

Abstract
Background
Parkinson's disease (PD) is a progressive disorder characterised by both motor and non‐motor problems. Glucagon‐like peptide‐1 (GLP‐1) receptor agonists, licensed for treatment of type 2 diabetes, work by stimulating GLP‐1 receptors in the pancreas, which triggers the release of insulin. GLP‐1 receptors have been found in the brain. Insulin signalling in the brain plays a key role in neuronal metabolism and repair and in synaptic efficacy, but insulin signalling is desensitised in the brain of people with PD. Researchers are exploring the neuroprotective effects of GLP‐1 receptor agonists in neurodegenerative disorders such as PD.
Objectives
To evaluate the effectiveness and safety of GLP‐1 receptor agonists for Parkinson's disease.
Search methods
We searched the Cochrane Movement Disorders Group trials register; the Cochrane Central Register of Controlled Trials (CENTRAL), in the Cochrane Library; and Ovid MEDLINE and Embase. We also searched clinical trials registries, and we handsearched conference abstracts. The most recent search was run on 25 June 2020.
Selection criteria
We included randomised controlled trials (RCTs) of adults with PD that compared GLP‐1 receptor agonists with conventional PD treatment, placebo, or no treatment.
Data collection and analysis
Two review authors independently assessed studies for inclusion, extracted data, and assessed risk of bias. We rated the quality of evidence using GRADE. We resolved discrepancies between the two data extractors by consultation with a third review author.
Main results
Through our searches, we retrieved 99 unique records, of which two met our inclusion criteria. One double‐blind study of exenatide versus placebo randomised 62 participants, who self‐administered exenatide or placebo for 48 weeks and were followed up at 60 weeks after a 12‐week washout. One single‐blind study of exenatide versus no additional treatment randomised 45 participants; participants in the intervention group self‐administered exenatide for 12 months, and all participants were followed up at 14 months and 24 months following absence of exenatide for 2 months and 12 months, respectively. These trials had low risk of bias, except risk of performance bias was high for Aviles‐Olmos 2013.
Exenatide versus placebo
Primary outcomes
We found low‐certainty evidence suggesting that exenatide improves motor impairment as assessed by the Movement Disorder Society‐Unified Parkinson's Disease Rating Scale (MDS‐UPDRS) Part III in the off‐medication state (mean difference (MD) ‐3.10, 95% confidence interval (CI) ‐6.11 to ‐0.09). The difference in scores was slightly greater when scores were adjusted for baseline severity of the condition (as reported by study authors) (MD ‐3.5, 95% CI ‐6.7 to ‐0.3), exceeding the minimum clinically important difference (MCID).
We found low‐certainty evidence suggesting that exenatide has little or no effect on health‐related quality of life (HRQoL) as assessed by the Parkinson's Disease Questionnaire (PDQ)‐39 Summary Index (SI) (MD ‐1.80, 95% CI ‐6.95 to 3.35), the EuroQol scale measuring health status in five dimensions (EQ5D) (MD 0.07, 95% CI ‐0.03 to 0.16), or the EQ5D visual analogue scale (VAS) (MD 5.00, 95% CI ‐3.42 to 13.42). Eight serious adverse events (SAEs) were recorded, but all were considered unrelated to the intervention. Low‐certainty evidence suggests that exenatide has little or no effect on weight loss (risk ratio (RR) 1.25, 95% CI 0.89 to 1.76).
Exenatide versus no treatment
Primary outcomes at 14 months
We found very low‐certainty evidence suggesting that exenatide improves motor impairment as assessed by MDS‐UPDRS Part III off medication (MD ‐4.50, 95% CI ‐8.64 to ‐0.36), exceeding the MCID. We are uncertain whether exenatide improves HRQoL as assessed by the PDQ‐39 SI (MD 3.50, 95% CI ‐2.75 to 9.75; very low‐quality evidence). We found very low‐certainty evidence suggesting that exenatide has little or no effect on the number of SAEs (RR 1.60, 95% 0.40 to 6.32). We found very low‐certainty evidence suggesting that exenatide may lead to weight loss (MD ‐2.40 kg, 95% CI ‐4.56 to ‐0.24).
Primary outcomes at 24 months
We found evidence as reported by study authors to suggest that exenatide improves motor impairment as measured by MDS‐UPDRS Part III off medication (MD 5.6 points, 95% CI 2.2 to 9.0). Exenatide may not improve HRQoL as assessed by the PDQ‐39 SI (P = 0.682) and may not result in weight loss (MD 0.1 kg, 95% CI 3.0 to 2.8).
Authors' conclusions
Low‐ or very low‐certainty evidence suggests that exenatide may improve motor impairment for people with PD. The difference in motor impairment observed between groups may persist for some time following cessation of exenatide. This raises the possibility that exenatide may have a disease‐modifying effect. SAEs were unlikely to be related to treatment. The effectiveness of exenatide for improving HRQoL, non‐motor outcomes, ADLs, and psychological outcomes is unclear. Ongoing studies are assessing other GLP‐1 receptor agonists.
Keywords: Humans, Bias, Double-Blind Method, Exenatide, Exenatide/administration & dosage, Exenatide/adverse effects, Exenatide/therapeutic use, Glucagon-Like Peptide-1 Receptor, Glucagon-Like Peptide-1 Receptor/agonists, Hypoglycemic Agents, Hypoglycemic Agents/administration & dosage, Hypoglycemic Agents/therapeutic use, Parkinson Disease, Parkinson Disease/drug therapy, Placebos, Placebos/administration & dosage, Quality of Life, Randomized Controlled Trials as Topic, Self Administration, Single-Blind Method
Plain language summary
Can exenatide, a diabetes drug, be used to treat Parkinson's disease?
Review question
To evaluate the effectiveness and safety of GLP‐1 receptor agonists for Parkinson's disease.
Background
People with Parkinson's disease (PD) have problems with movement, such as slow movement and shaking at rest. They may also have other problems such as depression, difficulty swallowing, and gastrointestinal dysfunction. Glucagon‐like peptide‐1 (GLP‐1) receptor agonists are used for treatment of type 2 diabetes. They work by stimulating GLP‐1 receptors in the pancreas, which causes the release of insulin. GLP‐1 receptors have also been found in the brain. Neurones in the brain send signals to and from the brain and the rest of the body. Insulin signalling in the brain is important for keeping neurones healthy, but it has been shown that insulin signalling does not work well in the brain of people with PD. Researchers are interested in finding out about the protective effects of GLP‐1 receptor agonists on neurones, and how these agonists might help people with disease affecting the brain, such as PD.
Study characteristics
We found two studies that provided data for a total of 104 patients (following dropout of three patients). One study compared exenatide (a GLP‐1 receptor agonist) versus placebo (a pretend medicine), and the other study compared exenatide versus no treatment (other than the usual treatment that people received). Evidence is current to June 2020.
Key results
We found low‐certainty evidence suggesting that people who took exenatide had better improvement in motor symptoms than people who took placebo. Movement was measured 12 weeks after patients had stopped taking exenatide. We found low‐certainty evidence suggesting that for people taking exenatide, there may be little or no difference in health‐related quality of life (HRQoL). Six serious adverse events (SAEs) were seen in people taking exenatide and two in people taking placebo, but all were considered by the study authors to be not related to the drug.
We found very low‐certainty evidence suggesting that people who took exenatide had better improvement in motor symptoms than people who received no treatment other than their usual care. Movement was measured two months after patients stopped taking exenatide. We found very low‐certainty evidence suggesting that exenatide compared to no treatment had little or no effect on HRQoL, and we found very low‐certainty evidence suggesting little or no difference in the number of SAEs among people taking exenatide.
Quality of evidence
The quality of evidence was low or very low. In one study, people not taking exenatide received their usual treatment only; thus, people in the study knew whether they were given extra treatment, and this may have changed the study results.
Conclusions
We are uncertain whether exenatide may improve motor symptoms for people with PD. The improvement in symptoms found in two small studies persisted for several weeks after people stopped taking the drug. This might mean that the drug has modified the disease process in some way. More studies with more people are needed so that we can be more sure whether GLP‐1 receptor agonists do help people with PD.
Summary of findings
Summary of findings 1. GLP‐1 receptor agonists compared to placebo for adults with Parkinson's disease.
| GLP‐1 receptor agonists compared to placebo for adults with Parkinson's disease | ||||||
|
Patient or population: adults with Parkinson's disease
Setting: research institutes, tertiary care facilities Intervention: GLP‐1 receptor agonists Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with GLP‐1 receptor agonists | |||||
| PD motor impairment Assessed with MDS‐UPDRS Part III Follow‐up: 60 weeks MDS‐UPDRS Part III: 33 scores based on 18 items due to left, right, and other body distributions, scored as 0 normal, 1 slight, 2 mild, 3 moderate, 4 severe. Scale from 0 to 132. MCID of ‐3.25 points for improvement and 4.63 points for worsening |
Mean PD motor impairment; change from baseline in MDS‐UPDRS Part III was 2.1 | MD 3.1 lower (6.11 lower to 0.09 lower) | ‐ | 60 (1 RCT) | ⊕⊕⊝⊝ Lowa,b | GLP‐1 receptor agonists produced a significant mean between‐group reduction in motor impairment score. MD of ‐3.1 not ≥ MCID of ‐3.25 for improvement |
| HRQoL Assessed with PDQ‐39 SI Follow‐up: 60 weeks PDQ‐39 SI: 39 items, 5‐point ordinal scoring system 0 = never, 1 = occasionally, 2 = sometimes, 3 = often, 4 = always. Lower score reflects better HRQoL |
Mean HRQoL; change from baseline in PDQ‐39 SI was 0.3 | MD 1.8 lower (6.95 lower to 3.35 higher) | ‐ | 60 (1 RCT) | ⊕⊕⊝⊝ Lowa,c | No significant difference in mean between‐group quality of life scores. MD of ‐1.8 not ≥ MCID of ‐4.72 for improvement |
| Serious adverse events Assessed with number of participants with an SAE Follow‐up: 48 weeks |
The included study did not report the number of participants with an SAE. The study reported the number of SAEs over all time points | |||||
| Adverse events ‐ weight loss Assessed with number of participants who lost weight Follow‐up: 48 weeks |
621 per 1000 | 776 per 1000 (552 to 1000) | RR 1.25 (0.89 to 1.76) | 60 (1 RCT) | ⊕⊕⊝⊝ Lowa,d | No significant difference in weight loss between groups receiving exenatide or placebo |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; GLP‐1: glucagon‐like peptide‐1; HRQoL: health‐related quality of life; MCID: minimal clinically important difference; MD: mean difference; MDS‐UPDRS: Movement Disorder Society‐Unified Parkinson's Disease Rating Scale; PDQ‐39 SI: Parkinson's Disease Questionnaire Summary Index; RA: receptor agonist; RCT: randomised controlled trial; RR: risk ratio; SAE: serious adverse event. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aWe downgraded for indirectness as the intervention was only one example of an RA.
bWe downgraded for imprecision due to a small sample size. CI excludes no effect and fails to exclude appreciable benefit (MCID = ‐3.25 for improvement).
cWe downgraded for imprecision due to a small sample size. CI includes no effect and fails to exclude appreciable benefit (MCID = ‐4.72 for improvement).
dWe downgraded for imprecision due to a small sample size. CI includes no effect and fails to exclude appreciable harm.
Summary of findings 2. GLP‐1 receptor agonists compared to no treatment for adults with Parkinson's disease.
| GLP‐1 receptor agonists compared to no treatment for adults with Parkinson's disease | ||||||
|
Patient or population: adults with Parkinson's disease
Setting: research institutes, tertiary care facilities Intervention: GLP‐1 receptor agonists Comparison: no treatment | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with no treatment | Risk with GLP‐1 receptor agonists | |||||
| PD motor impairment
Assessed with
MDS‐UPDRS Part III Follow‐up: 14 months |
Mean PD motor impairment; change from baseline in MDS‐UPDRS Part III was 2.8 | MD 4.5 lower (8.64 lower to 0.36 lower) | ‐ | 44 (1 RCT) | ⊕⊝⊝⊝ Very lowa,b,c | Exenatide produced a significant mean between‐group reduction in motor impairment score. MD of ‐4.5 > MCID of ‐3.25 for improvement |
| HRQoL Assessed with PDQ‐39 SI Follow‐up: 14 months |
Mean HRQoL; change from baseline in PDQ‐39 SI was 2.3 | MD 3.5 higher (2.75 lower to 9.75 higher) | ‐ | 44 (1 RCT) | ⊕⊝⊝⊝ Very lowa,b,d | No significant difference in mean between‐group quality of life scores. MCID = ‐4.72 for improvement and 4.22 for worsening |
| Serious adverse events.
Assessed with number of participants with an SAE Follow‐up: 14 months |
125 per 1000 | 200 per 1000 (50 to 790) | RR 1.60 (0.40 to 6.32) | 44 (1 RCT) | ⊕⊝⊝⊝ Very lowa,b,e | No significant difference in the number of serious adverse events reported between groups receiving exenatide or placebo |
| Adverse events ‐ weight loss Assessed with mean weight loss Follow‐up: 12 months |
Mean weight loss; change from baseline in kg was ‐3.2 | MD 2.4 kg lower (4.56 lower to 0.24 lower) | ‐ | 44 (1 RCT) | ⊕⊝⊝⊝ Very lowa,b,f | Participants in the exenatide group lost significantly more weight than participants in the no treatment group |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; GLP‐1: glucagon‐like peptide‐1; HRQoL: health‐related quality of life; MCID: minimal clinically important difference; MD: mean difference; MDS‐UPDRS: Movement Disorder Society‐Unified Parkinson's Disease Rating Scale; PD: Parkinson's disease; PDQ‐39 SI: Parkinson's Disease Questionnaire Summary Index; RA: receptor agonist; RCT: randomised controlled trial; RR: risk ratio; SAE: serious adverse event. | ||||||
| GRADE Working Group grades of evidence. High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate: the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited: the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate: the true effect is likely to be substantially different from the estimate of effect. | ||||||
aWe downgraded for study limitations of high risk of performance bias.
bWe downgraded for indirectness as the intervention was only one example of an RA.
cWe downgraded for imprecision due to a small sample. CI excludes no effect and fails to exclude appreciable benefit (MCID = ‐3.25 for improvement).
dWe downgraded for imprecision due to a small sample size. CI fails to exclude no effect and appreciable harm (MCID = 4.22 for worsening).
eWe downgraded for imprecision due to a small sample size. CI includes no effect and fails to exclude appreciable harm or benefit.
fWe downgraded for imprecision due to a small sample size. CI excludes no effect.
Background
Description of the condition
Parkinson’s disease (PD) is the second most common neurodegenerative disorder after Alzheimer’s disease (AD), affecting approximately 0.5% of the population over 60 years of age in industrialised countries (Pringhseim 2014). PD is caused by loss of dopamine‐producing nerve cells in the part of the brain called the substantia nigra. Dopamine functions as a neurotransmitter and plays a key role in motor control. It is not known what causes the loss of these dopamine‐producing nerve cells.
PD is a long‐term, progressive disorder that causes significant disability. Symptoms generally develop slowly, typically over 10 to 15 years. PD is characterised by motor features (problems with movement) that include slow movement, shaking at rest, muscular rigidity, and postural instability (Kalia 2015), along with a variety of non‐motor features that include loss of the sense of smell and sleep and psychiatric dysfunction, including depression, anxiety, and dementia. As the disease progresses and treatment‐resistant motor and non‐motor features dominate, falls, freezing gait, choking, urinary incontinence, and dementia are common (Hely 2005; Hely 2008).
PD shows increasing incidence with age and is more common among men than women (Hirsch 2016). Risk factors for PD include exposure to pesticides and other environmental chemicals (often experienced by agricultural workers), high consumption of dairy products, a diagnosis of melanoma, and traumatic brain injury (Ascherio 2016; de Lau 2006). Protective factors include use of tobacco; consumption of coffee, caffeine, and tea; higher plasma concentrations of urates (salts of uric acid); physical activity; and use of non‐steroidal anti‐inflammatory drugs (Ascherio 2016).
At present, no effective disease‐modifying or neuroprotective interventions are known; current therapies for PD are provided to treat symptoms only. Available therapies include levodopa, which is converted in the brain (as well as in the periphery) to dopamine, and dopamine receptor agonists, which stimulate dopamine receptors.
Typically, PD is defined pathologically by prominent loss of dopaminergic neurons and the presence in the brain of Lewy bodies containing α‐synuclein. It is increasingly recognised that the neurodegenerative process in PD is complex and multi‐factorial and is likely to involve mitochondrial dysfunction and oxidative stress (Abou‐Sleiman 2006), inflammation (Collins 2012), blood‐brain barrier dysfunction (Gray 2015), and neurovascular changes (Al‐Bachari 2017). Such factors are likely to have treatment and prognostic implications. Vascular comorbidity (including prior stroke, transient ischaemic attack (TIA), or more than two vascular risk factors), for instance, has been found to be significantly associated with cognitive and gait impairment in early PD (Malek 2016).
Description of the intervention
Glucagon‐like peptide‐1 (GLP‐1) receptor agonists are a class of drugs that are licensed for treatment of type 2 diabetes (Baggio 2007; Campbell 2013; Doyle 2003; Holst 2004). An agonist acts by binding to a receptor (a protein molecule that is the target for the drug), which causes some form of cellular response (Pleuvry 2004). For people with type 2 diabetes, GLP‐1 receptor agonists work by stimulating GLP‐1 receptors in the pancreas, which triggers the release of insulin. However, GLP‐1 receptors have also been found in the brain; thus GLP‐1 receptor agonists may also play a role in the treatment of PD. Insulin signalling in the brain plays a key role in neuronal metabolism and repair and in synaptic efficacy (Freiherr 2013; Ghasemi 2013; van der Heide 2006). Insulin activates growth factor receptors on neurones that control energy utilisation, cell repair, mitochondrial function, synapse growth, and functionality. Several classic second messenger cell signalling pathways are activated while apoptotic (programmed cell death) cell signalling is inhibited (Hölscher 2014). It has been shown that insulin signalling is desensitised in the brain of people with PD (Aviles‐Olmos 2013a; Moroo 1994; Morris 2011), which may explain why type 2 diabetes has been identified as a risk factor for development of PD (Hu 2007; Schernhammer 2011; Sun 2012; Wahlqvist 2012). GLP‐1 receptor agonists are administered by subcutaneous injection.
How the intervention might work
GLP‐1 activates the same key growth factor cell signalling cascades as insulin, and therefore compensates for loss of insulin signalling (Jalewa 2016). Protease‐resistant analogues of GLP‐1 have shown neuroprotective effects in animal models of AD (Bomfim 2012; Li 2010; McClean 2011), and they have been found to re‐sensitise insulin signalling in the brain (Long‐Smith 2013). Furthermore, previous studies found that GLP‐1 receptor agonists have neuroprotective effects in animal models of PD. The GLP‐1 mimetic (molecule resembling GLP‐1), exendin‐4, protected motor activity and dopamine levels in the striatum, and reduced chronic inflammation and oxidative stress (Harkavyi 2008; Li 2009; Liu 2015a; Zhang 2015). In the 1‐methyl‐4‐phenyl‐1,2,3,6‐tetrahydropyridine (MPTP) mouse model of PD, GLP‐1 mimetics protected the brain from aspects of MPTP‐induced pathology, such as motor impairment, increased α‐synuclein levels, chronic inflammation in the brain, loss of dopaminergic neurons, oxidative stress, and growth factor expression (Ji 2016; Li 2016; Liu 2015a; Liu 2015b; Zhang 2015). The GLP‐1 mimetics liraglutide and lixisenatide are more effective than the first‐generation drug exenatide. The newer GLP‐1 mimetics improve motor co‐ordination and activity, and both drugs rescue the expression of tyrosine hydroxylase, a key enzyme in dopamine synthesis (Liu 2015a).
A challenge for clinical trials for neurodegenerative diseases such as PD is to differentiate between the disease‐modifying effects and the symptomatic effects of any therapeutic agent. Scales used in clinical assessment of PD to measure changes in, for example, motor impairment or quality of life are unable to distinguish between symptomatic and disease‐modifying effects of a treatment; thus, for any novel potential treatment to demonstrate disease modification, there needs to be evidence that the drug, when administered for a period of time, stops or slows disease progression. This can be demonstrated in a clinical trial by the absence of deterioration in clinical outcome measures by comparison with a control or placebo group. Inclusion of a washout period in clinical trial design or long‐term follow‐up helps to confirm that any differences in clinical outcome measures observed between treatment groups are indeed evidence of disease modification rather than symptomatic effects (McGhee 2013). Changes in relevant biomarkers, such as presynaptic striatal dopamine transporter (DAT) binding as assessed by [¹²³I]FP‐CIT single‐photon emission computed tomography (DaTSCAN) examination, would provide additional evidence of disease modification. In addition, confounders may influence therapeutic effects. For example, although evidence suggests that GLP‐1 can cause weight loss (Vilsbøll 2012), it is known that the amount of levodopa and its maximum concentration in plasma are negatively correlated with body weight (Müller 2000), and consequently weight loss can lead to increased effectiveness of levodopa; thus awareness of potential changes in weight loss due to GLP‐1 and subsequent therapeutic effects on PD is essential in the study of GLP‐1 receptor agonists.
Why it is important to do this review
Recent advances in our understanding of the neuroprotective effects of incretin‐based therapies, including GLP‐1 receptor agonists, mean that there is considerable interest in their potential utility as re‐purposed treatment for several neurodegenerative disorders, including PD. People with PD treated with exenatide in an open‐label clinical trial showed clinical benefit (Aviles‐Olmos 2013b), with subsequent evidence of significant improvement in motor features 12 months after stopping exenatide (Aviles‐Olmos 2014). Similarly, a recent double‐blind clinical trial of people with PD found that those treated with exenatide showed improved motor features 60 weeks after coming off the medication, while motor features for those on placebo had worsened (Athauda 2017). It is therefore timely to undertake this review of GLP‐1 receptor agonists for PD, as this will provide a summary of the current state of the evidence and a platform for updating the evidence base as results of future studies become available.
Objectives
To evaluate the effectiveness and safety of GLP‐1 receptor agonists for Parkinson's disease.
Methods
Criteria for considering studies for this review
Types of studies
We included published and unpublished, parallel‐designed, randomised controlled trials (RCTs). We excluded cross‐over trials due to uncertainty about whether this type of study design is appropriate for studying people with PD (Higgins 2011).
Types of participants
We included trials in any setting with a study population of adults (i.e. ≥ 18 years of age) with a clinical diagnosis made by any physician, specialist, or otherwise of PD according to the UK Parkinson’s Disease Society Brain Bank diagnostic criteria (Hughes 1992), or other equivalent clinical diagnostic criteria, or on the basis of clinical neurological assessment. We included people at all stages of the disease. Participants may have medical conditions in addition to PD. We did not apply any restrictions based on the number of participants recruited to trials nor on the number of recruitment centres.
Types of interventions
We included studies that involved delivery of GLP‐1 receptor agonists with no restrictions on dosage or duration of treatment. We included studies in which experimental and comparator groups received an intervention (either active or inactive) in addition to conventional treatment. We planned to assess the following comparisons.
GLP‐1 receptor agonists versus conventional PD treatment.
GLP‐1 receptor agonists versus placebo intervention.
GLP‐1 receptor agonists versus no treatment.
Types of outcome measures
Primary outcomes
PD motor impairment as measured by the Movement Disorder Society‐Unified Parkinson's Disease Rating Scale (MDS‐UPDRS) subscale Part III (Fahn 1987)
-
Health‐related quality of life (HRQoL) as measured by a validated scale such as:
Parkinson’s Disease Questionnaire (PDQ)‐39 (Peto 1995), or short form PDQ‐8 (Jenkinson 1997);
Parkinson's Disease Quality of Life Questionnaire (PDQL) (de Boer 1996); or
36‐Item Short Form Health Survey (SF‐36) (Ware 1992)
Adverse events, including rapid weight loss (> 1.5 kg/week) (we have defined adverse events as serious based on information reported by individual trials)
Secondary outcomes
PD motor impairment as measured by a validated scale other than UPDRS subscale Part III (Fahn 1987), such as the Unified Dyskinesia Rating Scale (UDysRS) (Goetz 2008)
Non‐motor outcomes as measured by validated scales including UPDRS Part I (Fahn 1987), as well as the Non‐Motor Symptoms Questionnaire (NMSQuest) (Chaudhuri 2008)
Activities of daily living (ADLs) as measured by scales such as UPDRS Part II (Fahn 1987), as well as Schwab and England Activities of Daily Living (SEADL) (Schwab 1969)
Psychological outcomes such as dementia and depression as measured by validated scales (e.g. the Mattis Dementia Rating Scale (DRS) (Mattis 1976); the Montgomery‐Åsberg Depression Rating Scale (MADRS)) (Montgomery 1979)
These primary and secondary outcomes address key disease aspects identified as important by patients and clinicians; outcomes classified as related to participation, mobility, and motor functioning were considered the most important (Hammarlund 2012), and these are assessed by UPDRS Parts I and III, respectively. A minimal clinically important difference (MCID) for the motor examination portion of the MDS‐UPDRS is asymmetrical, with ‐3.25 points for detecting minimal but clinically important improvement, and 4.63 points for observing minimal but clinically important worsening (Horváth 2015). An estimate of a minimal clinically important improvement on the PDQ‐39 SI is ‐4.72, and an estimate for worsening is +4.22 (Horváth 2017). As we were keen to assess the neuroprotective effects of GLP‐1 receptor agonists, we reported outcomes assessed two to three months post cessation of treatment. Reporting one or more of the outcomes listed was not an inclusion criterion for trials considered for this review.
Search methods for identification of studies
Electronic searches
We searched the following databases from the date of inception to June 2020 for relevant studies.
Cochrane Movement Disorders Group trials register.
Cochrane Central Register of Controlled Trials (CENTRAL), in the Cochrane Library (Appendix 1).
MEDLINE (OVID) (1946 to present) (Appendix 2).
Embase (1974 to present) (Appendix 3).
Searching other resources
We searched the following clinical trials registers.
World Health Organization (WHO) Portal (covers ClinicalTrials.gov; International Standard Randomized Controlled Trials Number (ISRCTN); Australian and New Zealand Clincal Trial Registry; Chinese Clinical Trial Register; India Clinical Trials Registry; German Clinical Trials Register; Iranian Registry of Clinical Trials; Sri Lanka Clinical Trials Registry; The Netherlands National Trial Register): www.who.int/trialsearch. We searched up to May 2019.
UK Clinical Trials Gateway: www.ukctg.nihr.ac.uk/default.aspx. We searched up to June 2020.
We handsearched abstracts of the 16th to 21st International Congresses of the Parkinson's Disease and Movement Disorders conference (2012 to 2019) and meeting abstracts from the Association of British Neurologists (2012 to 2017). As this therapy is relatively new, we handsearched the more recent conference proceedings. In June 2020 we searched the clinical trials listed on the Journal of Parkinson's Disease website (https://www.journalofparkinsonsdisease.com/). We also screened the reference lists of included trials and review articles for potentially eligible studies.
Data collection and analysis
Selection of studies
We merged the results of our searches and removed duplicates. Two review authors (GD, JH) independently screened titles and abstracts of studies identified by our search for potential inclusion in the review. We searched for full‐text reports of all potentially relevant studies remaining after the initial assessment, and two review authors (GD, JH) independently assessed these for inclusion in the review. We resolved any disagreements between the two authors by consulting a third review author (CM). We excluded studies according to a hierarchy based on the inclusion criteria, that is, wrong study design, wrong patient population, and wrong comparator. We recorded the reason for study exclusion as the first criterion not met, and we have presented our reasons for excluding full‐text reports in the Characteristics of excluded studies table. We have produced a PRISMA flow chart (Figure 1) showing how we selected our studies for inclusion in the review (Liberati 2009), along with the reasons for study exclusion.
1.
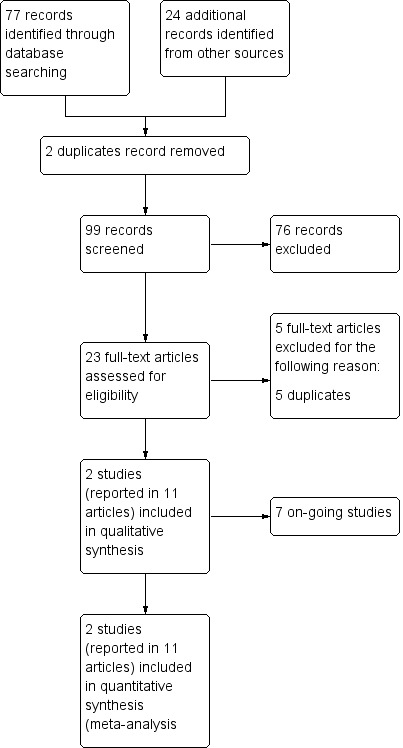
2 Study flow diagram.
Data extraction and management
Two review authors (CM, DE) independently extracted data from included studies using a standard data extraction form that we customised for use in this review. We pilot‐tested the form. We extracted the data detailed below.
Publication details.
Study eligibility criteria.
Study details (e.g. aim, study design, randomisation method, study location, start and end dates).
Participant characteristics (e.g. number of participants, age, sex, diagnostic criteria, study setting).
Description of intervention and comparator (e.g. duration of treatment, timing, delivery, numbers of participants randomised to groups).
Outcome data (e.g. numerical data such as means and standard deviations, instruments used to assess outcomes of interest, time points of outcome assessment, withdrawals).
Funding sources and any conflicts of interest for study authors.
We compared the extracted data and resolved any disagreements by consensus or by deferment to a third review author (JH). One review author (CM) input the data into Review Manager 5 (RevMan 2014), and a second review author (DE) checked these for accuracy.
Assessment of risk of bias in included studies
Two review authors (CM, DE) independently assessed each included study for risk of bias using the Cochrane tool for assessing risk of bias, as outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We assessed risk of bias by examining the following six domains.
Random sequence generation (checking for possible selection bias): we assessed the method used to generate the allocation sequence as being at low risk of bias (any truly random process, e.g. random number table; computer random number generator) or unclear risk of bias (method used to generate the sequence not clearly stated). We excluded studies that used a non‐random process (e.g. odd or even date of birth; hospital or clinic record number).
Allocation concealment (checking for possible selection bias): we assessed the method used to conceal allocation to interventions before assignment to determine whether intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment. We assessed these methods as being at low risk of bias (e.g. telephone or central randomisation; consecutively numbered sealed opaque envelopes); unclear risk of bias (method not clearly stated); or high risk of bias (e.g. open list).
Blinding of participants and personnel (checking for possible performance bias): we assessed methods used to blind study participants and personnel from knowledge of which intervention a participant received. We assessed these methods as being at low risk of bias (study states that it was blinded and describes the method used to achieve blinding, such as identical tablets matched in appearance or smell, or a double‐dummy technique) or unclear risk of bias (study states that it was blinded but does not provide an adequate description of how this was achieved). We considered studies that are not double‐blind as being at high risk of bias.
Blinding of outcome assessment (checking for possible detection bias): we assessed methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received. We assessed these methods as being at low risk of bias (study has a clear statement that outcome assessors were unaware of treatment allocation and ideally describes how this was achieved) or unclear risk of bias (study states that outcome assessors were blind to treatment allocation but lacks a clear statement on how this was achieved). We considered studies where outcome assessment was not blinded as having high risk of bias.
Selective reporting (checking for reporting bias): we aimed to assess whether reported primary and secondary outcome measures were pre‐specified in a protocol. If the trial protocol was available from a trial registry, the reported outcomes should be consistent with those listed in the protocol if the protocol was registered before or at the time the trial began. We assessed selective reporting as being at low risk of bias (studies reporting primary and secondary outcomes as specified in the original protocol) or high risk of bias (not all pre‐specified outcomes reported, or only for certain data collection time points).
Incomplete outcome data (checking for possible attrition bias due to the quantity, nature, and handling of incomplete outcome data): we assessed methods used to deal with incomplete data as being at low risk (< 10% of participants did not complete the study and/or used ‘baseline observation carried forward’ analysis) or high risk of bias (used 'last observation carried forward' analysis or 'completer' analysis).
We resolved disagreements by discussion or by deferment to a third review author (JH). When information was missing from the published papers, we contacted study authors. We have presented our judgements in the 'Risk of bias' tables for each study, and we have provided statements to justify our decisions, along with appropriate quotes from reports or personal communications to support our decisions. We have produced figures summarising the risk of bias for all included studies.
Measures of treatment effect
Continuous data
We analysed these data based on mean, standard deviation (SD), and numbers of participants assessed for both intervention and comparison groups to calculate mean difference (MD) and 95% confidence interval (CI). If more than one study measured the same outcome using different validated scales, we intended to calculate a standardised mean difference (SMD), SD, and 95% CI. We aimed to calculate SMD as the difference in mean outcomes between groups divided by the pooled SD of both groups. We used change from baseline scores for continuous data.
Dichotomous data
We analysed these data based on the numbers of events and the numbers of participants assessed in intervention and comparison groups. We intended to use these data to calculate the risk ratio (RR) and 95% CI.
Unit of analysis issues
The unit of analysis is the study participant with PD. For studies with more than two arms, we planned to include only arms that met the inclusion criteria of the review. For studies including multiple intervention groups of interest, we aimed to combine all arms for a single pair‐wise comparison (Higgins 2011).
Dealing with missing data
When data were missing, we intended to contact study authors to obtain the missing data. We planned to make two attempts to contact study authors, although this was not required. We examined reports of studies with missing data, and, when possible, we reported the reasons for missing data.
Assessment of heterogeneity
When we were able to undertake a meta‐analysis, we aimed to assess heterogeneity using the I² statistic that is included in the forest plot of a Cochrane Review. We planned to regard a level of heterogeneity above 50% as substantial or high, as explained in the Cochrane Handbook for Systematic Reviews of Interventions, Section 9.5.2 (Higgins 2011). If heterogeneity existed, we planned to examine study reports to identify possible reasons for it. If we identified sufficient studies, we aimed to undertake subgroup analysis according to possible identified reasons for heterogeneity.
Assessment of reporting biases
If we were able to pool 10 or more trials in a single analysis, we planned to create and examine a funnel plot to explore possible small‐study and publication biases. We intended to test for asymmetry using Egger's test (Egger 1997). When protocols were available, we compared outcomes reported in published trial reports with those listed in trial protocols to assess reporting bias.
Data synthesis
When two or more studies reported the same outcome and were sufficiently similar in terms of treatments and participants, we planned to undertake meta‐analyses using a fixed‐effect model. We intended to report pooled effect measures for dichotomous outcomes using Mantel‐Haenszel methods, and for continuous outcomes, the inverse variance method. If we noted considerable heterogeneity that could not readily be explained, we planned to use a random‐effects model. When it was not possible to pool findings from studies in a meta‐analysis, we aimed to present the results of each study and to provide a narrative synthesis of findings. We performed statistical analysis using Review Manager 5 (RevMan 2014).
Subgroup analysis and investigation of heterogeneity
If sufficient data were available, we aimed to conduct the following subgroup analyses.
Severity of PD: defined as severe, moderate, or mild as scored on the UDPRS subscale Part III (Fahn 1987).
Clinical subtypes: tremor dominant, mixed, akinetic/rigid.
Different dosages of GLP‐1 receptor agonists.
Different durations of treatment.
Sensitivity analysis
If we identified sufficient studies, we intended to repeat the analyses while excluding studies at high risk of bias.
Quality of the evidence
Independently, two review authors (CM, DE) assessed the quality of evidence for the three primary outcomes using the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach (Schünemann 2013). We used methods and recommendations as described in Section 8.5 and Chapter 12 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
To ensure consistency and reproducibility of GRADE judgements, we assessed key outcomes using the criteria below for each of the five domains.
Study limitations: if a study suffers from major limitations, such as lack of blinding, these are likely to result in a biased assessment of the intervention effect.
Indirectness of evidence: this may occur when the review intervention of interest is not compared directly with comparators of interest, or when trials that meet the inclusion criteria address a restricted version of the review question in terms of participants, intervention, comparator, or outcomes.
Consistency of effect: when studies show differing estimates of effects, we must look for explanations for heterogeneity.
Imprecision of results: this occurs if included studies have few participants or events and large confidence intervals.
Publication bias: this occurs if investigators do not report studies ‐ usually those with no effect, or outcomes ‐ typically harmful ones or those showing no effect.
The GRADE system uses the following criteria for assigning the grade of evidence.
High: we are very confident that the true effect lies close to that of the estimate of the effect.
Moderate: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of effect, but there is a possibility that it is substantially different.
Low: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect.
Very low: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect.
We downgraded the GRADE rating by one (‐1) or two (‐2) levels if we identified:
serious (‐1) or very serious (‐2) limitations to study quality;
important inconsistency (‐1);
some (‐1) or major (‐2) uncertainty about directness;
imprecise or sparse data (‐1); or
a high probability of reporting bias (‐1).
We have provided reasons for our decisions regarding grading of the quality of evidence.
'Summary of findings' tables
We created two ‘Summary of findings’ tables to summarise results for the three primary outcomes (PD motor impairment, health‐related quality of life, and adverse events) for two comparisons. For adverse events, we have presented separate data for serious adverse events and weight loss. We have presented in these tables our assessment of the quality of evidence. We used GRADEpro software to prepare the tables (GRADEpro 2015), importing data from Review Manager 5 (RevMan 2014).
Results
Description of studies
Results of the search
Our searches yielded 99 unique records. After screening titles and abstracts, we excluded 63 records and obtained the full text of the remaining 23. We assessed these 23 full‐text articles and excluded five that we determined to be duplicates. Two studies met our inclusion criteria and are described in 11 full‐text articles. Seven studies are ongoing (Characteristics of ongoing studies). See Figure 1 for a flowchart illustrating study selection.
Included studies
Two studies described in 11 papers are included in this review. In both studies, participants received exenatide. One study compared exenatide with placebo, with all participants continuing to receive their usual treatment (Athauda 2017), and the other study compared exenatide plus conventional treatment with conventional treatment (Aviles‐Olmos 2013b) (we have termed this comparison group 'no treatment'). Athauda 2017 is described in five papers. Three of these papers describe post‐hoc analyses of study data; thus their findings are not included in this review. Aviles‐Olmos 2013b is described in six papers. A total of 107 participants were randomised across the two studies, and there were three dropouts. Both studies were conducted in the UK.
Methods
Although both were randomised, controlled, parallel‐group studies, one study was double‐blind (Athauda 2017), and the other was single‐blind (Aviles‐Olmos 2013b). In the single‐blind study (Aviles‐Olmos 2013), study participants and investigators were unblinded to the assigned intervention, but assessments of the MDS‐UPDRS subscale Part III were video recorded and rated by clinicians who were blinded to randomisation. Both studies were based both in a research institute and in tertiary care.
Participants
Eligible participants were at Hoehn and Yahr stage 2.5 or less (Athauda 2017), or they were at Hoehn and Yahr stage 2 to 2.5 (Aviles‐Olmos 2013b), when on medication (Hoehn 1967). Athauda 2017 recruited participants aged 25 to 75 years, and Aviles‐Olmos 2013b recruited participants aged 45 to 70 years, with at least five years of symptoms. The mean age of study participants was similar in both studies, ranging from 57.8 years (control group) to 61.6 years (intervention group) in Athauda 2017, and ranging from 59.4 years (control group) to 61.4 years (intervention group) in Aviles‐Olmos 2013b.
Intervention
In the double‐blind study (Athauda 2017), participants were taught how to self‐administer weekly subcutaneous injections of exenatide 2 mg or placebo for 48 weeks, in addition to their usual medication. Participants were followed up at 60 weeks (i.e. after a 12‐week washout period). In the single‐blind study (Aviles‐Olmos 2013b), participants in the intervention group were taught how to self‐administer twice‐daily 5 µg exenatide injections for one month, then twice‐daily injections of 10 µg exenatide for 11 months (equivalent to a 0.14 mg weekly dose). Participants in both intervention and control groups continued to receive their conventional treatment. Participants were followed up at 14 months (i.e. after a 2‐month washout period) (Aviles‐Olmos 2013b), and again at 24 months (i.e. after a 12‐month absence of exenatide) (Aviles‐Olmos 2014).
Outcomes
The main outcome for both studies was PD motor impairment measured by the MDS‐UPDRS subscale Part III off medication (i.e. after withdrawal of levodopa overnight representing a period of at least eight hours). For secondary outcomes, both studies also assessed MDS‐UPDRS Parts I, II, III, and IV, as well as PDQ‐39, MATTIS DRS, MADRS, and the Dyskinesia Rating Scale, in the on‐medication state. These outcomes were reported as mean change from baseline. Studies also reported the numbers of adverse events, including change in weight. Athauda 2017 assessed outcomes at 12, 24, 36, 48, and 60 weeks post baseline. Aviles‐Olmos 2013b assessed outcomes at 6, 12, 14, and 24 months post baseline.
Excluded studies
All five excluded studies were duplicates.
Risk of bias in included studies
We have provided details of the risk of bias for each study in the Characteristics of included studies table. Figure 2 and Figure 3 present a summary of the risk of bias for both included studies.
2.

Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
We judged both studies to be at low risk of selection bias. Athauda 2017 used a commercial company to generate the random sequence and to allocate participants.
Blinding
In both studies, assessment of the primary outcome was undertaken by assessors blinded to treatment groups. We judged the single‐blind study to be at high risk of performance bias (Aviles‐Olmos 2013). These investigators reported that participants might detect their treatment allocation as a result of adverse events including injection site reactions.
Incomplete outcome data
We assessed the two studies as being at low risk of attrition bias. In Athauda 2017, 60 of the 62 randomised participants (97%) were included in the primary analysis, and in Aviles‐Olmos 2013, 44 of the 45 randomised participants (98%) were included in the analysis
Selective reporting
We judged both studies to be at low risk of selective reporting bias. Both studies were prospectively registered with clinical trials registries, and the outcomes they presented in their published reports matched those stated in the registries.
Other potential sources of bias
We considered the two studies to be at low risk of other potential biases. One study reported some imbalance between intervention and control groups at baseline in terms of MDS‐UPDRS Part III scores, but these were adjusted for in the analysis.
Effects of interventions
As we were keen to assess the neuroprotective effects of GLP‐1 receptor agonists, in our analyses, we have used data from outcomes assessed two to three months post cessation of treatment, unless stated otherwise. We have presented narratively the data from outcomes measured at a later follow‐up.
GLP‐1 receptor agonists versus placebo
One study with 60 participants included in primary analyses contributed data to all the following outcomes in this comparison (Athauda 2017). For the analyses, we used data from the 60 weeks post baseline assessment (unless stated otherwise) following a 12‐week washout period.
Primary outcomes
1. MDS‐UPDRS Part III
Data show a statistically significant difference in change from baseline in MDS‐UPDRS Part III off‐medication scores when groups receiving exenatide were compared with those receiving placebo (mean difference (MD) ‐3.10, 95% confidence interval (CI) ‐6.11 to ‐0.09) (unadjusted) (Analysis 1.1). Participants receiving exenatide reported a mean decrease in off‐medication scores representing improvement in motor impairment. However, 3.10 did not exceed the minimal clinically important difference (MCID) for improvement in motor impairment of ‐3.25 (Horváth 2015). We rated the quality of evidence as low. We downgraded the evidence once for indirectness as the intervention consisted of only one receptor agonist (RA), and once for imprecision due to a small sample size. Athauda 2017 adjusted the mean difference in change from baseline at 60 weeks for Hoehn and Yahr stage and baseline raw MDS‐UPDRS Part III scores (Hoehn 1967), revealing a difference of ‐3.5 (95% CI ‐6.7 to ‐0.3). This value exceeds the MCID of ‐3.25 points for detecting improvement (Horváth 2015).
1.1. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 1: PD motor impairment ‐ MDS‐UPDRS Part III (off medication)
2. Health‐related quality of life
Data show no statistically significant difference in change from baseline in HRQoL scores assessed by the PDQ‐39 scale when groups receiving exenatide were compared with those receiving placebo (MD ‐1.80, 95% CI ‐6.95 to 3.35) (Analysis 1.2). The MD did not exceed the MCID for improvement in HRQoL of ‐4.72 (Horváth 2017). Similarly, there was no statistically significant difference in change from baseline in HRQoL scores as assessed by the EQ5D scale when groups receiving exenatide were compared with those receiving placebo (MD 0.07, 95% CI ‐0.03 to 0.16) (Analysis 1.3), or by the EQ5D VAS (MD 5.00, 95% CI ‐3.42 to 13.42) (Analysis 1.4). We rated the quality of evidence as low. We downgraded the evidence once for indirectness as the intervention was only one RA, and once for imprecision due to a small sample size and to the CI including the null effect and risk of appreciable harm and benefit.
1.2. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 2: HRQoL ‐ PDQ‐39 SI
1.3. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 3: HRQoL ‐ EQ5D
1.4. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 4: HRQoL ‐ EQ5D‐VAS
3. Adverse events
Serious adverse events (SAEs)
Eight SAEs were reported (intervention (I) = 6, comparator (C) = 2). "Significant weight loss" was reported as a single SAE in the placebo group, but review authors considered that none of the SAEs were related to the intervention; thus we have not included the data in a meta‐analysis. It is not clear whether adverse events were assessed at 48 or 60 weeks.
Weight loss
Data show no statistically significant difference in the number of participants reporting weight loss at 48 weeks when groups receiving exenatide and those given placebo were compared (risk ratio (RR) 1.25, 95% CI 0.89 to 1.76) (Analysis 1.5). We rated the quality of evidence as low. We downgraded the evidence once for indirectness as the intervention was only one RA, and once for imprecision due to a small sample size and to the CI including the null effect and risk of appreciable harm and benefit. Study authors reported that at 48 weeks, a total of 21 participants had lost less than 2 kg (I = 11, C = 10), five participants had lost between 2 and 4 kg (I = 2, C = 3), and 16 participants had lost more than 4 kg (I = 11, C = 5). It is unclear whether any participants experienced rapid weight loss.
1.5. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 5: Adverse events ‐ weight loss (kg) (assessed at 48 weeks post baseline)
Athauda 2017 reported that participants in the exenatide group lost a mean of 2.6 kg (95% CI ‐4.0 to ‐1.2) compared to a mean weight loss in the control group of 0.6 kg (95% CI ‐1.9 to 0.8) at 48 weeks. However, study authors found no significant correlation between degree of weight loss and change in score for the primary outcome.
Other adverse events
Participants reported a range of adverse events. Athauda 2017 reported these data as the number of events experienced by individuals across all time points by group. In terms of gastrointestinal adverse events, participants reported the following.
Nausea: a total of 26 events (I = 16, C = 10).
Constipation: a total of 23 events (I = 12, C = 11).
Diarrhoea: a total of 14 events (I = 8, C = 6).
Abdominal pain: a total of 8 events (I = 5, C = 3).
Loss of appetite: a total of 4 events (I = 3, C = 1).
Vomiting: a total of 2 events (I = 2, C = 0).
Athauda 2017 reported that there was no statistically significant correlation between the presence or absence of weight loss, nausea, loss of appetite, or abdominal pain and treatment group (χ² = ‐0.388, P = 0.5330). Seven participants in the exenatide group and 11 in the placebo group reported weight gain.
Other adverse events reported by participants over all time points included the following.
Injection site reaction: a total of 53 events (I = 27, C = 26).
Increased time off medication: a total of 20 events (I = 8, C = 12).
Secondary outcomes
1. PD motor impairment assessed by a validated scale other than off medication MDS‐UPDRS Part III
There was no statistically significant difference in change from baseline in MDS‐UPDRS Part III scores on medication when groups receiving exenatide were compared with those receiving placebo (MD 0.52, 95% CI ‐2.34 to 3.38) (Analysis 1.6). Similarly, there was no statistically significant difference in change from baseline in UDysRS scores when groups receiving exenatide were compared with those receiving placebo (MD ‐0.90, 95% CI ‐4.29 to 2.49) (Analysis 1.7).
1.6. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 6: PD motor impairment ‐ MDS‐UPDRS Part III
1.7. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 7: PD motor impairment ‐ UDysRS
2. Non‐motor outcomes
Data show no statistically significant differences in change from baseline in MDS‐UPDRS Part I scores (MD ‐1.20, 95% CI ‐3.23 to 0.83) (Analysis 1.8) or NMSQuest scores (MD ‐0.80, 95% CI ‐10.83 to 9.23) (Analysis 1.9) when groups receiving exenatide were compared with those receiving placebo.
1.8. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 8: Non‐motor impairment ‐ MDS‐UPDRS Part I
1.9. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 9: Non‐motor impairment ‐ NMSQuest
3. Activities of daily living
Data show no statistically significant difference in change from baseline in MDS‐UPDRS Part II scores when groups receiving exenatide were compared with those receiving placebo (MD ‐1.00, 95% CI ‐3.04 to 1.04) (Analysis 1.10).
1.10. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 10: Activities of daily living ‐ MDS‐UPDRS Part II
4. Psychological outcomes
There was no statistically significant difference in change from baseline in Mattis DRS scores (MD 1.50, 95% CI ‐0.40 to 3.40) (Analysis 1.11) or MADRS scores (MD ‐0.70, 95% CI ‐2.41 to 1.01) (Analysis 1.12) when groups receiving exenatide were compared with those receiving placebo.
1.11. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 11: Psychological outcomes ‐ Mattis DRS
1.12. Analysis.

Comparison 1: Exenatide vs placebo (60 weeks post‐baseline), Outcome 12: Psychological outcomes ‐ MADRS
Subgroup analysis and sensitivity analysis
Data were insufficient for subgroup analyses or sensitivity analyses to be conducted for this comparison.
GLP‐1 receptor agonists versus no treatment
One study with 44 participants included in the primary analyses contributed data to all the following outcomes in this comparison (Aviles‐Olmos 2013). In the analyses, we used data from the 14 months post baseline assessment following a two‐month washout. We also report narratively data from 24 months post baseline.
Primary outcomes
1. MDS‐UPDRS Part III
At 14 months, data show a statistically significant difference in change from baseline in MDS‐UPDRS Part III off‐medication scores when groups receiving exenatide were compared with those receiving no treatment (MD ‐4.50, 95% CI ‐8.64 to ‐0.36) (Analysis 2.1). This value exceeds the MCID of ‐3.25 points for detecting improvement (Horváth 2015). We rated the quality of evidence as very low. We downgraded the evidence by one level for study limitations of unclear risk of selection bias and high risk of performance bias; we downgraded by one level for indirectness as the intervention was only one RA; and we downgraded by one level for imprecision due to a small sample size.
2.1. Analysis.

Comparison 2: Exenatide vs no treatment (14 months (60.8 weeks) post baseline), Outcome 1: PD motor impairment ‐ MDS‐UPDRS Part III (off medication)
Study authors report that when scores from the open‐label rating of rigidity scores were added to the blinded data, an MD of 7.2 points (95% CI 2.1 to 12.2; P = 0.006) was obtained, with participants in the exenatide group showing a mean improvement of 0.8 points (standard deviation (SD) 8.7) and those in the no treatment group showing a mean decline of 6.4 points (SD 7.8).
At 24 months, study authors report a statistically significant difference in change from baseline in MDS‐UPDRS Part III off‐medication scores measured blindly when groups receiving exenatide were compared with those receiving no treatment (MD 5.6, 95% CI 2.2 to 9.0; P = 0.002). Participants in the exenatide group showed a mean improvement of 1.1 points (SD 5.9), and those receiving no treatment showed a mean decline of 4.5 points (SD 5.3). Study authors report that when scores from the open‐label rating of rigidity scores were added to the blinded data, an MD of 8.0 points (95% CI 3.8 to 12.2; P < 0.001) was obtained, with participants in the exenatide group showing a mean decline of 0.5 points (SD 7.3) and those in the no treatment group showing a mean decline of 8.5 points (SD 6.3).
2. Health‐related quality of life
At 14 months, there was no statistically significant difference in change from baseline in HRQoL scores assessed by the PDQ‐39 SI when groups receiving no treatment were compared with those receiving placebo (MD 3.50, 95% CI ‐2.75 to 9.75) (Analysis 2.2). We rated the quality of evidence as very low. We downgraded evidence by one level for study limitations of unclear risk of selection bias and high risk of performance bias; we downgraded by one level for indirectness as the intervention was only one RA; and we downgraded by one level for imprecision due to a small sample and the CI including the null effect and risk of appreciable harm and benefit.
2.2. Analysis.

Comparison 2: Exenatide vs no treatment (14 months (60.8 weeks) post baseline), Outcome 2: HRQoL ‐ PDQ‐39 SI
At 24 months, study authors reported that there was a non‐significant difference in scores from baseline when the two groups were compared. Participants receiving exenatide showed a mean difference from baseline on the PDQ‐39 SI of ‐0.1 points (SD 12.3, 95% CI ‐5.9 to 5.6), and participants receiving no treatment reported a mean difference of 1.2 points (SD 9.3, 95% CI ‐2.7 to 5.1; P = 0.682).
3. Adverse events
A total of four SAEs were reported in both groups ‐ by four participants in the exenatide group and by three in the no treatment group. These events included sciatica, insomnia, possible transient ischaemic attack, prostatectomy, lymph node dissection, anxiety, and fractured radius. There was no significant difference in the number of all‐cause SAEs reported by those receiving exenatide compared to those receiving no treatment (RR 1.60, 95% CI 0.40 to 6.32) (Analysis 2.3). We rated the quality of evidence as very low. We downgraded the evidence by one level for study limitations of unclear risk of selection bias and high risk of performance bias; we downgraded by one level for indirectness as the intervention was only one RA; and we downgraded by one level for imprecision due to a small sample and the CI including the null effect and risk of appreciable harm and benefit.
2.3. Analysis.

Comparison 2: Exenatide vs no treatment (14 months (60.8 weeks) post baseline), Outcome 3: Serious adverse events
Aviles‐Olmos 2013 presents the total number of adverse events reported at 1, 3, and 9 months. Participants reported a range of adverse events. In terms of the total number of gastrointestinal adverse events, participants reported the following.
Nausea: a total of 21 events (I = 13, C = 8).
Constipation: a total of 32 events (I = 18, C = 14).
Diarrhoea: a total of 12 events (I = 7, C = 5).
Abdominal pain: a total of 12 events (I = 6, C = 6).
Loss of appetite: a total of 5 events (I = 5, C = 0).
Weight loss: a total of 27 events (I = 19, C = 8).
Other adverse events reported by participants included the following.
Injection site bruising: I = 2.
Increased time off medication: a total of 16 events (I = 4, C = 12).
There was a statistically significant difference in change from baseline in weight measured in kg when groups receiving exenatide were compared with those receiving no treatment (MD ‐2.40, 95% CI ‐4.56 to ‐0.24) (Analysis 2.4). We rated the quality of evidence as very low. We downgraded the evidence by one level for study limitations of unclear risk of selection bias and high risk of performance bias; we downgraded by one level for indirectness as the intervention was only one RA; and we downgraded by one level for imprecision due to a small sample and the CI including the null effect and risk of appreciable harm and benefit. Details are insufficient to show whether any participants experienced rapid weight loss.
2.4. Analysis.

Comparison 2: Exenatide vs no treatment (14 months (60.8 weeks) post baseline), Outcome 4: Adverse events ‐ weight loss (kg) (assessed at 12 months (52 weeks))
At 24 months, study authors reported a non‐significant between‐group difference in weight loss from baseline of 0.1 kg (95% CI 3.0 to 2.8; P = 0.93), with participants receiving exenatide experiencing a mean weight loss from baseline of 1.6 kg (SD 3.1) and participants receiving no treatment experiencing a mean wight loss of 1.7 kg (SD 5.8).
Secondary outcomes
1. PD motor impairment as assessed by a validated scale other than off medication MDS‐UPDRS Part III
Data show a significant difference in change from baseline in MDS‐UPDRS Part III scores on medication when receiving exenatide were compared with those receiving no treatment (MD ‐9.80, 95% CI ‐14.47 to ‐5.13) (Analysis 2.5). This value exceeds the MCID of ‐3.25 points for detecting improvement (Horváth 2015). There was no significant difference in change from baseline in Rush Dyskinesia Rating Scale scores when groups receiving exenatide were compared with those receiving no treatment (MD 0.90, 95% CI ‐1.49 to 3.29) (Analysis 2.6).
2.5. Analysis.

Comparison 2: Exenatide vs no treatment (14 months (60.8 weeks) post baseline), Outcome 5: PD motor impairment ‐ MDS‐UPDRS Part III
2.6. Analysis.

Comparison 2: Exenatide vs no treatment (14 months (60.8 weeks) post baseline), Outcome 6: PD motor impairment ‐ Rush Dyskinesia Rating Scale
At 24 months, study authors reported that participants receiving exenatide reported a mean difference from baseline in MDS‐UPDRS Part III scores on medication of ‐0.9 points (SD 6.9, 95% CI ‐4.2 to 2.3), and control participants reported a mean difference of 7.8 points (SD 6.7, 95% CI 5.0 to 10.7), representing a significant difference (P < 0.001).
At 24 months, participants receiving exenatide reported a mean difference from baseline in Rush Dyskinesia Rating Scale scores on medication of 0.8 points (SD 6.0, 95% CI ‐2.0 to 3.6), and control participants reported a mean difference of ‐0.6 points (SD 3.0, 95% CI ‐1.8 to 0.7), representing a non‐significant difference (P = 0.328).
2. Non‐motor outcomes
There was no significant difference in change from baseline in MDS‐UPDRS Part I scores when groups receiving exenatide were compared with those receiving no treatment (MD ‐3.70, 95% CI ‐7.45 to 0.05) (Analysis 2.7).
2.7. Analysis.

Comparison 2: Exenatide vs no treatment (14 months (60.8 weeks) post baseline), Outcome 7: Non‐motor impairment ‐ MDS‐UPDRS Part I
At 24 months, participants receiving exenatide reported a mean difference from baseline in MDS‐UPDRS Part I scores of 2.0 points (SD 4.2, 95% CI 0.0 to 4.0), and participants receiving no treatment reported a mean difference of 5.1 points (SD 5.5, 95% CI 2.8 to 7.4), representing a significant difference (P = 0.049).
Data on NMSQuest scores were not published for 14 months (Aviles‐Olmos 2013b). At 24 months, participants receiving exenatide reported a mean difference from baseline in NMSQuest scores of ‐0.8 points (SD 3.8, 95% CI ‐2.6 to 1.0), and control participants reported a mean difference of 0.2 points (SD 4.3, 95% CI ‐1.6 to 2.1), representing a non‐significant difference (P = 0.403).
3. Activities of daily living
There was no statistically significant difference in change from baseline in MDS‐UPDRS Part II scores when groups receiving exenatide were compared with those receiving no treatment (MD ‐2.90, 95% CI ‐6.41 to 0.61) (Analysis 2.8).
2.8. Analysis.

Comparison 2: Exenatide vs no treatment (14 months (60.8 weeks) post baseline), Outcome 8: Activities of daily living ‐ MDS‐UPDRS Part II
At 24 months, participants receiving exenatide reported a mean difference from baseline in MDS‐UPDRS Part II scores of 2.7 points (SD 5.4, 95% CI 0.2 to 5.3), and control participants reported a mean difference of 7.0 points (SD 5.0, 95% CI 4.9 to 9.1), representing a significant difference (P = 0.009).
4. Psychological outcomes
Data show a significant difference in change from baseline in Mattis DRS scores when groups receiving exenatide were compared with those receiving no treatment (MD 6.30, 95% CI 2.79 to 9.81) (Analysis 2.9). There was no statistically significant difference in change from baseline in MADRS scores (MD ‐2.20, 95% CI ‐5.40 to 1.00) (Analysis 2.10) when groups receiving exenatide were compared with those receiving no treatment.
2.9. Analysis.

Comparison 2: Exenatide vs no treatment (14 months (60.8 weeks) post baseline), Outcome 9: Psychological outcomes ‐ Mattis DRS
2.10. Analysis.

Comparison 2: Exenatide vs no treatment (14 months (60.8 weeks) post baseline), Outcome 10: Psychological outcomes ‐ MADRS
At 24 months, participants receiving exenatide reported a mean difference from baseline in Mattis DRS scores of 1.8 points (SD 6.5, 95% CI ‐1.2 to 4.8), and control participants reported a mean difference of ‐3.5 points (SD 6.4, 95% CI ‐0.8 to ‐6.3), representing a significant mean difference between groups (P = 0.006).
At 24 months, participants receiving exenatide reported a mean difference from baseline in MADRS scores of ‐1.9 points (SD 5.2, 95% CI 0.5 to ‐4.3), and control participants reported a mean difference of 1.5 points (SD 7.0, 95% CI ‐1.4 to 4.4), representing a non‐significant difference (P = 0.79).
Subgroup analysis and sensitivity analysis
We had insufficient data to undertake subgroup analyses or sensitivity analyses for this comparison.
Discussion
Summary of main results
Two studies are included in this review evaluating the effectiveness and safety of glucagon‐like peptide‐1 (GLP‐1) receptor agonists (RAs) for Parkinson's disease (PD). One study of data from 60 participants compared a GLP‐1 RA (exenatide) with placebo, and one study of data from 44 participants compared a GLP‐1 RA (exenatide) with no treatment (both groups continued to receive their usual medication). Seven studies are ongoing, assessing the effects of exenatide, liraglutide, lixisenatide and semaglutide.
GLP‐1 receptor agonists versus placebo
Outcomes were assessed at 60 weeks (i.e. after a 12‐week washout period following 48 weeks of intervention). In terms of primary outcomes, findings show there was significant improvement at 60 weeks in motor impairment assessed by the Movement Disorder Society‐Unified Parkinson's Disease Rating Scale (MDS‐UPDRS) Part III in the off‐medication state for participants receiving exenatide compared to those receiving placebo. The mean difference is below that of a minimal clinically important difference (MCID) of ‐3.25 points for detecting improvement (Horváth 2015); however, the adjusted mean difference of ‐3.5 points exceeded an MCID. The quality of the evidence was rated as low. There was no difference between groups in terms of health‐related quality of life (HRQoL) or the number of participants reporting weight loss; we rated the quality of evidence for these outcomes as low.
In terms of secondary outcomes, exenatide did not provide any improvement in motor impairment as assessed by MDS‐UPDRS Part III scores in the on‐medication state, nor in Unified Dyskinesia Rating Scale (UDysRS) scores. There was no improvement among participants receiving exenatide for non‐motor outcomes as assessed by MDS‐UPDRS Part I and the Non‐Motor Symptoms Questionnaire (NMSQuest). Similarly, participants receiving exenatide showed no improvement in activities of daily living as assessed by MDS‐UPDRS Part II, nor in psychological outcomes as assessed by the Mattis Dementia Rating Scale (DRS) or the Montgomery‐Åsberg Depression Rating Scale (MADRS), when compared to participants receiving placebo. Adjusted mean difference scores were not significantly different between the two groups.
GLP‐1 receptor agonists versus no treatment
Outcomes were assessed at 14 months (60.8 weeks) (i.e. after a two‐month washout period following 12 months of intervention) and at 24 months. In terms of primary outcomes, findings show there was significant improvement at 14 months in motor impairment assessed by MDS‐UPDRS Part III in the off‐medication state for participants receiving exenatide compared to those receiving no treatment. The mean difference is above that of an MCID of ‐3.25 points for detecting improvement (Horváth 2015). The quality of evidence was rated as very low. There was no difference between groups in terms of HRQoL; the quality of this evidence was rated as very low. Participants receiving exenatide were significantly more likely to report weight loss (quality of evidence was rated as very low); however, there was no significant difference in the number of participants reporting serious adverse events (SAEs) (quality of evidence was rated as very low).
In terms of secondary outcomes, there was significant improvement in motor impairment as assessed by MDS‐UPDRS Part III in the on‐medication state for participants receiving exenatide compared to those receiving no treatment. The mean difference is above that of an MCID of ‐3.25 points for detecting improvement (Horváth 2015). However, there was no significant difference in motor impairment as measured by the Rush Dyskinesia Rating Scale. Similarly there was no significant change in non‐motor outcomes as assessed by MDS‐UPDRS Part I scores, activities of daily living as measured by MDS‐UPDRS Part II, or psychological outcomes as assessed by the MADRS.
At 24 months post baseline (i.e. 12 months post cessation of exenatide), study participants receiving exenatide scored significantly lower MDS‐UPDRS Part III scores off medication than participants receiving no treatment. There was no difference between groups in terms of HRQoL or weight loss. In terms of secondary outcomes, there was a significant between‐group difference in MDS‐UPDRS Part III scores as assessed, but there was no difference in Rush Dyskinesia Rating Scale scores. For non‐motor impairment, there was a significant between‐group difference as assessed by MDS‐UPDRS Part I but not by NMSQuest. There was a significant between‐group difference for activities of daily living as assessed by MDS‐UPDRS Part II. There was a significant between‐group difference for psychological outcomes as assessed by the Mattis DRS but not by the MADRS.
Evidence of improvement in motor and non‐motor scores on the MDS‐UPDRS following a minimum washout period of three months suggests that exenatide may slow progression of the disease. It is difficult to draw conclusions regarding a symptomatic effect of exenatide, as studies were designed to measure long‐term rather than short‐term effects of exenatide. To assess symptomatic effects of a GLP‐1 RA would require outcomes to be assessed several times immediately after the start of the intervention.
Overall completeness and applicability of evidence
We found only two studies that met our inclusion criteria; they compared the effects of administering exenatide versus placebo and no treatment. Although it was not possible to pool data from these studies, there were similarities within the studies. Participants in both studies self‐administered exenatide for similar lengths of time, and the primary outcome for both studies was motor impairment as assessed by MDS‐UPDRS Part III while off medication and following a washout period. Improvements in assessments of PD disability using the MDS‐UPDRS have been reported among study participants receiving placebo (Goetz 2000). Both included studies assessed HRQoL, adverse events, non‐motor impairment, psychological impairment, and effects on activities of daily living. Although the sample size of these studies was small, attrition was low. Both studies reported some imbalance in baseline characteristics between intervention and control groups, including slight differences in disease duration and severity. One study undertook a pre‐specified adjustment in analyses to account for this imbalance. Another study was designed as a single‐blind study, with study authors reporting that this was due to the prohibitive costs of developing a placebo. This study was thus considered as a proof‐of‐concept study that would provide the opportunity to test whether findings in the laboratory could be replicated in people with PD. Both studies were single‐centre studies. Both studies collected data at multiple time points, but in our analyses, we used data collected at similar time points to increase the comparability of our analyses. One study did not report between‐group mean differences in score changes for some outcomes.
Quality of the evidence
According to GRADE criteria, we rated the certainty of evidence from Athauda 2017 as low, and from Aviles‐Olmos 2013 as very low. We rated Athauda 2017 at low risk for all biases. Aviles‐Olmos 2013 was designed as a proof‐of‐concept study. We judged this study to be at high risk of performance bias. In this study, participants self‐administered exenatide using a pen, and as the costs to produce a placebo pen were considered by study authors to be prohibitive, it was not possible to blind participants and personnel to treatment groups. The primary outcome for this study and many of the secondary outcomes assessed were rated by assessors blinded to treatment groups; thus we rated detection bias as low. We downgraded Aviles‐Olmos 2013 for study limitations. We downgraded both studies for indirectness, as this review is concerned with assessing the effectiveness of any RA, and both studies examined the effects of just one RA ‐ exenatide; thus our findings relate only to exenatide. We downgraded both studies for imprecision, with confidence intervals for some outcomes including the possibility of opposite effects.
Potential biases in the review process
We undertook a comprehensive search for relevant studies, including handsearching of conference abstracts. We followed the systematic review process as outlined by the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). Two review authors undertook each stage of the review process, thereby reducing the risk of introducing bias into the study selection process. We are confident that we have identified all relevant randomised controlled trials.
Agreements and disagreements with other studies or reviews
One recent systematic review assessing the neuroprotective role of GLP‐1 RAs concluded that their use for longer than three to six months led to improvement in motor symptoms of people with PD, while control groups experienced deterioration in symptoms (Erbil 2019). Much of the work done so far on GLP‐1 RAs has been undertaken in the laboratory with rodents.
Authors' conclusions
Implications for practice.
We found low‐ or very low‐certainty evidence suggesting that the GLP‐1 RA exenatide improves motor impairment as assessed by MDS‐UPDRS Part III in the off‐medication state following a minimum of 11 months of medication and a maximum washout period of 12 weeks. As improvement in motor impairment was seen at three months post cessation of exenatide, this suggests that exenatide may have a disease‐modifying effect on PD. No conclusions can be drawn about the symptomatic benefits of exenatide. We are uncertain whether exenatide improved HRQoL as assessed by the Parkinson's Disease Questionnaire Summary Index (PDQ‐39 SI). We found low‐ or very low‐certainty evidence suggesting that for people taking exenatide, there may be little or no difference in the number of SAEs. We found low‐ or very low‐certainty evidence suggesting that exenatide may lead to slight weight loss. The findings of our review are most applicable to adults with mild PD less than stage 2.5 as assessed by the Hoehn and Yahr Scale (Hoehn 1967).
Implications for research.
We found only two studies for inclusion in this review, and of these, one was a single‐blind study. Both studies had small sample sizes and some baseline imbalances between intervention and control groups. Studies with larger sample sizes would be less likely to show an imbalance in baseline characteristics. Both studies assessed effects of the intervention on the same primary and similar secondary outcomes. In addition, both studies used similar intervention and washout periods. Findings from future double‐blind, placebo‐controlled, multi‐centre trials adopting a similar protocol as the two included studies would contribute additional evidence to this review. Long‐term follow‐up of participants to establish longer‐term effects of the intervention is encouraged, while taking account of the progressive nature of PD. Both studies assessed the impact of exenatide; therefore studies of other GLP‐1 RAs are needed.
Seven ongoing studies are assessing the effects of exenatide, liraglutide, lixisenatide and semaglutide. Findings from these studies can be included in an update of this review. Novel dual GLP‐1/glucose‐dependent insulinotropic polypeptide (GIP) RAs also show neuroprotective effects in PD models (Hölscher 2018). Further clinical trials using newer drugs of this class are likely to be conducted in due course.
History
Protocol first published: Issue 3, 2018 Review first published: Issue 7, 2020
Acknowledgements
We thank Ema Roque, Managing Editor, Cochrane Movement Disorders Group, for assistance with this review.
We thank Dolores Matthews for her assistance with editing his review.
We thank Professor Christian Hölscher for assistance in writing the protocol.
Thank you to Professor Tom Foltynie, co‐author of the Athauda 2017 study, for responding to our questions on study design.
Appendices
Appendix 1. CENTRAL search strategy
#1 Parkinson Disease:TI,AB,KY
#2 Parkinson*:TI,AB,KY
#3 #1 OR #2
#4 MESH DESCRIPTOR Glucagon‐Like Peptide 1 EXPLODE ALL TREES WITH QUALIFIERS AA,AG
#5 ((glucagon like peptide* or GLP 1 or GLP1) ADJ3 (analog* or agonist*)):TI,AB,KY
#6 (exenatide or AC 2993 or ITCA 650):TI,AB,KY
#7 (liraglutide or NN 2211 or NN2211 or NNC 90 1170 or NNC90 1170):TI,AB,KY
#8 (albiglutide or GSK 716155):TI,AB,KY
#9 (elsiglutide):TI,AB,KY
#10 (lixisenatide or AVE 0010):TI,AB,KY
#11 (dulaglutide or LY2189265 or LY 2189265):TI,AB,KY
#12 (taspoglutide or BIM 51077 or BIM51077 or ITM 077 or ITM077 or R 1583 or R1583 or RO 5073031 or RO5073031):TI,AB,KY
#13 (semaglutide or NN 9535 or NN9535):TI,AB,KY
#14 (teduglutide or ALX 0600 or ALX0600):TI,AB,KY
#15 OR/#4‐#14
#16 #3 and #15 in Trials
Appendix 2. MEDLINE search strategy
#1 randomized controlled trial.pt.
#2 controlled clinical trial.pt.
#3 randomized.ab.
#4 placebo.ab.
#5 clinical trials as topic.sh.
#6 randomly.ab.
#7 trial.ti.
#8 1 or 2 or 3 or 4 or 5 or 6 or 7
#9 animals.mh. not humans.mh.
#10 #8 not #9
#11 Parkinson Disease.mh.
#12 Parkinson*.ti,ab.
#13 #11 or #12
#14 exp Glucagon‐Like Peptide 1/aa,ag [Analogs & Derivatives, Agonists]
#15 ((glucagon like peptide* or GLP 1 or GLP1) adj3 (analog* or agonist*)).tw.
#16 (exenatide or AC 2993 or ITCA 650).tw.
#17 (liraglutide or NN 2211 or NN2211 or NNC 90 1170 or NNC90 1170).tw.
#18 (albiglutide or GSK 716155).tw.
#19 (elsiglutide).tw.
#20 (lixisenatide or AVE 0010).tw.
#21 (dulaglutide or LY2189265 or LY 2189265).tw.
#22 (taspoglutide or BIM 51077 or BIM51077 or ITM 077 or ITM077 or R 1583 or R1583 or RO 5073031 or RO5073031).tw.
#23 (semaglutide or NN 9535 or NN9535).tw.
#24 (teduglutide or ALX 0600 or ALX0600).tw.
#25 OR/14‐24
#26 #10 and #13 and #25
Appendix 3. Embase search strategy
#1 random$.tw.
#2 clinical trial:.mp.
#3 placebo$.mp.
#4 double‐blind$.tw.
#5 1 or 2 or 3 or 4
#6 Parkinson*.ti,ab.
#7 Parkinson Disease/exp
#8 6 or 7
#9 ((glucagon like peptide* or GLP 1 or GLP1) adj3 (analog* or agonist*)).tw.
#10 (exenatide or AC 2993 or ITCA 650).tw.
#11 (liraglutide or NN 2211 or NN2211 or NNC 90 1170 or NNC90 1170).tw.
#12 (albiglutide or GSK 716155).tw.
#13 (elsiglutide).tw.
#14 (lixisenatide or AVE 0010).tw.
#15 (dulaglutide or LY2189265 or LY 2189265).tw.
#16 (taspoglutide or BIM 51077 or BIM51077 or ITM 077 or ITM077 or R 1583 or R1583 or RO 5073031 or RO5073031).tw.
#17 (semaglutide or NN 9535 or NN9535).tw.
#18 (teduglutide or ALX 0600 or ALX0600).tw.
#19 or/9‐37
#20 5 and 8 and 19
Data and analyses
Comparison 1. Exenatide vs placebo (60 weeks post‐baseline).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 PD motor impairment ‐ MDS‐UPDRS Part III (off medication) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.2 HRQoL ‐ PDQ‐39 SI | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.3 HRQoL ‐ EQ5D | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.4 HRQoL ‐ EQ5D‐VAS | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.5 Adverse events ‐ weight loss (kg) (assessed at 48 weeks post baseline) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.6 PD motor impairment ‐ MDS‐UPDRS Part III | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.7 PD motor impairment ‐ UDysRS | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.8 Non‐motor impairment ‐ MDS‐UPDRS Part I | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.9 Non‐motor impairment ‐ NMSQuest | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.10 Activities of daily living ‐ MDS‐UPDRS Part II | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.11 Psychological outcomes ‐ Mattis DRS | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 1.12 Psychological outcomes ‐ MADRS | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only |
Comparison 2. Exenatide vs no treatment (14 months (60.8 weeks) post baseline).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 PD motor impairment ‐ MDS‐UPDRS Part III (off medication) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.2 HRQoL ‐ PDQ‐39 SI | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.3 Serious adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 2.4 Adverse events ‐ weight loss (kg) (assessed at 12 months (52 weeks)) | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.5 PD motor impairment ‐ MDS‐UPDRS Part III | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.6 PD motor impairment ‐ Rush Dyskinesia Rating Scale | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.7 Non‐motor impairment ‐ MDS‐UPDRS Part I | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.8 Activities of daily living ‐ MDS‐UPDRS Part II | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.9 Psychological outcomes ‐ Mattis DRS | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only | |
| 2.10 Psychological outcomes ‐ MADRS | 1 | Mean Difference (IV, Fixed, 95% CI) | Subtotals only |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Athauda 2017.
| Study characteristics | ||
| Methods |
Study design: randomised, double‐blind, placebo‐controlled, parallel‐group study Duration of study: participants were followed up at 60 weeks Number of centres: 1 Location: London, UK Study setting: research/tertiary care facility Withdrawals: 2 participants before 12 weeks (I = 1, C = 1), both excluded from analysis Discontinued treatment: 3 participants (I = 1, C = 2). All continued follow‐up assessments as per protocol Compliance: assessed as "very high: 58 patients reported not missing a single dose" Dates of study: June 2014 to August 2016 (from ClinicalTrials.gov; https://clinicaltrials.gov/ct2/show/NCT01971242) |
|
| Participants |
Number of participants: 62 randomised, 2 withdrawals, so 60 included in analysis (I = 31, C = 29) Age in years, mean (SD): intervention: 61.6 (8.2), control: 57.8 (8.0) Gender, N (%): intervention: female 9 (29%), male 22 (71%); control: female 7 (24%), male 22 (76%) Duration of diagnosis at baseline, years (SD): intervention: 6.4 (3.3); control: 6.4 (3.3) Severity of condition, N (%): Hoehn and Yahr stage 1.0 to 2.0: intervention: 29 (94%); control: 29 (100%) Hoehn and Yahr stage 2.5: intervention: 2 (6%); control: 0 (0%) Study inclusion criteria:
Study exclusion criteria:
|
|
| Interventions |
Intervention: self‐administered exenatide 2 mg via subcutaneous injection once weekly for 48 weeks + regular drugs Comparator: self‐administered placebo via subcutaneous injection once weekly for 48 weeks + regular drugs |
|
| Outcomes |
Primary: MDS‐UPDRS part III scores in the practically defined off‐medication state at 60 weeks Secondary: differences at 48 and 60 weeks between groups for:
|
|
| Notes |
Funding: Michael J Fox Foundation for Parkinson Disease Interests: AJL reports grants from the Frances and Renee Hock Fund, consulting fees from Britannia Pharmaceuticals (Genus) and BIAL Portela, and honoraria from Profile Pharma, Teva, Lundbeck, BIAL, Roche, Britannia, UCB, Nordiclnfu Care, NeuroDerm, and Decision Resources TTW has received honoraria from Britannia Pharmaceuticals PL has received honoraria from Medtronic and St Jude Medical NHG is a named inventor on a National Institutes of Health patent describing the use of GLP‐1 RAs in neurodegenerative disorders TF has received honoraria from Profile Pharma, BIAL, AbbVie, Genus, Medtronic, and St Jude Medical |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “We used SealedEnvelope, an independent, commercial, Internet‐based randomisation service that generated the online randomisation list on the basis of guidance from the trial IT manager (SH) and trial statistician (SSS)” |
| Allocation concealment (selection bias) | Low risk | "SealedEnvelope will provide a unique trial identification code for each recruited participant. ……At the baseline visit, the clinical investigator will enter the patient’s initials, gender, date of birth, date of consent, criteria fulfilment, and PD severity strata into the SealedEnvelope.com secure website, which will then allocate the appropriate trial identification code to the patient…… All patients will be randomly assigned to treatment via the SealedEnvelope.com website" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "SealedEnvelope will provide the patient trial identification codes at randomisation. The trial drug kit identification code list will be prepared by the Trial Statistician and provided separately to SealedEnvelope and to the QP, who will ensure that labelling of trial drug packs occurs appropriately and so as to ensure complete blinding of the IMP to all investigators, participants, and the pharmacy staff on the study" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Triple blinding of investigators, participants, and pharmacy staff "To prevent the possibility of adverse events compromising rater blinding, all adverse events, biochemical results, blood pressure, heart rate, and weight were recorded separately by clinicians who were masked to treatment allocation" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition rate was low (< 5%) and balanced between groups |
| Selective reporting (reporting bias) | Low risk | None was identified |
| Other bias | Low risk | None was identified. Some baseline imbalances were noted (i.e. MDS‐UPDRS scores); however, these were adjusted for in the data analysis |
Aviles‐Olmos 2013.
| Study characteristics | ||
| Methods |
Study design: randomised, single‐blind, controlled, parallel‐group study Duration of study: participants were followed up at 14 and 24 months Number of centres: 1 Location: London, UK Study setting: research/tertiary care facility Withdrawals: 1 participant before 3 months (I = 1), excluded from analysis Discontinued treatment: 3 participants (I = 2, C = 1). All continued follow‐up assessments at 12 months. Data were missing from 2 at 14 months (I = 1, C = 1) Compliance: judged "to be very high for all participants" Date of study: July 2010 to March 2013 (from ClinicalTrials.gov; https://clinicaltrials.gov/ct2/show/NCT01174810) |
|
| Participants |
Number of participants: 45 randomised, 1 withdrawal, so 44 included in analysis (intervention = 20, control = 24) Age in years, mean (SD): intervention: 61.4 (6.0); control: 59.4 (8.4) Gender N (%): intervention: female 5 (25%), male 15 (75%); control: female 4 (17%), male 20 (83%) Duration of symptoms at baseline, years (SD): intervention: 9.6 (3.4); control: 11.0 (5.9) Severity of condition, N (%): Hoehn and Yahr stage 2:2.5 (intervention: 14:6, control: 16:8) Study inclusion criteria:
Study exclusion criteria:
|
|
| Interventions |
Intervention: self‐administered, twice‐daily exenatide added to conventional PD treatment. Participants were supplied with a 5 μg exenatide pen device (Byetta 5 μg) for 1 month, then were supplied with a 10 μg exenatide pen device (Byetta 10 μg) for the subsequent 11 months Comparator: continued to receive conventional PD treatment |
|
| Outcomes |
Primary: blinded video rating of MDS‐UPDRS Part III in the practically defined off‐medication condition; change from baseline to 12, 14, and 24 months Secondary: differences between groups in change from baseline to 12, 14, and 24 months on:
|
|
| Notes |
Funding: The Cure Parkinson’s Trust Interests: T. Foltynie is supported by the Parkinson's Appeal. He has received consultancies from Abbie Pharmaceuticals. He holds grants from the Michael J Fox Foundation and the Brain Research Trust and European Union FP7. He has received payment for lectures including service on speakers’ bureaus from St Jude Medical, Medtronic, Genus Pharmaceuticals, and Novartis |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "The randomisation list was computer generated by a senior statistician... using 2 strata according to patient PD severity" (Foltynie 2020 [pers comm]) |
| Allocation concealment (selection bias) | Low risk | "The randomisation list generated was kept by the trial pharmacist securely and completely separately from the trial team. She had no knowledge of which participants were attending, but notified the randomisation outcome to the team on request after the participants baseline visit was completed, and dispensed the active drug when indicated" (Foltynie 2020 [pers comm]) |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | “In view of the prohibitive costs associated with manufacture of (QP released) placebo versions of the exenatide pens, these were not available for the purposes of this trial, which was necessarily configured to be open‐label from the patient’s perspective” |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blinded to treatment groups. For the primary outcome, "assessments of PD severity using MDS‐UPDRS part III were made 'off‐medication' after an overnight period and were video recorded to allow objective rating of PD disability by observers blinded to randomisation outcomes. Each patient video was rated by the same blinded clinician at each time point" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition was low (< 10%) and was balanced between groups |
| Selective reporting (reporting bias) | Low risk | The pre‐specified primary outcome and most secondary outcomes were reported |
| Other bias | Low risk | None was identified |
AUT: autonomic dysfunction.
C: comparator.
COMT: catechol‐O‐methyl transferase.
CT: computed tomography.
DAT: dopamine transporter.
DaTSCAN: DAT binding as assessed by [¹²³I]FP‐CIT single‐photon emission computed tomography.
EQ‐5D: EuroQol scale measuring health status in five dimensions.
FSH: follicle‐stimulating hormone.
I: intervention.
MADRS: Montgomery‐Åsberg Depression Rating Scale.
MAO‐B: monoamine oxidase‐B.
MDS‐UPDRS: Movement Disorder Society‐Unified Parkinson's Disease Rating Scale.
MRI: magnetic resonance imaging.
NMSQuest: Non‐Motor Symptoms Questionnaire.
PD: Parkinson's disease.
SCOPA: Scales for Outcomes in Parkinson's Disease.
SD: standard deviation.
SPECT: single‐photon emission computed tomography.
UPDRS: Unified Parkinson's Disease Rating Scale.
Characteristics of ongoing studies [ordered by study ID]
Clinicaltrials.gov identifier: NCT04154072.
| Study name | Multicenter, randomized, double‐blind, placebo‐controlled study to evaluate the efficacy, safety, and tolerability of 36 weeks of treatment with NLY01 in early‐stage Parkinson’s disease |
| Methods | Multi‐centre, randomised, double‐blind, placebo‐controlled study |
| Participants | With early PD |
| Interventions |
Intervention treatment 1: NLY01 exenatide and polyethylene glycol (PEG) 2.5mg injection for 36 weeks Intervention treatment 2: NLY01 exenatide and polyethylene glycol (PEG) 5.0mg injection for 36 weeks Comparison: Placebo |
| Outcomes | Change in Unified Parkinson's Disease Rating Scale in combined score of Parts II and III from baseline |
| Starting date | February 2020 |
| Contact information | ParkinsonsStudy@cssienroll.com |
| Notes | www.PrismPDstudy.com |
Clinicaltrials.gov identifier: NCT04269642.
| Study name | Effects of exenatide on motor function and the brain |
| Methods | Multi‐centre, randomised, double‐blind, placebo‐controlled, parallel comparison, phase IIa clinical study |
| Participants | Early stages of PD |
| Interventions |
Intervention treatment 1: PT320 2.0mg injected subcutaneously once a week for 48 weeks Intervention treatment 2: PT320 2.5mg injected subcutaneously every two weeks for 48 weeks. (patients will be injected PT320 2.5 mg and placebo alternately once a week.) Comparison: placebo injected subcutaneously once a week for 48 weeks |
| Outcomes | Change of MDS‐UPDRS (Movement Disorder Society ‐Unified Parkinson's Disease Rating Scale ) part 3 score from baseline |
| Starting date | March 2020 |
| Contact information | ihmhjoy@peptron.co.kr |
| Notes |
Clinicaltrials.gov identifier: NCT02953665.
| Study name | A phase II, randomized, double‐blinded, placebo‐controlled trial of liraglutide in Parkinson’s disease |
| Methods | Single‐centre, double‐blind, placebo‐controlled study |
| Participants | Early stages of PD |
| Interventions |
Intervention: liraglutide 6 mg/ml once daily at a maximum dose of 1.8 mg for 52 weeks Comparison: placebo once daily for 52 weeks |
| Outcomes | Change in the motor (Part III) Movement Disorders Society‐Unified Parkinson Disease Rating Scale (MDS‐UPDRS) score |
| Starting date | April 2017 |
| Contact information | tina.wux@cshs.org |
| Notes |
ClinicalTrials.gov Identifier: NCT03439943.
| Study name | Study to evaluate the effect of lixisenatide in patients with Parkinson's disease (LixiPark) |
| Methods | Multi‐centre, randomised, placebo‐controlled, double‐blind, parallel‐arm proof‐of‐concept trial |
| Participants | With early PD |
| Interventions |
Intervention: lixisenatide (10 μg/d for 14 days, then 20 μg/d), once daily subcutaneously Comparator: placebo, once‐daily subcutaneous injection |
| Outcomes | Change from baseline to end point (M12) in MDS‐UPDRS III motor examination score, evaluated in the best ON condition in patients with early Parkinson's disease |
| Starting date | June 2018 |
| Contact information | olivier.rascol@univ-tlse3.fr |
| Notes |
ClinicalTrials.gov Identifier: NCT03659682.
| Study name | Effect of GLP1R stimulation on neuroprotection and inflammation in Parkinson's disease |
| Methods | Single‐centre, double‐blind, placebo‐controlled study |
| Participants | Newly diagnosed patients with PD, age 40 to 75 |
| Interventions |
Intervention: semaglutide subcutaneous, 1.0 mg weekly, 48 months Comparator: placebo for 4 months, then semaglutide for 24 months |
| Outcomes | MDS‐UPDRS Part III in OFF‐medication state |
| Starting date | January 2019 |
| Contact information | h.f.harbo@medisin.uio.no |
| Notes |
ClinicalTrials.gov Identifier: NCT04232969.
| Study name | Exenatide once weekly over 2 years as a potential disease modifying treatment for Parkinson's disease (Exenatide‐PD3) |
| Methods | Randomised, double‐blind, parallel‐group, placebo‐controlled, phase 3 trial |
| Participants | Newly diagnosed patients |
| Interventions |
Intervention: Exenatide extended release 2mg (Bydureon) once weekly for 96 weeks Comparison: Placebo once weekly for 96 weeks |
| Outcomes | MDS‐UPDRS part 3 motor sub‐score in the practically defined OFF medication |
| Starting date | January 2020 |
| Contact information | t.foltynie@ucl.ac.uk |
| Notes |
ClinicalTrials.gov Identifier: NCT04305002.
| Study name | Effect of exenatide on disease progression in early Parkinson’s disease |
| Methods | Single‐centre, double‐blind, placebo‐controlled study |
| Participants | Early stages of PD |
| Interventions |
Intervention: exenatide 2mg once weekly for 18 months Comparison: placebo once weekly for 18 months |
| Outcomes | Primary: FDG‐PET analysis Secondary: MDS‐UPDRS part 3 in OFF medication state |
| Starting date | January 2020 |
| Contact information | per.svenningsson@ki.se |
| Notes |
GLP1R: glucagon‐like peptide‐1 receptor.
MDS‐UPDRS: Movement Disorder Society‐Unified Parkinson's Disease Rating Scale.
PD: Parkinson's disease.
Differences between protocol and review
To clarify the interventions and comparators that we would include in this review, we added the sentence, "We included studies in which experimental and comparator groups received an intervention (either active or inactive) in addition to conventional treatment" to the Types of interventions section.
We modified our definition of weight loss as an adverse event to "Adverse events, including rapid weight loss (> 1.5 kg/week)". We modified our definition of serious adverse events to "we have defined adverse events as serious based on information reported by individual trials".
As we were keen to assess the neuroprotective effects of GLP‐1 RAs, we planned to report outcomes assessed two to three months post cessation of treatment rather than immediately following cessation of treatment as initially planned.
We had planned to assess adverse events as "the number of participants with an adverse event associated with the intervention". However, included studies reported the total number of adverse events reported by individuals over all time points.
Contributions of authors
The idea for this review was conceived by all review authors. The protocol was drafted by all review authors.
Sources of support
Internal sources
-
Internal sources of support, Other
No support was received to write this protocol.
External sources
-
External sources of support, Other
No support was received to write this protocol.
Declarations of interest
CAM: none known.
GSD: none known.
JH: none known.
DE: none known.
SM: none known.
RW: none known.
HCAE: none known.
New
References
References to studies included in this review
Athauda 2017 {published data only}
- Athauda D, Budnik N, Chowdhury K, Skene S, Foltynie T. The effect of exenatide on specific non-motor symptoms in Parkinson’s disease – a post-hoc analysis. In: European Journal of Neurology. Issue S2 edition. Vol. 25. Abstracts of the 4th Congress of the European Academy of Neurology, Lisbon, Portugal, June 2018, 2018:309.
- Athauda D, Maclagan K, Budnik N, Zampedri L, Hibbert S, Aviles‐Olmos I, et al. Post hoc analysis of the exenatide‐PD trial - factors that predict response. European Journal of Neuroscience 2019;49(3):410-21. [DOI: 10.1111/ejn.14096] [DOI] [PubMed] [Google Scholar]
- Athauda D, Maclagan K, Budnik N, Zampedri L, Hibbert S, Skene SS, et al. What effects might exenatide have on non-motor symptoms in Parkinson's disease: a post hoc analysis. Journal of Parkinson’s Disease 2018;8:247-58. [DOI: 10.3233/JPD-181329] [DOI] [PubMed] [Google Scholar]
- *.Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet 2017;390(10103):1664-75. [DOI: 10.1016/S0140-6736(17)31585-4] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athauda D, Wyse R, Brundin P, Foltynie T. Is exenatide a treatment for Parkinson's disease? Journal of Parkinson's Disease 2017;7(3):451-8. [DOI: 10.3233/JPD-171192] [DOI] [PubMed] [Google Scholar]
Aviles‐Olmos 2013 {published data only}
- *.Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, et al. Exenatide and the treatment of patients with Parkinson’s disease. Journal of Clinical Investigation 2013;123(6):2730-6. [DOI: 10.1172/JCI68295.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Kahan J, Ell P, et al. Exenatide and motor symptoms in Parkinson's disease (PD) [abstract]. In: Movement Disorders. Vol. 29 Suppl 1:611. 2014.
- Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Kahan J, Ell P, et al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson's disease. Journal of Parkinson's Disease 2015;4(3):337-44. [DOI: 10.3233/JPD-140364] [DOI] [PubMed] [Google Scholar]
- Aviles-Olmos I, Dickson J, Kefalopoupou Z, Djamshidian A, Kahan J, Ell P, et al. Exenatide and non motor symptoms In Parkinson's disease (PD) [abstract]. In: Movement Disorders. Vol. 29 Suppl 1 6:12. 2014.
- Aviles-Olmos I, Kefalopoulou Z, Djamshidian A, Limousin P, Dickson J, Lees A, et al. An open label, single site, 12 month, phase II, randomised controlled trial evaluating the safety and efficacy of Exendin-4 (exenatide) in the treatment of patients with moderate severity Parkinson's disease [abstract]. Movement Disorders 2012;27(Suppl 1:343). [Google Scholar]
- Foltynie T, Aviles-Olmos I. Exenatide as a potential treatment for patients with Parkinson's disease: first steps into the clinic. Alzheimer's and Dementia 2014;10(1 Suppl):S38-46. [DOI: 10.1016/j.jalz.2013.12.005] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
Clinicaltrials.gov identifier: NCT02953665 {published data only}
- A phase II, randomized, double-blinded, placebo-controlled trial of liraglutide in Parkinson’s disease. Ongoing study. April 2017. Contact author for more information.
ClinicalTrials.gov Identifier: NCT03439943 {published data only}
- Study to evaluate the effect of lixisenatide in patients with Parkinson's disease (LixiPark). Ongoing study. June 2018. Contact author for more information.
ClinicalTrials.gov Identifier: NCT03659682 {published data only}
- Effect of GLP1R stimulation on neuroprotection and inflammation in Parkinson's disease. Ongoing study. January 2019. Contact author for more information.
Clinicaltrials.gov identifier: NCT04154072 {published data only}
- Multicenter, randomized, double-blind, placebo-controlled study to evaluate the efficacy, safety, and tolerability of 36 weeks of treatment with NLY01 in early-stage Parkinson’s disease. Ongoing study. February 2020. Contact author for more information.
ClinicalTrials.gov Identifier: NCT04232969 {published data only}
- Exenatide once weekly over 2 years as a potential disease modifying treatment for Parkinson's disease (Exenatide-PD3). Ongoing study. January 2020. Contact author for more information.
Clinicaltrials.gov identifier: NCT04269642 {published data only}
- Effects of exenatide on motor function and the brain. Ongoing study. March 2020. Contact author for more information.
ClinicalTrials.gov Identifier: NCT04305002 {published data only}
- Effect of exenatide on disease progression in early Parkinson’s disease. Ongoing study. January 2020. Contact author for more information.
Additional references
Abou‐Sleiman 2006
- Abou-Sleiman PM, Muqit MM, Wood NW. Expanding insights of mitochondrial dysfunction in Parkinson's disease. Nature Reviews. Neuroscience 2006;7(3):207-19. [DOI: 10.1038/nrn1868] [DOI] [PubMed] [Google Scholar]
Al‐Bachari 2017
- Al-Bachari S, Vidyasagar R, Emsley HC, Parkes LM. Structural and physiological neurovascular changes in idiopathic Parkinson’s disease and its clinical phenotype. Journal of Cerebal Blood Flow & Metabolism 2017;37(10):3409-21. [DOI: 10.1177/0271678X16688919] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ascherio 2016
- Ascherio A, Schwarzschild MA. The epidemiology of Parkinson’s disease: risk factors and prevention. Lancet Neurology 2016;15:1257-72. [DOI: 10.1016/S1474-4422(16)30230-7] [DOI] [PubMed] [Google Scholar]
Aviles‐Olmos 2013a
- Aviles-Olmos I, Limousin P, Lees A, Foltynie T. Parkinson's disease, insulin resistance and novel agents of neuroprotection. Brain 2013;136:374-84. [DOI: 10.1093/brain/aws009] [DOI] [PubMed] [Google Scholar]
Aviles‐Olmos 2013b
- Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Ell P, Soderlund T, et al. Exenatide and the treatment of patients with Parkinson's disease. Journal of Clinical Investigation 2013;123(6):2730-6. [DOI: 10.1172/JCI68295] [DOI] [PMC free article] [PubMed] [Google Scholar]
Aviles‐Olmos 2014
- Aviles-Olmos I, Dickson J, Kefalopoulou Z, Djamshidian A, Kahan J, Ell P, et al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson's disease. Journal of Parkinson's Disease 2014;4(3):337-44. [DOI: 10.3233/JPD-140364] [DOI] [PubMed] [Google Scholar]
Baggio 2007
- Baggio LL, Drucker DJ. Biology of incretins: GLP-1 and GIP. Gastroenterology 2007;132:2131-57. [DOI: 10.1053/j.gastro.2007.03.054] [DOI] [PubMed] [Google Scholar]
Bomfim 2012
- Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer's disease-associated A-beta oligomers. Journal of Clinical Investigation 2012;122:1339-53. [DOI: 10.1172/JCI57256] [DOI] [PMC free article] [PubMed] [Google Scholar]
Campbell 2013
- Campbell JE, Drucker DJ. Pharmacology, physiology, and mechanisms of incretin hormone action. Cell Metabolism 2013;17:819-37. [DOI: 10.1016/j.cmet.2013.04.008] [DOI] [PubMed] [Google Scholar]
Chaudhuri 2008
- Chaudhuri KR, Martinez-Martin P. Quantitation of nonmotor symptoms in Parkinson's disease. European Journal of Neurology 2008;15 Suppl 2:2-7. [DOI: 10.1111/j.1468-1331.2008.02212.x] [DOI] [PubMed] [Google Scholar]
Collins 2012
- Collins LM, Toulouse A, Connor TJ, Nolan YM. Contributions of central and systemic inflammation to the pathophysiology of Parkinson's disease. Neuropharmacology 2012;62(7):2154-68. [DOI: 10.1016/j.neuropharm.2012.01.028] [DOI] [PubMed] [Google Scholar]
de Boer 1996
- Boer AGEM, Wijker W. Quality of life in patients with Parkinson's disease: development of a questionnaire. Journal of Neurology, Neurosurgery, and Psychiatry 1996;61:70-4. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
de Lau 2006
- Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurology 2006;5(6):525-35. [DOI: 10.1016/S1474-4422(06)70471-9] [DOI] [PubMed] [Google Scholar]
Doyle 2003
- Doyle ME, Egan JM. Pharmacological agents that directly modulate insulin secretion. Pharmacological Reviews 2003;55:105-31. [DOI: 10.1124/pr.55.1.7] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta-analysis detected by a simple, graphical test. BMJ 1997;315:629. [DOI: org/10.1136/bmj.315.7109.629 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Erbil 2019
- Erbila D, Eren CY, Demirela C, Küçükera MU, Solaroğlua I, Eser HY. GLP-1’s role in neuroprotection: a systematic review. Brain Injury 2019;33(6):734-819. [DOI: 10.1080/02699052.2019.1587000] [DOI] [PubMed] [Google Scholar]
Fahn 1987
- Fahn S, Elton R, Members of the UPDRS Development Committee. The Unified Parkinson’s Disease Rating Scale. In: Fahn S, Marsden CD, Calne DB, Goldstein M, editors(s). Recent Developments in Parkinson’s Disease. Vol. 2. Florham Park (NJ): Macmillan Health Care Information, 1987:153-163, 293-304. [Google Scholar]
Foltynie 2020 [pers comm]
- Foltynie T. Random sequence generation and allocation concealment used in our trial [personal communication]. Email to: C Mulvaney. 4 May 2020.
Freiherr 2013
- Freiherr J, Hallschmid M, Frey WH 2nd, Brünner YF, Chapman CD, Hölscher C, et al. Intranasal insulin as a treatment for Alzheimer's disease: a review of basic research and clinical evidence. CNS Drugs 2013;27(7):505-14. [DOI: 10.1007/s40263-013-0076-8] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ghasemi 2013
- Ghasemi R, Dargahi L, Haeri A, Moosavi M, Mohamed Z, Ahmadiani A. Brain insulin dysregulation: implication for neurological and neuropsychiatric disorders. Molecular Neurobiology 2013;47:1045-65. [DOI: 10.1007/s12035-013-8404-z] [DOI] [PubMed] [Google Scholar]
Goetz 2000
- Goetz CG, Leurgans S, Raman R, Stebbins GT. Objective changes in motor function during placebo treatment in PD. Neurology 2000;54(3):710-4. [DOI: 10.1212/wnl.54.3.710] [DOI] [PubMed] [Google Scholar]
Goetz 2008
- Goetz CG, Nutt JG, Stebbins GT. The Unified Dyskinesia Rating Scale: presentation and clinimetric profile. Movement Disorders 2008;23(6):2398-403. [DOI: 10.1002/mds.22341] [DOI] [PubMed] [Google Scholar]
GRADEpro 2015 [Computer program]
- GRADEpro GDT. Version accessed 18 October 2017. Hamilton (ON): McMaster University (developed by Evidence Prime, Inc.), 2015.
Gray 2015
- Gray MT, Woulfe JM. Striatal blood-brain barrier permeability in Parkinson's disease. Journal of Cerebral Blood Flow & Metabolism 2015;35(5):747-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hammarlund 2012
- Hammarlund CS, Nilsson MH, Hagell P. Measuring outcomes in Parkinson’s disease: a multi-perspective concept mapping study. Quality of Life Research 2012;21(3):453-63. [DOI: 10.1007/s11136-011-9995-3] [DOI] [PubMed] [Google Scholar]
Harkavyi 2008
- Harkavyi A, Abuirmeileh A, Lever R, Kingsbury AE, Biggs CS, Whitton PS. Glucagon-like peptide 1 receptor stimulation reverses key deficits in distinct rodent models of Parkinson's disease. Journal of Neuroinflammation 2008;5(19):11-9. [DOI: 10.1186/1742-2094-5-19] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hely 2005
- Hely MA, Morris JG, Reid WG, Trafficante R. Sydney multicenter study of Parkinson’s disease: non-L-dopa–responsive problems dominate at 15 years. Movement Disorders 2005;20(2):190-9. [DOI: 10.1002/mds.20324] [DOI] [PubMed] [Google Scholar]
Hely 2008
- Hely MA, Reid WG, Adena MA, Halliday GM, Morris JG. The Sydney multicenter study of Parkinson’s disease: the inevitability of dementia at 20 years. Movement Disorders 2008;23(6):837-44. [DOI: 10.1002/mds.21956] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hirsch 2016
- Hirsch L, Jette N, Frolkis A, Steeves T, Pringsheim T. The incidence of Parkinson’s disease:a systematic review and meta-analysis. Neuroepidemiology 2016;46:292-300. [DOI: 10.1159/000445751] [DOI] [PubMed] [Google Scholar]
Hoehn 1967
- Hoehn MM, Yahr MD. Parkinsonism: onset, progression, and mortality. Neurology 1967;17(5):427-42. [DOI: 10.1212/WNL.17.5.427] [DOI] [PubMed] [Google Scholar]
Holst 2004
- Holst JJ. Treatment of type 2 diabetes mellitus with agonists of the GLP-1 receptor or DPP-IV inhibitors. Expert Opinion on Emerging Drugs 2004;9:155-66. [DOI: 10.1517/eoed.9.1.155.32952] [DOI] [PubMed] [Google Scholar]
Horváth 2015
- Horváth K, Ascherman Z, Ács P, Deli G, Janszky J, Komoly S, et al. Minimal clinically important difference on the Motor Examination part of MDS-UPDRS. Parkinsonism & Related Disorders 2015;21(12):1421-6. [10.1016/j.parkreldis.2015.10.006] [DOI] [PubMed] [Google Scholar]
Horváth 2017
- Horváth K, Aschermann Z, Kovács M, Makkos A, Harmat M, Janszky J. Changes in quality of life in Parkinson's disease: how large must they be to be relevant? Neuroepidemiology 2017;48(1-2):1-8. [DOI: 10.1159/000455863] [DOI] [PubMed] [Google Scholar]
Hu 2007
- Hu G, Jousilahti P, Bidel S, Antikainen R, Tuomilehto J. Type 2 diabetes and the risk of Parkinson's disease. Diabetes Care 2007;30:842-7. [DOI: 10.2337/dc06-2011] [DOI] [PubMed] [Google Scholar]
Hughes 1992
- Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease. A clinico-pathological study of 100 cases. Journal of Neurology, Neurosurgery, and Psychiatry 1992;55:181-4. [DOI: 10.4103/0972-2327.83083] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hölscher 2014
- Hölscher C. Insulin, incretins and other growth factors as potential novel treatments for Alzheimer's and Parkinson's diseases. Biochemical Society Transactions 2014;42:593-9. [DOI: 10.1042/BST20140016] [DOI] [PubMed] [Google Scholar]
Hölscher 2018
- Hölscher C. Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer's and Parkinson's disease models. Neuropharmacology 2018;(136 (Pt B)):251-9. [DOI: 10.1016/j.neuropharm.2018.01.040] [DOI] [PubMed] [Google Scholar]
Jalewa 2016
- Jalewa J, Sharma MK, Holscher C. Novel incretin analogues improve autophagy and protect from mitochondrial stress induced by rotenone in SH-SY5Y cells. Journal of Neurochemistry 2016;139:55-67. [DOI: 10.1111/jnc.13736] [DOI] [PubMed] [Google Scholar]
Jenkinson 1997
- Jenkinson C, Fitzpatrick R, Peto V, Grenhall R, Hyman N. The PDQ-8: development and validation of a short-form Parkinson’s disease questionnaire. Psychology and Health 1997;12:805-14. [DOI: 10.1002/mds.10678] [DOI] [Google Scholar]
Ji 2016
- Ji C, Xue GF, Li G, Li D, Holscher C. Neuroprotective effects of glucose-dependent insulinotropic polypeptide in Alzheimer's disease. Reviews in the Neurosciences 2016;27:61-70. [DOI: 10.1515/revneuro-2015-0021] [DOI] [PubMed] [Google Scholar]
Kalia 2015
- Kalia LV, Lang AE. Parkinson’s disease. Lancet 2015;386:896-912. [DOI: 10.1016/S0140-6736(14)61393-3] [DOI] [PubMed] [Google Scholar]
Li 2009
- Li Y, Perry T, Kindy MS, Harvey BK, Tweedie D, Holloway HW, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proceedings of the National Academy of Sciences of the United States of America 2009;106:1285-90. [DOI: 10.1073/pnas.0806720106] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2010
- Li Y, Duffy K, Ottinger M, Ray B, Bailey J, Holloway H, et al. GLP-1 receptor stimulation reduces amyloid-beta peptide accumulation and cytotoxicity in cellular and animal models of Alzheimer's disease. Journal of Alzheimer's Disease 2010;19(4):1205-19. [DOI: 10.3233/JAD-2010-1314] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2016
- Li Y, Liu W, Li L, Holscher C. Neuroprotective effects of a GIP analogue in the MPTP Parkinson's disease mouse model. Neuropharmacology 2016;101:255-63. [DOI: 10.1016/j.neuropharm.2015.10.002] [DOI] [PubMed] [Google Scholar]
Liberati 2009
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gotzsche PC, Ioannidis JP, et al. The PRISMA statement for reporting systematic reviews and meta-analyses of studies that evaluate health care interventions: explanation and elaboration. BMJ 2009;339:b2700. [DOI: 10.1136/bmj.b2700] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liu 2015a
- Liu W, Jalewa J, Sharma M, Li G, Li L, Holscher C. Neuroprotective effects of lixisenatide and liraglutide in the MPTP mouse model of Parkinson’s disease. Neuroscience 2015;303:42-50. [DOI: 10.1016/j.neuroscience.2015.06.054] [DOI] [PubMed] [Google Scholar]
Liu 2015b
- Liu W, Li Y, Jalewa J, Saunders-Wood T, Li L, Hölscher C. Neuroprotective effects of an oxyntomodulin analogue in the MPTP mouse model of Parkinson's disease. European Journal of Pharmacology 2015;765:284-90. [DOI: 10.1016/j.ejphar.2015.08.038] [DOI] [PubMed] [Google Scholar]
Long‐Smith 2013
- Long-Smith CM, Manning S, McClean PL, Coakley MF, O'Halloran DJ, Holscher C, et al. The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-beta plaque and glial pathology in a mouse model of Alzheimer's disease. Neuromolecular Medicine 2013;15:102-14. [DOI: 10.1007/s12017-012-8199-5] [DOI] [PubMed] [Google Scholar]
Malek 2016
- Malek N, Lawton MA, Swallow DM, Grosset KA, Marrinan SL, Bajaj N, et al. Vascular disease and vascular risk factors in relation to motor features and cognition in early Parkinson's disease. Movement Disorders 2016;31(10):1518-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mattis 1976
- Mattis S. Geriatric Psychiatry: A Handbook for Psychiatrists and Primary Care Physicians. New York (NY): Grune and Stratton Inc, 1976. [Google Scholar]
McClean 2011
- McClean P, Parthsarathy V, Faivre E, Hölscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer's disease. Journal of Neuroscience 2011;31:6587-94. [DOI: 10.1523/JNEUROSCI.0529-11.2011] [DOI] [PMC free article] [PubMed] [Google Scholar]
McGhee 2013
- McGhee DJ, Royle PL, Thompson PA, Wright DE, Zajicek JP, Counsell CE. A systematic review of biomarkers for disease progression in Parkinson’s disease. BMC Neurology 2013;13:35. [DOI: 10.1186/1471-2377-13-35] [DOI] [PMC free article] [PubMed] [Google Scholar]
Montgomery 1979
- Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. British Journal of Psychiatry 1979;134:382-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Moroo 1994
- Moroo I, Yamada T, Makino H, Tooyama I, McGeer PL, McGeer EG, et al. Loss of insulin receptor immunoreactivity from the substantia nigra pars compacta neurons in Parkinson's disease. Acta Neuropathologica 1994;87:343-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Morris 2011
- Morris JK, Bomhoff GL, Gorres BK, Davis VA, Kim J, Lee PP, et al. Insulin resistance impairs nigrostriatal dopamine function. Experimental Neurology 2011;231:171-80. [DOI: 10.1016/j.expneurol.2011.06.005] [DOI] [PMC free article] [PubMed] [Google Scholar]
Müller 2000
- Müller T, Woitalla D, Saft C, Kuhn W. Levodopa in plasma correlates with body weight of parkinsonian patients. Parkinsonism & Related Disorders 2000;6(3):171-3. [PMID: PMID: 10817957] [DOI] [PubMed] [Google Scholar]
Peto 1995
- Peto V, Jenkinson C, Fitzpatrick R, Greenhall R. The development and validation of a short measure of functioning and well being for individuals with Parkinson’s disease. Quality of Life Research 1995;4:241-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Pleuvry 2004
- Pleuvry BJ. Receptors, agonists and antagonists. Anaesthesia & Intensive Care Medicine 2004;5(10):350-2. [DOI: ] [Google Scholar]
Pringhseim 2014
- Pringsheim T, Jette N, Frolkis A, Steeves TD. The prevalence of Parkinson’s disease: a systematic review and meta-analysis. Movement Disorders 29;13:1583-90. [DOI: 10.1002/mds.25945] [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Schernhammer 2011
- Schernhammer E, Hansen J, Rugbjerg K, Wermuth L, Ritz B. Diabetes and the risk of developing Parkinson's disease in Denmark. Diabetes Care 2011;34:1102-8. [DOI: 10.2337/dc10-1333] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schwab 1969
- Schwab RS, England AC. Projection technique for evaluating surgery in Parkinson’s disease. In: Gillingham FJ, Donaldson MC, editors(s). Third Symposium on Parkinson’s Disease. Edinburgh & London (UK): E & S Livingstone, 1969:152-7. [Google Scholar]
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s). Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach (updated October 2013). GRADE Working Group, 2013. Available from gdt.guidelinedevelopment.org/app/handbook/handbook.html.
Sun 2012
- Sun Y, Chang YH, Chen HF, Su YH, Su HF, Li CY. Risk of Parkinson disease onset in patients with diabetes: a 9-year population-based cohort study with age and sex stratifications. Diabetes Care 2012;35:1047-9. [DOI: 10.2337/dc11-1511] [DOI] [PMC free article] [PubMed] [Google Scholar]
van der Heide 2006
- Heide LP, Ramakers GM, Smidt MP. Insulin signaling in the central nervous system: learning to survive. Progress in Neurobiology 2006;79:205-21. [DOI: 10.1016/j.pneurobio.2006.06.003] [DOI] [PubMed] [Google Scholar]
Vilsbøll 2012
- Vilsbøll T, Christensen M, Junker AE, Knop FK, Gluud LL. Effects of glucagon-like peptide-1 receptor agonists on weight loss: systematic review and meta-analyses of randomised controlled trials. BMJ 2012;344:d7771. [DOI: 10.1136/bmj.d7771] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wahlqvist 2012
- Wahlqvist ML, Lee MS, Hsu CC, Chuang SY, Lee JT, Tsai HN. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson's disease occurring with type 2 diabetes in a Taiwanese population cohort. Parkinsonism & Related Disorders 2012;18:753-8. [DOI: 10.1016/j.parkreldis.2012.03.010] [DOI] [PubMed] [Google Scholar]
Ware 1992
- Ware JE Jr, Sherbourne CD. The MOS 36-item short-form health survey (SF-36). I. Conceptual framework and item selection. Medical Care 1992;30(6):473-83. [PMID: ] [PubMed] [Google Scholar]
Zhang 2015
- Zhang YF, Chen YM, LI L, Hölscher C. Neuroprotective effects of (Val8) GLP-1-Glu-PAL in the MPTP Parkinson’s disease mouse model. Behavioural Brain Research 2015;293:107-13. [DOI: 10.1016/j.bbr.2015.07.021] [DOI] [PubMed] [Google Scholar]