

Abstract
非酮性高甘氨酸血症(NKH)是一种罕见的先天性遗传代谢性疾病,该文报道1例GLDC基因突变所致NKH的中国患儿,就其临床经过、基因缺陷进行研究。患儿以早发性代谢性脑病以及大田原综合征起病,血、尿串联质谱分析均未见异常,颅脑MRI提示胼胝体发育欠佳,脑电图提示爆发抑制。目标基因捕获下代测序结合多重连接探针扩增发现,患儿存在GLDC基因的母源外显子15 c.1786 C > T(p.R596X)杂合无义突变及父源外显子4-15大片段杂合缺失,均为明确致病突变,确诊为NKH。经过促肾上腺皮质激素、托吡酯、右美沙芬治疗后,患儿病情无好转,4月龄死亡。NKH临床表型复杂,可通过代谢筛查以及分子遗传学分析获得确诊。
Keywords: 非酮性高甘氨酸血症, 大田原综合征, GLDC基因, 基因突变, 婴儿
Abstract
Nonketotic hyperglycinemia (NKH) is a rare, inborn error of metabolism. In this case report, a Chinese male infant was diagnosed with NKH caused by GLDC gene mutation. The clinical characteristics and genetic diagnosis were reported. The infant presented with an onset of early metabolic encephalopathy and Ohtahara syndrome. Both blood and urinary levels of metabolites were in the normal range. Brain MRI images indicated a poor development of corpus callosum, and a burst suppression pattern was found in the EEG. Results of target gene sequencing technology combined with multiplex ligation-dependent probe amplification (MLPA) indicated a heterozygous missense mutation of c.1786 C > T (p.R596X) in maternal exon 15 and a loss of heterozygosity of 4-15 exon gross deletions in paternal GLDC gene. These definite pathogenic mutations confirmed the diagnosis of NKH. The infant's clinical condition was not improved after treatment with adreno-cortico-tropic-hormone, topiramate and dextromethorphan, and he finally died at 4 months of age. Patients with NKH often exhibit complicated clinical phenotypes and are lack of specific symptoms. NKH could be diagnosed by metabolic screening and molecular genetic analysis.
Keywords: Nonketotic hyperglycinemia, Ohtahara syndrome, GLDC gene, Gene mutation, Infant
非酮性高甘氨酸血症(nonketotic hyperglycinemia, NKH, MIM #605899)为常染色体隐性遗传,为一种罕见的先天性遗传代谢性疾病,据估计其发病率为1/250 000[1]。但加拿大西部发病率约为1/63 000,芬兰北部为1:12 000[2];我国大陆地区发病率不详、仅有1例报道[3],台湾地区发生率7.2:1 000 000[4]。NKH的特征性表现为血和脑脊液甘氨酸增高伴难治性癫癎、肌张力低下、发育迟缓,是早发性癫癎性脑病的重要原因。随着基因测序技术的发展和应用,目前已经报道的NKH致病基因主要包括AMT(MIM 238310)、GLDC(MIM 238300)和GCSH(MIM 238330)。另有文献报道,GLRX5(MIM 609588)、BOLA3(MIM 613183)和LIAS(MIM 607031)基因缺陷也可引起本病[5]。其中GLDC基因突变所致的占70%~75%。至今全球共报道210种GLDC基因缺陷导致的NKH,我国尚未见文献报道,本文就首例中国GLDC基因突变所致NKH患儿的临床诊疗经过、基因突变特点进行研究。
1. 资料与方法
1.1. 病例介绍
患儿,男,28 d,第1胎第1产,足月顺产出生,出生体重3 120 g,出生时羊水、脐带正常,无窒息史。患儿生后11 h因精神反应差、纳差住院。查体:体温正常、呼吸稍促、节律规整、嗜睡、强刺激后哭声低、肌张力减低,余无特殊。实验室检查:肝、肾功能,血氨、乳酸未见异常;TORCH阴性;血、尿串联质谱分析未见异常;颅脑MRI提示胼胝体发育欠佳,双侧内囊、中脑、脑桥背侧弥散像高信号。住院期间出现呼吸衰竭,无发热、无哭声尖直及抽搐,予呼吸机辅助通气等治疗后呼吸好转出院。出院后于12天龄出现抽搐,为频繁的点头抱团样发作。28天龄时因反复抽搐16 d入院。查体:体重5.2 kg,发育正常,营养可,神清,反应欠佳,对声光刺激反应弱,面容、皮肤、毛发无特殊,无特殊体味,呼吸平稳,双瞳等大等圆、对光反射存在,心肺无异常,肝脾未触及,四肢肌张力低,余神经系统查体未见异常。实验室检查:肝肾功能,血氨、乳酸未见异常;血、尿串联质谱分析无异常;脑电图提示爆发-抑制。诊断大田原综合征。予促肾上腺皮质激素肌注,每次25 IU,每日1次,治疗4周,无效;换用托吡酯,每次12.5 mg,2次/日,治疗4周,发作较前减少50%~60%。复查脑电图仍为爆发-抑制。3月龄时基因诊断明确,加用右美沙芬,每次10 mg,3次/日,治疗1周,效果不佳,4月龄死亡。患儿父母体健、非近亲结婚,家族中无遗传病史可寻。
1.2. 分子遗传学分析
签署知情同意书,分别采集患儿及父母静脉血5 mL(EDTA抗凝),提取DNA。并构建DNA文库。
测序原始数据用NextGene Ⅴ2.3.4软件与UCSC数据库提供的人类基因组hg19参考序列进行比对。通过人类基因突变数据库HGMD、千人基因组数据库、dbSNP数据库信息对检出的变异位点进行注释,并用SIFT、PolyPhen2等软件预测单核苷酸改变对蛋白功能的影响。
Sanger测序验证:对患儿突变区域进行Sanger测序验证,并进行家系传递分析。GLDC基因的15号外显子测序引物:5'-GCCAACCATACCTGAA GCAAC-3 ',5'-TGTGAGGGAGAACACGATGC-3'(上海生工合成),产物长度633 bp。PCR扩增。单一条带者可进行Sanger测序。
采用多重连接探针扩增(multiplex ligationdependent probe amplification, MLPA)试剂盒P209检测GLDC基因,并以正常DNA作对照,数据采用Coffalyser软件分析。
2. 结果
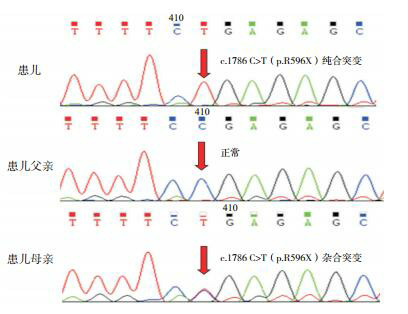
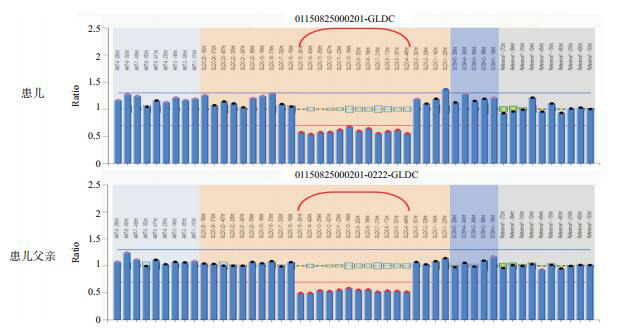
目标基因捕获下代测序发现,患儿GLDC基因外显子15存在c.1786 C>T(p.R596X)纯合无义突变,并经Sanger测序验证(图 1);其父母同片段序列分析显示,母亲存在c.1786C>T(p.R596X)杂合突变,而父亲正常。考虑患儿可能存在父源性大片段缺失,MLPA证实患者及其父亲均存在GLDC基因外显子4-15区域大片段杂合缺失(图 2)。该两种突变均为明确致病突变。
1.
患者GLDC基因突变验证及其父母的一代测序
患儿存在c.1786 C>T(p.R596X)纯合突变(突变位点如箭头所示);患儿父亲正常;患儿母亲存在c.1786 C>T(p.R596X)杂合突变(箭头所示)。
2.
患者及其父亲GLDC基因的MLPA验证
患儿及其父亲均存在外显子4-15的缺失(括号内示缺失位点)。
3. 讨论
NKH是一种罕见的遗传代谢性疾病,为常染色体隐性遗传,是由甘氨酸裂解酶系统(glycinelyase system, GCS)缺陷使甘氨酸堆积,直接导致中枢神经系统受累,故称为甘氨酸脑病(glycine encephalopathy, GCE)[6]。甘氨酸是人体非必需氨基酸,在体内代谢主要是通过GCS进行。GCS存在于肝、肾、脑和胎盘的线粒体膜上,由4个蛋白(P,H,T和L)组成。Kanno等[7]对56例新生儿型NKH患儿行基因检测,证实位于9p24的GLDC基因缺陷是NKH的主要病因,而GLDC为编码GCS中P蛋白的基因。目前已知NKH中70%~75%是由GLDC基因缺陷所致,20%由AMT基因缺陷所致,GCSH基因缺陷所占比例 < 1%[8]。
NKH的临床表现不一,分为经典型和非经典型。经典型即新生儿型,常于生后48 h内呈进行性脑病表现,如反应低下、嗜睡,肌张力减低、呕吐、肌阵挛癫癎、呃逆或呼吸暂停,多数需要呼吸机支持,约30%在新生儿期死亡,大部分在1岁以内死亡,幸存者多存在严重脑发育障碍和难治性癫癎[9-10]。非经典型包括6月龄左右发病的婴儿型和2~33岁发病的晚发型。婴儿型酷似新生儿型,但症状较轻,智力低下不如新生儿型显著;晚发型轻症表现为轻度智力低下、癫癎或舞蹈手足徐动症,重症表现为肺动脉高血压、进行性痉挛性瘫痪、视神经萎缩[11]。NKH的诊断除依据临床表现,还需满足下列条件:血和脑脊液甘氨酸增高,脑脊液/血甘氨酸比值>0.08,无酮症酸血症,尿有机酸正常;进一步确诊需行肝穿检查GCS活性以明确何种蛋白缺陷,或进行基因检测。
本例患儿生后12 d出现点头、抱团样发作,脑电图显示爆发-抑制,符合大田原综合征诊断。大田原综合征病因众多,以大脑结构异常最常见,NKH、吡哆醇依赖症、细胞色素C氧化酶缺乏等代谢异常也是重要病因。该患儿生后11 h即有嗜睡、昏迷、肌张力减低以及呼吸暂停之后难以控制的惊厥发作,考虑存在早发性代谢性脑病可能。先天代谢紊乱是本类疾病最常见的原因,结合患儿无宫内缺氧窒息史,无宫内感染,无围产期异常证据,多次血、尿串联质谱分析无异常,考虑特殊类型的遗传代谢病可能性大。本例患儿血甘氨酸不高,虽未查脑脊液甘氨酸,但是综合临床以及分子遗传学结果,明确患儿为GLDC基因突变所致NKH患者。
本例患者进行了目标基因捕获下代测序,发现GLDC基因外显子15存在c.1786 C>T(p.R596X)纯合无义突变,并经Sanger测序进行了验证。但传递分析发现,患者母亲携带GLDC基因c.1786 C>T(p.R596X)杂合无义突变,而父亲正常。鉴于已报道的GLDC基因突变类型中约20%为大片段缺失或重复,考虑患者父源等位基因存在大片段缺失的可能性较大,且该缺失涵盖外显子15区域,从而导致一代测序及Sanger测序仅检测到c.1786 C>T纯合突变。经MLPA证实,患者及父亲GLDC基因均存在外显子4-15大片段杂合缺失,即患儿实为母源GLDC基因c.1786 C>T(p.R596X)杂合突变及父源GLDC基因外显子4-15大片段杂合缺失。本例患者GLDC基因的突变特点应引起重视,对于高度疑似的NKH病例,如果仅检测到GLDC基因一个杂合突变,应进一步行MLPA检测以明确其他区域是否存在大片段缺失或重复。
迄今为止,NKH仍缺乏有效的治疗手段,低甘氨酸饮食并不能改善症状和预后[12];而且需要注意慎用丙戊酸钠,因为丙戊酸钠可干扰肝脏甘氨酸裂解酶系统而导致血甘氨酸浓度增高,加重病情[13]。
尽管NKH发病率很低,但我国人口众多,应该有相当数量的NKH病例,而仅见个别文献报道,这可能与目前国内大部分地区无法检测脑脊液甘氨酸水平有关;另外,可能与部分新生儿病情变化迅速,在确诊前已死亡有关。NKH缺乏特异的临床表现,因此如果发现新生儿反应低下、嗜睡、肌张力减低、呕吐、惊厥、呃逆或呼吸暂停等症状,要考虑早发性代谢性脑病的可能,及早进行血、尿串联质谱分析等一系列相关检查,并尽可能进行脑脊液的代谢物检测。
本研究首次报道1例GLDC基因突变所致以大田原综合征为主要表现的NKH患儿,患儿经目标基因捕获测序并经Sanger法DNA测序以及MLPA,证实为GLDC基因母源c.1786 C>T(p.R596X)杂合无义突变和父源外显子4-15区域大片段杂合缺失,明确了该家系的致病基因突变。通过本研究希望能使儿科医师及神经科医师提高对NKH的认识,为NKH的早期诊断提供理论基础。
志谢
感谢北京信诺佰世医学检验所徐忠尧博士对本研究的大力协助。感谢上海诺华贸易有限公司在本研究病例资料收集中给予的支持。
Biography
高志杰, 男, 硕士研究生, 主治医师
References
- 1.Applegarth DA, Toone JR, Lowry RB. Incidence of inborn errors of metabolism in British Columbia, 1969-1996. Pediatrics. 2000;105(1):e10. doi: 10.1542/peds.105.1.e10. [DOI] [PubMed] [Google Scholar]
- 2.von Wendt L, Hirvasniemi A, Similä S. Nonketotic hyperglycinemia. A genetic study of 13 Finnish families. https://www.researchgate.net/publication/22696141_Nonketotic_hyperglycinemia_A_genetic_study_of_13_Finnish_families. Clin Genet. 1979;15(5):411–417. doi: 10.1111/j.1399-0004.1979.tb01773.x. [DOI] [PubMed] [Google Scholar]
- 3.张 蓉, 陈 超, 曹 云, et al. 新生儿非酮症性高甘氨酸血症1例. http://www.cnki.com.cn/Article/CJFDTOTAL-XZEK200902027.htm 中国循证儿科杂志. 2009;4(2):153–155. [Google Scholar]
- 4.Chiu CF, Lin JL, Lin JJ, et al. Nonketotic hyperglycinemia of infants in Taiwan. https://www.researchgate.net/profile/Jainn_Jim_Lin/publication/292677887_Nonketotic_Hyperglycinemia_of_Infants_in_Taiwan/links/5720d9cf08aead26e721325d.pdf?origin=publication_detail. Pediatr Neonatol. 2016;S1875-9572(16):21–28. doi: 10.1016/j.pedneo.2015.10.008. [DOI] [PubMed] [Google Scholar]
- 5.Baker 2nd PR, Friederich MW, Swanson MA, et al. Variant nonketotic hyperglycinemia is caused by mutations in LIAS, BOLA3 and the novel gene GLRX5. https://www.researchgate.net/publication/259319338_Variant_non_ketotic_hyperglycinemia_is_caused_by_mutations_in_LIAS_BOLA3_and_the_novel_gene_GLRX5. Brain. 2014;137(Pt 2):366–379. doi: 10.1093/brain/awt328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iqbal M, Prasad M, Mordekar SR. Nonketotic hyperglycinemia case series. J Pediatr Neurosci. 2015;10(4):355–358. doi: 10.4103/1817-1745.174445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanno J, Hutchin T, Kamada F, et al. Genomic deletion within GLDC is a major cause of non-ketotic hyperglycinaemia. https://www.researchgate.net/publication/6446245_Genomic_deletion_within_GLDC_is_a_major_cause_of_non-ketotic_hyperglycinaemia. J Med Genet. 2007;44(3):e69. doi: 10.1136/jmg.2006.043448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beijer P, Lichtenbelt KD, Hofstede FC, et al. A known and a novel mutation in the glycine decarboxylase gene in a newborn with classic nonketotic hyperglycinemia. Neuropediatrics. 2012;43(3):164–167. doi: 10.1055/s-0032-1313914. [DOI] [PubMed] [Google Scholar]
- 9.Gencpinar P, Çavuşoğlu D, Özbeyler Ö, et al. Nonketotic hyperglycinemia:novel mutation in the aminomethyl transferase gene. Case report. https://www.researchgate.net/publication/302909942_Nonketotic_hyperglycinemia_Novel_mutation_in_the_aminomethyl_transferase_gene_Case_report. Arch Argent Pediatr. 2016;(3):e142–146. doi: 10.5546/aap.2016.eng.e142. [DOI] [PubMed] [Google Scholar]
- 10.Bjoraker KJ, Swanson MA, Coughlin 2nd CR, et al. Neurodevelopmental outcome and treatment efficacy of benzoate and dextromethorphan in siblings with attenuated nonketotic hyperglycinemia. J Pediatr. 2016;170:234–239. doi: 10.1016/j.jpeds.2015.12.027. [DOI] [PubMed] [Google Scholar]
- 11.Menéndez Suso JJ, Del Cerro Marín MJ, Dorao Martínez-Romillo P, et al. Nonketotic hyperglycinemia presenting as pulmonary hypertensive vascular disease and fatal pulmonary edema in response to pulmonary vasodilator therapy. J Pediatr. 2012;161(3):557–559. doi: 10.1016/j.jpeds.2012.04.044. [DOI] [PubMed] [Google Scholar]
- 12.Krieger I, Hart ZH. Valine-sensitive nonketotic hyperglycinemia.Case report. J Pediatr. 1974;85(1):43–48. doi: 10.1016/S0022-3476(74)80283-0. [DOI] [PubMed] [Google Scholar]
- 13.Subramanian V, Kadiyala P, Hariharan P, et al. A rare case of glycine encephalopathy unveiled by valproate therapy. J Pediatr Neurosci. 2015;10(2):143–145. doi: 10.4103/1817-1745.159200. [DOI] [PMC free article] [PubMed] [Google Scholar]