

Abstract
In the post-genomic era, genetics has led to limited clinical applications in the diagnosis and treatment of major depressive disorder (MDD). Variants in genes coding for cytochrome enzymes are included in guidelines for assisting in antidepressant choice and dosing, but there are no recommendations involving genes responsible for antidepressant pharmacodynamics and no consensus applications for guiding diagnosis or prognosis. However, genetics has contributed to a better understanding of MDD pathogenesis and the mechanisms of antidepressant action, also thanks to recent methodological innovations that overcome the challenges posed by the polygenic architecture of these traits. Polygenic risk scores can be used to estimate the risk of disease at the individual level, which may have clinical relevance in cases with extremely high scores (e.g. top 1%). Genetic studies have also shed light on a wide genetic overlap between MDD and other psychiatric disorders. The relationships between genes/pathways associated with MDD and known drug targets are a promising tool for drug repurposing and identification of new pharmacological targets. Increase in power thanks to larger samples and methods integrating genetic data with gene expression, the integration of common variants and rare variants, are expected to advance our knowledge and assist in personalized psychiatry.
Keywords: antidepressant, exome sequencing, gene, genome-wide association studies, major depressive disorder, polygenic
Introduction
Historically, psychiatrists always had few or no tools to interrogate their patients’ bodies on the pathophysiology of their symptoms. Patterns of symptoms that often occur together have been classified in disorders in order to have a standard nosology that guides diagnosis and treatment. Attempts to give more space to dimensional over categorical classifications have mostly failed, because of the complexity of that kind of approach. As a matter of fact, the possible combinations of symptoms, their respective intensity and fluctuations cannot easily be captured in a way that is applicable in routine clinical practice. For example, 1030 unique depressive symptom profiles were identified in one sample only (Fried and Nesse, 2015). This represents an obstacle to personalized psychiatry and leads to the delivery of relatively homogeneous treatments within diagnostic categories. A dimensional approach can be more easily applied to objectively measurable quantitative parameters, such as blood protein levels, neuroimaging brain measures, or genetic variants. These biomarkers can help in finding the biological underpinnings of clinical manifestations, distinguish different dimensions within and across diagnostic categories and tailor treatment prescription (Strawbridge et al., 2017). The idea of implementing this approach has led to the term precision psychiatry, which implies that each patient has a distinctive profile of biological dysfunctions, which interacting with environmental factors is responsible for the clinical presentation (Fernandes et al., 2017). The knowledge needed to implement precision psychiatry is still partial, but recent rapid technological and methodological improvements are making it more and more feasible. A central part of this process is the postgenomic revolution: the cost/time for sequencing a human genome dropped from $100 millions/several years in 2001 to $1000/2 days in 2017, while genome-wide common variant genotyping can be done for ~ $25–50 per subject (National Human Genome Research Institute, 2018). This has made possible the genotyping of large samples with major depressive disorder (MDD), as well as other psychiatric disorders and healthy controls, and testing the influence of genetic variants on the risk of disease and treatment response (Howard et al., 2019). The existing literature shows that genetic variants explain very small variance individually and the cumulative effect of many variants (hundreds or thousands) is responsible for the genetic susceptibility to these traits (Zhang et al., 2018). Specific analysis approaches have been applied to capture this polygenic architecture, such as pathway analysis and polygenic risk scores (PRS). These methodologies have provided promising results in identifying the genetic contribution to MDD and antidepressant response, which have rapidly expanding clinical applications, and they can have an important potential in contributing to the development of new drugs for depression or drug repositioning, as discussed in the next paragraphs.
Pathway analysis: insights into the biological mechanisms of depression
The analysis of genetic pathways instead of individual genetic variants as unit of analysis is a way to unravel the complexity of MDD pathogenesis and the corresponding mechanisms mediating antidepressant response. Genetic pathways are groups of genes functionally related among each other, which mediate a distinct cellular or molecular process or reflect interactions among proteins or molecules.
Pathway analysis led to the identification of several biological mechanisms that mediate depression and antidepressant action. These can be grouped in some main clusters: axonal development, neuron differentiation and morphogenesis, neural-plasticity, excitatory neurotransmission, cytokines, immune response and regulation of gene expression (Zeng et al., 2017; Wray et al., 2018; Howard et al., 2018; Fabbri et al., 2019a).
These findings suggest that part of the genetic predisposition to depression manifests during brain development, as exemplified by the involvement of the NETRIN1 signaling pathway. Key proteins of this pathway affect axon guidance, the process by which neurons send out axons to reach the correct targets during neural development (Zeng et al., 2017). Consistently, genetic variation in the NETRIN1 pathway was demonstrated to affect white matter integrity in MDD patients, particularly in the superior longitudinal fasciculus (a tract connecting the frontal, temporal, parietal and occipital lobes), in the inferior longitudinal fasciculus (a tract connecting the temporal and occipital lobes) and in the thalamic radiations tract. These tracts were implicated in MDD by independent studies and they go across regions relevant to MDD pathogenesis such as the amygdala and hippocampus (Cole et al., 2012; Whalley et al., 2013; Shen et al., 2017). The hypothesis that neurodevelopmental mechanisms may be implicated in MDD is supported by an overlap between the biological pathways involved in MDD and schizophrenia (Wray et al., 2018). This finding may be explained in the perspective of the continuity model of psychiatric disorders, with severe MDD cases at the end of the spectrum, in the line with the observation of extensive genetic overlap among major psychiatric disorders (Cross-Disorder Group of the Psychiatric Genomics Consortium, 2013; Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium, 2015; Amare et al., 2019). Clinically, severe MDD patients may show symptoms relatively rare in this disorder and more typical of schizophrenia and other neurodevelopmental disorders, such as early onset, cognitive deficits, social difficulties and psychotic symptoms, and higher risk of childhood attention deficit hyperactivity disorder (ADHD) diagnosis (e.g. 6.3% in early onset vs. 0.9% in later onset MDD cases) (Rice et al., 2019). Some of these symptoms are relatively common in MDD, for example cognitive and psychosocial deficits were described in 30–50% of patients in partial or complete remission (Lam et al., 2014), but they are distributed on a continuum, and clinically it is not straightforward to identify a threshold for distinguishing patients having high risk of a negative prognosis (e.g. incomplete functional recovery between episodes and high disease recurrence). Genetics may help in reaching this objective, through the identification of genetic risk factors in specific genomic areas (genes or pathways) or general genetic risk factors (e.g. by using PRS). For example, higher PRS for schizophrenia or ADHD was associated with the risk of early onset MDD, psychotic symptoms and social communication difficulties (Rice et al., 2019). At treatment level, patients sharing more genetic risk factors with schizophrenia may have higher risk of poor treatment response, as demonstrated in bipolar disorder [International Consortium on Lithium Genetics (ConLi+Gen) et al., 2018], and possibly need treatment with drugs having alternative mechanisms of action.
Despite a probable neurodevelopmental component in a subgroup of cases, genetic studies provided quite convincing evidence that MDD pathogenesis and antidepressant action mostly involve modifications of neural-plasticity and neurotransmission, which are controlled by changes in gene expression patterns and influenced by the activity of the immune system (Calabrese et al., 2014). In terms of specific pathways, association signals came from those modulating long-term potentiation, a persistent increase in synaptic strength following high-frequency synaptic stimulation, and second messengers mediating the cellular events activated by neurotrophins such as brain-derived neurotrophic factor (BDNF) (Hunter et al., 2013; Fabbri et al., 2019a). Excitatory neurotransmission is a key regulator of neural plasticity and neural survival, and in the central nervous system, the most common excitatory neurotransmitter is glutamate. The glutamatergic genes mostly associated with MDD within this pathway were sortilin-related VPS10 domain containing receptor 3 (SORCS3), glutamate metabotropic receptor 5 (GRM5), dopamine receptor D2 (DRD2) and calcium binding protein 1 (CABP1) (Howard et al., 2018).
Neurotrophins stimulate neural survival, neurogenesis in specific brain areas and development of new synapses, while inflammatory factors such as a number of cytokines have the opposite effect. The impact of genetic pathways modulating immune response and inflammation was indeed demonstrated for both MDD risk and antidepressant response, involving for example, the antigen processing and presentation pathway, the tumor necrosis factor pathway and the B cell receptor signaling pathway (Hunter et al., 2013; Fabbri et al., 2014; Wray et al., 2018).
Finally, no change in neural plasticity or persistent change in neurotransmission would be possible without a proper modulation of gene expression, which, for example, determines the level of neurotransmitter receptors and transporters, or the microtubule reorganization needed to develop new synaptic connections. Chromatin (i.e. the structure formed by DNA and proteins that constitutes chromosomes) has a tridimensional structure that varies over time in response to a variety of stimuli, such as neurotransmitters, growth factors, neuropeptides (Ou et al., 2017), which can be modulated by antidepressant treatments. Pathways modulating chromatin structure have been associated with depression, antidepressant response and animal models showed that downregulation of histone deacetylase (an enzyme modulating chromatin structure) in the hippocampus has antidepressant-like effect (Tsankova et al., 2006; Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium, 2015; Fabbri et al., 2019a).
It is possible to hypothesize that not all MDD patients have the same pattern of biological dysfunctions, or in other words specific pathways may contribute to a different extent to the clinical manifestations observed in different patients. For example, in part of the subjects there may be genetic alterations prevalently in pathways responsible for immune response and inflammation, in others in pathways controlling glutamatergic neurotransmission, while impaired ability to regulate and change chromatin structure/gene expression in response to certain stimuli may be the main mechanism in other cases. As reported above, some severe cases may have variations in neurodevelopmental pathways. Genetics can theoretically serve the purpose of identifying the group each patient belongs to, assisting in diagnosis and treatment. However, pathway analysis has not been applied to study the heterogeneity among MDD subtypes to the best of our knowledge, while other methods such as PRS showed very interesting results for atypical depression, suggesting that it is genetically correlated with obesity-related traits and treatments effectively targeting immune-metabolic dysregulations may benefit this subgroup of patients (Milaneschi et al., 2017). Pathways and genes associated with MDD can be also a tool for drug repositioning, another valuable clinical application of genetics.
Genetic analysis for drug repositioning and development of new drugs for depression
Bioinformatic approaches based on matching genetic findings with known drug targets can also help to perform drug repositioning. Alternatively, genetic findings may suggest new drug targets and guide the development of drugs with alternative mechanisms of action compared to the ones currently available.
Conventional drug development is a very long and expensive process (13–15 years and US$2–3 billion) with only 10% chance of being approved by regulatory agencies (Nosengo, 2016; Smietana et al., 2016). It was estimated that repositioned drugs could have approval in less than half of the time and at one-quarter of the cost, because they usually have already passed the early phases of development and clinical testing (Nosengo, 2016).
‘Druggability’ is a mutable concept; however, it is usually referred to those genes that encode protein targets of approved or clinical trial-phase drug candidates, genes with sequence similarity to them or genes that encode secreted and extracellular proteins (Gaspar et al., 2019). Pathways are more druggable than single genes, since they provide opportunities of pharmacological modulation at different levels (Breen et al., 2016). Genes/pathways associated with MDD or antidepressant response can be matched with druggable genes/pathways to identify existing drugs acting on these targets, which could be repositioned for treating MDD. This approach is based on the integration of multiple data sources and uses the known interactions between drugs and proteins and drug-associated changes in gene expression, systematically reported in publicly available resources such as DSigDB and Connectivity Map (Finan et al., 2017; Subramanian et al., 2017). When applied to genes and pathways associated with MDD, drug-target networks suggested that the following modes of action may be useful in MDD treatment: dopamine receptor D2 antagonism/agonism (DRD2), serotonin receptor 5-HT1D antagonism/agonism (HTR1D), calcium channels (particularly CACNA1C) modulation and antagonism, and estrogen receptor ER-α (ESR1) and ER-β (ESR2) modulation (Gaspar et al., 2019). These findings confirm the results of other studies showing the relevance of these modes of action in the treatment of depression. For example, L-type voltage-dependent calcium channels (L-VDCC) were demonstrated to mediate the effect of rapid-acting antidepressants such as ketamine, and L-VDCC plays a critical role in the release of BDNF and synaptic plasticity (Jourdi et al., 2009). It was recently demonstrated that selective serotonin reuptake inhibitors also have an effect on L-VDCC, which is independent from the blockage of the serotonin transporter (Normann et al., 2018). Fendiline, a calcium channel blocker, was among the top drugs suggested for repositioning in MDD by an independent study, as well as 4-hydroxyestrone, an endogenous estrogen (So et al., 2019). In rats, co-administration of 17β-estradiol improved escitalopram-induced antidepressant effect altering its effects on the gene expressions of serotonin receptor 1A, estrogen receptors alpha and beta (Ibrahim et al., 2016). In line with this, the synthetic selective agonist of ER-β WAY-200070 was suggested to act as an anxiolytic and antidepressant in mice (Hughes et al., 2008). However, the study of this compound did not progress to the clinical phase, while tibolone, a synthetic steroid acting on ER-β but preferably ER-α and having also progestogenic and androgenic effects, has shown preliminary evidence of improving depressive symptoms during the menopause transition (Kulkarni et al., 2018).
Other pharmacological mechanisms of action suggested for drug repurposing in MDD include the antagonism of alpha-2 and beta-2 adrenergic receptors, the inhibition of the enzyme histone deacetylase, phosphodiesterase inhibition and GABA-A receptor modulation (So et al., 2017, 2019; Gaspar et al., 2019). Interestingly, this last mode of action is responsible for the antidepressant effect of brexanolone, the first drug approved for post-partum depression (Meltzer-Brody et al., 2018). Drugs that inhibit cell proliferation (e.g. mitoxantrone) have also been suggested (So et al., 2019), but the direction of the effect seems dubious in this case, since an increase in neurogenesis has been associated with the antidepressant effect (Harmer et al., 2017).
The available findings (Table 1) support the fact that genetics is a valuable resource for drug repositioning as well as for the identification of potential pharmacological targets. The growth of large repositories of genetic data through biobanks and consortia gives the opportunity to exploit these data not only for finding predictors of treatment response but also for helping in the development of new drugs for depression. Based on the hypothesis that different genetic pathways may be responsible for disease pathogenesis in different patients, drug repositioning could be applied in a more selective way, looking at the genetic profile of subgroups of patients who are treatment-resistant and show dysfunctions in pathways not directly targeted by the available antidepressant drugs. The genetics of treatment response or resistance has still not been used for drug repurposing to the best of our knowledge, mainly because of the lower sample size (and power) of pharmacogenetic genome-wide association studies (GWASs).
Table 1.
Drugs identified as potentially effective for repurposing in major depressive disorder
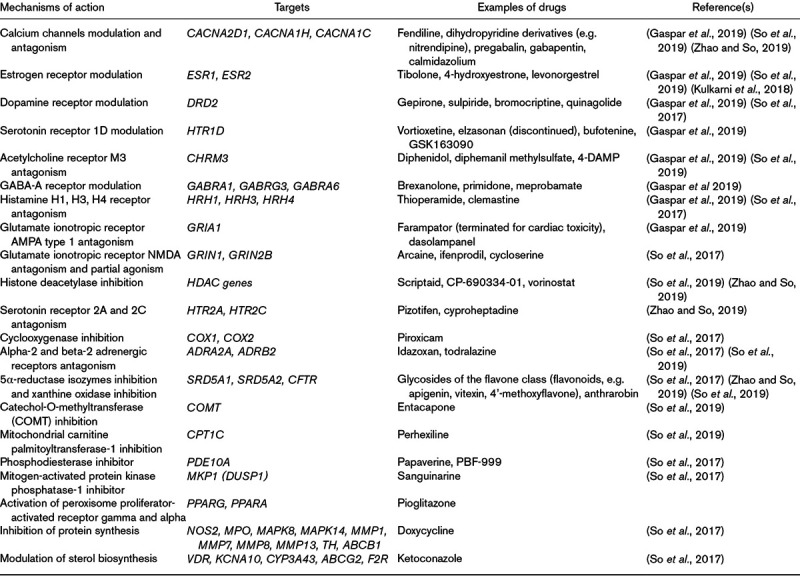
Despite the promising results of genetics applied to drug repurposing in MDD, only a few studies were published, all in the last couple of years, and a longer time is needed to expand and refine the results, in order to translate them in the development/approval of new drugs for MDD. The current clinical applications of genetics consist in indications for drug prescription (endorsed by established international guidelines) and for disease risk estimation (a consensus is lacking, but commercial use is spreading), which are discussed in the next paragraph.
Risk of depression and treatment nonresponse: a genetic risk score for each patient?
The current established clinical applications of genetics in the treatment of MDD consist in prescribing recommendations based on cytochrome 2D6 and 2C19 (CYP2D6 and CYP2C19) genetic variants, as described in guidelines by the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetic Working Group (Pharmacogenomics Knowledge Base, 2019). These genes code for enzymes involved in the metabolism of most antidepressants (pharmacokinetics) and different levels of enzymatic activity are predicted based on the genetic variants carried by an individual. The clinical indications provided by guidelines include drug choice and dose, based on the genetically predicted enzymatic activity, as exemplified for some antidepressants in Table 2. Although this information can be helpful in complementing clinical judgment, it captures only a small fraction of the interindividual differences in antidepressant response through the variation in their metabolism. There are indeed no genes mediating antidepressant action (pharmacodynamics) having prescribing indications in current guidelines. The failure to consistently replicate the effect of genetic variants in pharmacodynamic candidate genes led to the development of new methodological approaches, which take into account the complex polygenic architecture of antidepressant efficacy. PRSs aim to fulfill this purpose by summing the risk alleles carried by each subject, weighted for their effect size on the trait (i.e. the estimated magnitude of effect on the trait). Ideally, PRSs could provide an estimation of the genetic risk of an individual to develop a certain trait (e.g. MDD or antidepressant nonresponse or treatment resistance). PRSs have some key advantages: they take into account the cumulative impact of all the variants associated with the trait and they avoid missing the contribution of variants having a weak effect on the trait. However, PRSs show also relevant limitations: they include only common genetic variants (found in >1% of the population), they do not incorporate information on possible interactions among variants and they assume they have addictive effects. Rare variants indeed are scarcely captured by genome-wide arrays, which represent the most commonly used genotyping technique, and the estimation of rare variant effect size on treatment response would not possible or would be instable in relatively small samples. The largest MDD samples currently available [807 553 individuals in total (Howard et al., 2019)] were actually estimated to not provide adequate power for the identification of all the common variants involved in MDD either. MDD genetic risk was indeed estimated to be highly polygenic and to involve a continuum of very small effects, with odds ratio very close to one. Thus, up to 10 million individuals are needed to explain 80% of SNP-based heritability of MDD, while between 0.7 and 1.5 million for most of the other psychiatric diseases (Zhang et al., 2018). On the contrary, most nonpsychiatric chronic diseases such as type 2 diabetes and coronary artery disease show greater numbers of susceptibility SNPs with larger effects and PRSs are able to explain a meaningful proportion of variance in these traits in samples of hundreds of thousand subjects (Zhang et al., 2018). This is exemplified by the PRS of cardiovascular disease risk that was shown to improve prediction of disease compared to clinical risk factors only (Knowles and Ashley, 2018). For psychiatric traits, the variance explained by PRSs on the liability scale was 4% for bipolar disorder, 3% for MDD and 7% for schizophrenia (Ripke et al., 2014; Wray et al., 2018; Howard et al., 2019; Stahl et al., 2019), while heritability estimated by twin studies was considerably higher (70%, 37% and 80%, respectively) (Sullivan et al., 2000; Smoller and Finn, 2003; Sullivan et al., 2003). Despite the clearly limited performance of PRS in the available sample sizes, direct to consumer genetic services are rapidly expanding, including access to individual genetic profiles from genotyping microarrays. Education of the public to a correct interpretation of these results, including PRSs of psychiatric traits, is a challenge taken by open source tools such as Impute.me, which allows users to upload consumer genetics data and receive evidence-based information about more than 2000 traits (Folkersen et al., 2019). The PRS of antidepressant response is even at an earlier stage of development compared to the PRS of MDD and other psychiatric disorders, since the sample size of the published studies did provide adequate power for PRS estimation (García-González et al., 2017).
Table 2.
Examples of clinical indications provided by guidelines curated by the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetic Working Group

In the described scenario, there are two possible alternatives to move forward: (1) the recruitment of larger MDD samples characterized in terms of antidepressant response; (2) the use of alternative analysis approaches, which improve power. The first option is doable with time and money, and it seems feasible by joining the efforts of many research groups. It could also be facilitated by the use of self-reported data, bearing in mind the known limitations of this approach (Cai et al., 2019). In any case, the second option should be pursued as well, and a promising strategy seems to be the combination of common variant genome-wide genotyping with complementary or adjunctive information. In terms of complementary information, a possible strategy is the integration in the analysis of the impact of genetic variants on gene expression levels, which is not considered in GWASs and standard PRSs, an approach called transcriptome-wide association study (TWAS) (Gusev et al., 2016). TWAS was showed to significantly increase power compared to GWAS, through a better detection of causal variants (Gusev et al., 2016). A number of different methodological approaches have been developed to perform TWAS. The most commonly used methods apply multi-SNP prediction (MP) analysis that directly model linkage disequilibrium when causal variants are not genotyped, by imputing gene expression based on a reference set of individuals for whom both gene expression and genetic variation are available (Gamazon et al., 2015; Gusev et al., 2016). TWAS-SMR (summary-based Mendelian Randomization) instead uses expression-genotype and genotype-phenotype summary-statistics to determine whether the effect of the genotype on the trait is mediated by alternations in gene expression (Zhu et al., 2016). Differently from TWAS-MP, TWAS-SMR can distinguish when gene expression mediates the association SNP-trait (causality) and when a SNP has direct and independent effects on gene expression as well as the phenotype (pleiotropy).
An example of information that can integrate GWASs is represented by rare variants in coding regions, obtained by exome sequencing. GWASs cover mostly noncoding regions of the genome and common variants; however, coding regions are pivotal in determining gene expression and protein functionality; for this reason rare variants were hypothesized to be at least partly responsible for the proportion of trait heritability not captured by GWAS (Zuk et al., 2014). Whole exome sequencing (WES) requires ~3 days and costs ~500 USD (National Human Genome Research Institute, 2018), thus it is relatively affordable despite costing ~10 times compared to genome-wide genotyping. In the psychiatric field, WES was performed mainly in neurodevelopmental disorders, such as schizophrenia, and for identifying genes carrying damaging variants associated with the disease (Singh et al., 2017). Few studies used WES in MDD, in relatively small samples (e.g. Tombácz et al., 2017; Zhang et al., 2019), and only two studies are available for antidepressant response, of which one was performed on 10 subjects (Tammiste et al., 2013), while the other on ~1200 subjects (Fabbri et al., 2019b). Thus, the contribution of rare variants to these traits is largely unknown and difficult to explore since the relatively smaller samples analyzed compared to other psychiatric traits. A possible method to increase the power of detecting the contribution of rare variants works similarly to PRSs, being calculated as a weighted sum of variant effects, but instead of using variant effect size it uses the predicted functional impact or pathogenicity of each variant (Curtis, 2018). The functional impact of a variant can be estimated using a number of available functional scores and/or the frequency of the alternative allele (the rarest a variant is, the most detrimental it is expected to be). Functional scores are based on sequence homology, physical properties of amino acids (to determine if an amino acid change is expected to alter protein structure/function), annotations of protein families and domains, 3D protein structure, conservation. Different types of variant annotations were combined to create more complex scores reflecting allelic diversity and pathogenicity, such as Combined Annotation Dependent Depletion (Kircher et al., 2014) and Eigen scores (Ionita-Laza et al., 2016). A genetic risk score for rare variants would add information that currently PRSs do not include and putatively increase the performance of predictive models of MDD risk and antidepressant response. A cumulative genetic score reflecting the burden of rare and common variants could be used to estimate the individual genetic risk of unfavorable disease progression or treatment outcome (Fig. 1). We are currently testing this approach to predict antidepressant response and resistance using variants obtained through WES and genome-wide genotyping in a multi-centric MDD sample recruited by the European Group for the Study of treatment-Resistant Depression. Preliminary results showed encouraging prediction of treatment-resistant depression (TRD) by using the burden of rare and common genetic variants in genes or pathways as predictors, which was improved by the addition of clinical risk factors (Fabbri et al., 2019b). The combination of the effect of common and rare variants and other future improvements of genetic methods are expected to improve the performance of genetic factors in predicting psychiatric traits. However, even if with future improvements, clinical risk factors may be easier to assess and perform similar or better than genetic predictors, except in two main scenarios: (1) obviously, in patients with low or no clinical risk factors, who are also the ones most likely to benefit from prognostic or treatment outcome prediction, because of higher chances of effective preventive and therapeutic strategies; (2) in patients having genetic risk factors at the highest extreme of the distribution, in line to what suggested for the clinical application of PRS (Lewis and Vassos, 2017) (Fig. 1). Pathway (or gene)-based scores can also be used for cluster analysis to identify homogeneous groups of patients in terms of distribution of genetic risk factors. This could facilitate the matching of each genetic profile with personalized treatments and the development of new treatments acting on pathways not targeted by the available antidepressants.
Fig. 1.
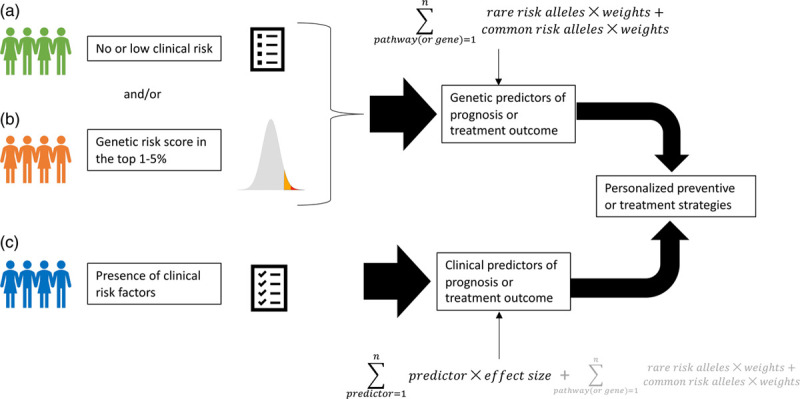
In the scenarios A and B, genetic risk factors are hypothesized to be the most useful to predict disease prognosis and/or treatment outcome and guide the prescription of personalized clinical interventions. A and B can co-exist in the same subject. In scenario C, when the patient shows known clinical risk factors, these probably represent the simplest and most effective way to guide clinical interventions. However, genetic predictors may still add helpful information in case C. Genetic predictors may be pathway- or gene-based or genome-wide, and they should ideally include the contribution of rare variants.
Discussion
The clinical applications of genetics are still limited to the use of variants in pharmacokinetic genes (CYP2D6 and CYP2C19) to guide antidepressant choice and dosing (Table 2). Advances in our knowledge of the pathogenetic processes responsible for MDD and the mechanisms of antidepressant action were achieved, thanks to GWAS and multi-marker tests such as pathway analysis. Genetics also provided new opportunities for drug repositioning. However, the improvements in genotyping technologies and analysis methods were not good enough to explain the hypothesized contribution of genetic variants to MDD and antidepressant response. Twin studies estimated that MDD has an heritability of 37% (Sullivan et al., 2000), but the largest GWAS meta-analysis estimated a heritability of ~9% on the liability scale (Howard et al., 2019), suggesting that the inclusion in the analysis of common variants only and/or our current methodological approach is not able to get close to the theoretical heritability. The variance in MDD and antidepressant response estimated by PRSs was also much lower compared to the expected values, as discussed in section ‘Risk of depression and treatment nonresponse: a genetic risk score for each patient?’. Several complementary strategies can be put in place to address the power limitation of previous studies, as well as a progressive increase in sample size. The use of minimal phenotyping, typically based on self-reported information, has been increasingly applied for this purpose, with the limitation that minimal phenotypes of MDD were demonstrated to have higher genetic overlap with other psychiatric traits and lower heritability compared to DSM-based MDD (Cai et al., 2019). When evaluating antidepressant response, the collection of detailed phenotypic information is probably more relevant, since the risk of imprecision on multiple levels (diagnosis, treatment and symptom longitudinal variation). The balance between sample size and level of phenotyping remains problematic. Among the possible methodological approaches to improve the power of genetic studies, we discussed the integration of information from rare variants, which has been poorly tested in MDD and antidepressant response (Fig. 1). In studies of other complex traits such as BMI and height, whole genome sequencing was shown to recover the expected heritability, suggesting that standard GWASs miss a relevant part of the genetic contribution to polygenic traits (Wainschtein et al., 2019). Methods able to combine the effects of rare and common genetic variants across relevant genes and pathways, and to take into account possible interactions, would be theoretically ideal to uncover the genetic factors involved in MDD and antidepressant response, through predictive modeling or machine learning. A few studies applied these approaches to antidepressant response prediction, with encouraging findings, but the issue of independent replication remains (Iniesta et al., 2018; Fabbri et al., 2019b).
The progress of GWAS and related methods in uncovering the genetics of MDD and other depressive traits may seem relatively unsatisfying on one side, since the low genetic variance explained, but it has already generated a number of direct to consumer products that provide a wide range of information based on microarray genotyping, including disease risk calculated using PRSs (Folkersen et al., 2019). A PRS can be converted into a standardized score that follows a normal distribution, with higher PRS corresponding to higher risk, in a way that could be used to determine an individual’s risk of the corresponding trait based on his/her position on this distribution. However, it is unclear if there is a threshold able to identify subjects having a clinically meaningful increase in risk and at which point of the curve this threshold should be set. It was speculated that a PRS in the top 1–5% of the population would warrant feedback (Lewis and Vassos, 2017), but the best threshold is uncertain as well the possible consequences for individuals predicted to be at high risk, in terms of availability of preventive strategies and risk of stigma/discrimination. Interestingly, there are significant genetic correlations between MDD and other psychiatric but also nonpsychiatric disorders according to GWASs, thus the same person may have increased risk for a number of diseases according to PRSs, and this would make difficult to plan preventive interventions. The uncareful communication of this information may also result in disproportionate worries and other negative consequences. Numerous studies have indeed demonstrated that the genetic predisposition to depression is correlated with the genetics of schizophrenia, bipolar disorder, anorexia nervosa, ADHD, autism spectrum disorder, other than a number of nonpsychiatric traits such as coronary artery disease, inflammatory bowel disease and lung cancer (Howard et al., 2019) (Fig. 2). Currently, the genetic variants specifically conferring risk for MDD are poorly known.
Fig. 2.
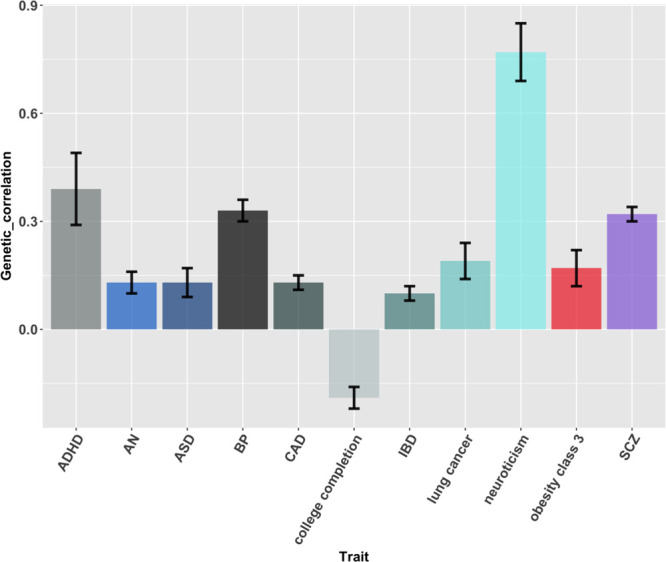
Genetic correlations between depression (including DSM-diagnosed MDD and self-reported major depression) and other psychiatric and nonpsychiatric traits, according to the results reported by Howard et al. Genetic correlation of depression with other traits was reported as well and this figure exemplifies part of the most significant findings. Bars represent standard errors. ADHD, attention deficit hyperactivity disorder; AN, anorexia nervosa; ASD, autism spectrum disorder; BP, bipolar disorder; CAD, coronary artery disease; IBD, inflammatory bowel disease; SCZ, schizophrenia.
In conclusion, future studies should aim not only to identify the missing heritability of MDD and related traits, but also to provide a deeper understanding of the shared and specific genetic risk factors for MDD and other psychiatric disorders, in order to accurately predict disease risk and avoid unspecific genetic risk prediction. There are not univocal strategies to accomplish these objectives but complementary approaches should be applied.
Acknowledgements
C.F. is supported by a Marie Skłodowska-Curie Actions Individual Fellowship funded by the European Community (EC Grant agreement number: 793526; project title: Exome Sequencing in stages of Treatment Resistance to Antidepressants – ESTREA). C.M.L. is part-funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. A.S. received funding from Miur Prin: Progetti Di Ricerca Di Rilevante Interesse Nazionale – Bando 2017, Prot. 2017K2NEF4, Dopamine - Dysbindin Genetic Interaction: A Multidisciplinary Approach to Characterize Cognitive Phenotypes of Schizophrenia and Develop Personalized Treatments.
Conflicts of interest
A.S. is or has been consultant/speaker for: Abbott, Abbvie, Angelini, Astra Zeneca, Clinical Data, Boheringer, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Innovapharma, Italfarmaco, Janssen, Lundbeck, Naurex, Pfizer, Polifarma, Sanofi, Servier. S.M. has been a consultant or served on Advisory boards: AstraZeneca, Bristol Myers Squibb, Forest, Johnson & Johnson, Leo, Lundbeck, Medelink, Neurim, Pierre Fabre, Richter. The other authors declare no potential conflicts of interest.
References
- Amare AT, Vaez A, Hsu YH, Direk N, Kamali Z, Howard DM, et al. Bivariate genome-wide association analyses of the broad depression phenotype combined with major depressive disorder, bipolar disorder or schizophrenia reveal eight novel genetic loci for depression. Mol Psychiatry. 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breen G, Li Q, Roth BL, O’Donnell P, Didriksen M, Dolmetsch R, et al. Translating genome-wide association findings into new therapeutics for psychiatry. Nat Neurosci. 2016; 19:1392–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai N, Revez JA, Adams MJ, Andlauer TFM, Breen G, Byrne EM, et al. Minimal phenotyping yields GWAS hits of reduced specificity for major depression. bioRxiv. 2019. Available at: http://biorxiv.org/lookup/doi/10.1101/440735. [Accessed 07/12/19]
- Calabrese F, Rossetti AC, Racagni G, Gass P, Riva MA, Molteni R. Brain-derived neurotrophic factor: a bridge between inflammation and neuroplasticity. Front Cell Neurosci. 2014; 8:430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole J, Chaddock CA, Farmer AE, Aitchison KJ, Simmons A, McGuffin P, Fu CH. White matter abnormalities and illness severity in major depressive disorder. Br J Psychiatry. 2012; 201:33–39 [DOI] [PubMed] [Google Scholar]
- Cross-Disorder Group of the Psychiatric Genomics Consortium Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013; 381:1371–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis D. Construction of an exome-wide risk score for schizophrenia based on a weighted burden test. Ann Hum Genet. 2018; 82:11–22 [DOI] [PubMed] [Google Scholar]
- Fabbri C, Marsano A, Albani D, Chierchia A, Calati R, Drago A, et al. PPP3CC gene: a putative modulator of antidepressant response through the B-cell receptor signaling pathway. Pharmacogenomics J. 2014; 14:463–472 [DOI] [PubMed] [Google Scholar]
- Fabbri C, Kasper S, Kautzky A, Bartova L, Dold M, Zohar J, et al. Genome-wide association study of treatment-resistance in depression and meta-analysis of three independent samples. Br J Psychiatry. 2019; 214:36–41 [DOI] [PubMed] [Google Scholar]
- Fabbri C, Kasper S, Kautzky A, Zohar J, Montgomery S, Albani D, et al. A polygenic predictor of treatment-resistant depression using whole exome sequencing and genome-wide genotyping. medRxiv. 2019. Available at: http://medrxiv.org/lookup/doi/10.1101/19007161. [Accessed 23/09/19] [DOI] [PMC free article] [PubMed]
- Fernandes BS, Williams LM, Steiner J, Leboyer M, Carvalho AF, Berk M. The new field of ‘precision psychiatry’. BMC Med. 2017; 15:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finan C, Gaulton A, Kruger FA, Lumbers RT, Shah T, Engmann J, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med. 2017; 9:eaag1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkersen L, Pain O, Ingasson A, Werge T, Lewis CM, Austin J. Impute.me: an open source, non-profit tool for using data from DTC genetic testing to calculate and interpret polygenic risk scores. bioRxiv. 2019. Available at: http://biorxiv.org/lookup/doi/10.1101/861831. [Accessed 07/12/19] [DOI] [PMC free article] [PubMed]
- Fried EI, Nesse RM. Depression is not a consistent syndrome: an investigation of unique symptom patterns in the STAR*D study. J Affect Disord. 2015; 172:96–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, et al. ; GTEx Consortium. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet. 2015; 47:1091–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-González J, Tansey KE, Hauser J, Henigsberg N, Maier W, Mors O, et al. ; Major Depressive Disorder Working Group of the Psychiatric Genomic Consortium. Pharmacogenetics of antidepressant response: a polygenic approach. Prog Neuropsychopharmacol Biol Psychiatry. 2017; 75:128–134 [DOI] [PubMed] [Google Scholar]
- Gaspar HA, Gerring Z, Hübel C, Middeldorp CM, Derks EM, Breen G; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Using genetic drug-target networks to develop new drug hypotheses for major depressive disorder. Transl Psychiatry. 2019; 9:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BW, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016; 48:245–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmer CJ, Duman RS, Cowen PJ. How do antidepressants work? New perspectives for refining future treatment approaches. Lancet Psychiatry. 2017; 4:409–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M, et al. ; 23andMe Research Team; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019; 22:343–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard DM, Adams MJ, Shirali M, Clarke TK, Marioni RE, Davies G, et al. ; 23andMe Research Team. Genome-wide association study of depression phenotypes in UK biobank identifies variants in excitatory synaptic pathways. Nat Commun. 2018; 9:1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ZA, Liu F, Platt BJ, Dwyer JM, Pulicicchio CM, Zhang G, et al. WAY-200070, a selective agonist of estrogen receptor beta as a potential novel anxiolytic/antidepressant agent. Neuropharmacology. 2008; 54:1136–1142 [DOI] [PubMed] [Google Scholar]
- Hunter AM, Leuchter AF, Power RA, Muthén B, McGrath PJ, Lewis CM, et al. A genome-wide association study of a sustained pattern of antidepressant response. J Psychiatr Res. 2013; 47:1157–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim WW, Safar MM, Khattab MM, Agha AM. 17β-estradiol augments antidepressant efficacy of escitalopram in ovariectomized rats: neuroprotective and serotonin reuptake transporter modulatory effects. Psychoneuroendocrinology. 2016; 74:240–250 [DOI] [PubMed] [Google Scholar]
- Iniesta R, Hodgson K, Stahl D, Malki K, Maier W, Rietschel M, et al. Antidepressant drug-specific prediction of depression treatment outcomes from genetic and clinical variables. Sci Rep. 2018; 8:5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amare AT, Schubert KO, Hou L, Clark SR, Papiol S, Heilbronner U, et al. ; International Consortium on Lithium Genetics (ConLi+Gen). Association of polygenic score for schizophrenia and HLA antigen and inflammation genes with response to lithium in bipolar affective disorder: a Genome-Wide Association Study. JAMA psychiatry. 2018; 75:65–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita-Laza I, McCallum K, Xu B, Buxbaum JD. A spectral approach integrating functional genomic annotations for coding and noncoding variants. Nat Genet. 2016; 48:214–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdi H, Hsu YT, Zhou M, Qin Q, Bi X, Baudry M. Positive AMPA receptor modulation rapidly stimulates BDNF release and increases dendritic mRNA translation. J Neurosci. 2009; 29:8688–8697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014; 46:310–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles JW, Ashley EA. Cardiovascular disease: the rise of the genetic risk score. Plos Med. 2018; 15:e1002546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni J, Gavrilidis E, Thomas N, Hudaib AR, Worsley R, Thew C, et al. Tibolone improves depression in women through the menopause transition: a double-blind randomized controlled trial of adjunctive tibolone. J Affect Disord. 2018; 236:88–92 [DOI] [PubMed] [Google Scholar]
- Lam RW, Kennedy SH, Mclntyre RS, Khullar A. Cognitive dysfunction in major depressive disorder: effects on psychosocial functioning and implications for treatment. Can J Psychiatry. 2014; 59:649–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis CM, Vassos E. Prospects for using risk scores in polygenic medicine. Genome Med. 2017; 9:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meltzer-Brody S, Colquhoun H, Riesenberg R, Epperson CN, Deligiannidis KM, Rubinow DR, et al. Brexanolone injection in post-partum depression: two multicentre, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet. 2018; 392:1058–1070 [DOI] [PubMed] [Google Scholar]
- Milaneschi Y, Lamers F, Peyrot WJ, Baune BT, Breen G, Dehghan A, et al. ; CHARGE Inflammation Working Group and the Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Genetic association of major depression with atypical features and obesity-related immunometabolic dysregulations. JAMA Psychiatry. 2017; 74:1214–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Human Genome Research Institute DNA Sequencing Costs: Data. 2018. Available at: https://www.genome.gov/about-genomics/fact-sheets/DNA-Sequencing-Costs-Data. [Accessed 11/03/19]
- Network and Pathway Analysis Subgroup of Psychiatric Genomics Consortium Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat Neurosci. 2015; 18:199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normann C, Frase S, Haug V, von Wolff G, Clark K, Münzer P, et al. Antidepressants rescue stress-induced disruption of synaptic plasticity via serotonin transporter-independent inhibition of L-type calcium channels. Biol Psychiatry. 2018; 84:55–64 [DOI] [PubMed] [Google Scholar]
- Nosengo N. Can you teach old drugs new tricks? Nature. 2016; 534:314–316 [DOI] [PubMed] [Google Scholar]
- Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, O’Shea CC. ChromEMT: visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science. 2017; 357:eaag0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pharmacogenomics Knowledge Base Drug labels and clinical guideline annotations. 2019. Available at: https://www.pharmgkb.org. [Accessed 11/03/19]
- Rice F, Riglin L, Thapar AK, Heron J, Anney R, O’Donovan MC, Thapar A. Characterizing developmental trajectories and the role of neuropsychiatric genetic risk variants in early-onset depression. JAMA Psychiatry. 2019; 76:306–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014; 511:421–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Reus LM, Cox SR, Adams MJ, Liewald DC, Bastin ME, et al. Subcortical volume and white matter integrity abnormalities in major depressive disorder: findings from UK biobank imaging data. Sci Rep. 2017; 7:5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh T, Walters JTR, Johnstone M, Curtis D, Suvisaari J, Torniainen M, et al. ; INTERVAL Study; UK10K Consortium. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat Genet. 2017; 49:1167–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smietana K, Siatkowski M, Møller M. Trends in clinical success rates. Nat Rev Drug Discov. 2016; 15:379–380 [DOI] [PubMed] [Google Scholar]
- Smoller JW, Finn CT. Family, twin, and adoption studies of bipolar disorder. Am J Med Genet C Semin Med Genet. 2003; 123C:48–58 [DOI] [PubMed] [Google Scholar]
- So HC, Chau CK, Chiu WT, Ho KS, Lo CP, Yim SH, Sham PC. Analysis of genome-wide association data highlights candidates for drug repositioning in psychiatry. Nat Neurosci. 2017; 20:1342–1349 [DOI] [PubMed] [Google Scholar]
- So HC, Chau CK, Lau A, Wong SY, Zhao K. Translating GWAS findings into therapies for depression and anxiety disorders: gene-set analyses reveal enrichment of psychiatric drug classes and implications for drug repositioning. Psychol Med. 2019; 49:2692–2708 [DOI] [PubMed] [Google Scholar]
- Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, et al. ; eQTLGen Consortium; BIOS Consortium; Bipolar Disorder Working Group of the Psychiatric Genomics Consortium. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet. 2019; 51:793–803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strawbridge R, Young AH, Cleare AJ. Biomarkers for depression: recent insights, current challenges and future prospects. Neuropsychiatr Dis Treat. 2017; 13:1245–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017; 171:1437–1452.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan PF, Neale MC, Kendler KS. Genetic epidemiology of major depression: review and meta-analysis. Am J Psychiatry. 2000; 157:1552–1562 [DOI] [PubMed] [Google Scholar]
- Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003; 60:1187–1192 [DOI] [PubMed] [Google Scholar]
- Tammiste A, Jiang T, Fischer K, Mägi R, Krjutškov K, Pettai K, et al. Whole-exome sequencing identifies a polymorphism in the BMP5 gene associated with SSRI treatment response in major depression. J Psychopharmacol. 2013; 27:915–920 [DOI] [PubMed] [Google Scholar]
- Tombácz D, Maróti Z, Kalmár T, Csabai Z, Balázs Z, Takahashi S, et al. High-coverage whole-exome sequencing identifies candidate genes for suicide in victims with major depressive disorder. Sci Rep. 2017; 7:7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat Neurosci. 2006; 9:519–525 [DOI] [PubMed] [Google Scholar]
- Wainschtein P, Jain DP, Yengo L, Zheng Z, Cupples LA, Shadyab AH, et al. Recovery of trait heritability from whole genome sequence data. bioRxiv. 2019. Available at: http://biorxiv.org/lookup/doi/10.1101/588020. [Accessed 23/07/19]
- Whalley HC, Sprooten E, Hackett S, Hall L, Blackwood DH, Glahn DC, et al. Polygenic risk and white matter integrity in individuals at high risk of mood disorder. Biol Psychiatry. 2013; 74:280–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. ; eQTLGen; 23andMe; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018; 50:668–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Navarro P, Fernandez-Pujals AM, Hall LS, Clarke TK, Thomson PA, et al. ; Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. A combined pathway and regional heritability analysis indicates NETRIN1 pathway is associated with major depressive disorder. Biol Psychiatry. 2017; 81:336–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li M, Wang Q, Hsu JS, Deng W, Ma X, et al. A joint study of whole exome sequencing and structural MRI analysis in major depressive disorder. Psychol Med. 20191–12 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Qi G, Park JH, Chatterjee N. Estimation of complex effect-size distributions using summary-level statistics from genome-wide association studies across 32 complex traits. Nat Genet. 2018; 50:1318–1326 [DOI] [PubMed] [Google Scholar]
- Zhao K, So HC. Drug repositioning for schizophrenia and depression/anxiety disorders: a machine learning approach leveraging expression data. IEEE J Biomed Health Inform. 2019; 23:1304–1315 [DOI] [PubMed] [Google Scholar]
- Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eqtl studies predicts complex trait gene targets. Nat Genet. 2016; 48:481–487 [DOI] [PubMed] [Google Scholar]
- Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, et al. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci U S A. 2014; 111:E455–E464 [DOI] [PMC free article] [PubMed] [Google Scholar]