

Abstract
Inhibition of Hsp90 is associated with anti-inflammatory effects. We employed human lung microvascular endothelial cells to investigate the effects of the Hsp90 inhibitors 17-AAG, AUY-922 and 17-DMAG in the unfolded protein response (UPR) and viability of lung cells. Our observations indicate that moderate doses of those compounds trigger the activation of the UPR without inducing lethal effects in vitro. Indeed, AUY-922 triggered UPR activation in the lungs of C57BL/6 mice. UPR has been previously involved in the enhancement of the lung endothelial barrier function. Thus, the present study suggests that the barrier protective effects of Hsp90 inhibition in the lung microvasculature are highly probable to be associated with the activation of the UPR. Hence, the development of novel compounds which stochastically capacitate the repairing elements of UPR, may deliver new therapeutic possibilities against the severities of the acute respiratory distress syndrome.
Keywords: Inflammation, P53, Acute Lung Injury, Acute Respiratory Distress Syndrome
1. Introduction
Heat shock protein 90 (Hsp90) is a highly conserved molecular chaperone, which supports the folding and maturation of several proteins, including transcription factors. Those client proteins are essential elements in the regulation of crucial cellular processes, including proliferation, differentiation and defense [1]. Malignancies employ Hsp90 to hyper-activate signaling cascades, which in turn promote metastasis and cancer prevalence. Thus, the concept of developing Hsp90 inhibitors to counteract tumors appear to be a promising therapeutic strategy in oncology [2].
Cancer and inflammation are highly interrelated pathological conditions, which co-exist very frequently in human and other tissues. Inflammation contributes towards the induction and sustenance of dysregulated cellular proliferation, promoting tumor development and metastasis. Hence, agents with anti-inflammatory properties may exert tumor suppressor activities [3].
P53 is a 53 kDa protein, destined to protect the cells against extracellular and intracellular threats. It was first considered to be an oncogene, but it was later discovered that it is a tumor suppressor protein, capable of opposing environmental challenges. Depending on the severity of those stimuli, it dictates a wide range of cellular responses, including growth arrest and apoptosis [4,5]. Indeed, P53 propels anti-inflammatory effects in diseased tissues, including the lungs [6,7].
In human and bovine lung microvascular cells the endothelial defender [4] was shown to orchestrate the anti-inflammatory effects of Hsp90 inhibitors in the LPS-inflicted Acute Lung Injury (ALI) [8]. Those compounds augmented P53 expression levels, which in turn suppressed the inflammatory RhoA-MLCII pathway [9], and induced the deactivation of cofilin [10]. Moreover, P53 suppressed the APE1/Ref1 [11], as well as the LPS-induced P53 phosphorylation [8,12]. AUY-922 suppressed the generation of reactive oxidant species in the lungs, by inducing P53 [13].
UPR is a molecular machinery which activates survival mechanisms in support of cellular adaption under various environmental conditions. Hence, it may either repair or eliminate the cells, depending on the severity of the injury; and the intensity of the corresponding endoplasmic reticulum (ER) stress. Three different proteins are key UPR components. The activating transcription factor 6 (ATF6), the protein kinase RNA-like ER kinase (PERK), and the inositol-requiring enzyme-1α (IRE1α) [14]. Those branches are highly associated with the binding immunoglobulin protein (BiP), as well as with the C/EBP homologous protein (CHOP). Increased ER stress may activate ATF6, PERK, and IRE1α [15,16]. Our recent studies demonstrated that UPR activation is affecting the abundance of P53. In bovine pulmonary arterial endothelial cells (BPAEC); treatment with UPR activators induced P53, while UPR inhibition produced the opposite effects [17].
The present study explores the effects of Hsp90 inhibition in the UPR activation of commercially available human lung cells and mice lungs. Furthermore, it examines the effects of Hsp90 inhibition in lung cell proliferation, to identify possible toxic effects of Hsp90 inhibition in the lungs. Our observations suggest that moderate doses of Hsp90 inhibitors induce UPR, and that those effects are not associated with lethal effects in vitro. Hence, it is highly probable, that UPR induction facilitates the anti-inflammatory responses of those compounds in the pulmonary micro-vasculature. That may be due to the fact that UPR has been shown to repair the inflamed lung endothelium [15,18]. However, the exacts mechanisms which may be involved in those events are to be elucidated in future studies, by employing genetically modified experimental material.
2. Materials and methods
2.1. Reagents
17-AAG (AAJ66960-EX3), AUY-922 (101756–820),17-DMAG (102513–662), RIPA buffer (AAJ63306-AP), Trypan Blue (76180–676), sheep anti-mouse IgG HRP-linked polyclonal antibody (95017–554), donkey anti-rabbit IgG HRP-linked polyclonal antibody (95017–556), EZBlock™ protease inhibitor cocktail (75837–938) and nitrocellulose membranes (10063–173) were obtained from VWR (Radnor, PA). The ATF-6 (65,880 s), PERK (5683 s), Phospho-PERK (Thr980) (3179 s), IRE-1α (3294 s), BiP (3183 s), CHOP (5554 s), PDI (2446 s), and Ero1-Lα (3264 s) antibodies were purchased from Cell Signaling (Danvers, MA). The β-actin (A5441–0.5 ml) antibody was purchased from Sigma-Aldrich (St Louis, MO). The phospho-IRE1α antibody (Ser724) (16927) was obtained from Thermo Fisher Scientific (Waltham, MA).
2.2. Cell Culture
Human Lung Microvascular Endothelial Cells (HuLEC-5a) (CRL-3244) were purchased from the American Type Culture Collection (Manassas, VA). Those cells were maintained at 37 °C in a humidified atmosphere of 5% CO2/95% air in PromoCell Endothelial Cell Growth Medium MV (10172–280), supplemented with 1× penicillin/streptomycin (97063–708). All reagents were purchased from VWR (Radnor, PA).
2.3. Animals
Seven-week old C57BL/6 (male) mice were purchased from Envigo (Indianapolis, IN). They were maintained in a 12:12-h light/dark cycle, in pathogen free conditions. The temperature was controlled (22–24°C), as well as the humidity (50–60%). All experimental procedures were approved by the University of Louisiana Monroe Institutional Animal Care and Use Committee (IACUC), and were in line with the principles of humane animal care adopted by the American Physiological Society.
2.4. In vivo treatments
C57BL/6 mice were treated with either vehicle (10% DMSO in saline) or AUY-922 (10 mg/kg each) via an intra-peritoneal injection. The animals were sacrificed by cervical dislocation after 48 h of the treatment. All procedures were approved by the Committee on Animal Research at University of Louisiana Monroe.
2.5. Western Blot Analysis
Proteins were isolated from the cells or tissues using RIPA buffer, and the EZBlock™ protease inhibitor cocktail. Protein concentration was calculated by employing the BCA protein assay method. Equal amounts of protein samples were separated onto sodium dodecyl sulfate polyacrylamide gel electrophoresis. Wet transfer was used to transfer the proteins onto the nitrocellulose membranes. After incubating the membranes for one hour at room temperature using 5% non-fat dry milk, the blots were exposed overnight (4 °C) to appropriate primary antibodies (1:1000). The following day, the membranes were incubated with the appropriate HRP-linked secondary antibody (1:2000) for 2 h at room temperature. Protein signal was detected using chemiluminescent substrate (Thermo Scientific). All the images were captured using ChemiDoc™ Touch Imaging System from Bio-Rad (Hercules, CA). The β-actin was used as a loading control, unless indicated otherwise. All reagents were purchased from VWR (Radnor, PA).
2.6. Cell Proliferation Assay
The number of cells was determined by seeding 10,000 cells onto 24-well plates. After 96 h of treatment with the Hsp90 inhibitors, the cells were counted under light microscope with an hemocytometer, and employing the trypan blue assay.
2.7. Densitometry and Statistical Analysis
The densitometry of the immunoblots was performed by using Image J software (NIH). All values are expressed as the mean ± SEM (standard error of mean). Graphpad Prism (version 5.01) was used to analyze the data. Values of P <.05 were considered as an indication of statistical significance. The number of experimental repeats is indicated by the letter n.
3. Results
3.1. 17-AAG (1 μM) induces the UPR in HuLEC
Human lung endothelial cells were seeded onto a 6-well plate and were exposed to either vehicle (0.1% DMSO), or 17-AAG (1 μM) for 4, 6, 8, 16 and 48 h. This Hsp90 inhibitor activated the UPR branches in all treatments, as reflected in the expression levels of cATF6 (Fig. 1A), pIRE1α (Fig. 1B), and pPERK (Fig. 1C). BiP (Fig. 1D), ERO1-Lα (Fig. 1E), and PDI (Fig. 1F) were also induced after 4, 6, 8, 16 and 48 h of treatment with this compound.
Fig. 1. Activation of UPR by 17-AAG (1μM) in HuLEC.
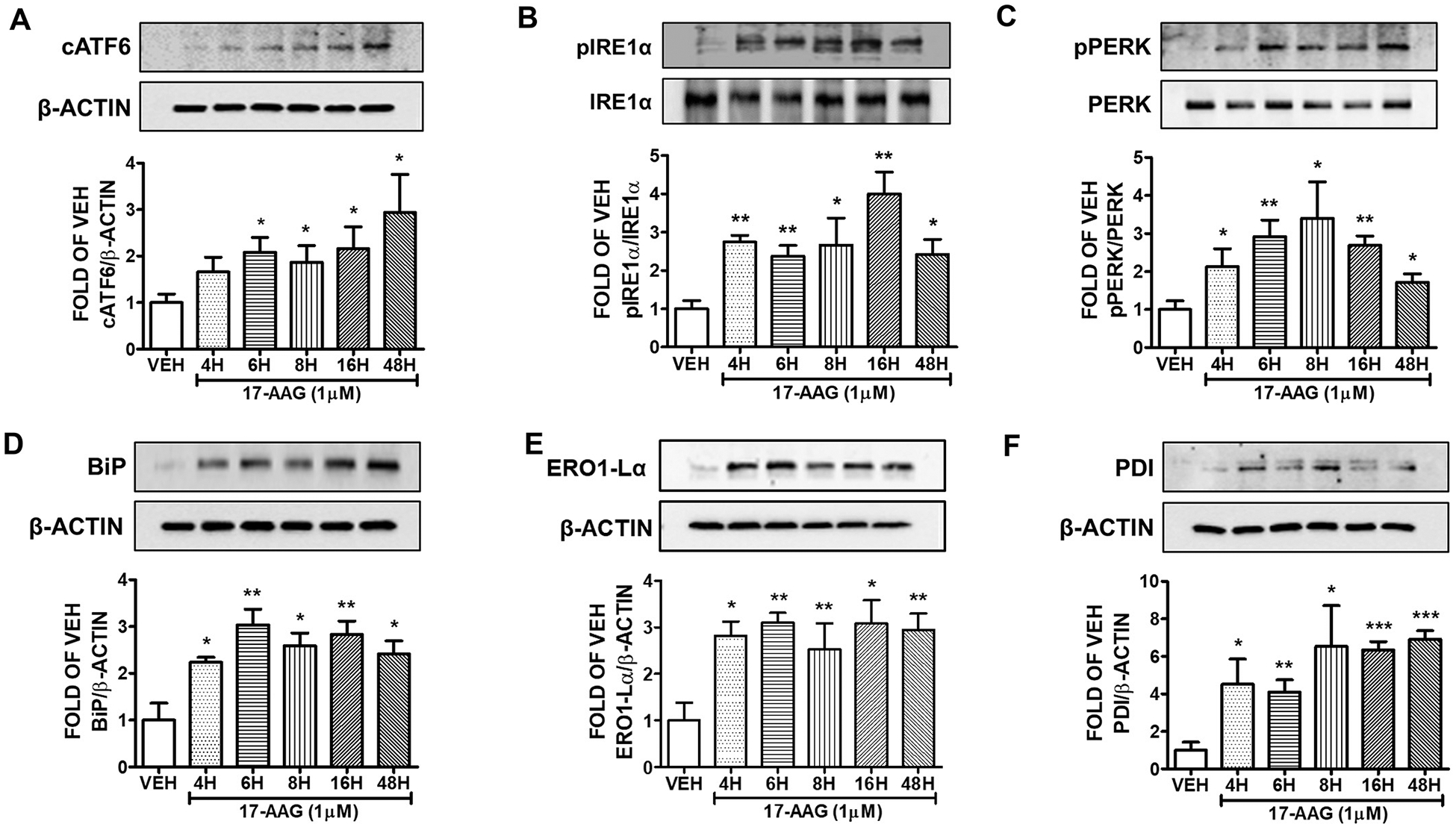
Western Blot analysis of (A) cATF6 and β-actin (B) pIRE1α and IRE1α (C) pPERK and PERK (D) BiP and β-actin (E) ERO1-Lα and β-actin (F) PDI and β-actin after treatment of HuLEC with VEH (DMSO) or 17-AAG (1μM). The blots represent three independent experiments. Densitometric analysis was performed to evaluate the signal intensity of cATF6, pIRE1α, pPERK, BiP, ERO1-Lα and PDI. B-actin was used for the normalization of the protein bands, unless otherwise stated. *P < .05, **P < .01, ***P < .001 vs vehicle (VEH). Means ± SEM.
3.2. Effects of 17-AAG (2 μM) in the UPR of HuLEC
The cells were treated with either vehicle (0.1% DMSO) or 2 μM 17-AAG for 4, 6, 8, 16 and 48 h. 17-AAG increased the expression levels of pIRE1α (Fig. 2A) and pPERK (Fig. 2B) in all treatments. BiP (Fig. 2C), ERO1-Lα (Fig. 2E), and PDI (Fig. 2F) were also induced after 4, 6, 8, 16 and 48 h treatment. Indeed, the ER stress marker CHOP was significantly induced after 16 and 48 h of exposure (Fig. 2D).
Fig. 2. Activation of UPR by 17-AAG (2μM) in HuLEC.
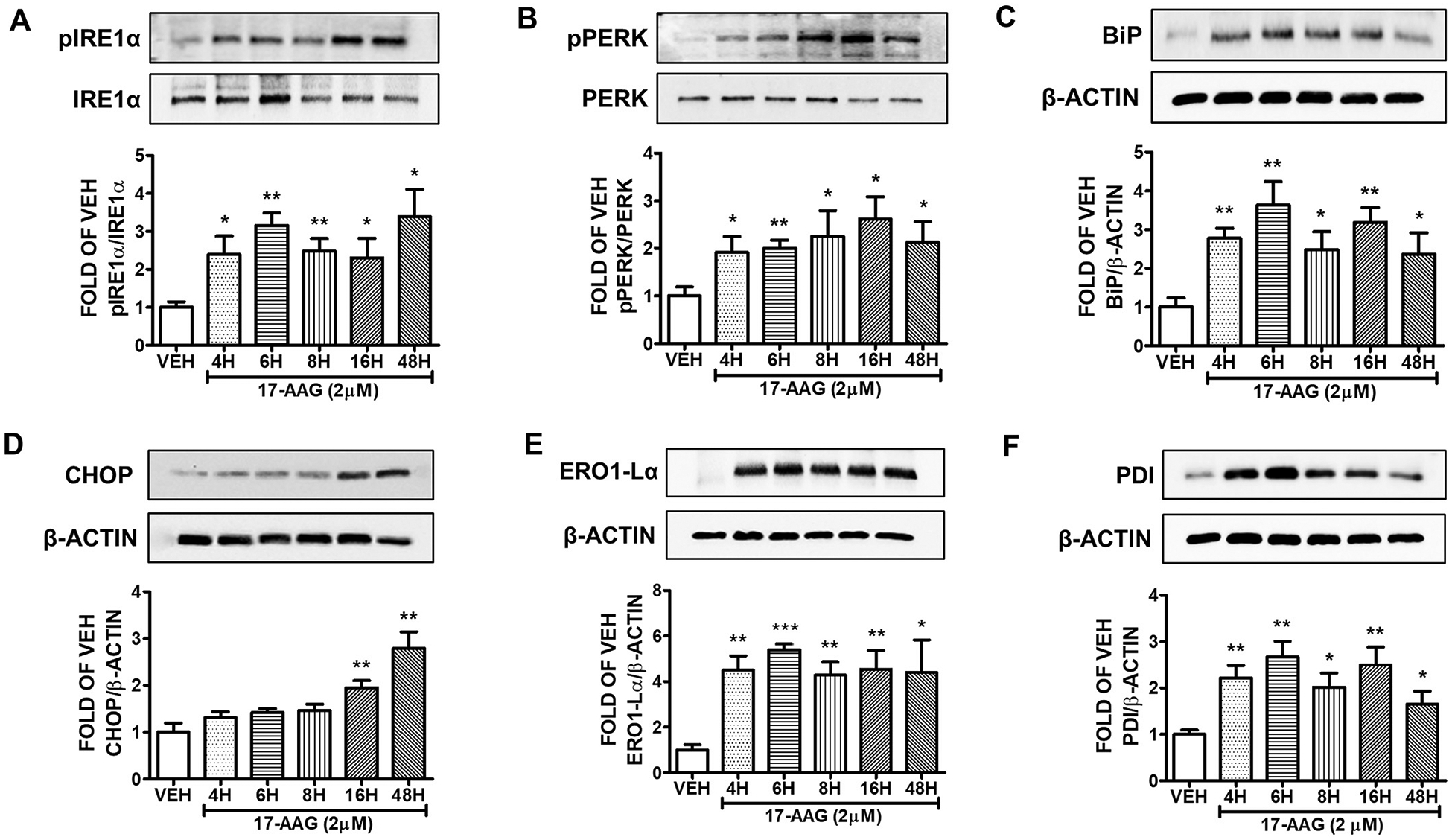
Western Blot analysis of (A) pIRE1α and IRE1α (B) pPERK and PERK (C) BiP and β-actin (D) CHOP and β-actin (E) ERO1-Lα and β-actin (F) PDI and β-actin after treatment of HuLEC with VEH (DMSO) or 17-AAG (2μM). The blots represent three independent experiments. Densitometric analysis was performed to evaluate the signal intensity of the proteins of interest. B-actin was used for the normalization of the protein bands, unless otherwise indicated. *P < .05, **P < .01, ***P < .001 vs vehicle (VEH). Means ± SEM.
3.3. AUY-922 (1 μM) induces UPR in human lung cells
HuLEC were exposed to1 μM AUY-922 or vehicle (0.1% DMSO) for 4, 6, 8, 16 and 48 h. This Hsp90 inhibitor induced the expression of cATF6 (Fig. 3A), pIRE1α (Fig. 3B), and pPERK (Fig. 3C) in all tretaments. BiP (Fig. 3D), ERO1-Lα (Fig. 3E), and PDI (Fig. 3F) expression levels were also elevated due to Hsp90 inhibition.
Fig. 3. Activation of UPR by AUY-922 (1μM) in HuLEC.
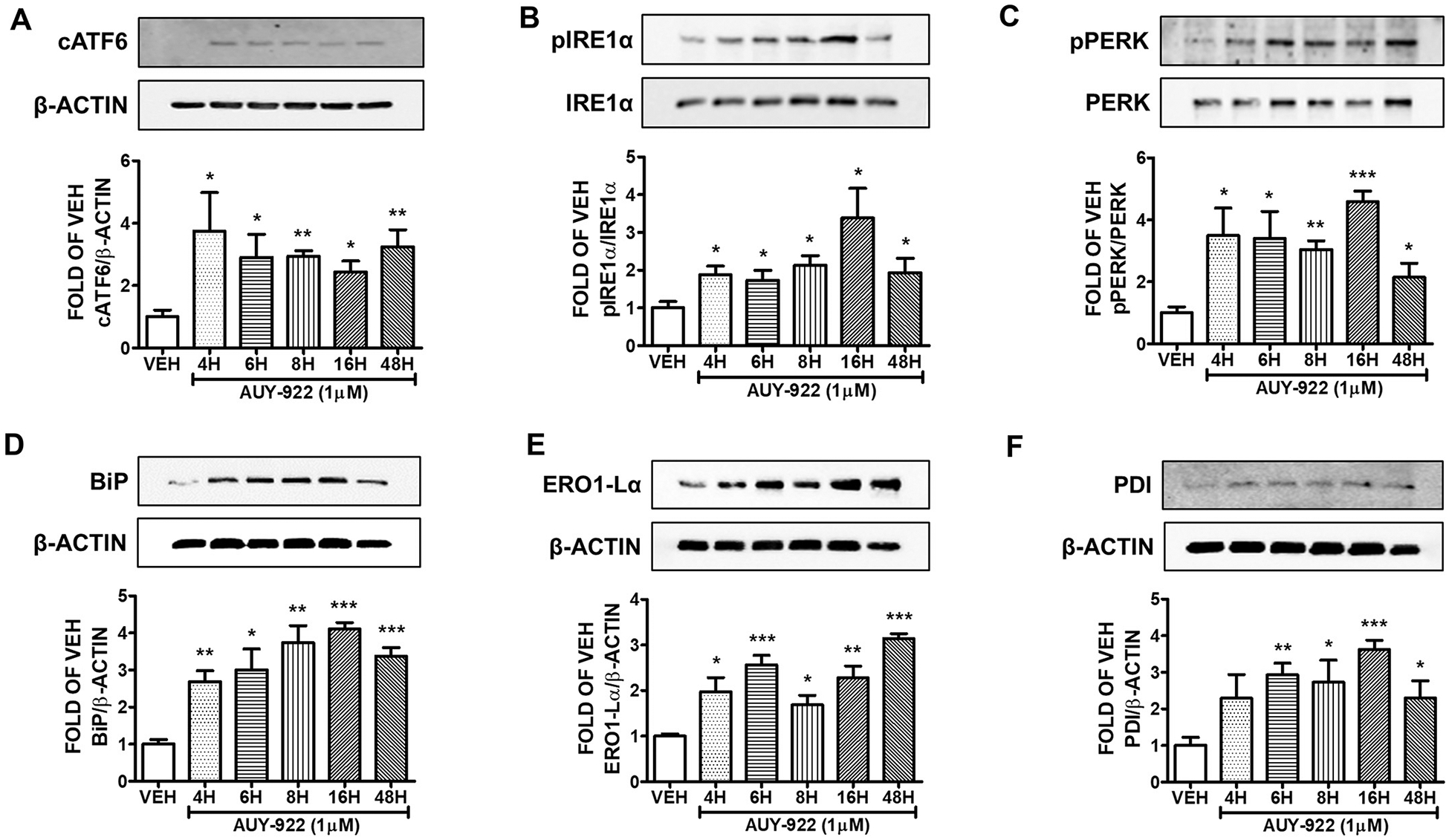
Western Blot analysis of (A) cATF6 and β-actin (B) pIRE1α and IRE1α (C) pPERK and PERK (D) BiP and β-actin (E) ERO1-Lα and β-actin (F) PDI and β-actin after treatment of HuLEC with VEH (DMSO) or AUY-922 (1μM). The blots represent three independent experiments. Densitometric analysis was performed to evaluate the signal intensity of the proteins of interest. B-actin was used for the normalization of the protein bands, unless otherwise indicated. *P < .05, **P < .01, ***P < .001 vs vehicle (VEH). Means ± SEM.
3.4. AUY-922 (2μM) activates UPR in HuLEC
Human lung cells were exposed to 2 μM of AUY-922 or vehicle (0.1% DMSO) for 4, 6, 8, 16 and 48 h. The expression levels of cATF6 (Fig. 4A), pIRE1α (Fig. 4B), and pPERK (Fig. 4C) were induced due to that treatment. Fig. 4A indicates that the highest induction of cATF6 occurred after 8 and 16 h of exposure. BiP (Fig. 4D), ERO1-Lα (Fig. 4E), and PDI (Fig. 4F) expression levels indicate the induction of the UPR machinery due to AUY-922 treatment.
Fig. 4. Activation of UPR by AUY-922 (2 μM) in HuLEC.
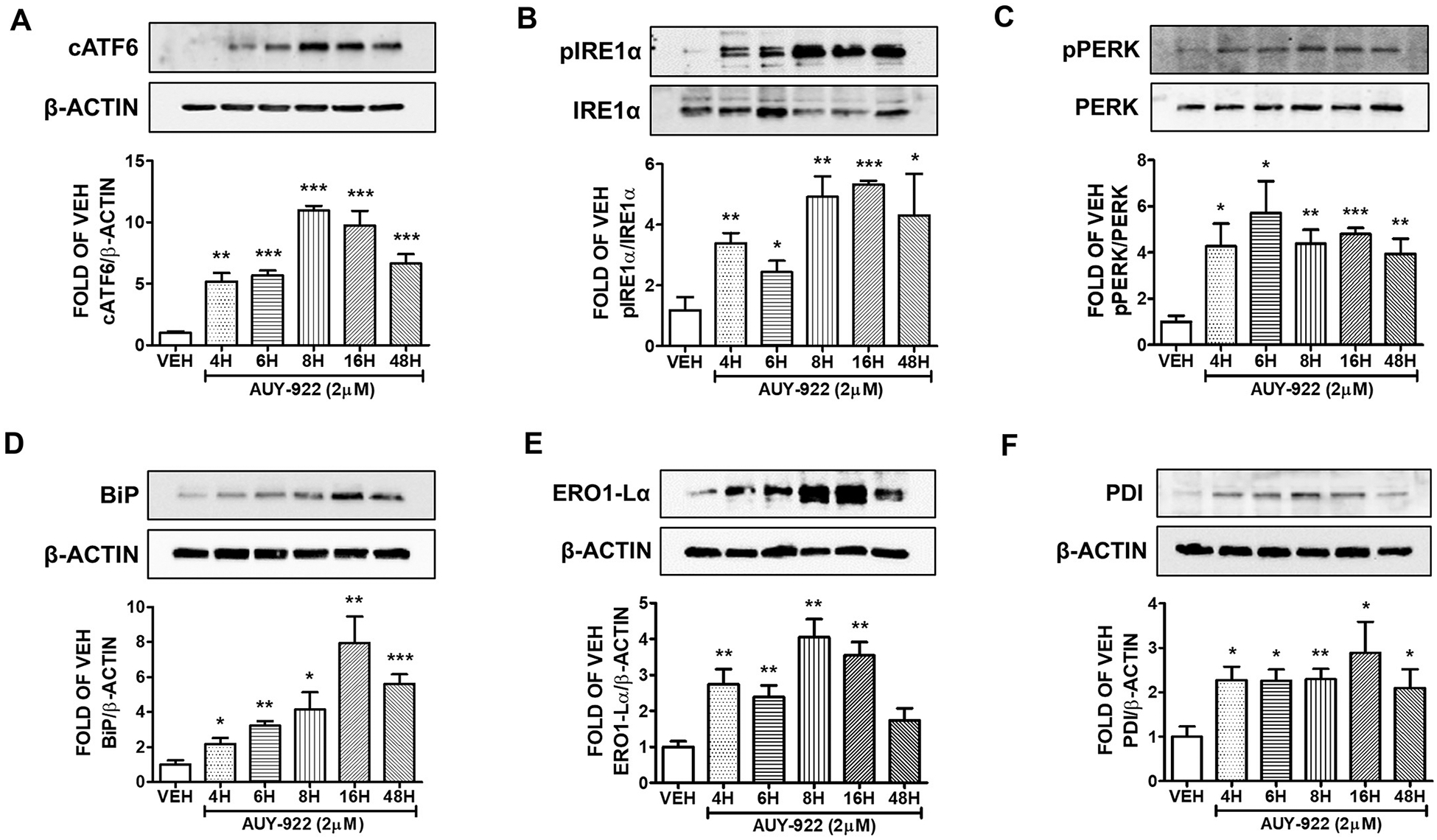
Western Blot analysis of (A) cATF6 and β-actin (B) pIRE1α and IRE1α (C) pPERK and PERK (D) BiP and β-actin (E) ERO1-Lα and β-actin (F) PDI and β-actin after treatment of HuLEC with VEH (DMSO) or AUY-922 (2 μM). The blots represent three independent experiments. Densitometric analysis was performed to evaluate the signal intensity of the proteins of interest. B-actin was used for the normalization of the protein bands, unless otherwise stated. *P < .05, **P < .01, ***P < .001 vs vehicle (VEH). Means ± SEM.
3.5. Effects of AUY-922 (10 μM) in the UPR of human lung cell
Human pulmonary microvascular cells were treated with either 10 μM of AUY-922 or vehicle (0.1%DMSO) for 4, 6, 8, 16 and 48 h. AUY-922 triggered UPR, since it induced the expression levels of cATF6 (Fig. 5A), pIRE1α (Fig. 5B), and pPERK (Fig. 5C) in all time periods. The expression levels of the UPR marker BiP (Fig. 5D) and PDI (Fig. 5F) were also induced after 4, 6, 8, 16 and 48 h of AUY-922 treatment. CHOP expression was induced after 8, 16 and 48 h (Fig. 5E).
Fig. 5. Activation of UPR by AUY-922 (10 μM) in HuLEC.
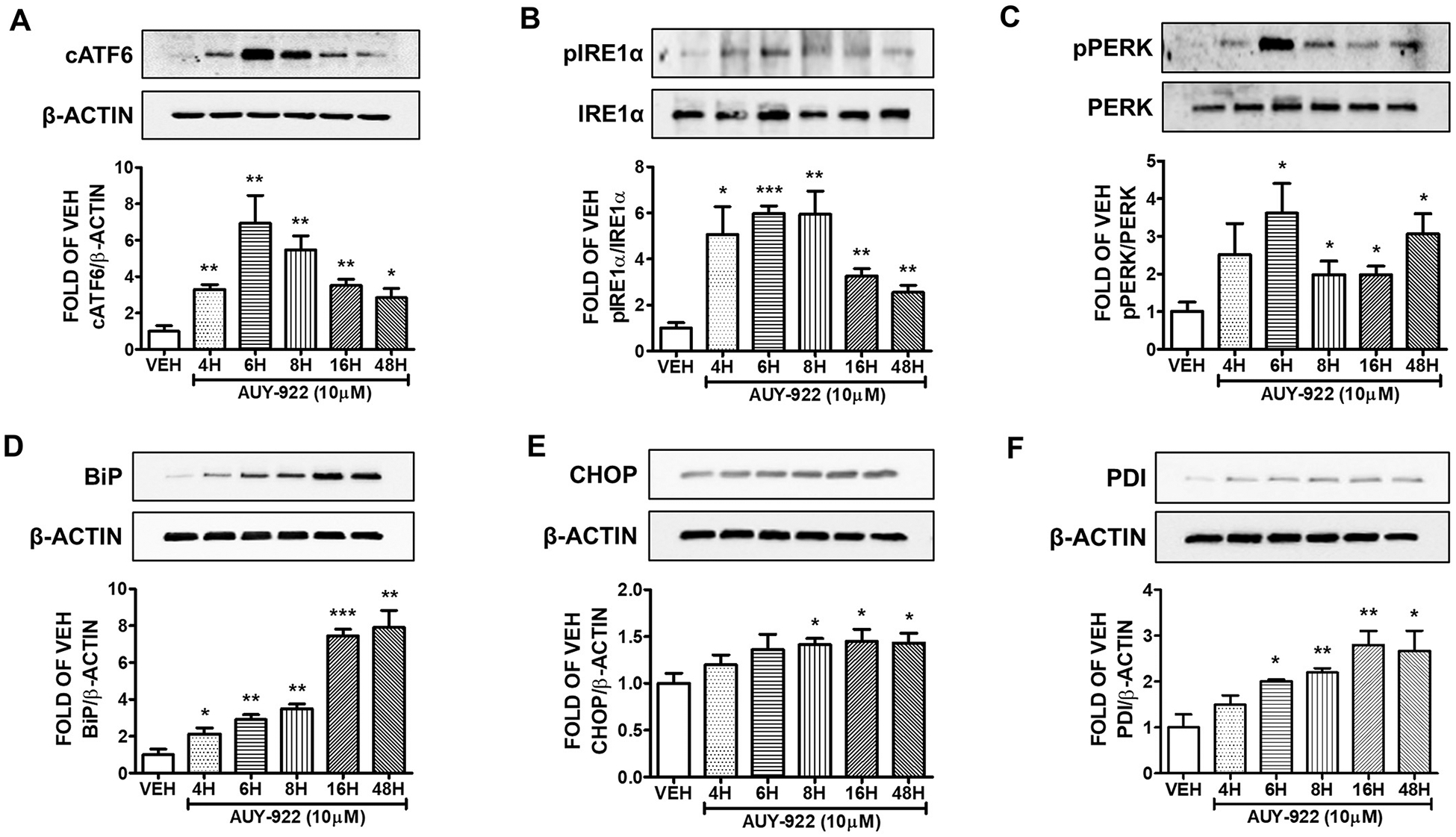
Western Blot analysis of (A) cATF6 and β-actin (B) pIRE1α and IRE1α (C) pPERK and PERK (D) BiP and β-actin (E) CHOP and β-actin (F) PDI and β-actin after treatment of HuLEC with VEH (DMSO) or AUY-922 (10 μM). The blots represent three independent experiments. Densitometric analysis was performed to evaluate the signal intensity of the proteins of interest. B-actin was used for the normalization of the protein bands, unless otherwise stated. *P < .05, **P < .01, ***P < .001 vs vehicle (VEH). Means ± SEM.
3.6. 17-DMAG (2 μM) activates the UPR of the lungs
The lung endothelial cells were incubated with either 2 μM of 17-DMAG or vehicle (0.1% DMSO) for 4, 6, 8, 16 and 48 h. That compound elevated the expression of cATF6 (Fig. 6A), pIRE1α (Fig. 6B), and pPERK (Fig. 6C) in all treatments. The increased expression of BiP (Fig. 6D), CHOP (Fig. 6E), and ERO1-Lα (Fig. 6F) after this exposure manifest the UPR activation.
Fig. 6. Activation of UPR by 17-DMAG (2 μM) in HuLEC.
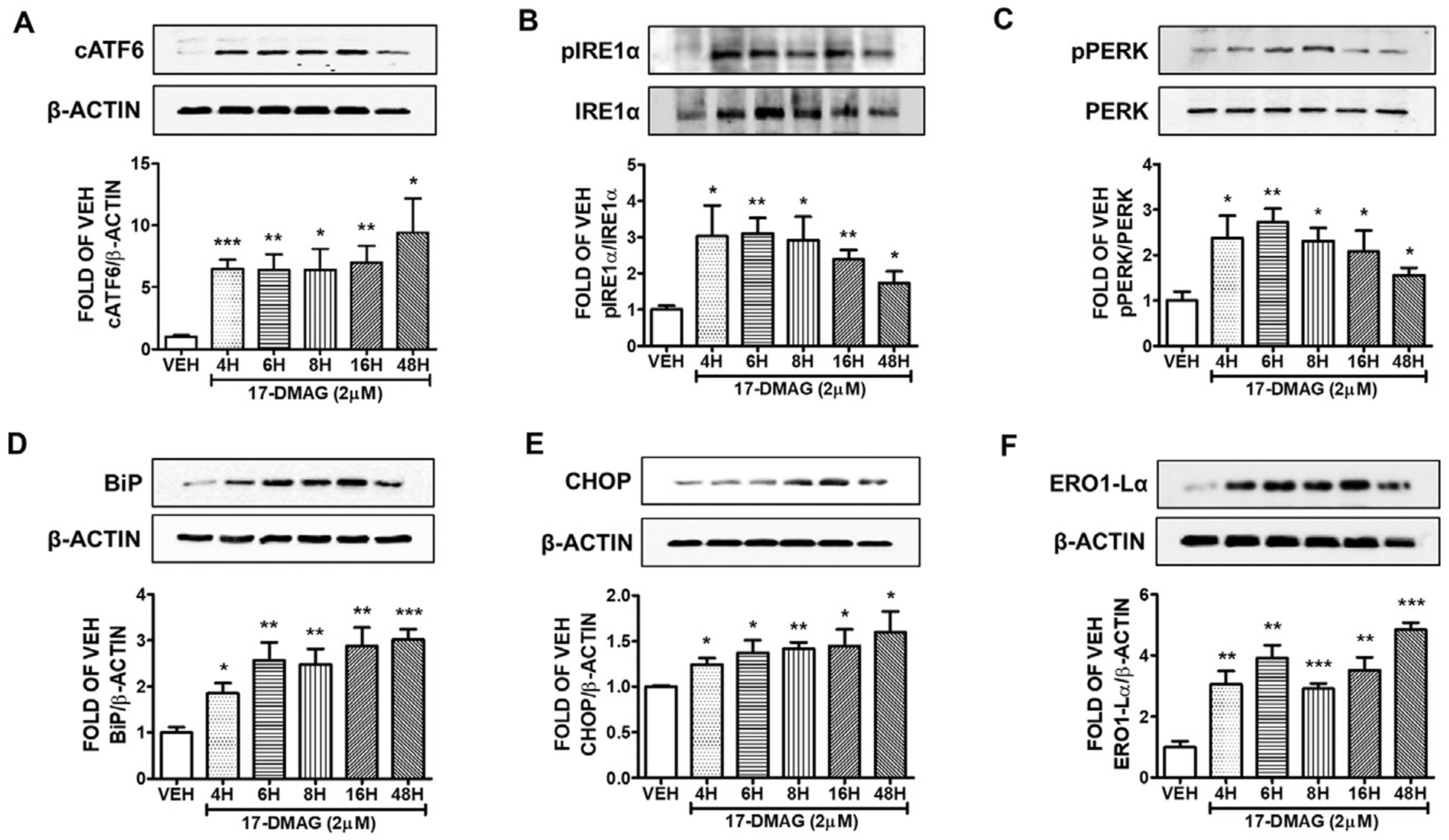
Western Blot analysis of (A) cATF6 and β-actin (B) pIRE1α and IRE1α (C) pPERK and PERK (D) BiP and β-actin (E) CHOP and β-actin (F) ERO1-Lα and β-actin after treatment of HuLEC with VEH (DMSO) or 17-DMAG (2 μM). The blots represent three independent experiments. Densitometric analysis was performed to evaluate the signal intensity of the proteins of interest. B-actin was used for the normalization of the protein bands, unless otherwise stated. *P < .05, **P < .01, ***P < .001 vs vehicle (VEH). Means ± SEM.
3.7. Effects of AUY-922 in the UPR activation in vivo
C57BL/6 mice were subjected to an intra-peritoneal injection of vehicle (10% DMSO) or AUY-922 (10 mg/kg) and were sacrificed 48 h after those treatments. The results shown in Fig. 7 demonstrate that the Hsp90 inhibition due to AUY-922 induced the BiP and CHOP expression levels in the mice lungs.
Fig. 7. Activation of UPR by AUY-922 in vivo.
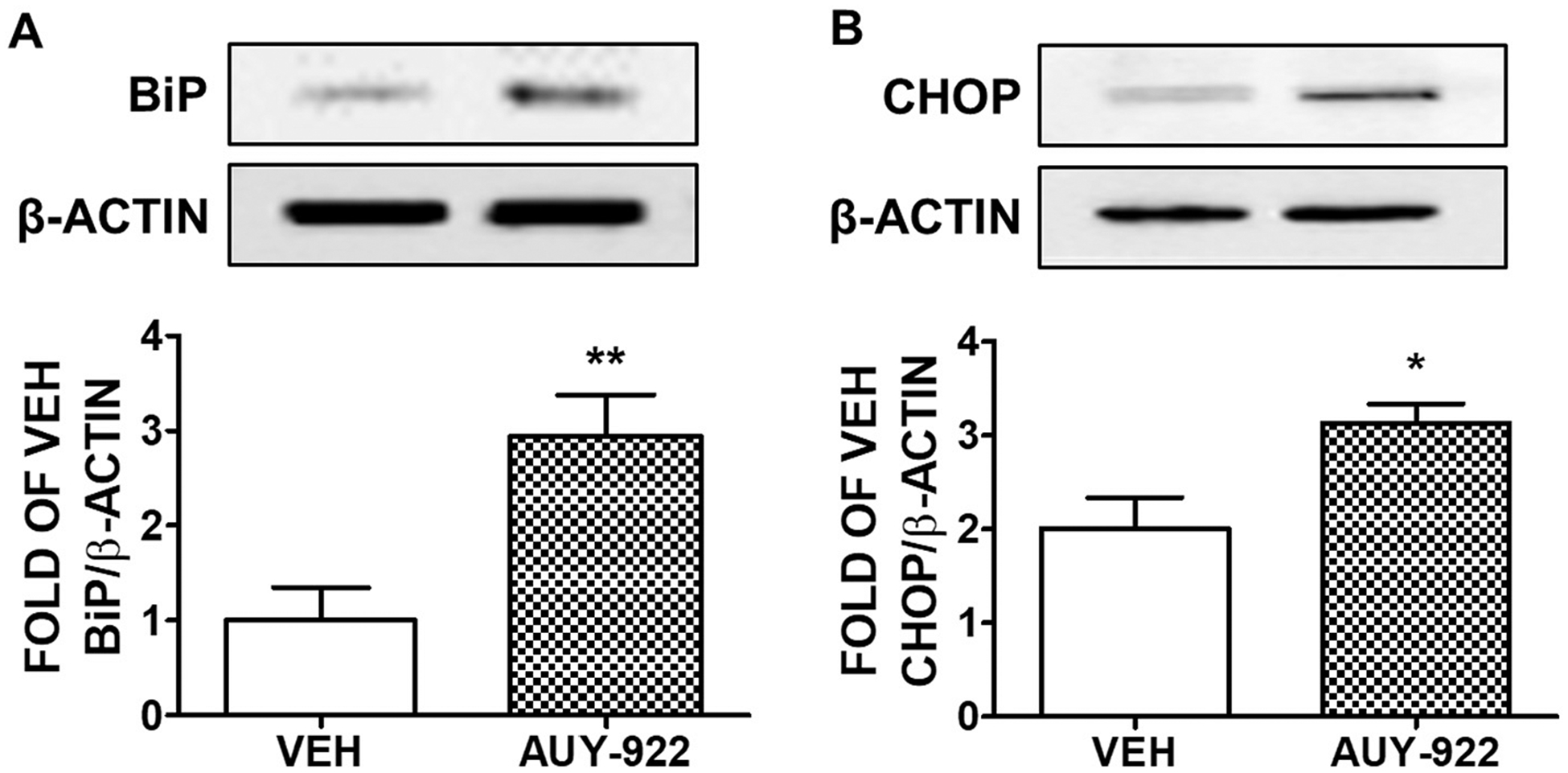
Western blot analysis of (A) BiP and β-actin (B) CHOP and β-actin in C57BL/6 mice lungs treated with either vehicle (10% DMSO in saline) or AUY-922 (10 mg/kg each) via an intra-peritoneal injection for 48 h. Densitometric analysis was performed to measure the BiP and CHOP expression levels. The β-actin was used as a loading control. *P < .05, **P < .01 vs vehicle (VEH), n = 4. Means ± SEM.
3.8. Effects of Hsp90 inhibitors in the proliferation of human lung microvascular endothelial cells
Human lung endothelial cells (10,000) were seeded in each well of a 24-well plate, and were treated with vehicle (0.1% DMSO), or 17-AAG (1, 10, 25, 50, 100 μM) (Fig. 8A), or 17-DMAG (1, 10, 25, 50, 100 μM) (Fig. 8B), or AUY-922 (1, 10, 25, 50, 100 μM) (Fig. 8C) for 96 h. moderate doses (1, 10 μM) of Hsp90 inhibitors did not affect the viability of the pulmonary endothelial cells. However, 25, 50, and 100 μM of those compounds suppressed the proliferation of those cells, suggesting induction of lethal effects.
Fig. 8. Effects of Hsp90 inhibitors in the proliferation of human lung microvascular endothelial cells.
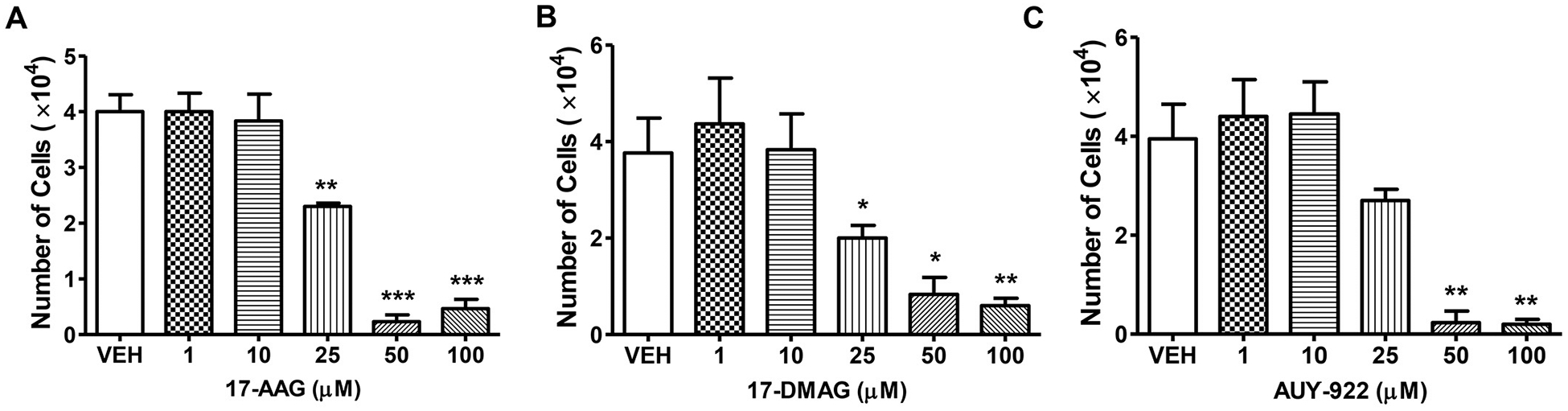
HuLEC seeded in 24-well plates (10,000 cells in each well) were treated with (A) 17-AAG (1, 10, 25, 50, 100 μM) (B) 17-DMAG (1, 10, 25, 50, 100 μM) (C) AUY-922 (1, 10, 25, 50, 100 μM) for 96 h. In all cases DMSO was used as the vehicle (VEH). *P < .05, **P < .01, ***P < .001 vs VEH, n = 3. Means ± SEM.
4. Discussion
The current work investigates the effects of three Hsp90 inhibitors (17-AAG, AUY-922, 17-DMAG) in the activation of UPR. Those compounds represent three different generations on the development of this class of compounds. AUY-922 is the most advanced inhibitor, and it is associated with less side effects as compared to the 17-AAG and 17-DMAG [19]. 17-AAG belongs to the benzoquinone ansamycin class of Hsp90 inhibitors, and the first one to have entered clinical trial. It inhibits the Hsp90 function by binding to its NH2-terminal ATP-binding domain [20]. On the other hand, AUY-922 is a C-terminal domain–based Hsp90 inhibitor which has already shown promising results in a phase I clinical trial [21].
In our study, the inhibition of Hsp90 resulted to the activation of PERK, ATF6 and IRE1α [22]. Those three elements are considered the hallmark of UPR activation, and are induced upon ER stress. PERK phosphorylates the α subunit of eukaryotic translation initiation factor 2 to suppress protein synthesis, hence it is acting to reduce ER stress. ATF6α is an ER-localized transmembrane transcription factor, which upon ER stress increases is transported; and consequently cleaved; to the Golgi apparatus, so to restore ER homeostasis. IRE1α is a transmembrane kinase/endonuclease, which propels the splicing of XBP1 mRNA. This kinase may also reduce protein synthesis by the IRE1α-dependent mRNA decay [23].
We also examined the expression levels of Endoplasmic Reticulum Oxidoreductin-1alpha (Ero1-Lα) and Protein Disulfide Isomerase (PDI). ERO1-La is a hypoxia-inducible ER-resident oxidase, which is activated due to ER stress, and it is s essential for the formation of disulfide bonds in protein synthesis [24]. PDI is a ER chaperone, plays a crucial role in catalyzing disulfide bond formation, reduction, and isomerization [25]. Both proteins reflect increases of ER stress [26].
An emerging body of evidence suggest that a mild UPR activation is involved in tissue repair and that Hsp90 inhibition induces UPR [15,27] via IRE1 stabilization, as well as by triggering GRP94- and ER stress-dependent transcription [28]. It was recently revealed that IRE1α in intestinal epithelial cells protects against colitis, thus protecting against inflammation in bowel disease [29]. It was also reported that pretreatment of mesangial cells with the ER stress inducer Tunicamycin strongly counteracted the induction of IL-6, MCP-1, TNF-α and CD40 due to LDL [30].
It was shown that PERK exerts protective activities in pigs and mice and protects against heart failure and lung remodeling in pigs [31]. In a murine model of lung fibrosis, knock out mice that did not express PERK exerted a more severe phenotype of lung fibrosis compared to the wild type littermates [32]. Moreover, BIP and CHOP were involved in the protection against respiratory implications. Mice expressing mutant BiP were subjected to lung failure due to non-physiological secretion of pulmonary surfactant [33]. Interestingly, CHOP-KO mice presented increased pulmonary edema and lung permeability, suggesting that CHOP protects the lungs in prolonged hyperoxia [34]. In human endothelial cells the suppression of the ERp57 augmented BiP induction; and counteracted the hyperoxia and tunicamycin-induced apoptotic cell death [35].
The release of Growth Hormone (GH) is regulated by the hypothalamic Growth Hormone Releasing Hormone (GHRH) [3,36]. It was recently shown that GHRH antagonists induce both UPR and P53 [37,38]. Moreover, the most advanced generation of those peptides enhance the lung endothelial barrier [39,40], and exert robust protective effects against the bleomycin-induced lung inflammation and fibrosis [41]. It was also demonstrated that P53 mediates the protective effects of Hsp90 inhibition in the lungs [8] by modulating the Rac1/RhoA signaling [10]; and by suppressing ROS production [13]. Moreover; UPR affects pulmonary P53 expression in a positive manner; and P53 induces UPR [4,7,17]. Since Hsp90 inhibition induces UPR, it is possible that P53 may represent the functional link between Hsp90 inhibition and UPR induction. Future experiments in endothelial specific P53-KO mice will investigate the importance of P53 into the possible anti-inflammatory effects of the UPR activation in the lungs.
A robust UPR induction due to ER stress, will inevitably cause cellular death [42,43]. On the other hand, a mild induction of this molecular machinery contributes to tissue hemostasis [18,44]. Thus, we decided to investigate the effects of Hsp90 inhibitors in the UPR induction and cell proliferation. Commercially available lung cells were treated with different concentrations of Hsp90 inhibitors, to evaluate their effects towards UPR activation and cellular proliferation. Our results suggest that the Hsp90 inhibition in moderate doses induce UPR, and that effect is not associated with toxicity. However, at higher doses of Hsp90 inhibitors, the numbers of lung cells were strongly reduced. Our in vivo experimentations substantiated our in vitro observations, suggesting that the effects of AUY-922 in the UPR activation are not limited in human lung cells. That compound induced both CHOP and BiP (ER stress markers) in mice lungs, in line with previous observations reporting that Hsp90 inhibition increases the pulmonary abundance of PDI and ERO1-Lα in mice [27].
Interestingly, malignancies are more sensitive to Hsp90 inhibitors compared to the non-cancer cells. Cancers rely on Hsp90 to proliferate and infiltrate the surrounding tissues [45], and Hsp90 inhibitors have been shown to exert a higher affinity towards the activated (inflamed) Hsp90, as compared to that of the non-inflamed tissues [46]. Inflammatory agents, such as LPS, have been shown to activate this molecular chaperone [47,48]. Thus, a certain dose of Hsp90 inhibitor in cancers, may exert different results in non-malignant tissues.
The current study shows that Hsp90 inhibition activates the UPR machinery in human and mice lung cells. Thus, future endeavors, may investigate the exact UPR-related signaling cascades involved in the protective activities of Hsp90 inhibitors in the impaired lungs, and reveal whether P53 is the functional link between Hsp90 inhibition and UPR induction. Those studies shall contribute to the development of promising therapeutic approaches against the lethal severe Acute Respiratory Distress Syndrome.
Acknowledgment of funding source
This research work was funded by the R&D, Research Competitiveness Subprogram (RCS) of the Louisiana Board of Regents through the Board of Regents Support Fund LEQSF(2019-22)-RD-A-26 to Nektarios Barabutis (Principal Investigator).
Footnotes
Competing Interest Statement
The authors declare no competing interests.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- [1].Barabutis N Heat shock protein 90 inhibition in the inflamed lungs. Cell Stress Chaperones. 2020;25(2):195–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Abbasi M, et al. New heat shock protein (Hsp90) inhibitors, designed by pharmacophore modeling and virtual Screening: Synthesis, Biological evaluation and molecular dynamics studies. J Biomol Struct Dyn. 2019:1–16. [DOI] [PubMed] [Google Scholar]
- [3].Barabutis N, Schally AV, Siejka A. P53, GHRH, inflammation and cancer. EBioMedicine. 2018;37:557–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Uddin MA, Barabutis N. P53: The endothelium defender. J Cell Biochem. 2019;120(7): 10952–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Levine AJ. The many faces of p53: something for everyone. J Mol Cell Biol. 2019;11(7): 524–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gudkov AV, Komarova EA. p53 and the Carcinogenicity of Chronic Inflammation. Cold Spring Harb Perspect Med. 2016:6(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kubra KT, et al. P53 versus inflammation: an update. Cell Cycle. 2020;19(2):160–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Barabutis N, et al. p53 protects against LPS-induced lung endothelial barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2015;308(8):L776–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Barabutis N P53 in RhoA regulation. Cytoskeleton (Hoboken). 2020. 10.1002/cm.21604. [DOI] [PubMed] [Google Scholar]
- [10].Barabutis N, et al. Wild-type p53 enhances endothelial barrier function by mediating RAC1 signalling and RhoA inhibition. J Cell Mol Med. 2018;22(3):1792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Uddin MA, et al. P53 supports endothelial barrier function via APE1/Ref1 suppression. Immunobiology. 2019;224(4):532–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Barabutis N, Uddin MA, Catravas JD. Hsp90 inhibitors suppress P53 phosphorylation in LPS - induced endothelial inflammation. Cytokine. 2019;113:427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Akhter MS, Uddin MA, Barabutis N. P53 Regulates the Redox Status of Lung Endothelial Cells. Inflammation. 2020;43(2):686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Maguire JA, Mulugeta S, Beers MF. Multiple ways to die: delineation of the unfolded protein response and apoptosis induced by Surfactant Protein C BRICHOS mutants. Int J Biochem Cell Biol. 2012;44(1):101–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Barabutis N Unfolded Protein Response supports endothelial barrier function. Biochimie. 2019;165:206–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Xie Y, et al. Knockdown of IRE1a suppresses metastatic potential of colon cancer cells through inhibiting FN1-Src/FAK-GTPases signaling. Int J Biochem Cell Biol. 2019;114:105572. [DOI] [PubMed] [Google Scholar]
- [17].Akhter MS, Uddin MA, Barabutis N. Unfolded protein response regulates P53 expression in the pulmonary endothelium. J Biochem Mol Toxicol. 2019;33(10):e22380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Barabutis N Unfolded Protein Response in Acute Respiratory Distress Syndrome. Lung. 2019;197(6):827–8. [DOI] [PubMed] [Google Scholar]
- [19].Zuehlke AD, Moses MA, and Neckers L, Heat shock protein 90: its inhibition and function. Philos Trans R Soc Lond B Biol Sci, 2018. 373(1738). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Mayor-Lopez L, et al. Comparative Study of 17-AAG and NVP-AUY922 in Pancreatic and Colorectal Cancer Cells: Are There Common Determinants of Sensitivity? Transl Oncol. 2014;7(5):590–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Sessa C, et al. First-in-human phase I dose-escalation study of the HSP90 inhibitor AUY922 in patients with advanced solid tumors. Clin Cancer Res. 2013;19(13):3671–80. [DOI] [PubMed] [Google Scholar]
- [22].Glembotski CC, Rosarda JD, Wiseman RL. Proteostasis and Beyond: ATF6 in Ischemic Disease. Trends Mol Med. 2019;25(6):538–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Sundaram A, et al. Dynamic changes in complexes of IRE1alpha, PERK, and ATF6alpha during endoplasmic reticulum stress. Mol Biol Cell. 2018;29(11):1376–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yang S, et al. Endoplasmic reticulum resident oxidase ERO1-Lalpha promotes hepatocellular carcinoma metastasis and angiogenesis through the S1PR1/STAT3/VEGF-A pathway. Cell Death Dis. 2018;9(11):1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kim JY, et al. PDI regulates seizure activity via NMDA receptor redox in rats. Sci Rep. 2017;7:42491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tu BP, et al. Biochemical basis of oxidative protein folding in the endoplasmic reticulum. Science. 2000;290(5496):1571–4. [DOI] [PubMed] [Google Scholar]
- [27].Kubra KT, et al. Hsp90 inhibitors induce the unfolded protein response in bovine and mice lung cells. Cell Signal. 2020;67:109500. [DOI] [PubMed] [Google Scholar]
- [28].Marcu MG, et al. Heat shock protein 90 modulates the unfolded protein response by stabilizing IRE1alpha. Mol Cell Biol. 2002;22(24):8506–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Zhang HS, et al. The Endoplasmic Reticulum Stress Sensor IRE1alpha in Intestinal Epithelial Cells Is Essential for Protecting against Colitis. J Biol Chem. 2015;290(24):15327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yu Y, et al. Endoplasmic reticulum stress preconditioning antagonizes low-density lipoprotein-induced inflammation in human mesangial cells through upregulation of XBP1 and suppression of the IRE1alpha/IKK/NF-kappaB pathway. Mol Med Rep. 2015;11(3):2048–54. [DOI] [PubMed] [Google Scholar]
- [31].Chen WY, et al. Genotype 2 Strains of Porcine Reproductive and Respiratory Syndrome Virus Dysregulate Alveolar Macrophage Cytokine Production via the Unfolded Protein Response. J Virol. 2018;92(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu X, et al. Endoplasmic reticulum stress sensor protein kinase R-like endoplasmic reticulum kinase (PERK) protects against pressure overload-induced heart failure and lung remodeling. Hypertension. 2014;64(4):738–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mimura N, et al. Aberrant quality control in the endoplasmic reticulum impairs the biosynthesis of pulmonary surfactant in mice expressing mutant BiP. Cell Death Differ. 2007;14(8):1475–85. [DOI] [PubMed] [Google Scholar]
- [34].Lozon TI, et al. PKR-dependent CHOP induction limits hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2011;300(3):L422–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xu D, et al. Knockdown of ERp57 increases BiP/GRP78 induction and protects against hyperoxia and tunicamycin-induced apoptosis. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L44–51. [DOI] [PubMed] [Google Scholar]
- [36].Munoz-Moreno L, et al. Stimulation of neuroendocrine differentiation in prostate cancer cells by GHRH and its blockade by GHRH antagonists. Invest New Drugs. 2020;38(3): 746–54. [DOI] [PubMed] [Google Scholar]
- [37].Barabutis N Growth hormone releasing hormone in the unfolded protein response context. Endocrine. 2020;67(2):291–3. [DOI] [PubMed] [Google Scholar]
- [38].Barabutis N P53 In Lung Vascular Barrier Dysfunction. Vascul Biol. 2020. 10.1530/VB-20-0004. https://vb.bioscientifica.com/view/journals/vb/aop/vb-20-0004/vb-20-0004.xml. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Uddin MA, et al. GHRH antagonists support lung endothelial barrier function. Tissue Barriers. 2019;7(4):1669989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Barabutis N, Siejka A. A Glimpse at the Highly Interrelated GHRH, P53 and Hsp90 Cosmos. Cell Biol Int. 2020. 10.1002/cbin.11356. [DOI] [PubMed] [Google Scholar]
- [41].Zhang C, et al. Growth Hormone-Releasing Hormone Receptor Antagonist Modulates Lung Inflammation and Fibrosis due to Bleomycin. Lung. 2019;197(5):541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Thiebaut PA, et al. Protein tyrosine phosphatase 1B regulates endothelial endoplasmic reticulum stress; role in endothelial dysfunction. Vascul Pharmacol. 2018;109: 36–44. [DOI] [PubMed] [Google Scholar]
- [43].Ampem PT, Smedlund K, Vazquez G. Pharmacological evidence for a role of the transient receptor potential canonical 3 (TRPC3) channel in endoplasmic reticulum stress-induced apoptosis of human coronary artery endothelial cells. Vascul Pharmacol. 2016;76:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Barabutis N Regulation of lung endothelial permeability by NEK kinases. IUBMB Life. 2020;72(4):801–4. [DOI] [PubMed] [Google Scholar]
- [45].Butler LM, et al. Maximizing the Therapeutic Potential of HSP90 Inhibitors. Mol Cancer Res. 2015;13(11):1445–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Echeverria PC, et al. The sensitivity to Hsp90 inhibitors of both normal and oncogenically transformed cells is determined by the equilibrium between cellular quiescence and activity. PLoS One. 2019;14(2):e0208287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Barabutis N, Verin A, Catravas JD. Regulation of pulmonary endothelial barrier function by kinases. Am J Physiol Lung Cell Mol Physiol. 2016;311(5):L832–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Barabutis N, et al. LPS induces pp60c-src-mediated tyrosine phosphorylation of Hsp90 in lung vascular endothelial cells and mouse lung. Am J Physiol Lung Cell Mol Physiol. 2013;304(12):L883–93. [DOI] [PMC free article] [PubMed] [Google Scholar]