

Abstract
On the occasion of Professor Frances H. Arnold’s recent acceptance of the 2018 Nobel Prize in Chemistry, we honor her numerous contributions to the fields of directed evolution and biocatalysis. Arnold pioneered the development of directed evolution methods for engineering enzymes as biocatalysts. Her highly interdisciplinary research has provided a ground not only for understanding the mechanisms of enzyme evolution but also for developing commercially viable enzyme biocatalysts and biocatalytic processes. In this Account, we highlight some of her notable contributions in the past three decades in the development of foundational directed evolution methods and their applications in the design and engineering of enzymes with desired functions for biocatalysis. Her work has created a paradigm shift in the broad catalysis field.
Keywords: directed evolution, biocatalysis, enzyme engineering, P450s, abiological functions, carbene transfer reactions, nitrene transfer reaction, C–H functionalization
Introduction
Enzymes are the workhorses for nearly all biological processes on earth. Largely thanks to their exquisite specificity and selectivity, enzymes have been increasingly used as catalysts for synthesis of chemicals and materials. However, compared to chemical catalysts, enzymes often have limited stability, substrate scope and/or productivity, which significantly hinders their practical applications in industry. To address these key limitations in biocatalysis, in the early 1990s Frances Arnold took inspiration from nature and developed a simple yet powerful algorithm, called ‘directed evolution’, for evolving enzymes with tailored features.1 In essence, this algorithm mimics the process of natural Darwinian evolution in the test tube and involves subjecting a gene encoding an enzyme to random mutagenesis, followed by screening or selection of the resulting library for enzyme variants that show improvement of the desired function. The improved enzyme variant is then used as the ‘parent’ for the following round of mutagenesis and screening or selection, and the process is repeated iteratively until the desired goal is attained (Figure 1). Around the same time as this algorithm was reduced to practice by Arnold,2 another pioneer of directed enzyme evolution, Willem P. C. Stemmer at Maxygen, developed and applied DNA shuffling methods to engineer enzymes with tailored functions.3, 4 In the subsequent years, Arnold, Stemmer, and several other protein engineers around the world, including Donald Hilvert,5 Manfred Reetz,6 and Uwe Bornscheuer,7 have applied this algorithm in combination with state-of-the-art tools from molecular biology, genetic engineering, analytical chemistry, and computational biology, to evolve enzymes with improved or novel functions and investigate the protein structure-function relationships and evolutionary mechanisms, resulting in a new field of directed evolution.
Figure 1.

Workflow of a typical directed evolution experiment.
Arnold is a card-carrying engineer who was trained as a mechanical engineer as an undergraduate student at Princeton University and a chemical engineer as a graduate student at the University of California at Berkeley. Since she started her independent academic career at the Division of Chemistry and Chemical Engineering at Caltech in the mid-1980s, she has been exploring enzymes for biocatalytic applications and fundamental research. In this Account, we will first highlight some of her pioneering contributions in the development and application of directed evolution tools for enzyme engineering in the 1990s, which laid the foundation for the directed evolution field. We will then survey her work on the synthetically versatile P450 monooxygenases as an example to illustrate her group’s key contributions in engineering enzymes with improved catalytic efficiency or new substrate specificity, and understanding the evolutionary mechanisms and protein structure-function relationships. Finally, we will highlight her recent contributions in evolving enzymes with abiological function, which has contributed to expand the boundaries of traditional biocatalysis. With the Arnold group continuing to be innovative and highly productive, this Account only serves as a mid-point review of Frances Arnold’s long and illustrious career in biocatalysis.
I. Early Days in Directed Enzyme Evolution
Thanks to the advances in recombinant DNA technologies and protein crystallography, the mainstream school of thought in enzyme engineering in the 1980s was to use rational design approaches based on structural analysis and site-directed mutagenesis. Frances Arnold was among the first chemical engineers who adopted rational design approaches for enzyme engineering. For example, the Arnold group introduced rationally designed new metal binding sites into enzymes to improve their stability.8, 9 However, Arnold quickly recognized the limitations of rational design which was mainly due to our limited understanding of enzyme structure, function, and dynamics as well as catalytic mechanisms. Therefore, in the late 1980s, inspired by the early studies of John Maynard Smith10 and Manfred Eigen11, Arnold decided to explore molecular evolutionary strategies for engineering enzymes with improved stability and activity.
In her seminal work2, Arnold used iterative rounds of random mutagenesis coupled with screening to improve the activity of subtilisin E in 60% dimethylformamide by 256-fold. This work not only described the directed evolution algorithm and all the key steps in a directed evolution experiment, but also demonstrated that beneficial mutations could be accumulated gradually to yield drastic changes in protein functions and that many of these beneficial mutations were unexpected. This work inspired many other research groups to use directed evolution as an enzyme engineering strategy. However, one limitation with this random mutagenesis based directed evolution strategy was that deleterious mutations may be accumulated too, which resulted in less optimal enzymes. To address this limitation, Arnold group developed a staggered extension process (StEP) in vitro recombination method12, which not only accumulates the beneficial mutations but also removes the deleterious mutations by randomly shuffling the improved genes during each round of directed evolution. In addition to StEP recombination, which was inspired by Stemmer’s ‘DNA shuffling’ idea3, 4, Arnold’s group developed a range of directed evolution tools and strategies, such as the optimized DNA shuffling method13, in vitro random priming recombination method14, digital imaging based on high throughput screening methods for oxygenases15, 16, and SCHEMA structure-guided recombination, which enables extension to more distantly-related genes17.
Apart from developing new directed evolution tools and strategies, the Arnold group also applied them to solve some practical challenges in biocatalysis. For example, a para-nitrobenzyl esterase was engineered with improved stability18, as well as new substrate specificity and improved activity in aqueous-organic solvents19. Para-nitrobenzyl alcohol is used to protect carboxylic acid functionalities during the synthesis of cephalosporin-derived antibiotics, and the engineered para-nitrobenzyl esterase can be used to remove the para-nitrobenzyl alcohol protecting group during the synthesis of loracarbef, a fourth-generation cephalosporin-derived antibiotic. In addition, the Arnold group engineered a subtilisin E variant with drastically improved stability, which showed nearly identical thermal stability as a naturally occurring thermostable subtilisin, thermitase, yet was highly active at a much broader spectrum of temperatures than the thermitase20.
II. P450 BM3: a model enzyme for directed evolution
Starting from the late 1990s, protein engineering efforts in the Arnold group began to focus on P450 monooxygenases.21, 22 Cytochrome P450 monooxygenases make up a large class of heme-dependent enzymes23 which have evolved across all kingdoms of life to carry out a myriad of oxidation reactions, while sharing a common three-dimensional fold.24–27 This enzyme superfamily provides an impressive example of how nature can generate a broad range of oxidation catalysts from a common protein scaffold. Whereas all these enzymes share a cysteine-ligated heme cofactor that is responsible for their activity, the widely diverse substrate specificity of P450s is dictated by their protein sequences, which have diverged as much as 90% over the course of millions of years of natural evolution.24–26
Inspired by the functional pliability of natural P450s, the Arnold group identified cytochrome P450 from Bacillus megaterium (P450 BM3) a particularly attractive target for directed evolution experiments. At that time, P450 BM3 was one of very few known P450s in which the heme domain is fused to a diflavin (FMN/FAD) reductase domain in a single polypeptide chain.28–30 This structural arrangement makes P450 BM3 catalytically ‘self-sufficient’ and able to support the oxidation of a substrate with the only requirement of air oxygen and a source of reducing equivalents in the form of the nicotinamide adenine dinucleotide phosphate (NADPH) cofactor. In addition, P450 BM3 is soluble, it can be readily expressed in Escherichia coli, an excellent host for laboratory evolution experiments, and it is one of the most active hydroxylases known, supporting the hydroxylation of its preferred substrates, long-chain (C12–C20) fatty acids, with rates exceeding 1,000 turnovers per minute.29
While in those years protein engineering efforts with BM3 or other P450s were being pursued using structure-based mutagenesis,31–36 the Arnold group was interested in applying directed evolution to create P450-based catalysts with altered substrate specificity and/or increased stability. One of the earliest goals in the group was to evolve P450 BM3 to accept linear alkanes as substrates for hydroxylation, including the short-chain gaseous alkenes (propane, ethane, methane) found in natural gas. Such biocatalysts were envisioned to provide a potential approach for the biotransformation of natural gas into liquid alcohols as more convenient and easily transportable fuels. In nature, gaseous alkanes are hydroxylated by multi-component non-heme iron monooxygenase enzymes such as methane monooxygenases. Whereas the structural complexity of these enzymes complicates their use or engineering for biotechnological applications, they are also structurally and mechanistically distinct from P450s, begging the question of whether a P450 could be evolved to mimic their function. A challenge inherent to this goal was the fact the P450 BM3 had no detectable activity on propane or smaller alkanes, providing no obvious path for the improvement of such activity. The Arnold group thus decided to initially improve its hydroxylation activity on octane, which is a poor but viable substrate for this P450,22, 37 with the underlying idea that BM3 variants with enhanced octane activity would eventually acquire the ability to accept and hydroxylate shorter alkanes. After five generations of random mutagenesis targeted to the heme domain, followed by recombination of beneficial mutations, the group was able to identify a P450 BM3 variant (139–3) with significantly improved octane activity (30 → 1,000 total turnovers (TTN)) as a result of 11 amino acid substitutions accumulated over the course of this process.22 Importantly, such enhancement in octane activity was accompanied by the acquisition of activity on the smaller alkane substrate propane (0 → 500 TTN).38 Further rounds of mutagenesis and screening, using first an octane surrogate substrate and then a propane surrogate substrate, led to P450 BM3 variants with progressively increased activity toward the conversion of propane to 2-propanol and even modest activity on ethane (up to 250 TTN with variant 35E11).38, 39 Out of the 17 mutations accumulated in 35E11, only two were found to be located in the active site, illustrating the power of directed evolution in identifying beneficial mutations far from the active site. Integral part of this evolutionary process was the development of high-throughput assays for enabling the rapid screening of P450 libraries for activity on chromogenic substrates as surrogates for octane (p-nitrophenyl octyl ether and hexyl methyl ether) and propane (dimethyl ether).37, 38
Despite this progress, the catalytic efficiency of 35E11 as an alkane hydroxylase was significantly inferior compared to that of wild-type P450 BM3 on its preferred substrates. The function of this multi-domain P450 relies on finely regulated conformational changes and electron transfer mechanisms, which ensure efficient coupling of fatty acid hydroxylation with NADPH oxidation.29 These mechanisms are disrupted in the presence of non-native substrates and/or as a result of mutations, resulting in an ‘uncoupling’ process and production of reactive oxygen species (ROS) which inactivates the enzyme. To obtain a more efficient biocatalyst for propane hydroxylation, variant 35E11 was further evolved using a ‘domain engineering strategy’, in which beneficial mutations within the heme-, FMN-, and FAD-domain of this P450 were first identified via random and site-saturation mutagenesis and then combined in a final step.40 This process generated a proficient propane monooxygenase, named P450PMO, possessing high catalytic efficiency and coupling efficiency for this reaction (Figure 2), i.e. catalytic properties comparable to those exhibited by wild-type P450BM3 with its preferred substrates.40, 41 The laboratory-evolved P450PMO could then be applied for the efficient conversion of propane to 2-propanol in metabolically engineered E. coli cells.42
Figure 2. Directed evolution of a cytochrome P450 propane monooxygenase.
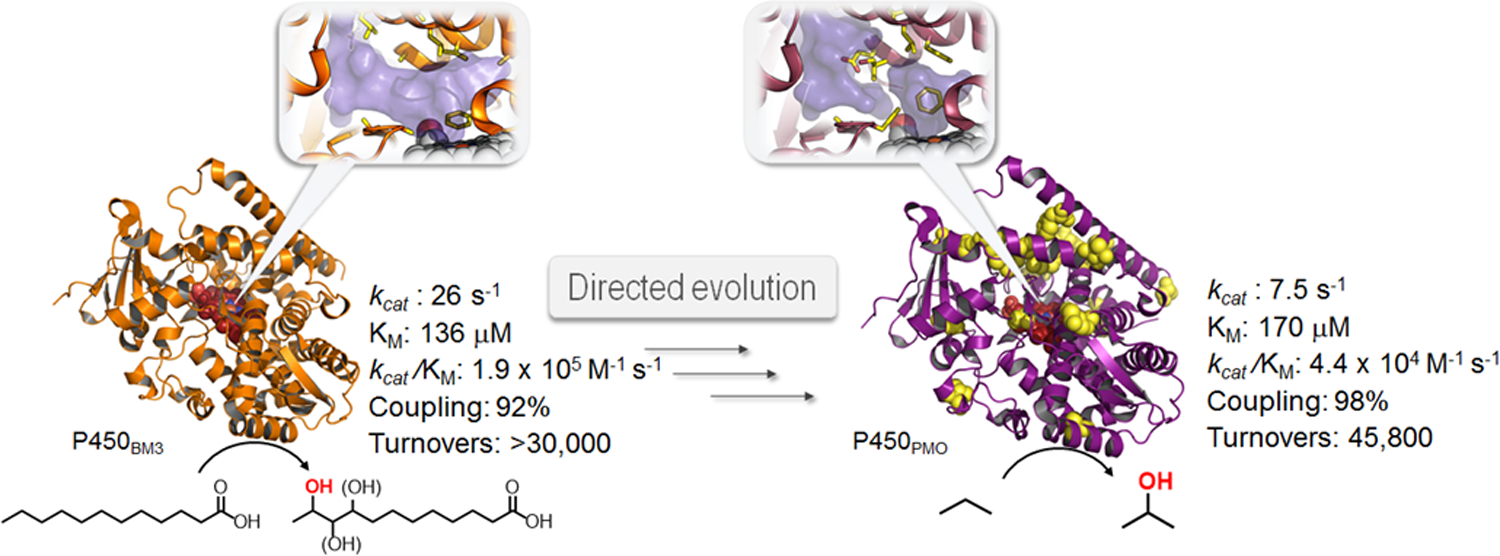
The long-chain fatty acid hydroxylase P450 BM3 from Bacillus megaterium was converted into a highly efficient propane monooxygenase (P450PMO) over 13 rounds of directed evolution.22, 38, 40 The evolved enzyme accumulated 3 beneficial mutations in the reductase domain and 20 in the heme domain (yellow, sphere models), many of which are distant from the heme cofactor (red, sphere model). Refinement of catalytic efficiency for propane oxidation, an activity not exhibited by wild-type P450 BM3, also involved a profound reshaping and partitioning of the substrate access pathway (inserts).41
Lessons from directed evolution experiments
The conversion of P450 BM3 into an efficient propane monooxygenase not only demonstrated the remarkable plasticity of P450s toward adopting new functions but also the effectiveness of directed evolution in progressively adapting an enzyme to execute a new function. This ‘substrate walk’ strategy has proven effective in the laboratory evolution of a variety of other enzymes and proteins.43–48 In addition to altered substrate specificity, the Arnold group demonstrated over the years how a variety of other properties of P450s, including the ability to operate as peroxygenases, i.e., catalyze hydroxylation reactions driven by hydrogen peroxide,49 and their stability against thermal50 and chemical denaturation51 could be improved through directed evolution. These studies, along with a growing number of examples from other groups, notably in the case of enantioselective transformations,52, 53 contributed to formulate the now broadly accepted notion that the fitness of a protein with respect to a given functional or structural property can be gradually enhanced in the laboratory, often through a series of small mutational steps,54 as long as the desired property remains feasible and properly chosen screening/selection tools are applied.
Important lessons on how proteins can evolve were also derived from a detailed characterization of the evolutionary intermediates along the lineage of P450PMO (Figure 3A).41 These studies revealed that the adaptive walk from the long-chain fatty acid hydroxylase P450BM3 to the propane monooxygenase P450PMO proceeded through a series of ‘promiscuous’ variants (e.g. 139–3, 35E11) that are characterized by a broad substrate profile, as determined based on their activity profile across the series of C1-C10 alkanes and other substrates.41 However, further optimization of propane activity was accompanied by ‘re-specialization’ of the P450 for the new function (Figure 3B), resulting in a variant (P450PMO) that features a very narrow substrate profile centered around propane and has no residual activity on fatty acids, i.e. no remnants of the enzyme native function.41 On the one hand, these experiments showed that, starting from a specialized enzyme, the acquisition of a new substrate activity can proceed via a ‘generalist’ enzyme, with modest activity on multiple substrates in addition to the native one(s). On the other hand, given that no negative selection was applied throughout the directed evolution of P450PMO, these studies also showed that positive selection alone (i.e. propane activity) was sufficient for evolving a ‘specialist’ enzyme from another specialized one. Prior to this work, it was generally assumed that a combination of positive and negative selection pressure was necessary to reinforce the desired phenotype in an enzyme/protein while removing undesirable native (or non-native) activities, as supported by a number of protein engineering experiments.55–57
Figure 3.
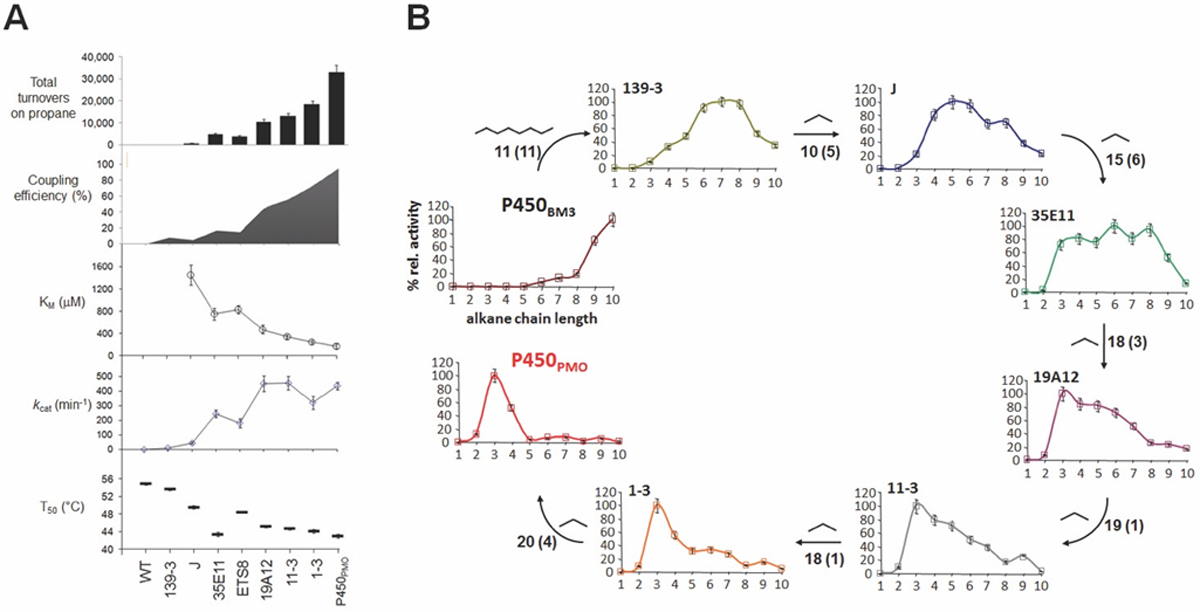
Catalytic and stability properties (a) and substrate profile (b) of selected variants along the P450PMO lineage.41 Total turnover numbers (TTN), coupling efficiency (mol oxidized product/mol oxidized NADPH), and kinetic properties (KM, kcat) refer to propane oxidation. Thermal stability is shown as T50 (half-maximal inactivation temperature after 10-min incubation). Substrate profile is reported as relative activity on C1–C10 alkane series. Adapted with permission from ref. 41. Copyright 2008, Elsevier.
The P450PMO ‘story’ also illustrated the importance of addressing the activity/stability trade-off often observed in directed evolution experiments. Indeed, at a point during the evolution of P450PMO – specifically at the level of 35E11 (Figure 3A), thermostabilization of the protein (via function-neutral mutations) was necessary to enable the accumulation of additional beneficial mutations to enhance propane activity.41 This finding reiterated the important relationship between protein stability and evolvability, a notion nicely outlined in a previous study of the Arnold group.58 Via a comparative study with a marginally stable vs. a highly stable variant of BM3, they indeed demonstrated how the thermostable P450 showed a significantly higher propensity to acquire activity on new substrates upon random mutations when compared to the less stable enzyme. This study thus illustrated the key role of stability-based epistasis in adaptive evolution,59 an important implication of which is that, in the realm of natural proteins, function-neutral yet stabilizing mutations may accumulate by genetic drift to permit the evolution of new functions.60 In another study involving P450 BM3 as a model enzyme, Arnold and coworkers further demonstrated that promiscuous enzymatic activities (e.g. activity on a new substrate) can arise via a neutral path, i.e., through mutations that are neutral to the property under selective pressure,61 and these can provide the starting points for the evolution of new functional proteins.
The Arnold group’s interest toward investigating the impact of mutational events on emergence of new functions has also extended to recombination, a key mechanism occurring in the context of natural protein evolution. In particular, the group introduced a new strategy for directed evolution via recombination of homologous enzymes, called SCHEMA, which relies on the structure-based identification of crossover points for minimizing structural disruption in the resulting chimeras. Using the β-lactamase scaffold as a testbed, a large set of chimeric proteins was generated by SCHEMA-guided recombination of the homologous β-lactamase enzymes TEM-1 and PSE-2 and analyzed for retention of function (Figure 4A). This study showed that recombination is conservative (much more so than random mutagenesis) and results in a higher frequency of folded and functional variants despite of the introduction of multiple amino acid mutations compared to the parent enzymes (Figure 4B).62 To address the question of whether these mutational changes are associated with the acquisition of new functional traits, similar recombination experiments were conducted using three homologs of P450 BM3 sharing ~65% sequence identity.63 This process generated a new family of ~6,000 chimeric P450s carrying an average of 70 mutations from the closet parent, approximately half of which folded properly and a third of which display enzymatic activity. Characterization of these chimeric P450s revealed that functional members of these libraries acquired significant functional diversity, including the ability to accept substrates not accepted by the parent enzymes.64 In addition, a fraction of the chimeric P450s were found to exhibit higher stability compared to the parent proteins (Figure 4C) and, based on structure-stability correlations, highly thermostable variants could be generated through the targeted recombination of stabilizing fragments.65 The same overall approach later proved valuable toward generating thermostabilized and functional variants of fungal cellobiohydrolases, which find utility in the degradation of cellulose into fermentable sugars for biofuel applications.66 Overall, these studies contributed to demonstrate the value of structure-based recombination as a strategy to navigate the functional space in enzymes and provide valuable starting points for further functional evolution.
Figure 4. Directed evolution of enzymes using structure-based recombination.
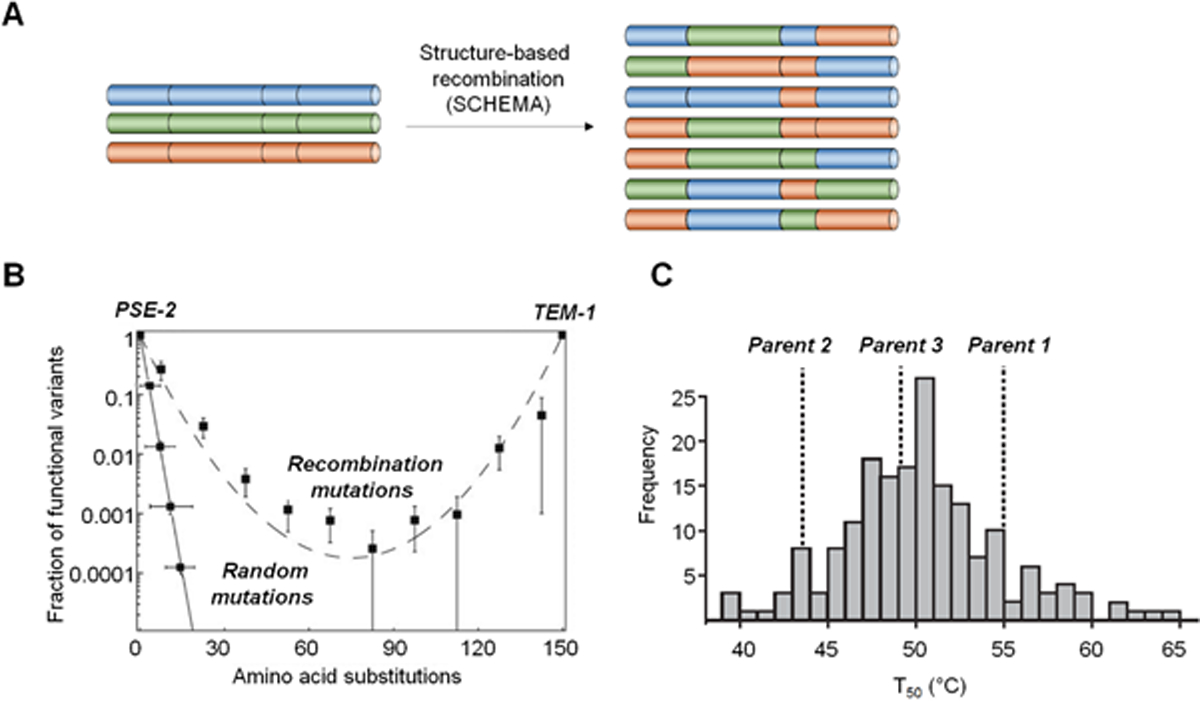
(a) Generation of chimeric enzymes via SCHEMA-guided recombination of fragments derived from homologous parent sequences and chosen to minimize structural disruption.17 (b) SCHEMA-guided recombination of β-lactamase PSE-2 and TEM-1 produces a higher fraction of functional chimeras compared to random mutagenesis.62 (c) Chimeric P450s obtained via SCHEMA-guided recombination of P450 BM3 (CYP102A1) with its homologous enzymes CYP102A2 and CYP102A3 can exhibit higher stability against thermal denaturation compared to any of the parent P450s.65 Adapted with permission from ref. 62. Copyright 2005, National Academy of Sciences.
Laboratory-evolved P450s for biocatalytic oxidations
Oxidation catalysts are valuable tools for a variety of synthetic applications,67 and the synthetic potential of (engineered) cytochrome P450s in this regard has not been overlooked by Frances Arnold, along with a number of other groups.68–71 In particular, Arnold and coworkers demonstrated how substrate promiscuous P450 BM3 variants isolated during the evolution of alkane oxidases could be useful for the selective hydroxylation of C6-C10 alkanes at the subterminal (ω−1) position (up to 86% regioselectivity and 83% ee)38, the epoxidation of terminal C5-C8 alkenes (up to 94% regioselectivity and 83% ee; Scheme 1A)72, and the α-hydroxylation of 2-aryl-acetic acid esters to give valuable mandelic acid derivatives (up to 99% regioselectivity and 94% ee; Scheme 1B)73. In another contribution, it was shown that P450-catalyzed hydroxylation could be combined with chemical fluorination to enable the rapid and site-selective installation of one or more fluorine substituents in a variety of target small-molecule drugs and building blocks.74 As the C–H sites targeted by engineered bacterial P450s often overlap with those oxidized by human liver P450s, this strategy is valuable toward improving the metabolic stability of drugs or other bioactive molecules.74, 75
Scheme 1.
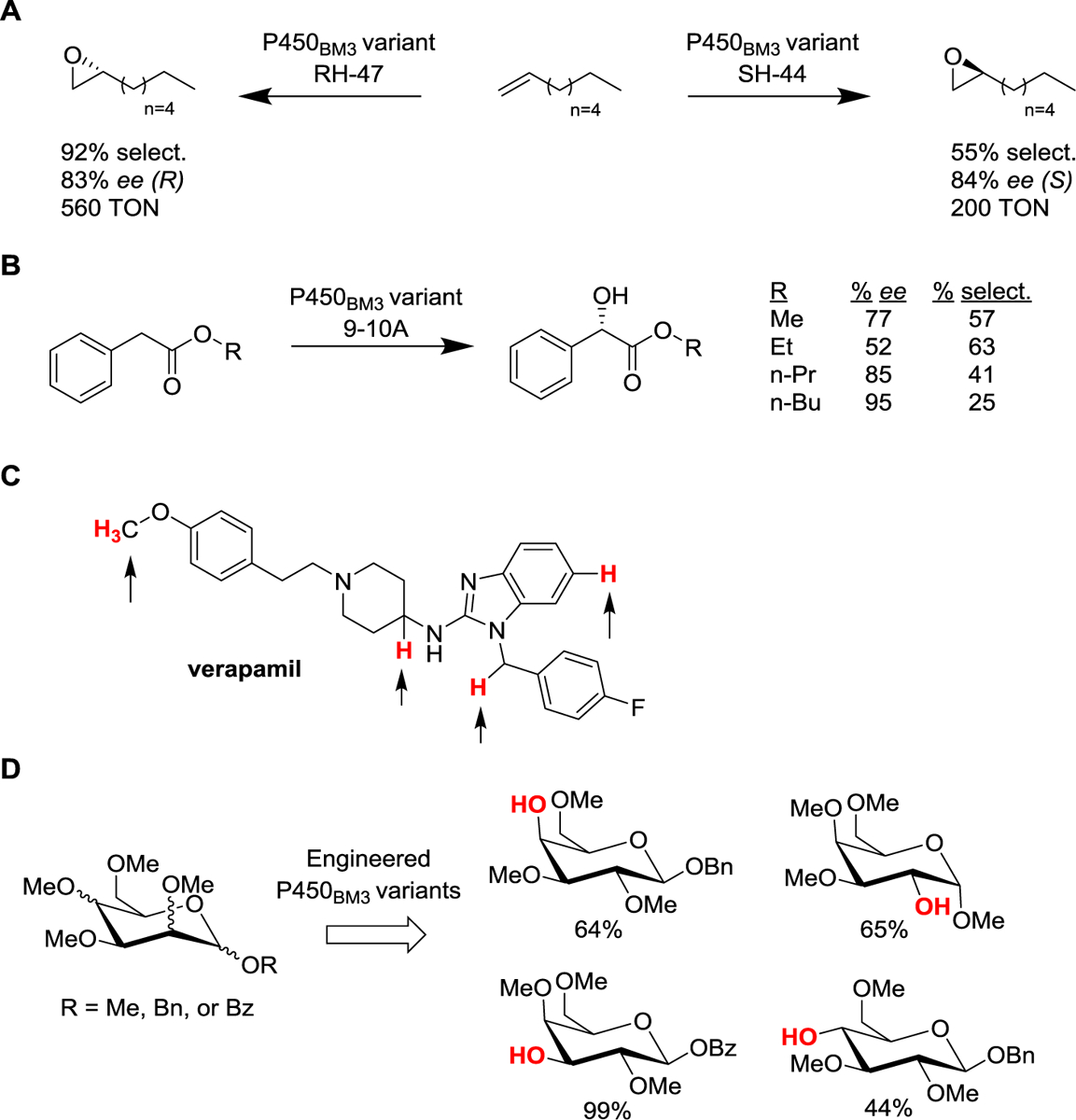
Laboratory-evolved P450 BM3 variants for synthetic applications: (a) selective epoxidation of terminal alkenes;72 (b) stereoselective synthesis of mandelic acid derivatives;73 (c) generation of human drug metabolites (sites hydroxylated by the engineered P450 BM3 variants are indicated by arrows);83 and (d) regioselective demethylation of permethylated monosaccharides.85 TON = turnover number; ee = enantiomeric excess; select. = selectivity.
Since hepatic P450s account for the majority of Phase I metabolic transformations of drugs in vivo, an important application of cytochrome P450s (human and non-human) lies in the production of the authentic human metabolites of drugs and drug candidates,76–79 as characterization of the pharmacological activity and toxicity of these compounds represents a critical step toward their approval for use as pharmaceuticals. Along with others,80, 81 the Arnold group showed that laboratory-evolved variants of P450 BM3, including substrate-promiscuous variants from the aforementioned P450PMO lineage and recombination libraries, can prove useful to generate human metabolites of marketed drugs.82, 83 In collaborative effort with Eli Lilly, Arnold and coworkers showed the successful application of a small collection of these engineered P450BM3 variants (~120) for producing nearly all of the human metabolites (12/13) of the drugs verapamil and astemizole.83 They also reported the directed evolution of a P450BM3 variant for the oxidation of acidic drugs such as naproxen, which are typically metabolized by human CYP2C9.84
In another important synthetic application, P450 BM3 variants were engineered by the Arnold group to enable the regioselective deprotection of single methyl groups from permethylated monosaccharides.85 The deprotected monosaccharides could be then converted in a chemoenzymatic process to deoxy- and fluorinated monosaccharides and disaccharides, thus providing a new approach to access valuable building blocks for the synthesis of polysaccharides and other sugar-functionalized molecules. While P450-mediated chemoenzymatic synthesis has recently emerged as an attractive strategy for the late-stage functionalization of complex bioactive molecules,75, 86, 87 a collaboration between the Arnold and Stoltz groups recently demonstrated the utility of engineered P450s for the total synthesis of the natural product nigelladine A.88 In this case, a key allylic oxidation step, which had proven inefficient using chemical methods, could be realized using an engineered P450 BM3 variant, enabling further elaboration of the synthetic intermediate to the desired natural product.
III. Evolving enzymes for new-to-nature biotransformations
Starting in 2010, protein engineering efforts in the Arnold group began to focus on the evolution of cytochrome P450s for abiological functions such as carbene and nitrene transfer reactions. These are synthetically valuable transformations not known in naturally-occurring enzymes. The contributions of the Arnold group and others89–100 in this area have greatly expanded the repertoire of enzymes as biocatalysts for synthetic applications.
The preparation of cyclopropanes by carbene transfer to olefins, though well known in transition metal catalysis,101, 102 is novel to the biological world. In 2013, the Arnold group showed that heme proteins such as engineered cytochrome P450 BM3 enzymes could function as cyclopropanation biocatalysts, promoting the cyclopropanation of styrenyl olefins in water.103 By mutating the proximal axial ligand from cysteine to serine, variants of P450BM3 (termed P411BM3 because of the signature ferrous CO Soret peak at 411 nm rather than 450 nm for cysteine-ligated P450) also function efficiently in whole-cell E. coli and catalyze carbene transfer styrene cyclopropanation in vivo with up to 67,800 turnovers.104
Enzymes are sustainable catalysts for the preparation of pharmaceuticals and other high-value compounds,105, 106 and new-to-nature enzymatic activities can significantly expand the biocatalytic repertoire available for this purpose.107 Taking advantage of the versatility of engineered cytochromes P45098, 100, 108, 109 as cyclopropanation enzymes, the Arnold group showed their potential for producing industrially important pharmaceutical building blocks such as the cyclopropane core of the antidepressant levomilnacipran, which was prepared on a preparative scale using P450BM3-Hstar, an evolved His-ligated cytochrome P450BM3 variant (Scheme 2A).110 Stereocomplementary biocatalysts are key assets for the synthesis of drugs and complex molecules.94, 111–119 Upon exploring a collection of heme enzymes, the Arnold group was able to identify a set of biocatalysts useful for producing the four possible stereoisomers of a cyclopropanation reaction with ethyl diazoacetate and an unactivated alkene (Scheme 2B).120 Notably, no iron-based catalyst was previously known for this transformation and heme alone only catalyzed this reaction with < 1 turnover.
Scheme 2.
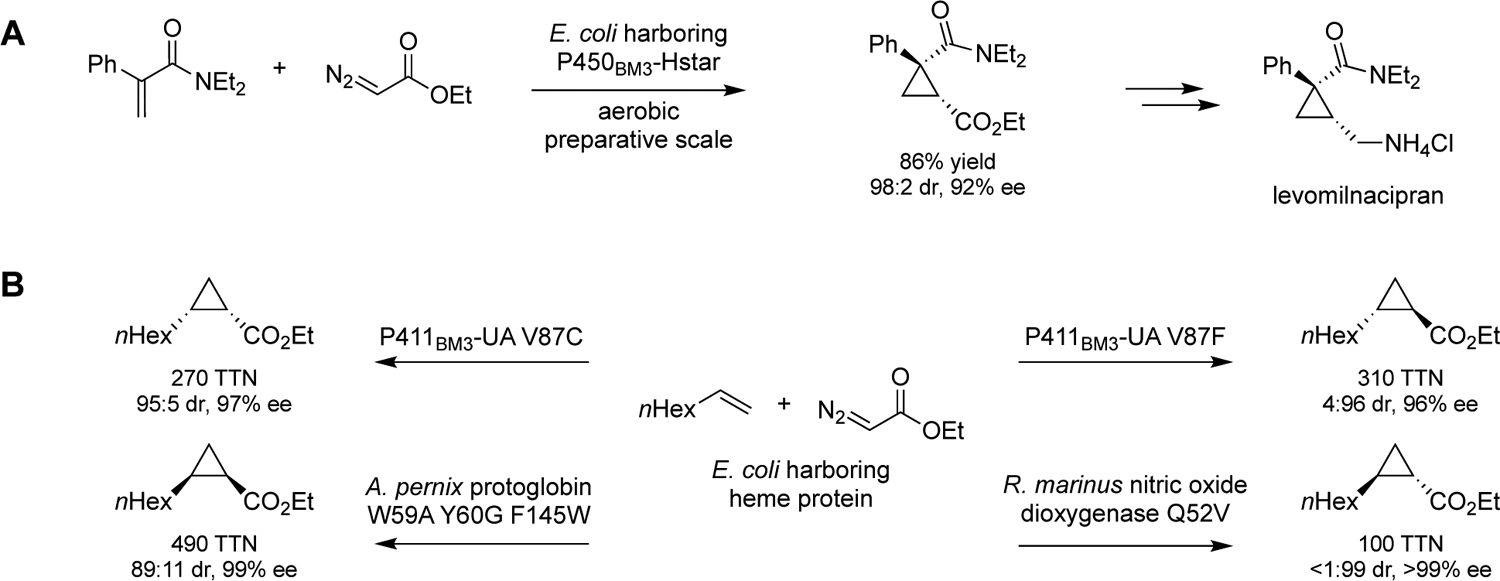
Engineered heme protein-catalyzed new-to-nature transformations: (a) synthesis of the cyclopropane core of antidepressant levomilnacipran;110 (b) enzymatic stereodivergent cyclopropanation of octene catalyzed by a suite of engineered cytochrome P411 proteins and globins.120
Highly strained rings
Bicyclobutanes are puckered carbocycles that are difficult to assemble owning to their high ring strain. Once synthesized, however, they manifest interesting properties that make them uniquely suited for a variety of applications, such as strain-release chemistry121 and stimulus-responsive materials.122 In a recent study, the Arnold group described the directed evolution of a P411 variant, called P411BM3-E10-FA, capable of accepting phenylacetylene and ethyl diazoacetate as substrates to make bicyclobutane product as a single stereoisomer (Scheme 3).123 Key to this success is the enzyme’s ability to stabilize the putative reactive cyclopropene intermediate and exert precise stereocontrol over two carbene transfer events. P411BM3 variants that yield cyclopropenes as the final products were also identified, such that each enantiomer of a cyclopropene can be made enzymatically with thousands of turnovers and high enantioselectivity (Scheme 3).
Scheme 3.

Cytochrome P411BM3-catalyzed bicyclobutanation and cyclopropenation.123
New chemical bonds
Carbon–silicon and carbon–boron bonds are absent in biology but commonly found in man-made molecules and materials in our modern world. By engineering cytochrome c protein from the thermohalophilic bacterium Rhodothermus marinus (Rma cyt c), the Arnold group described carbene insertion into Si–H and B–H bonds as a means to coax bacteria to forge carbon–silicon and carbon–boron bonds in living systems for the first time.124, 125 With only three mutations, directed evolution turned wild-type Rma cyt c into a silylation enzyme that exhibits up to 8,200 turnovers (Scheme 4A), capable of making 20 different organosilicon products with 95–99% ee.122 The mechanism of biological carbon–silicon bond formation was elucidated by the Arnold and Houk groups by capturing the catalytic iron-carbene intermediate in the active site of Rma cyt c crystallographically (Scheme 4A) and by probing experimentally and computationally how the iron-carbene electronic state and protein conformational dynamics affect the reaction.126 These efforts are complemented by the mechanistic work of other groups, further our understanding of how iron porphyrins and heme proteins catalyze carbene transfer to olefins, N–H, and C–H bonds.127–133
Scheme 4.

Engineered R. marinus cytochrome c-catalyzed carbon–silicon124 and carbon–boron bond formation.125, 135
Genetically programmable chiral organoborane synthesis was also realized by the laboratory evolution of Rma cyt c.123 This platform successfully yields structurally distinct organoboranes through divergent directed evolution (Scheme 4B), providing bacterial borylation catalysts that are suitable for gram-scale biosynthesis, and offering up to 15,300 turnovers and excellent selectivity (99:1 e.r., 100% chemoselectivity). Moreover, these enzymes are readily tunable to accommodate the synthesis of lactone-based organoboranes134 and a broad range of chiral α-trifluoromethylated organoboranes (Scheme 4C).135 The latter demonstration is particularly appealing as it represents a means to introduce fluorine-containing motifs to small molecules,95 orthogonal to existing strategies for organofluorine biosynthesis using engineered fluorinase enzymes or precursor-directed biosynthesis of fluorine-containing polyketides, peptides, and alkaloids.136, 137
C–H functionalization
Evolving heme proteins to functionalize C–H bonds in new ways is of interest to the Arnold group and others in the biocatalytic community.138 While native P450 enzymes are well versed in oxidizing even unactivated C–H bonds through a reactive, high-valent iron-oxene intermediate (Compound I),139 other than oxygen atoms, examples of heme proteins inserting other groups or functionalities into C–H bonds are sporadic in the natural and unnatural worlds. Inspired by a 1985 report describing a C–H amination reaction catalyzed by rabbit liver cytochrome P450,140 the Arnold and Fasan group independently discovered that engineered P411 (P411BM3-T268A and P411BM3-CIS)141 and P450BM3 variants,89 as well as other hemoproteins142, are able to catalyze and intramolecular nitrene insertion into benzylic C–H bonds on aryl sulfonyl azides. By further evolving these P411 variants through active site site-saturation mutagenesis and recombination of beneficial mutations, the regioselectivity of the cyclization process could be altered to yield either five-membered (benzylic C–H insertion) or six-membered ring (homobenzylic C–H insertion) products, representing a powerful example of how directed evolution can tune an enzyme active site to favor functionalization of higher-energy C–H bonds (Scheme 5A).143
Scheme 5.
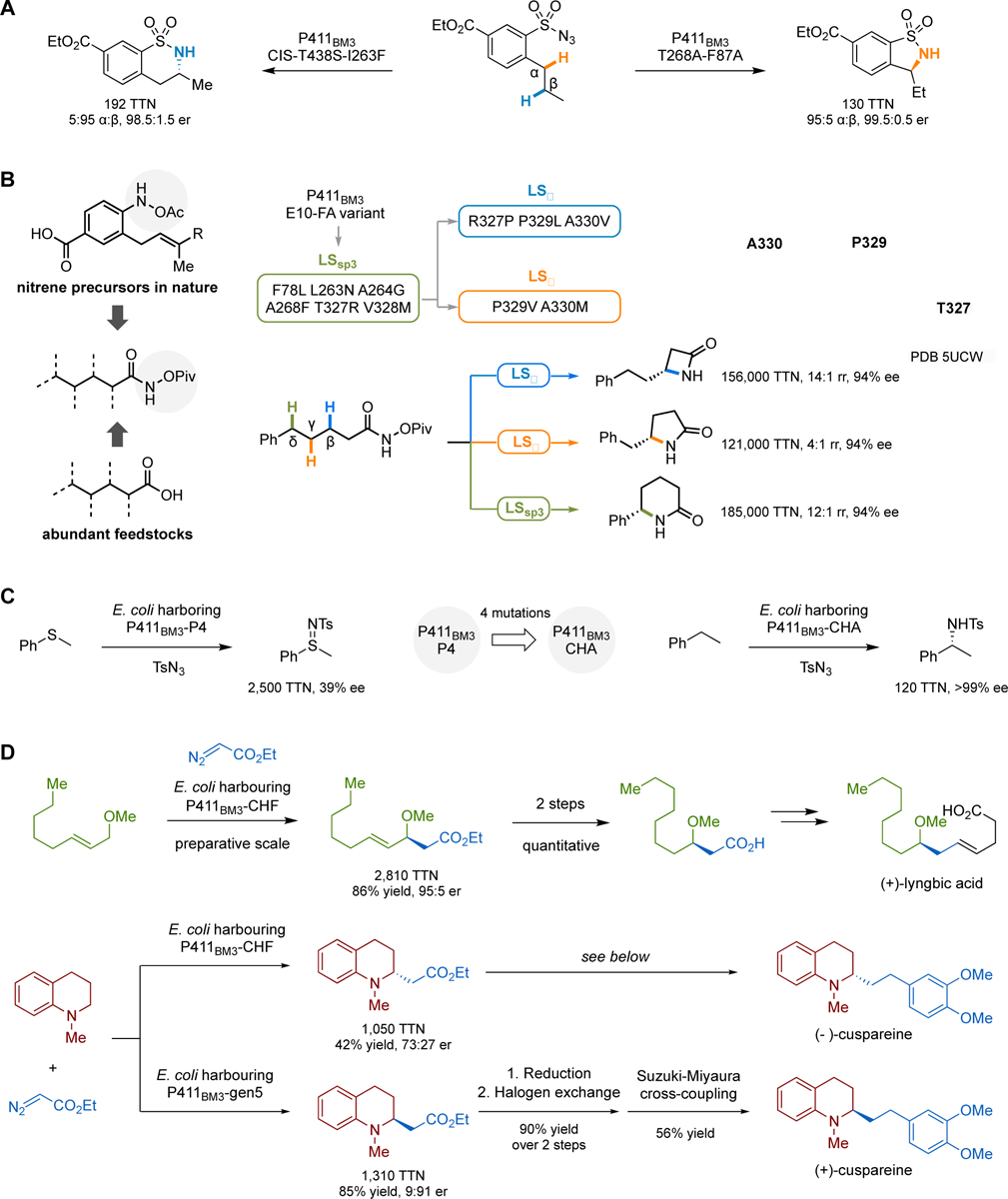
C–H functionalization enabled by enzymatic nitrene and carbene transfer C–H insertion: (a) regioselective cyclization via intramolecular nitrene insertion into benzylic C–H bonds;143 (b) divergent synthesis of β-, γ-, and δ-lactams by P411BM3-catalyzed intramolecular C–H amidation;144 (c) P411BM3-catalyzed intermolecular C–H amination;147 (d) P411BM3-catalyzed intermolecular C–H alkylation.148
Recently, this concept was further expanded and showcased in the divergent synthesis of β-, γ-, and δ-lactams by P411BM3-catalyzed intramolecular C–H amidation.144 Using acyl-protected hydroxamates as carbonyl nitrene precursors90, 145, a collection of P411BM3-derived lactam synthases with complementary cyclization modes was obtained (Scheme 5B). Remarkably, these biocatalysts can override intrinsic substrate bias from sterics, electronic preference, and ring strain to deliver lactams of different ring sizes with excellent selectivities and up to a million total turnovers.
Intermolecular C–H amination is a significantly challenging transformation and few catalytic systems are known to be both efficient and selective across diverse substrates. To overcome this, many heme proteins were tested in the Arnold group for intermolecular nitrene C–H insertion activity: none was found to be active until they tested a cytochrome P411BM3 variant, called P4, engineered for the nitrene transfer to sulfides and allylic sulfides.146, 147 The promiscuous activity observed may arise from transition state similarities between sulfimidation and nitrene C–H insertion. Introducing four mutations to P4 (A82L, A78V, F263L, and E267D) led to P411CHA, a highly enantioselective benzylic C–H amination biocatalyst capable of catalyzing a reaction which the iron cofactor alone cannot perform (Scheme 5C). Interestingly, P4 A82L also exhibits intermolecular C–H alkylation activity, unlocking a whole new world of opportunities for developing enzymatic C–H functionalization methods by carbene transfer.148 After 13 rounds of mutagenesis and screening and removal of the enzyme FAD domain, P411-CHF was created from P4 A82L, which demonstrates 140-fold turnover increase relative to the parent protein in carbene transfer C–H insertion reaction of p-methoxybenzyl methyl ether. P411-CHF is broad in scope: it functionalizes benzylic, allylic, and α-amino C–H bonds with up to 3750 turnovers and >99:1 er, and is applicable to streamlining the synthesis of natural products (Scheme 5D). Because this biocatalyst is fully genetically encoded, produced in E. coli, and functions at room temperature, it offers a desirable alternative to noble-metal catalysts for intermolecular C–H functionalization chemistry.
Summary
Frances Arnold pioneered the development of directed evolution for enzyme engineering. Her career has not only had a profound impact in the field of enzyme engineering and biocatalysis but also deeply influenced the lives of many of her students and postdocs, including the three of us. This account articles revisit some of her inspiring accomplishments over the past three decades and we are sure that more groundbreaking contributions will be forthcoming in the coming years, as a direct result of her fearless and tireless pursuit of excellence in research.
Acknowledgements
This work was supported by the U.S. National Science Foundation (CHE-1609550 grant to R.F.), the Rothenberg Innovation Initiative (RI2) Program and Jacobs Institute for Molecular Engineering for Medicine at Caltech (S.B.J.K.); U.S. Department of Energy (DE-SC0018260 and DE-SC0018420 grants to H.Z.), and U.S. National Institutes of Health (R01GM098628 grant to R.F.; AI144967 grant to H.Z.).
Footnotes
The authors declare no competing financial interest.
References
- 1.Bornscheuer UT; Hauer B; Jaeger KE; Schwaneberg U, Directed Evolution Empowered Redesign of Natural Proteins for the Sustainable Production of Chemicals and Pharmaceuticals. Angew Chem Int Edit 2019, 58, 36–40. [DOI] [PubMed] [Google Scholar]
- 2.Chen KQ; Arnold FH, Tuning the Activity of an Enzyme for Unusual Environments - Sequential Random Mutagenesis of Subtilisin-E for Catalysis in Dimethylformamide. P Natl Acad Sci USA 1993, 90, 5618–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stemmer WP, DNA Shuffling by Random Fragmentation and Reassembly: in vitro Recombination for Molecular Evolution. Proc Natl Acad Sci U S A 1994, 91, 10747–10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stemmer WP, Rapid Evolution of a Protein in vitro by DNA Shuffling. Nature 1994, 370, 389–391. [DOI] [PubMed] [Google Scholar]
- 5.Kast P; Asif-Ullah M; Jiang N; Hilvert D, Exploring the Active Site of Chorismate Mutase by Combinatorial Mutagenesis and Selection: the Importance of Electrostatic Catalysis. Proc Natl Acad Sci U S A 1996, 93, 5043–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reetz MT; Zonta A; Schimossek K; Liebeton K; Jaeger KE, Creation of Enantioselective Biocatalysts for Organic Chemistry by in vitro Evolution. Angew Chem Int Edit 1997, 36, 2830–2832. [Google Scholar]
- 7.Bornscheuer UT; Altenbuchner J; Meyer HH, Directed Evolution of an Esterase for the Stereoselective Resolution of a Key Intermediate in the Synthesis of Epothilones. Biotechnol Bioeng 1998, 58, 554–559. [DOI] [PubMed] [Google Scholar]
- 8.Suh SS; Haymore BL; Arnold FH, Characterization of His-X3-His Sites in Alpha-Helices of Synthetic Metal-Binding Bovine Somatotropin. Protein Eng 1991, 4, 301–305. [DOI] [PubMed] [Google Scholar]
- 9.Arnold FH; Haymore BL, Engineered Metal-Binding Proteins - Purification to Protein Folding. Science 1991, 252, 1796–1797. [DOI] [PubMed] [Google Scholar]
- 10.Smith JM, Natural Selection and the Concept of a Protein Space. Nature 1970, 225, 563–564. [DOI] [PubMed] [Google Scholar]
- 11.Eigen M; Gardiner W, Evolutionary Molecular Engineering Based on RNA Replication. Pure Appl Chem 1984, 56, 967–978. [Google Scholar]
- 12.Zhao HM; Giver L; Shao ZX; Affholter JA; Arnold FH, Molecular Evolution by Staggered Extension Process (StEP) in vitro Recombination. Nat Biotechnol 1998, 16, 258–261. [DOI] [PubMed] [Google Scholar]
- 13.Zhao HM; Arnold FH, Optimization of DNA Shuffling for High Fidelity Recombination. Nucleic Acids Res 1997, 25, 1307–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shao ZX; Zhao HM; Giver L; Arnold FH, Random-priming in vitro Recombination: an Effective Tool for Directed Evolution. Nucleic Acids Res 1998, 26, 681–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joo H; Arisawa A; Lin ZL; Arnold FH, A High-throughput Digital Imaging Screen for the Discovery and Directed Evolution of Oxygenases. Chem Biol 1999, 6, 699–706. [DOI] [PubMed] [Google Scholar]
- 16.Joern JM; Sakamoto T; Arisawa AA; Arnold FH, A Versatile High Throughput Screen for Dioxygenase Activity using Solid-phase Digital Imaging. J Biomol Screen 2001, 6, 219–223. [DOI] [PubMed] [Google Scholar]
- 17.Voigt CA; Martinez C; Wang ZG; Mayo SL; Arnold FH, Protein Building Blocks Preserved by Recombination. Nat Struct Biol 2002, 9, 553–558. [DOI] [PubMed] [Google Scholar]
- 18.Giver L; Gershenson A; Freskgard PO; Arnold FH, Directed evolution of a thermostable esterase. P Natl Acad Sci USA 1998, 95, 12809–12813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore JC; Arnold FH, Directed evolution of a para-nitrobenzyl esterase for aqueous-organic solvents. Nat Biotechnol 1996, 14, 458–467. [DOI] [PubMed] [Google Scholar]
- 20.Zhao HM; Arnold FH, Directed evolution converts subtilisin E into a functional equivalent of thermitase. Protein Eng 1999, 12, 47–53. [DOI] [PubMed] [Google Scholar]
- 21.Joo H; Lin ZL; Arnold FH, Laboratory evolution of peroxide-mediated cytochrome P450 hydroxylation. Nature 1999, 399, 670–673. [DOI] [PubMed] [Google Scholar]
- 22.Glieder A; Farinas ET; Arnold FH, Laboratory evolution of a soluble, self-sufficient, highly active alkane hydroxylase. Nat Biotechnol 2002, 20, 1135–1139. [DOI] [PubMed] [Google Scholar]
- 23.Nelson DR, Cytochrome P450 diversity in the tree of life. Bba-Proteins Proteom 2018, 1866, 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denisov IG; Makris TM; Sligar SG; Schlichting I, Structure and chemistry of cytochrome P450. Chem Rev 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]
- 25.Pylypenko O; Schlichting I, Structural aspects of ligand binding to and electron transfer in bacterial and fungal p450s. Annu Rev Biochem 2004, 73, 991–1018. [DOI] [PubMed] [Google Scholar]
- 26.de Montellano PRO, Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem Rev 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guengerich FP; Yoshimoto FK, Formation and Cleavage of C-C Bonds by Enzymatic Oxidation Reduction Reactions. Chem Rev 2018, 118, 6573–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Miura Y; Fulco AJ, (Omega - 2) Hydroxylation of Fatty-Acids by a Soluble System from Bacillus-Megaterium. J Biol Chem 1974, 249, 1880–1888. [PubMed] [Google Scholar]
- 29.Munro AW; Girvan HM; McLean KJ, Variations on a (t)heme - novel mechanisms, redox partners and catalytic functions in the cytochrome P450 superfamily. Nat Prod Rep 2007, 24, 585–609. [DOI] [PubMed] [Google Scholar]
- 30.Hannemann F; Bichet A; Ewen KM; Bernhardt R, Cytochrome P450 systems - biological variations of electron transport chains. Bba-Gen Subjects 2007, 1770, 330–344. [DOI] [PubMed] [Google Scholar]
- 31.Manchester JI; Ornstein RL, Rational approach to improving reductive catalysis by cytochrome P450cam. Biochimie 1996, 78, 714–722. [DOI] [PubMed] [Google Scholar]
- 32.Bottner B; Schrauber H; Bernhardt R, Engineering a mineralocorticoid- to a glucocorticoid-synthesizing cytochrome P450. J Biol Chem 1996, 271, 8028–8033. [DOI] [PubMed] [Google Scholar]
- 33.Oliver CF; Modi S; Primrose WU; Lian LY; Roberts GCK, Engineering the substrate specificity of Bacillus megaterium cytochrome P-450 BM3: hydroxylation of alkyl trimethylammonium compounds. Biochem J 1997, 327, 537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Noble MA; Miles CS; Chapman SK; Lysek DA; Mackay AC; Reid GA; Hanzlik RP; Munro AW, Roles of key active-site residues in flavocytochrome P450 BM3. Biochem J 1999, 339, 371–379. [PMC free article] [PubMed] [Google Scholar]
- 35.Harford-Cross CF; Carmichael AB; Allan FK; England PA; Rouch DA; Wong LL, Protein engineering of cytochrome P450(cam) (CYP101) for the oxidation of polycyclic aromatic hydrocarbons. Protein Eng 2000, 13, 121–128. [DOI] [PubMed] [Google Scholar]
- 36.Li QS; Schwaneberg U; Fischer M; Schmitt J; Pleiss J; Lutz-Wahl S; Schmid RD, Rational evolution of a medium chain-specific cytochrome P-450 BM-3 variant. Bba-Protein Struct M 2001, 1545, 114–121. [DOI] [PubMed] [Google Scholar]
- 37.Farinas ET; Schwaneberg U; Glieder A; Arnold FH, Directed evolution of a cytochrome P450 monooxygenase for alkane oxidation. Adv Synth Catal 2001, 343, 601–606. [Google Scholar]
- 38.Peters MW; Meinhold P; Glieder A; Arnold FH, Regio- and enantioselective alkane hydroxylation with engineered cytochromes P450 BM-3. J Am Chem Soc 2003, 125, 13442–13450. [DOI] [PubMed] [Google Scholar]
- 39.Meinhold P; Peters MW; Chen MMY; Takahashi K; Arnold FH, Direct conversion of ethane to ethanol by engineered cytochrome P450BM3. Chembiochem 2005, 6, 1765–1768. [DOI] [PubMed] [Google Scholar]
- 40.Fasan R; Chen MM; Crook NC; Arnold FH, Engineered alkane-hydroxylating cytochrome P450(BM3) exhibiting nativelike catalytic properties. Angew Chem Int Edit 2007, 46, 8414–8418. [DOI] [PubMed] [Google Scholar]
- 41.Fasan R; Meharenna YT; Snow CD; Poulos TL; Arnold FH, Evolutionary History of a Specialized P450 Propane Monooxygenase. J Mol Biol 2008, 383, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fasan R; Crook NC; Peters MW; Meinhold P; Buelter T; Landwehr M; Cirino PC; Arnold FH, Improved Product-Per-Glucose Yields in P450-Dependent Propane Biotransformations Using Engineered Escherichia coli. Biotechnol Bioeng 2011, 108, 500–510. [DOI] [PubMed] [Google Scholar]
- 43.Chen ZL; Zhao HM, Rapid creation of a novel protein function by in vitro coevolution. J Mol Biol 2005, 348, 1273–1282. [DOI] [PubMed] [Google Scholar]
- 44.Savile CK; Janey JM; Mundorff EC; Moore JC; Tam S; Jarvis WR; Colbeck JC; Krebber A; Fleitz FJ; Brands J; Devine PN; Huisman GW; Hughes GJ, Biocatalytic Asymmetric Synthesis of Chiral Amines from Ketones Applied to Sitagliptin Manufacture. Science 2010, 329, 305–309. [DOI] [PubMed] [Google Scholar]
- 45.Li H; Liao JC, Development of an NADPH-Dependent Homophenylalanine Dehydrogenase by Protein Engineering. ACS Synth Biol 2014, 3, 13–20. [DOI] [PubMed] [Google Scholar]
- 46.Payne JT; Poor CB; Lewis JC, Directed Evolution of RebH for Site-Selective Halogenation of Large Biologically Active Molecules. Angew Chem Int Edit 2015, 54, 4226–4230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Adeniran A; Stainbrook S; Bostick JW; Tyo KEJ, Detection of a Peptide Biomarker by Engineered Yeast Receptors. Acs Synth Biol 2018, 7, 696–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Affaticati PE; Dai SB; Payongsri P; Hailes HC; Tittmann K; Dalby PA, Structural Analysis of an Evolved Transketolase Reveals Divergent Binding Modes. Sci Rep 2016, 6:35716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cirino PC; Arnold FH, A self-sufficient peroxide-driven hydroxylation biocatalyst. Angew Chem Int Edit 2003, 42, 3299–3301. [DOI] [PubMed] [Google Scholar]
- 50.Salazar O; Cirino PC; Arnold FH, Thermostabilization of a cytochrome P450 peroxygenase. Chembiochem 2003, 4, 891–893. [DOI] [PubMed] [Google Scholar]
- 51.Wong TS; Arnold FH; Schwaneberg U, Laboratory evolution of cytochrome P450BM-3 monooxygenase for organic cosolvents. Biotechnol Bioeng 2004, 85, 351–358. [DOI] [PubMed] [Google Scholar]
- 52.Reetz MT; Jaeger KE, Enantioselective enzymes for organic synthesis created by directed evolution. Chem-Eur J 2000, 6, 407–412. [DOI] [PubMed] [Google Scholar]
- 53.Reetz MT, Laboratory Evolution of Stereoselective Enzymes: A Prolific Source of Catalysts for Asymmetric Reactions. Angew Chem Int Edit 2011, 50, 138–174. [DOI] [PubMed] [Google Scholar]
- 54.Tracewell CA; Arnold FH, Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr Opin Chem Biol 2009, 13, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Varadarajan N; Rodriguez S; Hwang BY; Georgiou G; Iverson BL, Highly active and selective endopeptidases with programmed substrate specificities. Nat Chem Biol 2008, 4, 290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Loughlin TL; Greene DN; Matsumura I, Diversification and specialization of HIV protease function during in vitro evolution. Mol Biol Evol 2006, 23, 764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nair NU; Zhao HM, Evolution in reverse: Engineering a D-xylose-specific xylose reductase. Chembiochem 2008, 9, 1213–1215. [DOI] [PubMed] [Google Scholar]
- 58.Bloom JD; Labthavikul ST; Otey CR; Arnold FH, Protein stability promotes evolvability. P Natl Acad Sci USA 2006, 103, 5869–5874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arnold FH, How proteins adapt: lessons from directed evolution. Cold Spring Harbor Symposia 2009, LXXIV, 41–46. [DOI] [PubMed] [Google Scholar]
- 60.Bloom JD; Arnold FH, In the light of directed evolution: Pathways of adaptive protein evolution. P Natl Acad Sci USA 2009, 106, 9995–10000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bloom JD; Romero PA; Lu ZY; Arnold FH, Neutral genetic drift can alter promiscuous protein functions, potentially aiding functional evolution. Biol Direct 2007, 2:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Drummond DA; Silberg JJ; Meyer MM; Wilke CO; Arnold FH, On the conservative nature of intragenic recombination. P Natl Acad Sci USA 2005, 102, 5380–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Otey CR; Landwehr M; Endelman JB; Hiraga K; Bloom JD; Arnold FH, Structure-guided recombination creates an artificial family of cytochromes P450. Plos Biol 2006, 4, 789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Landwehr M; Carbone M; Otey CR; Li YG; Arnold FH, Diversification of catalytic function in a synthetic family of chimeric cytochrome P450s. Chem Biol 2007, 14, 269–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li YG; Drummond DA; Sawayama AM; Snow CD; Bloom JD; Arnold FH, A diverse family of thermostable cytochrome P450s created by recombination of stabilizing fragments. Nat Biotechnol 2007, 25, 1051–1056. [DOI] [PubMed] [Google Scholar]
- 66.Heinzelman P; Snow CD; Wu I; Nguyen C; Villalobos A; Govindarajan S; Minshull J; Arnold FH, A family of thermostable fungal cellulases created by structure-guided recombination. P Natl Acad Sci USA 2009, 106, 5610–5615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Newhouse T; Baran PS, If C-H Bonds Could Talk: Selective C-H Bond Oxidation. Angew Chem Int Edit 2011, 50, 3362–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Whitehouse CJC; Bell SG; Wong LL, P450(BM3) (CYP102A1): connecting the dots. Chem Soc Rev 2012, 41, 1218–1260. [DOI] [PubMed] [Google Scholar]
- 69.Fasan R, Tuning P450 Enzymes as Oxidation Catalysts. Acs Catal 2012, 2 (4), 647–666. [Google Scholar]
- 70.Bernhardt R; Urlacher VB, Cytochromes P450 as promising catalysts for biotechnological application: chances and limitations. Appl Microbiol Biot 2014, 98, 6185–6203. [DOI] [PubMed] [Google Scholar]
- 71.Reetz MT, Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future. J Am Chem Soc 2013, 135, 12480–12496. [DOI] [PubMed] [Google Scholar]
- 72.Kubo T; Peters MW; Meinhold P; Arnold FH, Enantioselective epoxidation of terminal alkenes to (R)- and (S)-epoxides by engineered cytochromes P450BM-3. Chem-Eur J 2006, 12, 1216–1220. [DOI] [PubMed] [Google Scholar]
- 73.Landwehr M; Hochrein L; Otey CR; Kasrayan A; Backvall JE; Arnold FH, Enantioselective alpha-hydroxylation of 2-arylacetic acid derivatives and buspirone catalyzed by engineered cytochrome P450BM-3. J Am Chem Soc 2006, 128, 6058–6059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rentmeister A; Arnold FH; Fasan R, Chemo-enzymatic fluorination of unactivated organic compounds. Nat Chem Biol 2009, 5, 26–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhang KD; Shafer BM; Demars MD; Stern HA; Fasan R, Controlled Oxidation of Remote sp(3) C-H Bonds in Artemisinin via P450 Catalysts with Fine-Tuned Regio- and Stereoselectivity. J Am Chem Soc 2012, 134, 18695–18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guengerich FP, Cytochrome P450 enzymes in the generation of commercial products. Nat Rev Drug Discov 2002, 1, 359–366. [DOI] [PubMed] [Google Scholar]
- 77.Schroer K; Kittelmann M; Lutz S, Recombinant Human Cytochrome P450 Monooxygenases for Drug Metabolite Synthesis. Biotechnol Bioeng 2010, 106, 699–706. [DOI] [PubMed] [Google Scholar]
- 78.Orhan H; Vermeulen NPE, Conventional and Novel Approaches in Generating and Characterization of Reactive Intermediates from Drugs/Drug Candidates. Curr Drug Metab 2011, 12, 383–394. [DOI] [PubMed] [Google Scholar]
- 79.Caswell JM; O’Neill M; Taylor SJC; Moody TS, Engineering and application of P450 monooxygenases in pharmaceutical and metabolite synthesis. Curr Opin Chem Biol 2013, 17, 271–275. [DOI] [PubMed] [Google Scholar]
- 80.van Vugt-Lussenburg BMA; Damsten MC; Maasdijk DM; Vermeulen NPE; Commandeur JNM, Heterotropic and homotropic cooperativity by a drug-metabolising mutant of cytochrome P450BM3. Biochem Bioph Res Co 2006, 346, 810–818. [DOI] [PubMed] [Google Scholar]
- 81.van Vugt-Lussenburg BMA; Stjernschantz E; Lastdrager J; Oostenbrink C; Vermeulen NPE; Commandeur JNM, Identification of critical residues in novel drug metabolizing mutants of cytochrome P450BM3 using random mutagenesis. J Med Chem 2007, 50, 455–461. [DOI] [PubMed] [Google Scholar]
- 82.Otey CR; Bandara G; Lalonde J; Takahashi K; Arnold FH, Preparation of human metabolites of propranolol using laboratory-evolved bacterial cytochromes P450. Biotechnol Bioeng 2006, 93, 494–499. [DOI] [PubMed] [Google Scholar]
- 83.Sawayama AM; Chen MMY; Kulanthaivel P; Kuo MS; Hemmerle H; Arnold FH, A Panel of Cytochrome P450 BM3 Variants to Produce Drug Metabolites and Diversify Lead Compounds. Chem-Eur J 2009, 15, 11723–11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rentmeister A; Brown TR; Snow CD; Carbone MN; Arnold FH, Engineered Bacterial Mimics of Human Drug Metabolizing Enzyme CYP2C9. Chemcatchem 2011, 3, 1065–1071. [Google Scholar]
- 85.Lewis JC; Bastian S; Bennett CS; Fu Y; Mitsuda Y; Chen MM; Greenberg WA; Wong CH; Arnold FH, Chemoenzymatic elaboration of monosaccharides using engineered cytochrome P450BM3 demethylases. Proc Natl Acad Sci U S A 2009, 106, 16550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kolev JN; O’Dwyer KM; Jordan CT; Fasan R, Discovery of Potent Parthenolide-Based Antileukemic Agents Enabled by Late-Stage P450-Mediated C-H Functionalization. ACS Chem Biol 2014, 9, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lowell AN; DeMars MD; Slocum ST; Yu FA; Anand K; Chemler JA; Korakavi N; Priessnitz JK; Park SR; Koch AA; Schultz PJ; Sherman DH, Chemoenzymatic Total Synthesis and Structural Diversification of Tylactone-Based Macrolide Antibiotics through Late-Stage Polyketide Assembly, Tailoring, and C-H Functionalization. J Am Chem Soc 2017, 139, 7913–7920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Loskot SA; Romney DK; Arnold FH; Stoltz BM, Enantioselective Total Synthesis of Nigelladine A via Late-Stage C-H Oxidation Enabled by an Engineered P450 Enzyme. J Am Chem Soc 2017, 139, 10196–10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Singh R; Bordeaux M; Fasan R, P450-catalyzed intramolecular sp3 C—H amination with arylsulfonyl azide substrates. ACS Catal. 2014, 4, 546–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Singh R; Kolev JN; Sutera PA; Fasan R, Enzymatic C(sp(3))-H Amination: P450-Catalyzed Conversion of Carbonazidates into Oxazolidinones. ACS Catal. 2015, 5, 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bordeaux M; Tyagi V; Fasan R, Highly Diastereoselective and Enantioselective Olefin Cyclopropanation Using Engineered Myoglobin-Based Catalysts. Angew Chem Int Edit 2015, 54, 1744–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tyagi V; Bonn RB; Fasan R, Intermolecular carbene S-H insertion catalysed by engineered myoglobin-based catalysts. Chem Sci 2015, 6, 2488–2494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tyagi V; Fasan R, Myoglobin-Catalyzed Olefination of Aldehydes. Angew. Chem. Int. Ed. 2016, 55, 2512–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bajaj P; Sreenilayam G; Tyagi V; Fasan R, Gram-Scale Synthesis of Chiral Cyclopropane-Containing Drugs and Drug Precursors with Engineered Myoglobin Catalysts Featuring Complementary Stereoselectivity. Angew Chem Int Edit 2016, 55, 16110–16114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tinoco A; Steck V; Tyagi V; Fasan R, Highly Diastereo- and Enantioselective Synthesis of Trifluoromethyl-Substituted Cyclopropanes via Myoglobin-Catalyzed Transfer of Trifluoromethylcarbene. J Am Chem Soc 2017, 139, 5293–5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sreenilayam G; Moore EJ; Steck V; Fasan R, Stereoselective Olefin Cyclopropanation under Aerobic Conditions with an Artificial Enzyme Incorporating an Iron-Chlorin e6 Cofactor. Acs Catal 2017, 7, 7629–7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moore EJ; Steck V; Bajaj P; Fasan R, Chemoselective Cyclopropanation over Carbene Y-H Insertion Catalyzed by an Engineered Carbene Transferase. J Org Chem 2018, 83, 7480–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gober JG; Rydeen AE; Gibson-O’Grady EJ; Leuthaeuser JB; Fetrow JS; Brustad EM, Mutating a Highly Conserved Residue in Diverse Cytochrome P450s Facilitates Diastereoselective Olefin Cyclopropanation. Chembiochem 2016, 17, 394–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Key HM; Dydio P; Clark DS; Hartwig JF, Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Key HM; Dydio P; Liu ZN; Rha JYE; Nazarenko A; Seyedkazemi V; Cark DS; Hartwig JF, Beyond Iron: Iridium-Containing P450 Enzymes for Selective Cyclopropanations of Structurally Diverse Alkenes. ACS Central Sci 2017, 3, 302–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lebel H; Marcoux JF; Molinaro C; Charette AB, Stereoselective cyclopropanation reactions. Chem Rev 2003, 103, 977–1050. [DOI] [PubMed] [Google Scholar]
- 102.Wu WQ; Lin ZM; Jiang HF, Recent advances in the synthesis of cyclopropanes. Org Biomol Chem 2018, 16, 7315–7329. [DOI] [PubMed] [Google Scholar]
- 103.Coelho PS; Brustad EM; Kannan A; Arnold FH, Olefin Cyclopropanation via Carbene Transfer Catalyzed by Engineered Cytochrome P450 Enzymes. Science 2013, 339, 307–310. [DOI] [PubMed] [Google Scholar]
- 104.Coelho PS; Wang ZJ; Ener ME; Baril SA; Kannan A; Arnold FH; Brustad EM, A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat Chem Biol 2013, 9, 485–U33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bornscheuer UT; Huisman GW; Kazlauskas RJ; Lutz S; Moore JC; Robins K, Engineering the third wave of biocatalysis. Nature 2012, 485, 185–194. [DOI] [PubMed] [Google Scholar]
- 106.Tucker JL; Faul MM, Drug companies must adopt green chemistry. Nature 2016, 534, 27–29. [DOI] [PubMed] [Google Scholar]
- 107.Brandenberg OF; Fasan R; Arnold FH, Exploiting and engineering hemoproteins for abiological carbene and nitrene transfer reactions. Curr Opin Biotech 2017, 47, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Renata H; Wang ZJ; Kitto RZ; Arnold FH, P450-catalyzed asymmetric cyclopropanation of electron-deficient olefins under aerobic conditions. Catal Sci Technol 2014, 4, 3640–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Heel T; McIntosh JA; Dodani SC; Meyerowitz JT; Arnold FH, Non-natural Olefin Cyclopropanation Catalyzed by Diverse Cytochrome P450s and Other Hemoproteins. Chembiochem 2014, 15, 2556–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang ZJ; Renata H; Peck NE; Farwell CC; Coelho PS; Arnold FH, Improved Cyclopropanation Activity of Histidine-Ligated Cytochrome P450 Enables the Enantioselective Formal Synthesis of Levomilnacipran. Angew Chem Int Edit 2014, 53, 6810–6813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Reetz MT, Biocatalysis in Organic Chemistry and Biotechnology: Past, Present, and Future. J. Am. Chem. Soc 2013, 135, 12480–12496. [DOI] [PubMed] [Google Scholar]
- 112.Li GY; Wang JB; Reetz MT, Biocatalysts for the pharmaceutical industry created by structure-guided directed evolution of stereoselective enzymes. Bioorg. Med. Chem 2018, 26, 1241–1251. [DOI] [PubMed] [Google Scholar]
- 113.Turner NJ, Deracemisation methods. Curr. Opin. Chem. Biol 2010, 14, 115–121. [DOI] [PubMed] [Google Scholar]
- 114.Mugford PF; Wagner UG; Jiang Y; Faber K; Kazlauskas RJ, Enantiocomplementary Enzymes: Classification, Molecular Basis for Their Enantiopreference, and Prospects for Mirror-Image Biotransformations. Angew. Chem. Int. Ed 2008, 47, 8782–8793. [DOI] [PubMed] [Google Scholar]
- 115.Feske BD; Kaluzna IA; Stewart JD, Enantiodivergent, biocatalytic routes to both taxol side chain antipodes. J. Org. Chem 2005, 70, 9654–9657. [DOI] [PubMed] [Google Scholar]
- 116.Wu Q; Soni P; Reetz MT, Laboratory Evolution of Enantiocomplementary Candida antarctica Lipase B Mutants with Broad Substrate Scope. J. Am. Chem. Soc 2013, 135, 1872–1881. [DOI] [PubMed] [Google Scholar]
- 117.Zhang K; Shafer BM; Demars MD 2nd; Stern HA; Fasan R, Controlled oxidation of remote sp3 C-H bonds in artemisinin via P450 catalysts with fine-tuned regio- and stereoselectivity. J. Am. Chem. Soc 2012, 134, 18695–18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Koszelewski D; Grischek B; Glueck SM; Kroutil W; Faber K, Enzymatic Racemization of Amines Catalyzed by Enantiocomplementary omega-Transaminases. Chem. Eur. J 2011, 17, 378–383. [DOI] [PubMed] [Google Scholar]
- 119.France SP; Aleku GA; Sharma M; Mangas-Sanchez J; Howard RM; Steflik J; Kumar R; Adams RW; Slabu I; Crook R; Grogan G; Wallace TW; Turner NJ, Biocatalytic Routes to Enantiomerically Enriched Dibenz[c,e]azepines. Angew. Chem. Int. Ed 2017, 56, 15589–15593. [DOI] [PubMed] [Google Scholar]
- 120.Knight AM; Kan SBJ; Lewis RD; Brandenberg OF; Chen K; Arnold FH, Diverse Engineered Heme Proteins Enable Stereodivergent Cyclopropanation of Unactivated Alkenes. Acs Central Sci 2018, 4, 372–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Walczak MAA; Krainz T; Wipf P, Ring-Strain-Enabled Reaction Discovery: New Heterocycles from Bicyclo[1.1.0]butanes. Accounts Chem Res 2015, 48, 1149–1158. [DOI] [PubMed] [Google Scholar]
- 122.Chen ZX; Mercer JAM; Zhu XL; Romaniuk JAH; Pfattner R; Cegelski L; Martinez TJ; Burns NZ; Xia Y, Mechanochemical unzipping of insulating polyladderene to semiconducting polyacetylene. Science 2017, 357, 475–478. [DOI] [PubMed] [Google Scholar]
- 123.Chen K; Huang XY; Kan SBJ; Zhang RK; Arnold FH, Enzymatic construction of highly strained carbocycles. Science 2018, 360, 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kan SBJ; Lewis RD; Chen K; Arnold FH, Directed evolution of cytochrome c for carbon-silicon bond formation: Bringing silicon to life. Science 2016, 354, 1048–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kan SBJ; Huang XY; Gumulya Y; Chen K; Arnold FH, Genetically programmed chiral organoborane synthesis. Nature 2017, 552, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lewis RD; Garcia-Borras M; Chalkley MJ; Buller AR; Houk KN; Kan SBJ; Arnold FH, Catalytic iron-carbene intermediate revealed in a cytochrome C carbene transferase. P Natl Acad Sci USA 2018, 115, 7308–7313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Khade RL; Fan WC; Ling Y; Yang L; Oldfield E; Zhang Y, Iron Porphyrin Carbenes as Catalytic Intermediates: Structures, Mossbauer and NMR Spectroscopic Properties, and Bonding. Angew Chem Int Edit 2014, 53, 7574–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Khade RL; Zhang Y, Catalytic and Biocatalytic Iron Porphyrin Carbene Formation: Effects of Binding Mode, Carbene Substituent, Porphyrin Substituent, and Protein Axial Ligand. J Am Chem Soc 2015, 137, 7560–7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Khade RL; Zhang Y, C-H Insertions by Iron Porphyrin Carbene: Basic Mechanism and Origin of Substrate Selectivity. Chem-Eur J 2017, 23, 17654–17658. [DOI] [PubMed] [Google Scholar]
- 130.Wei Y; Tinoco A; Steck V; Fasan R; Zhang Y, Cyclopropanations via Heme Carbenes: Basic Mechanism and Effects of Carbene Substituent, Protein Axial Ligand, and Porphyrin Substitution. J Am Chem Soc 2018, 140, 1649–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Renata H; Lewis RD; Sweredoski MJ; Moradian A; Hess S; Wang ZJ; Arnold FH, Identification of Mechanism-Based Inactivation in P450-Catalyzed Cyclopropanation Facilitates Engineering of Improved Enzymes. J Am Chem Soc 2016, 138, 12527–12533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sharon DA; Mallick D; Wang BJ; Shaik S, Computation Sheds Insight into Iron Porphyrin Carbenes’ Electronic Structure, Formation, and N-H Insertion Reactivity. J Am Chem Soc 2016, 138, 9597–9610. [DOI] [PubMed] [Google Scholar]
- 133.Hayashi T; Tinzl M; Mori T; Krengel U; Proppe J; Soetbeer J; Klose D; Jeschke G; Reiher M; Hilvert D, Capture and characterization of a reactive haem-carbenoid complex in an artificial metalloenzyme. Nat Catal 2018, 1, 578–584. [Google Scholar]
- 134.Chen K; Huang XY; Zhang SQ; Zhou AZ; Kan SBJ; Hong X; Arnold FH, Engineered Cytochrome c -Catalyzed Lactone-Carbene B-H Insertion. Synlett 2019, 30, 378–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Huang XY; Garcia-Borras M; Miao K; Kan SBJ; Zutshi A; Houk KN; Arnold FH, A Biocatalytic Platform for Synthesis of Chiral alpha-Trifluoromethylated Organoborons. Acs Central Sci 2019, 5, 270–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Walker MC; Chang MCY, Natural and engineered biosynthesis of fluorinated natural products. Chem Soc Rev 2014, 43, 6527–6536. [DOI] [PubMed] [Google Scholar]
- 137.O’Hagan D; Deng H, Enzymatic Fluorination and Biotechnological Developments of the Fluorinase. Chem Rev 2015, 115, 634–649. [DOI] [PubMed] [Google Scholar]
- 138.Zhang RJK; Huang XY; Arnold FH, Selective C-H bond functionalization with engineered heme proteins: new tools to generate complexity. Curr Opin Chem Biol 2019, 49, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rittle J; Green MT, Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 2010, 330, 933–937. [DOI] [PubMed] [Google Scholar]
- 140.Svastits EW; Dawson JH; Breslow R; Gellman SH, Functionalized Nitrogen Atom Transfer Catalyzed by Cytochrome-P-450. J Am Chem Soc 1985, 107, 6427–6428. [Google Scholar]
- 141.McIntosh JA; Coelho PS; Farwell CC; Wang ZJ; Lewis JC; Brown TR; Arnold FH, Enantioselective Intramolecular C-H Amination Catalyzed by Engineered Cytochrome P450 Enzymes In Vitro and In Vivo. Angew Chem Int Edit 2013, 52, 9309–9312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Bordeaux M; Singh R; Fasan R, Intramolecular C(sp(3))H amination of arylsulfonyl azides with engineered and artificial myoglobin-based catalysts. Bioorg. Med. Chem 2014, 22, 5697–5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hyster TK; Farwell CC; Buller AR; McIntosh JA; Arnold FH, Enzyme-Controlled Nitrogen-Atom Transfer Enables Regiodivergent C-H Amination. J Am Chem Soc 2014, 136, 15505–15508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cho I; Jia ZJ; Arnold FH, Site-selective enzymatic C-H amidation for synthesis of diverse lactams. Science 2019, 364, 575–578. [DOI] [PubMed] [Google Scholar]
- 145.Tsutsumi H; Katsuyama Y; Izumikawa M; Takagi M; Fujie M; Satoh N; Shin-ya K; Ohnishi Y, Unprecedented Cyclization Catalyzed by a Cytochrome P450 in Benzastatin Biosynthesis. J Am Chem Soc 2018, 140, 6631–6639. [DOI] [PubMed] [Google Scholar]
- 146.Prier CK; Hyster TK; Farwell CC; Huang A; Arnold FH, Asymmetric Enzymatic Synthesis of Allylic Amines: A Sigmatropic Rearrangement Strategy. Angew Chem Int Edit 2016, 55, 4711–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Prier CK; Zhang RJK; Buller AR; Brinkmann-Chen S; Arnold FH, Enantioselective, intermolecular benzylic C-H amination catalysed by an engineered iron-haem enzyme. Nat Chem 2017, 9, 629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Zhang RJK; Chen K; Huang XY; Wohlschlager L; Renata H; Arnold FH, Enzymatic assembly of carbon-carbon bonds via iron-catalysed sp(3) C-H functionalization. Nature 2019, 565, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]