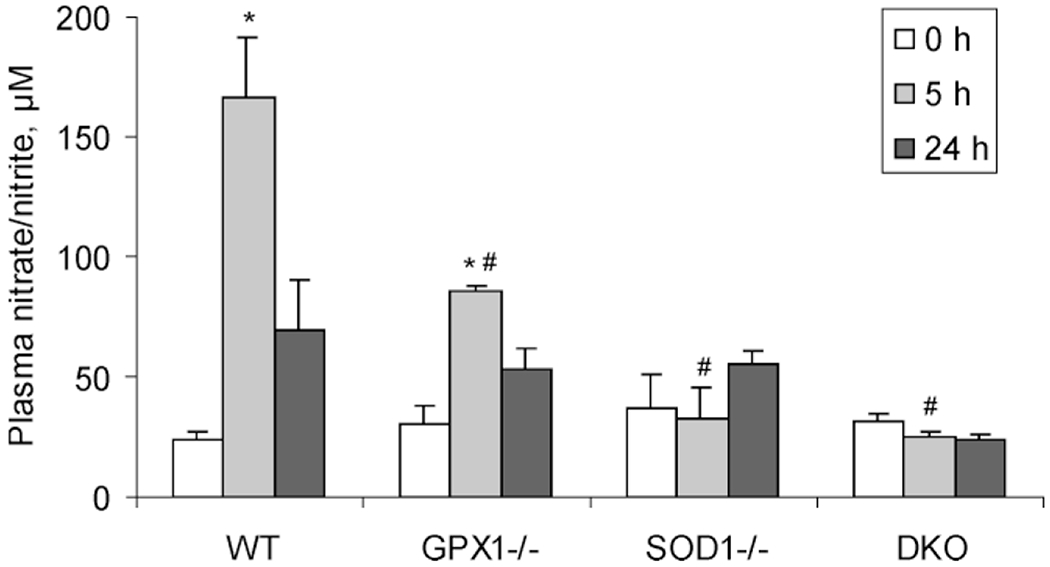
Plasma nitrate/nitrite concentrations in different genotypes of mice at 5 and 24 h following the injection of 300 mg of APAP/kg. The baseline (0 h) values were from mice that were injected (ip) with PBS and killed immediately after the injection. Values are means ± SE (n = 5). Asterisk (*) indicates the APAP-treatment effect (P < 0.05) as compared to the PBS treated controls (0 h) within the same genotype, whereas # indicates the genotype effect (P < 0.05) within the same treatment/time-point compared with the WT mice.
Impacts of the APAP Treatment on Hepatic Antioxidant Enzymes Were Altered by GPX1 and SOD1 Knockouts.
In the WT mice, hepatic GPX1 activity was reduced by 61% (P < 0.05) at 5 h compared with the initial activity (0 h), but elevated by 26% at 24 h (Fig. 4A). SOD activity was gradually decreased over time (Fig. 4B) and the difference between 0 and 24 h was significant (P < 0.05). In the SOD1−/− mice, a lower (P < 0.05) baseline hepatic GPX1 activity was detected compared with the WT mice, but the activity was not changed by the APAP treatment over time. There was no genotype or APAP-treatment effect on hepatic GST activity in SOD1−/− or DKO mice as compared to WT mice (Fig. 4C). In the GPX1−/− mice, APAP treatment resulted in a 47% (P < 0.05) decrease of SOD activity at 5 h, but resumed their initial values at 24 h. Hepatic GST activity was 25% greater (P < 0.05) at 0 h, but 49% lower (P < 0.05) at 5 h than those of the WT mice. However, the APAP-mediated changes in hepatic GPX1, SOD, and GST activities were not accompanied by respective protein alterations of GPX1, SOD1, or GST-π (See Supplement I in the online journal.)
Figure 4.
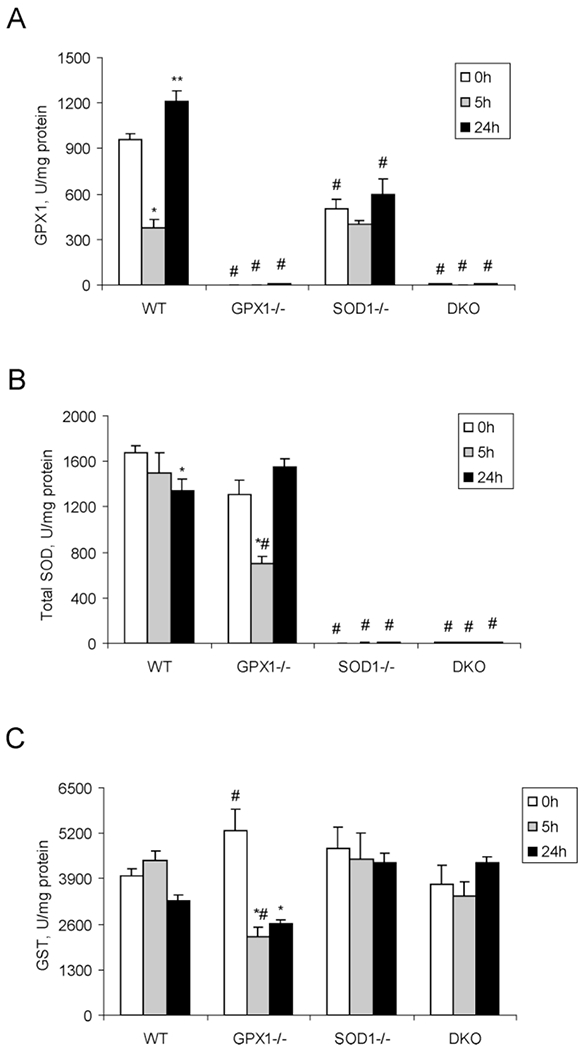
Hepatic activities of GPX1 (A), total SOD (B), and GST (C) in different genotypes of mice treated with PBS or APAP (300 mg/kg) at various time points. Mice were injected (ip) with PBS and killed immediately after the injection (0 h) or with APAP and euthanized at 5 and 24 h after the injection. Values are means ± SE (n = 5 to 10). Asterisk (*) indicates the APAP-induced decreases (P < 0.05) and ** indicates the APAP-induced increases (P < 0.05) as compared to the PBS-treated controls (0 h) within genotypes, whereas indicates the genotype effect (P < 0.05) within the same treatment/time-point compared with the WT mice.
Discussion
Knockout of SOD1 alone or in combination with GPX1 protected mice against the increases in plasma ALT activity, hepatic GSH depletion, and hepatic protein nitration induced by the ip injection of 300 mg APAP/kg. Meanwhile, GPX1 null produced only a modest attenuation of plasma ALT activity increase. Because the genotype differences associated with this low dose of APAP correspond well with our earlier findings with the higher dose (600 mg/kg) (17), the greater protection by the SOD1 deletion than by that of GPX1 against the APAP-induced hepatotoxicity does not depend on the APAP dose.
We have previously discovered the differential regulation of APAP-induced cell death and related signaling exerted by knockout of GPX1 or SOD1 (24). In the present study, we focused on the aspects of APAP metabolism and antioxidant defense to investigate the mechanism of differential responses in GPX1−/−, SOD1−/−, and a newly included DKO mice. In our earlier study with 600 mg APAP/kg (17), we have shown that the protection conferred by the SOD1 deletion was associated with a 50% reduction in activity of CYP2E1, a key APAP metabolism enzyme, and corresponding decreases in plasma and liver concentrations of APAP-cysteine at 5 h following the APAP injection. Indeed, similar responses of CYP2E1 were detected in liver of SOD1−/− and DKO, but not the GPX1−/− mice at 5 h in the present study (See Supplement II in the online journal.) Also concentrations of APAP or APAP-cysteine were decreased in plasma, liver, and urine of the SOD1−/− compared with the WT during the first 5 h after the APAP injection (See Supplement III in the online journal.) However, the relative percentage of urine APAP-cysteine to the total metabolites over the 24-h collection was very similar between the SOD1−/− and the other three genotypes. This observation implies two possibilities. Firstly, the decreased APAP-cysteine concentrations in plasma and liver of the SOD1−/− mice at the early phase (5 h following the injection) were the key for their resistance to APAP toxicity. Secondly, the resistance of SOD1−/− to the APAP toxicity did not correlate to the formation of APAP-cysteine. The second possibility was reported by another group using the CYP2E1 knockout mouse with enhanced resistance to APAP toxicity (23). It is unclear to us how the increased APAP-N-acetylcysteine and the decreased APAP-glucuronide in urine of the SOD1−/− mice affected their resistance to the APAP toxicity. In addition, the DKO mice showed similar resistance or biochemical responses to the APAP overdose, but without such metabolite shifts. Thus, down-regulation of CYP2E1 activity in the SOD1−/− or DKO mice at most might explain partially the attenuated hepatic GSH depletion and liver injury. The involvement of the comprehensive anti-oxidative/oxidative systems, such as GSH, nitration, and antioxidant enzymes, was also likely contributing to the protection in SOD1−/− or DKO mice.
Hepatic protein nitration was induced by the low dose of APAP in the WT and GPX1−/− mice, but not in the SOD1−/− and DKO mice. In our previous study (17, 25), we have found that the presence of SOD1 was necessary for the formation of APAP-induced liver protein nitration. In this study, we illustrated a similarly diminished protein nitration in DKO mice. Apparently, the blocked protein nitration in the DKO mice was most likely due to the SOD1 deletion because GPX1 null showed no effect at 5 h. The blocked protein nitration in the SOD1−/− and DKO mice was consistent with no increase in plasma nitrate/nitrite concentrations in these animals after the APAP treatment. Thus, less NO was available for the spontaneous reaction with O2• to form PN in these mice than in the WT mice. Nevertheless, the loss of SOD1 activity in these two genotypes might be the primary cause for the lack of the induced hepatic protein nitration because SOD1 has been shown to catalyze the PN-mediated protein nitration in in vitro and in vivo systems (25, 26). Interestingly, the APAP-induced hepatic protein nitration at 24 h was undetectable in the GPX1−/− mice and greatly attenuated in the WT mice compared with that at 5 h. This attenuation correlated well with the declines in plasma nitrate/nitrite concentrations and hepatic GSH restoration in the two genotypes. Because plasma ALT activities in these two genotypes still remained high at 24 h, hepatic protein nitration and GSH depletion apparently peaked earlier.
The ability of SOD1 to sensitize liver to APAP-induced protein nitration is of clinical interest in treating not only APAP toxicity, but also other diseases (27–30). The impact of GPX1 null on the APAP-induced protein nitration was somewhat intriguing. Using a cell-free system, Sies et al. (31) reported that GPX1 was a PN reductase. In contrast, our laboratory showed an increased resistance of hepatocytes of GPX1−/− mice to the PN-induced protein nitration and cell death (32). However, results from the present study and Knight et al. (11) showed little effect of GPX1 knockout on the APAP-induced hepatic protein nitration at the early phase, despite a diminishing effect at 24 h.
The systemic activity and protein analyses of GPX, SOD, and GST in the four genotypes of mice at three time points help in understanding the metabolic inter-relationships among these enzymes under the APAP-induced oxidative stress. In the WT mice with full expressions of both enzymes, the APAP treatment caused much greater and quicker alterations of hepatic GPX1 activities than SOD activities. Consistently, cytoplasmic and mitochondrial GPX1 have been identified as protein targets of reactive metabolites of APAP in liver (33). Furthermore, reductions in GPX activity by ROS and RNS treatments have been reported (9, 34). As shown previously (35, 36), hepatic GPX1 activity in the SOD1−/− mice was approximately 50% lower than the WT mice. However, that activity was not further decreased by the APAP-treatment in SOD1−/− mice. Likely, the SOD1 deficiency protected against covalent binding of APAP metabolites to the GPX1 protein (33). The greater decrease in hepatic SOD activity in the GPX1−/− mice, compared with the WT mice, at 5 h post the APAP injection, indicates that the full expression of GPX1 activity was protective against the APAP-induced SOD inactivation. The GPX1−/− mice also had elevated baseline and greater reductions of hepatic GST activities, compared with the WT, which provided alternative mechanisms for the moderate impacts of GPX1 null on APAP toxicity (37). Overall, these changes in antioxidant enzymes may contribute to the differential protection against APAP toxicity in different genotypes. Because there was no genotype difference in APAP treatment effect on protein levels of any of these three enzymes in liver, the observed activity changes were regulated beyond transcription or translation. Instead, it might be related to the functional coordination of intracellular ROS and RNS metabolisms (35).
Taking all our findings from a series of studies into account (17, 24, 25, 38), the decreased CYP2E1 activity, the attenuated GSH depletion, the diminished NO production and the prevention of protein nitration, the reduced cell death-related signaling, and the coordination of antioxidant enzymes may collectively contribute to the protection against APAP toxicity in the SOD1−/− and DKO mice (Fig. 5). These factors most likely overweigh the loss of SOD1 activity and help explain paradoxical results by other groups (13, 39). In comparison, knockout of GPX1 exerted a differential role and affected only part of these factors with a less potency, which consequently led to a moderate protection against the APAP toxicity. While metabolic implications of the increased APAP-N-acetylcysteine and the decreased APAP-glucuronide in urine of the SOD1−/− remain to be explored, our results do not validate the urinary APAP-cysteine formation as a key factor of APAP toxicity (23).
Figure 5.
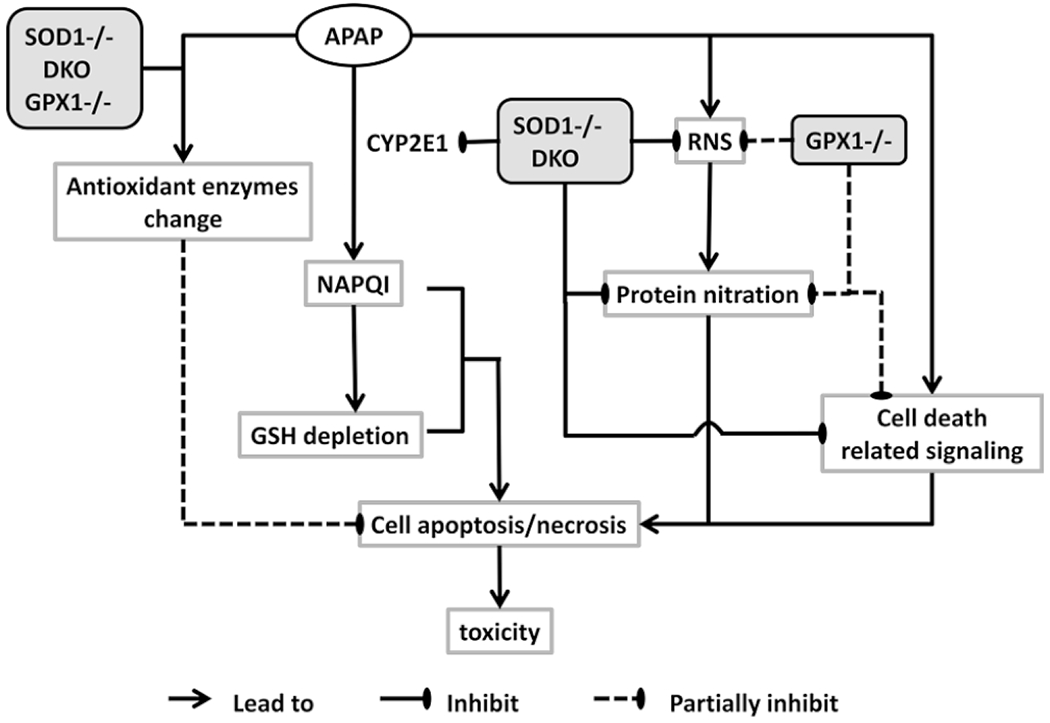
Schematic illustration of the summarized mechanisms against APAP toxicity by knockout of SOD1 and GPX1 alone or in combination.
Acknowledgments
This study was supported by the National Institute of Health grant DK53108 to XGL.
References
- 1.Jollow DJ, Thorgeirsson SS, Potter WZ, Hashimoto M, Mitchell JR. Acetaminophen-induced hepatic necrosis. Pharmacology 12:251–271, 1974. [DOI] [PubMed] [Google Scholar]
- 2.Dahlin DC, Miwa GT, Lu AYH, Nelson SD. N-acetyl-p-benzoquinone imine: a cytochrome P-450-mediated oxidation product of acetaminophen. Proc Natl Acad Sci U S A 81:1327–1331, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hinson JA, Reid AB, McCullough SS, James LP. Acetaminophen-induced hepatotoxicity: role of metabolic activation, reactive oxygen/nitrogen species, and mitochondrial permeability transition. Drug Metab Rev 36:805–822, 2004. [DOI] [PubMed] [Google Scholar]
- 4.Mitchell JR, Jollow DJ, Potter WZ, Gillette JR, Brodie BB. Acetaminophen-induced hepatic necrosis. IV. Protective role of glutathione. J Pharmacol Exp Ther 187:211–217, 1973. [PubMed] [Google Scholar]
- 5.Vermeulen NP, Bessems JG, Van de Straat R. Molecular aspects of paracetamol-induced hepatotoxicity and its mechanism-based prevention. Drug Metab Rev 24:367–407, 1992. [DOI] [PubMed] [Google Scholar]
- 6.Lee SST, Buters JTM, Pineau T, Fernandez-Salguero P, Gonzalez FJ. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J Biol Chem 271:12063–12067, 1996. [DOI] [PubMed] [Google Scholar]
- 7.Henderson CJ, Wolf CR, Kitteringham N, Powell H, Otto D, Park BK. Increased resistance to acetaminophen hepatotoxicity in mice lacking glutathione S-transferase Pi. Proc Natl Acad Sci U S A 97:12741–12745, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roberts DW, Bucci TJ, Benson RW, Warbritton AR, McRae TA, Pumford NR, Hinson JA. Immunohistochemical localization and quantification of the 3-(cystein-S-yl)-acetaminophen protein adduct in acetaminophen hepatotoxicity. Am J Pathol 138:359–371, 1991. [PMC free article] [PubMed] [Google Scholar]
- 9.Lores Arnaiz S, Llesuy S, Cutrin JC, Boveris A. Oxidative stress by acute acetaminophen administration in mouse liver. Free Radic Biol Med 19:303–310, 1995. [DOI] [PubMed] [Google Scholar]
- 10.Knight TR, Kurtz A, Bajt ML, Hinson JA, Jaeschke H. Vascular and hepatocellular peroxynitrite formation during acetaminophen toxicity: role of mitochondrial oxidant stress. Toxicol Sci 62:212–220, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Knight TR, Ho Y-S, Farhood A, Jaeschke H. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther 303:468–475, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Michael SL, Mayeux PR, Bucci TJ, Warbritton AR, Irwin LK, Pumford NR, Hinson JA. Acetaminophen-induced hepatotoxicity in mice lacking inducible nitric oxide synthase activity. Nitric Oxide 5:432–441, 2001. [DOI] [PubMed] [Google Scholar]
- 13.Mirochnitchenko O, Weisbrot-Lefkowitz M, Reuhl K, Chen L, Yang C, Inouye M. Acetaminophen toxicity. Opposite effects of two forms of glutathione peroxidase. J Biol Chem 274:10349–10355, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Ho Y-S, Gargano M, Cao J, Bronson RJ, Heimler I, Hutz RJ. Reduced fertility in female mice lacking copper-zinc superoxide dismutase. J Biol Chem 273:7765–7769, 1998. [DOI] [PubMed] [Google Scholar]
- 15.Fu Y, Cheng WH, Porres JM, Ross DA, Lei XG. Knockout of cellular glutathione peroxidase gene renders mice susceptible to diquat-induced oxidative stress. Free Radic Biol Med 27:605–611, 1999. [DOI] [PubMed] [Google Scholar]
- 16.Cheng WH, Ho Y-S, Valentine BA, Ross DA, Combs GF Jr, Lei XG. Cellular glutathione peroxidase is the mediator of body selenium to protect against paraquat lethality in transgenic mice. J Nutr 128:1070–1076, 1998. [DOI] [PubMed] [Google Scholar]
- 17.Lei XG, Zhu JH, McClung JP, Aregullin M, Roneker CA. Mice deficient in Cu,Zn-superoxide dismutase are resistant to acetaminophen toxicity. Biochem J 399:455–461, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng W-H, Ho Y-S, Ross DA, Valentine BA, Combs GF Jr, Lei XG. Cellular glutathione peroxidase knockout mice express normal levels of selenium-dependent plasma and phospholipid hydroperoxide glutathione peroxidases in various tissues. J Nutr 127:1445–1450, 1997. [DOI] [PubMed] [Google Scholar]
- 19.Anderson ME. Tissue glutathione In: Greenwald RA, Ed. CRC Handbook of Method for Oxygen Radical Research. Boca Raton, FL: CRC Press, pp 317–320, 1985. [Google Scholar]
- 20.Habig WH, Pabst MJ, Jakoby WB. Glutathione S-transferase: the first enzymatic step in mercapturic acid formation. J Biol Chem 249:7130–7139, 1974. [PubMed] [Google Scholar]
- 21.Pollock JS, Forstermann U, Mitchell JA, Warner TD, Schmidt HH, Nakane M, Murad F. Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc Natl Acad Sci USA 88:10480–10484, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275, 1951. [PubMed] [Google Scholar]
- 23.Chen C, Krausz KW, Idle JR, Gonzalez FJ. Identification of novel toxicity-associated metabolites by metabolomics and mass isotopomer analysis of acetaminophen metabolism in wild-type and Cyp2e1-null mice. J Biol Chem 283:4543–4559, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu JH, Zhang X, McClung JP, Lei XG. Impact of Cu, Zn-superoxide dismutase and Se-dependent glutathione peroxidase-1 knockouts on acetaminophen-induced cell death and related signaling in murine liver. Exp Biol Med 231:1726–1732, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Zhu J-H, Zhang X, Roneker CA, McClung JP, Zhang S, Thannhauser TW, Ripoll DR, Sun Q, Lei XG. Role of copper,zinc-superoxide dismutase in catalyzing nitrotyrosine formation in murine liver. Free Radic Biol Med 45:611–618, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys 298:431–437, 1992. [DOI] [PubMed] [Google Scholar]
- 27.Banks BA, Ischiropoulos H, McClelland M, Ballard PL, Ballard RA. Plasma 3-nitrotyrosine is elevated in premature infants who develop bronchopulmonary dysplasia. Pediatrics 101:870–874, 1998. [DOI] [PubMed] [Google Scholar]
- 28.Shishehbor MH, Aviles RJ, Brennan ML, Fu X, Goormastic M, Pearce GL, Gokce N, Keaney JF Jr, Penn MS, Sprecher DL, Vita JA, Hazen SL. Association of nitrotyrosine levels with cardiovascular disease and modulation by statin therapy. JAMA 289:1675–1680, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Ischiropoulos H Biological selectivity and functional aspects of protein tyrosine nitration. Biochem Biophys Res Commun 305:776–783, 2003. [DOI] [PubMed] [Google Scholar]
- 30.Adams ML, Pierce RH, Vail ME, White CC, Tonge RP, Kavanagh TJ, Fausto N, Nelson SD, Bruschi SA. Enhanced acetaminophen hepatotoxicity in transgenic mice overexpressing BCL-2. Mol Pharmacol 60:907–915, 2001. [DOI] [PubMed] [Google Scholar]
- 31.Sies H, Sharov VS, Klotz L-O, Briviba K. Glutathione peroxidase protects against peroxynitrite-mediated oxidations. A new function for selenoproteins as peroxynitrite reductase. J Biol Chem 272:27812–27817, 1997. [DOI] [PubMed] [Google Scholar]
- 32.Fu Y, Sies H, Lei XG. Opposite roles of selenium-dependent glutathione peroxidase-1 in superoxide generator diquat- and peroxynitrite-induced apoptosis and signaling. J Biol Chem 276:43004–43009, 2001. [DOI] [PubMed] [Google Scholar]
- 33.Qiu Y, Benet LZ, Burlingame AL. Identification of the hepatic protein targets of reactive metabolites of acetaminophen in vivo in mice using two-dimensional gel electrophoresis and mass spectrometry. J Biol Chem 273:17940–17953, 1998. [DOI] [PubMed] [Google Scholar]
- 34.Fujii J, Taniguchi N. Down regulation of superoxide dismutases and glutathione peroxidase by reactive oxygen and nitrogen species. Free Radic Res 31:301–308, 1999. [DOI] [PubMed] [Google Scholar]
- 35.Kessova IG, Ho YS, Thung S, Cederbaum AI. Alcohol-induced liver injury in mice lacking Cu, Zn-superoxide dismutase. Hepatology 38: 1136–1145, 2003. [DOI] [PubMed] [Google Scholar]
- 36.Elchuri S, Oberley TD, Qi W, Eisenstein RS, Jackson Roberts L, Van Remmen H, Epstein CJ, Huang TT. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene 24:367–380, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Burk RF, Lane JM. Ethane production and liver necrosis in rats after administration of drugs and other chemicals. Toxicol Appl Pharmacol 50:467–478, 1979. [DOI] [PubMed] [Google Scholar]
- 38.Zhu J-H, Lei XG. Double null of selenium-glutathione peroxidase-1 and copper, zinc-superoxide dismutase enhances resistance of mouse primary hepatocytes to acetaminophen toxicity. Exp Biol Med (Maywood) 231:545–552, 2006. [DOI] [PubMed] [Google Scholar]
- 39.Nakae D, Yamamoto K, Yoshiji H, Kinugasa T, Maruyama H, Farber JL, Konishi Y. Liposome-encapsulated superoxide dismutase prevents liver necrosis induced by acetaminophen. Am J Pathol 136:787–795, 1990. [PMC free article] [PubMed] [Google Scholar]