

Abstract
钠牛磺胆酸共转运多肽(NTCP)缺陷病是由于SLC10A1基因突变,肝细胞基侧膜转运蛋白NTCP的胆汁酸盐摄取功能受损面形成的一种遗传代谢病。该文患儿因发现皮肤巩膜黄染5.5个月(生后第2天出现黄疸)、肝功能异常4月余就诊。肝功能示总胆红素、直接胆红素、间接胆红素和总胆汁酸均明显上升。曾按胆汁淤积性肝病内科治疗,疗效欠佳,于2月龄时行剖腹探查+胆囊造瘘+胆道造影术,术中发现胆汁粘稠但胆道通畅。术后黄疸消退,但转氨酶和总胆汁酸水平逐渐升高。患儿母亲亦发现有轻微高胆汁酸血症。患儿未予特殊治疗,目前已门诊随访两年余,转氨酶逐渐恢复正常,总胆汁酸波动于23.3~277.7 μmol/L。患儿2岁9个月行SLC10A1基因分析,结果证实患儿及其母均为致病性变异c.800C > T(p.S267F)的纯合子,从而确诊NTCP缺陷病。该研究提示,NTCP缺陷病成人患者仅有轻微高胆汁酸血症,但儿科患者胆汁酸升高明显而且持续,且部分病例在婴儿早期可表现为胆汁淤积性黄疸。
Keywords: 胆汁淤积症, NTCP缺陷病, SLC10A1基因, 突变, 儿童
Abstract
Sodium taurocholate cotransporting polypeptide (NTCP) deficiency is caused by SLC10A1 mutations impairing the NTCP function to uptake plasma bile salts into the hepatocyte. Thus far, patients with NTCP deficiency were rarely reported. The patient in this paper was a 5-month-19-day male infant with the complaint of jaundiced skin and sclera for 5.5 months as well as abnormal liver function revealed over 4 months. His jaundice was noticed on the second day after birth, and remained visible till his age of 1 month and 13 days, when a liver function test unveiled markedly elevated total, direct and indirect bilirubin as well as total bile acids (TBA). Cholestatic liver disease was thus diagnosed. Due to unsatisfactory response to medical treatment, the patient underwent exploratory laparotomy, cholecystostomy and cholangiography when aged 2 months. This revealed inspissated bile but unobstructed bile ducts. Thereafter, his jaundice subsided, but the aminotransferases and TBA levels gradually rose. Of note, his mother also had mildly elevated plasma TBA. Since the etiology was unclear, no specific medication was introduced. The infant has been followed up over 2 years. The aminotransferases recovered gradually, but TBA levels fluctuated within 23.3-277.7 μmol/L (reference range:0-10 μmol/L). On SLC10A1 genetic analysis at 2 years and 9 months, both the infant and his mother proved to be homozygous for a pathogenic variant c.800C > T (p.S267F), and NTCP deficiency was thus definitely diagnosed. The findings suggest that, although only mildly increased plasma TBA is presented in adults with NTCP deficiency, pediatric patients with this disorder exhibit persistent and remarkable hypercholanemia, and some patients might manifest as cholestatic jaundice in early infancy.
Keywords: Cholestasis, Sodium taurocholate cotransporting polypeptide deficiency, SLC10A1 gene, Mutation, Child
1. 概述
钠牛磺胆酸共转运多肽(sodium taurocholate cotransporting polypeptide, NTCP)缺陷病是定位于染色体14q24.2的基因SLC10A1发生突变而导致的一种遗传病。SLC10A1基因于1994年被克隆[1],NTCP的分子功能也得以深入研究[2-3]。NTCP是一种表达于肝细胞基侧膜的转运蛋白,主要功能是作为参与胆汁酸肠肝循环的重要载体,将血浆中的结合胆汁酸盐摄取入肝细胞[4-5]。近年研究证实,NTCP还是乙肝/丁肝病毒感染肝细胞的特异性受体[6]。然而,作为一种胆汁酸代谢障碍性疾病,NTCP缺陷病长期以来罕见报道,其遗传方式、表现度和穿透度也不完全清楚。目前有关本病的国外文献仅两篇,分别发表于2015年[7]和2016年[8],而国内文献中尚未见NTCP缺陷病的研究报道。
1.1. 病因与发病机制
作为胆汁中最主要的固体成分,胆汁酸在肝细胞内由胆固醇合成,经胆道系统排入肠腔,在回肠末端,超过90%的胆汁酸盐被重吸收入血,再经过NTCP摄取入肝。以上步骤循环进行,构成胆汁酸的肠肝循环。肝细胞分泌的胆汁酸盐仅有不到10%是由肝细胞从头合成,绝大部分来自再循环池。
NTCP缺陷病由于SLC10A1基因突变,影响NTCP从血浆中摄取胆汁酸盐的功能,导致胆汁酸在血液中大量堆积,形成显著而顽固的高胆汁酸血症。但胆汁酸在肝细胞内的合成、在毛细胆管的跨膜分泌、在肝内外胆道中的流动,在小肠肠腔内生理作用的发挥,以及回肠末端的重吸收等环节均未受到直接影响,因此NTCP缺陷患者除了严重的高胆汁酸血症,其他临床表现可能不甚明显。但是,这种显著的高胆汁酸血症是否可能导致肝脏组织学改变或其它胆汁成份的淤积?患者是否存在生长落后和神经认知问题等肝外表现?其远期临床结局如何?均有待深入研究[9]。
1.2. 临床表现
目前,国内外有关NTCP缺陷病的报道相当有限,因此其临床表现仍有待观察、积累和总结。
2015年,荷兰学者Vaz等[7]报道了国际首例NTCP缺陷病。该患儿是一名5岁阿富汗裔女孩,以1岁前出现的持续而显著的高胆烷血症(hypercholanemia)为突出实验室特点,血清总胆汁酸水平峰值高达1 531 μmol/L(参考值0.8~16.3 μmol/L)、以结合型胆汁酸为主,但肝功正常,除轻微肌张力低下和生长发育落后外,没有胆汁淤积性黄疸、瘙痒、脂肪泻等临床表现;SLC10A1基因测序发现患儿系突变c.755G > A(p.R252H)的复合杂合子。该患儿的确诊,确定了长期以来理论推测的NTCP在肝细胞清除血浆胆汁酸中的主要作用,支持肝细胞摄取胆汁酸的其它替代机制作用非常有限的观点,同时深化了对于肝脏胆汁酸合成调节机制的认识,并提示结合胆汁酸以外的其它因素(如溶血磷脂酸)才是胆汁淤积症患者瘙痒的原因[9]。
2016年,本课题组报道了国际第二例NTCP缺陷病患儿和首例成人患者[8]。该患儿2岁半时经SLC10A1基因分析,并经全基因组测序排除其它遗传学病因后确诊本病,以显著而顽固的高胆汁酸血症(峰值达738.7 μmol/L)为突出特征。该患儿除了轻微维生素D缺乏、尘螨性皮炎和婴儿早期一过性胆汁淤积性黄疸,未发现其它临床症状体征;成人患者为一30岁女性,仅有轻微血清胆汁酸升高(19.3 μmol/L),也缺乏阳性临床表现;两名患者均为SLC10A1基因变异c.800C > T(p.S267F)的纯合子。
1.3. 诊断与鉴别诊断
NTCP缺陷病儿童患者往往缺乏明显的临床症状体征,而以血清胆汁酸升高为主要实验室改变。但是,部分患儿在婴儿期,尤其是新生儿期表现为胆汁淤积性黄疸,往往难以与其它遗传性胆汁淤积症区分开来,个别患儿甚至因此接受剖腹探查,或胆道造影/冲洗等创伤性检查/治疗。
本病婴儿早期/新生儿期要与单纯G6PD缺陷病和/或地中海贫血等所致的高胆红素血症相鉴别,婴幼儿则应与其他胆汁淤积症相区分。但G6PD缺陷病和地中海贫血所致的高胆红素血症以间接胆红素增高为主。NTCP缺陷病患儿SLC10A1基因分析可发现致病性突变,生化检查胆汁酸升高明显而持续,且与其它肝功指标变化趋势不同步、不平行,这是其有别于其它胆汁淤积症的显著特征。必要时可通过二代测序等手段,与其它遗传性胆汁淤积症相鉴别。
1.4. 治疗与预后
NTCP缺陷病目前缺乏特异性治疗手段,在密切随访基础上给予对症支持治疗是当前主要管理措施。本病患儿高胆汁酸血症显著而持续,但除了部分患儿婴儿早期黄疸,未发现其它明确与高胆汁酸血症相关的明显症状、体征,因此,本病的高胆汁酸血症是否需要积极处理,以及是否有必要开展产前诊断等等问题,均有待进一步研究总结。
2. 诊断要点
(1)最突出的特征是缺乏瘙痒和肝大等胆汁淤积性肝病的表现,而血浆总胆汁酸显著性、持续性升高(以结合型为主),且与其他肝功指标变化趋势不同步、不平行。(2)部分患者在婴儿期尤其是新生儿期表现为胆汁淤积性黄疸,即总胆红素 > 85 μmol/L,且直接胆红素占总胆红素比例超过20%[10]。(3)成人期除了轻微高胆汁酸血症,可能缺乏其它临床表现。(4)SLC10A1基因分析发现双等位基因致病突变是最可靠的确诊依据。
3. 病例报告
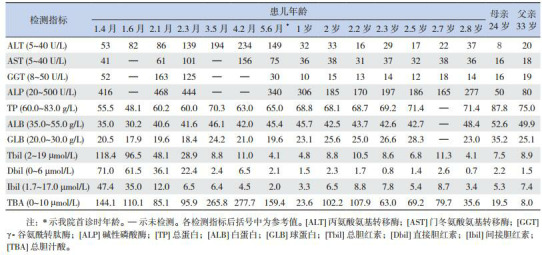
患儿,男,5个月19天,因发现皮肤巩膜黄染5.5个月(生后第2天即出现黄染)、肝功异常4月余就诊。患儿新生儿期即出现黄染,经光疗后好转,出院后黄疸一直未消退。生后1个月13天时因皮肤巩膜黄染、大便色浅、小便深黄就诊于当地医院,查肝功能(表 1)发现总胆红素118.4 μmol/L(参考值:2~19 μmol/L),直接胆红素71.0 μmol/L(参考值:0~6 μmol/L), 间接胆红素47.4 μmol/L(参考值:1.7~17.1 μmol/L),总胆汁酸144.1 μmol/L(参考值:0~10 μmol/L)。以“胆汁淤积症”收住院。住院期间经G6PD酶活性分析、地中海贫血基因突变筛查(阴性),诊断为G6PD缺陷病。予还原型谷胱甘肽护肝、益生菌调整肠道微生态等治疗,黄疸无消退、肝功能无好转。2月龄时行剖腹探查+胆囊造瘘+胆道造影术,术中发现肝脏呈淤胆样改变、表面未见明显结节、质地软,胆囊充盈欠佳、内积少量粘稠胆汁;造影显示胆道通畅。诊断“浓缩胆汁综合征”。术后黄疸逐渐消退,总胆红素和直接胆红素逐渐降至正常,但转氨酶和总胆汁酸水平反而进行性升高(表 1)。为明确病因转我院。发病以来食欲、睡眠可,无皮肤瘙痒症状。
1.
患儿肝功能动态变化及其父母肝功能结果
|
患儿系第一胎第一产,足月顺产,出生体重3.4 kg,身长50 cm。否认乙肝等传染病接触史。父母体健,非近亲婚配,否认遗传病家族史。其母体检发现肝功能总胆汁酸19.5 μmol/L(参考值:0~10 μmol/L),余项正常;其父肝功能各项指标正常,见表 1。
体格检查:头围41.5 cm,体重7.5 kg,身长67.5 cm。皮肤巩膜无黄染。头颅五官无畸形。双肺呼吸音清,心音有力,各瓣膜区未闻及病理性杂音。右上腹壁可见一约3 cm长手术疤痕,腹平软,肝右肋下2 cm,质软,脾肋下未触及。四肢肌张力正常,腹壁、膝腱和跟腱反射可引出,克、布、巴氏征均阴性。
辅助检查:肝功能(表 1)丙氨酸氨基转移酶、天冬氨酸氨基转移酶和总胆汁酸升高,余项正常。血清锌8.8 μmol/L(参考值:11.5~25.5 μmol/L)。
25-羟维生素D 23.7 ng/mL(参考值:30~100 ng/mL)。
尿液气相色谱-质谱法分析及纸片血串联质谱检测均未发现特定遗传代谢病依据。SLC25A13基因高频突变筛查未检测到c.851_854del4,c.1638-1660dup,IVS6+5G > A和IVS16ins3kb等突变类型,不支持希特林缺陷病。
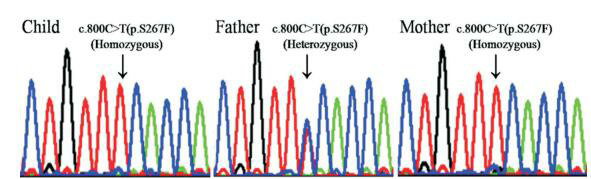
诊断:1.胆汁酸、转氨酶升高查因;2.G6PD缺陷病;3.锌缺乏症;4.维生素D缺乏症。予维生素D和锌剂治疗,并定期门诊随访。目前随访已2年余,维生素D缺乏和锌缺乏逐渐纠正;转氨酶亦逐渐恢复正常,总胆汁酸虽呈下降趋势,但一直高于正常(表 1)。患儿2岁9个月时行SLC10A1基因分析,证实患儿及其母亲均为变异c.800C > T(p.S267F)的纯合子,而其父为携带者(图 1)。该变异已被证实为一个严重影响NTCP功能的致病性SNP[2, 8, 11]。因此,母子二人均确诊患有NTCP缺陷病。
1.
患儿及其父母SLC10A1基因测序图
患儿及其母亲均为变异c.800C > T (p.S267F) 的纯合子,而其父为该变异携带者。
4. 诊断思维
本文患儿及其母亲分别为国内首次报道的NTCP缺陷病儿童和成人患者。患儿以新生儿期早发性、迁延性黄疸为主要临床特征,符合肝内胆汁淤积症的黄疸特点。黄疸消退后高胆汁酸血症仍持续存在,但无瘙痒症状。患儿胆汁酸升高与其他肝功指标变化趋势不同步、不平行的特点,强烈提示NTCP缺陷病。在此基础上,经SLC10A1基因分析,NTCP缺陷病最终确诊。其母仅有轻微胆汁酸升高,缺乏阳性症状、体征。本研究及国外文献[7-8]所报道的NTCP缺陷病患者,均为SLC10A1基因致病性变异的纯合子或复合杂合子,而携带者血清胆汁酸水平正常,符合常染色体隐性遗传病特征。
本文患儿胆汁淤积性黄疸发生机制不明,可能与NTCP缺陷影响肝细胞胆红素摄取有关。有机阴离子转运多肽(organic anion transporting polypeptides, OATPs)是一个包含多种蛋白质的超级家族,其中两个成员OATP1B1(OATP-C)和OATP1B3(OATP8)以异二聚体形式表达在肝细胞基侧膜,主要负责摄取血浆胆红素,包括直接和间接胆红素,尤其是直接胆红素,同时也具备胆汁酸摄取功能[12]。因此,NTCP缺陷病患儿明显升高的血浆胆汁酸将竞争性抑制OATP1B1/OATP1B3摄取胆红素的功能,导致胆红素(尤其直接胆红素)继发性升高,不仅可形成胆汁淤积症的生化改变,而且可引起或加重原发病(如本文患儿的G6PD缺陷病)黄疸程度,尤其是在胆红素来源丰富而代谢、排泄能力不足的新生儿期。胆汁淤积症致病基因种类繁多,病因诊断流程复杂,治疗具有一定难度。本文患儿2月龄时接受了剖腹探查+胆囊造瘘+胆道造影术,病因仍未明确。因此,在疑诊NTCP缺陷病的患儿中开展SLC10A1基因分析,可避免一些不必要的检查和治疗,尤其是创伤性操作。
另一方面,虽然NTCP是肝细胞摄取血浆胆汁酸的主要转运蛋白,但是肝细胞基侧膜存在胆汁酸摄取的替代分子机制。首先,OATP1B1/OATP1B3异二聚体摄取和清除血浆中多种兼性生物质,包括胆酸、甘胆酸和牛磺胆酸等胆汁酸盐[12]。其次,另一种表达于肝细胞膜的异二聚体OSTα/OSTβ是双向有机溶质转运系统,也可以摄取胆汁酸[13]。另外,双功能蛋白mEH也可定向表达于肝细胞基侧膜并具有一定的胆汁酸摄取功能[14]。由于上述胆汁酸摄取替代分子机制的存在和发育成熟,NTCP缺陷病患儿的胆汁酸可能不会无限制升高,甚至可随年龄增长而呈下降趋势,到成人期接近正常[7-8]。本文患儿胆汁酸在4月龄后出现下降趋势,而其母胆汁酸仅轻微升高,支持以上观点。
值得注意的是,SLC10A1基因c.800C > T(p.S267F)的等位基因频率在广州小样本健康志愿者中为4.7%(7/150)[8],在广州另一医院的健康志愿者中高达10.9%(400/3656)[15],而在中国人群中也达到7.4%(23/312)[3]。另外,本课题组近期研究发现,除了c.800C > T(p.S267F),SLC10A1基因还存在其它致病性突变类型(待发表)。以上发现提示,NTCP缺陷病在我国,至少在广东人群中可能并不罕见,值得关注。而本病长期以来罕见报道,可能与婴儿早期的临床表现缺乏特异性,而成人期仅有轻微实验室异常有关。
5. 评论
NTCP缺陷病是新近发现的一种以儿童期显著而持续的高胆汁酸血症为主要特征的遗传代谢病,在我国可能并不罕见。本文结果提示,NTCP缺陷病可能是造成婴儿早期胆汁淤积性黄疸的重要原因之一。在疑诊患儿中积极开展SLC10A1基因突变分析,有助于患儿及时确诊,并避免一些不必要的检查和治疗。但是,本病是否有肝外表现?患者远期结局如何?是否有必要进行产前诊断?诸多问题,均有待今后深入研究总结。
Biography
宋元宗, 男, 主任医师, 教授。Email:songyuanzong@vip.tom.com
Funding Statement
国家自然科学基金(81270957,81570793)
References
- 1.Hagenbuch B, Meier PJ. Molecular cloning, chromosomal localization, and functional characterization of a human liver Na+/bile acid cotransporter. J Clin Invest. 1994;93(3):1326–1331. doi: 10.1172/JCI117091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ho RH, Leake BF, Roberts RL, et al. Ethnicity-dependent polymorphism in Na+-taurocholate cotransporting polypeptide (SLC10A1) reveals a domain critical for bile acid substrate recognition. J Biol Chem. 2004;279(8):7213–7222. doi: 10.1074/jbc.M305782200. [DOI] [PubMed] [Google Scholar]
- 3.Pan W, Song IS, Shin HJ, et al. Genetic polymorphisms in Na+-taurocholate co-transporting polypeptide (NTCP) and ileal apical sodium-dependent bile acid transporter (ASBT) and ethnic comparisons of functional variants of NTCP among Asian populations. Xenobiotica. 2011;41(6):501–510. doi: 10.3109/00498254.2011.555567. [DOI] [PubMed] [Google Scholar]
- 4.Hagenbuch B, Dawson P. The sodium bile salt cotransport family SLC10. Pflugers Arch. 2004;447(5):566–570. doi: 10.1007/s00424-003-1130-z. [DOI] [PubMed] [Google Scholar]
- 5.Anwer MS, Stieger B. Sodium-dependent bile salt transporters of the SLC10A transporter family:more than solute transporters. Pflugers Arch. 2014;466(1):77–89. doi: 10.1007/s00424-013-1367-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yan H, Zhong G, Xu G, et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vaz FM, Paulusma CC, Huidekoper H, et al. Sodium taurocholate cotransporting polypeptide (SLC10A1) deficiency:conjugated hypercholanemia without a clear clinical phenotype. Hepatology. 2015;61(1):260–267. doi: 10.1002/hep.27240. [DOI] [PubMed] [Google Scholar]
- 8.Deng M, Mao M, Guo L, et al. Clinical and molecular study of a pediatric patient with sodium taurocholate cotransporting polypeptide deficiency. Exp Ther Med. 2016;12(5):3294–3300. doi: 10.3892/etm.2016.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karpen SJ, Dawson PA. Not all (bile acids) who wander are lost:the first report of a patient with an isolated NTCP defect. Hepatology. 2015;61(1):24–27. doi: 10.1002/hep.27294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrianov MG, Azzam RK. Cholestasis in infancy. Pediatr Ann. 2016;45(12):e414–e419. doi: 10.3928/19382359-20161118-01. [DOI] [PubMed] [Google Scholar]
- 11.Yan H, Peng B, Liu Y, et al. Viral entry of hepatitis B and D viruses and bile salts transportation share common molecular determinants on sodium taurocholate cotransporting polypeptide. J Virol. 2014;88(6):3273–3284. doi: 10.1128/JVI.03478-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Biochim Biophys Acta. 2003;1609(1):1–18. doi: 10.1016/s0005-2736(02)00633-8. [DOI] [PubMed] [Google Scholar]
- 13.Ballatori N, Christian WV, Lee JY, et al. OST alpha-OST beta:a major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology. 2005;42(6):1270–1279. doi: 10.1002/hep.20961. [DOI] [PubMed] [Google Scholar]
- 14.Zhu QS, Xing W, Qian B, et al. Inhibition of human m-epoxide hydrolase gene expression in a case of hypercholanemia. Biochim Biophys Acta. 2003;1638(3):208–216. doi: 10.1016/S0925-4439(03)00085-1. [DOI] [PubMed] [Google Scholar]
- 15.Peng L, Zhao Q, Li Q, et al. The p.Ser267Phe variant in SLC10A1 is associated with resistance to chronic hepatitis B. Hepatology. 2015;61(4):1251–1260. doi: 10.1002/hep.27608. [DOI] [PubMed] [Google Scholar]