

Abstract
Cellular homeostasis is dependent on a balance between DNA damage and DNA repair mechanisms. Cells are constantly assaulted by both exogenous and endogenous stimuli leading to high levels of reactive oxygen species (ROS) that cause oxidation of the nucleotide dGTP to 8-oxodGTP. If this base is incorporated into DNA and goes unrepaired, it can result in G>T transversions, leading to genomic DNA damage. MutT Homolog 1 (MTH1) is a nucleoside diphosphate X (Nudix) pyrophosphatase that can remove 8-oxodGTP from the nucleotide pool before it is incorporated into DNA by hydrolyzing it into 8-oxodGMP. MTH1 expression has been shown to be elevated in many cancer cells and is thought to be a survival mechanism by which a cancer cell can stave off the effects of high ROS that can result in cell senescence or death. It has recently become a target of interest in cancer because it is thought that inhibiting MTH1 can increase genotoxic damage and cytotoxicity. Determining the role of MTH1 in normal and cancer cells is confounded by an inability to reliably and directly measure its native enzymatic activity. We have used the chimeric ATP-releasing guanine-oxidized (ARGO) probe that combines 8-oxodGTP and ATP to measure MTH1 enzymatic activity in colorectal cancer (CRC), non-small cell lung cancer (NSCLC) and pancreatic ductal adenocarcinoma (PDAC) along with patient-matched normal tissue. MTH1 8-oxodGTPase activity is significantly increased in tumors across all three tissue types, indicating that MTH1 is a marker of cancer. MTH1 activity measured by ARGO assay was compared to mRNA and protein expression measured by RT-qPCR and Western blot in the CRC tissue pairs, revealing a positive correlation between ARGO assay and Western blot, but little correlation with RT-qPCR in these samples. The adoption of the ARGO assay will help in establishing the level of MTH1 activity in model systems and in assessing the effects of MTH1 modulation in the treatment of cancer.
Keywords: MTH1, DNA repair, Cancer, Genetic Instability, Reactive Oxygen Species, 8-oxodGTP
Graphical Abstract
1. Introduction
Genomic instability is a feature in almost all cancers, but it is unknown when exactly it arises in the process of tumorigenesis [1]. External and endogenous stimuli constantly attack DNA leading to mutations and therefore, the opposing functions of DNA damage and DNA repair must be balanced to maintain genetic stability. The classic mutator hypothesis proposes that a defect in the DNA repair machinery of a cell will lead to mutations in other genes and result in genomic instability [2]. Yet, even in repair proficient cells, certain stressors can overpower the repair machinery.
Endogenous processes such as metabolism create oxidative stress in a cell and the formation of ROS. Sources of ROS include mitochondrial electron transport, NADPH oxidase activity, ß-oxidation of fatty acids and flavin oxidase activity in peroxisomes and protein oxidation in the endoplasmic reticulum [3] [4]. Increased ROS is detected in almost all cancers and plays a role in regulating tumor proliferation, development and metastasis [5]. Oncogenic signaling, hypoxic tumor environments and the high metabolic rate and oxygen consumption needed to fuel tumor cells lead to sustained mitochondrial respiration and high ROS (reviewed in [6]). There is evidence that ROS can be either tumorigenic or tumor suppressive. At low or moderate levels, ROS can be tumorigenic by acting as a signaling molecule to stimulate the phosphorylation of mitogen-activated protein kinase, induce cyclin D1 expression and activate Jun N-terminal kinase, all of which are linked to tumor growth and survival [3, 7]. On the other hand, high levels of ROS can lead to damage of cellular components including DNA, proteins and lipids and can promote cell senescence or death. In cancer cells, there is an interplay of high ROS counteracted by up-regulation of antioxidant mechanisms regulated by the NRF2 transcription factor [8]. Although this helps to mitigate ROS induced oxidative stress in cancer cells, these cells still have higher levels of ROS than normal cells. Many chemotherapy drugs, as well as radiation, act in part to further increase ROS to a level that is toxic to the cells.
One of the major oxidatively damaged DNA lesions resulting from increased ROS is 8-oxo-7,8-dihydroguanine (8-oxoG), which can aberrantly pair with adenine during replication. If this lesion is not detected and excised before replication, this can result in G>T base transversions [9]. Incomplete repair of the lesion can also lead to single-strand breaks (SSBs). Of the ~70,000 DNA lesions generated per day in each human cell, approximately 75% are SSBs which can arise from oxidative damage during metabolism or base hydrolysis [10]. Cells have base excision repair mechanisms to counteract this type of damage, with 8-oxoguanine DNA Glycosylase (OGG1) excising the 8-oxoG and a second enzyme, MutY DNA Glycosylase, excising adenines that have been misincorporated opposite 8-oxoG during replication. However, an additional enzyme, MTH1, can remove the 8-oxodGTP moieties from the nucleotide pool even before incorporation into DNA by hydrolyzing 8-oxodGTP to 8-oxodGMP and diphosphate [11, 12]. MTH1, a member of the Nudix phosphohydrolase enzyme family, is found in both normal and cancer cells, but there is some debate as to its role and importance in cellular homeostasis and at different stages of tumorigenesis. MTH1 expression is often increased in cancer cells and is thought to play a vital role in the survival of tumor cells by protecting them from the cytotoxic effects of high ROS [13]. Studies have shown an increased expression of MTH1 in osteosarcoma, ulcerative colitis with neoplasm, gastric cancer, lung cancer, renal cell carcinoma, breast cancer and CRC [14–20]. Further evidence that MTH1 may play a role in tumorigenesis is that in NSCLC, high malignant potential and worse prognosis is associated with higher MTH1 protein expression [21] and in CRC, increased MTH1 expression correlates with lower overall survival and more advanced stage[22]. MTH1 inhibitors have recently been explored as an anti-cancer strategy to suppress the proliferation of tumor cells and induce apoptosis, and at least one clinical trial is underway to test this strategy [23]. However, with multiple MTH1 inhibitors being tested, there have been some confounding findings in which inhibition of MTH1 by small drug molecules does not always give the same results as with siRNA or shRNA-mediated MTH1 knock-down or MTH1 knock-out (KO) using CRISPR-Cas9 approaches, suggesting that some of these inhibitors may have off-target effects [reviewed in [24]]. Another issue is that the effect on cellular proliferation of a particular MTH1 inhibitor or MTH1-targeted siRNA/shRNA has given opposing results in different cell lines and among different researchers, indicating that there may be important cell-specific contexts at play or simply that there needs to be more consistency in the methods used to assess the effects of MTH1 inhibition.
Coming to a consensus on the role of MTH1 in both normal and cancer cells has been complicated by the use of surrogate measurements to assess MTH1 function in cells and tissues that are cumbersome and in certain cases, have low sensitivity or specificity and do not measure enzyme activity. The most common approaches include measuring levels of MTH1 mRNA by RT-qPCR and protein by Western blot and immunohistochemistry. Pyrophosphate release assays and HPLC measurement of 8-oxodGMP that measure enzymatic activity are also used, but are not specific to the MTH1 pathway and are not sensitive enough for use in native cells and tissues [25]. To address these concerns, we have recently described the development of the ARGO assay, a simple, sensitive assay to measure MTH1 activity in native cells and tissues utilizing the ARGO probe, a two-headed chimeric MTH1 substrate comprised of 8-oxodGTP linked to ATP. When cleaved by MTH1, the ARGO probe releases ATP that can be quantified by luminescence in a standard ATP measurement assay using firefly luciferase [25]. Here we report on the use of the ARGO assay to measure MTH1 function in paired normal and tumor tissues from CRC, NSCLC and PDAC patients.
2. Materials and Methods
2.1. HAP1 parental and MTH1 KO cell lines
C631 Human HAP1 parental control cell line and two different HAP1 MTH1 KO cell lines (HZGHC000753c002, 8 bp deletion exon 2; HZGHC000753c003, 10 bp deletion exon 2) were obtained from Horizon Discovery Limited. Cells were maintained in Iscove’s Modified Dulbecco’s Medium with 10% FBS and 1% Penicillin/Streptomycin/Fungizone as directed by the manufacturer.
2.2. Tissue samples
All tissues were flash frozen in liquid nitrogen and maintained at −80C until processing and extraction. For each tumor tissue, the corresponding patient-matched adjacent normal tissue was also obtained. Ten CRC and patient-matched normal tissue pairs were obtained from the Stanford Cancer Institute Tissue Bank. NSCLC (12 pairs) and PDAC (8 pairs) tumor/matched normal were obtained from the Sylvester Comprehensive Cancer Center, Miami, FL, USA. Tumor percentage in the CRC samples were determined by pathological examination (Supplemental Table 1) and representative haemotoxylin and eosin (H&E)-stained slides of fresh-frozen colon tumors are presented in Supplemental Fig. 1. Slides and/or excess tissue was not available for the NSCLC and PDAC samples and therefore, tumor percentage was not determined for these. However, a representative H&E slide of a PDAC tumor sample obtained from the Stanford Cancer Institute Tissue Bank that was not used in this study is presented in the bottom right panel of Supplemental Fig. 1 as an example.
2.3. Preparation of cell and tissue protein lysates in hypotonic buffer for ARGO assay
HAP1 parental or HAP1 MTH1KO cell lines were grown as described in section 2.1 in 10 cm tissue culture dishes until approximately 75% confluency. Medium was aspirated and plates were rinsed with ice-cold PBS which was also removed by aspiration. With the plates on ice, 0.5 ml of ice-cold Hypotonic buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 1 x EDTA-free Halt™ protease inhibitor (Thermo Scientific), 1 mM Na3V04) was added and cells were scraped into a 1.5 ml centrifuge tube. The crude lysate was incubated on ice for 30 minutes with vortexing every 10 minutes for 10 seconds after which cell debris was pelleted by centrifugation at 14,000 x g for 10 minutes at 4°C. The whole cell lysate was removed to a new 1.5 ml pre-chilled centrifuge tube. To deplete ATP, the lysate was transferred to an Amicon 3K centrifugal filter (EMD Millipore) and centrifuged at 14,000 x g for 10–15 minutes at 4°C until concentrate volume was approximately 75 μl. Sample volume was brought back up to 500 μl with ice-cold hypotonic buffer and the centrifugation was repeated for a total of six spins. ATP-depleted lysate was recovered by inverting the column into a fresh pre-chilled collection tube and centrifuging at 2,000 x g for 2 minutes. Protein concentration was determined using the Qubit Protein Assay Kit (Molecular Probes Life Technology) and lysates were stored at −20°C until further use.
For tissue lysate preparation, the tissues were kept frozen on dry ice while 20 mg of tissue was dissected, weighed and placed in pre-chilled 1.5 ml microcentrifuge tubes. Tubes were then placed on ice and 500 ul of ice-cold Hypotonic buffer was added. The tissue was thoroughly homogenized on ice using a handheld battery-operated Kimble Kontes Pellet Pestle® using 1 second bursts for a total of 10–20 seconds. At this point, the crude tissue lysate was processed as described for cells above.
2.4. Preparation of cell and tissue protein lysates in RIPA buffer for Western blot
HAP1 parental or HAP1 MTH1KO cell lines were grown as described in section 2.1 in 10 cm tissue culture dishes until approximately 75% confluency. Medium was aspirated and plates were rinsed with ice-cold PBS which was also removed by aspiration. With the plates on ice, 0.25 ml ice-cold RIPA buffer (150 mM Sodium Chloride, 50 mM Tris pH 8.0, 1% Triton X-100, 0.5% Sodium Deoxycholate, 0.1% Sodium Dodecyl Sulfate, 1 x EDTA-free HALT™ protease inhibitor cocktail) was added. Cells were scraped into a 1.5 ml centrifuge tube and the crude lysate was incubated on ice for 30 minutes with vortexing every 10 minutes for 10 seconds after which cell debris was pelleted by centrifugation at 14,000 x g for 10 minutes at 4°C. The whole cell lysate was removed to a new 1.5 ml pre-chilled centrifuge tube and protein was quantitated by Bio-Rad microassay protein assay according to manufacturer’s protocol. Whole cell lysates were stored at −20°C until further use.
For tissue lysate preparation, tissue (10 mg) was dissected as described above in section 2.3 and then 0.5 ml of ice-cold RIPA buffer was added. Tissue was homogenized on ice using a Tekmar Tissumizer for 2 × 10 sec bursts. Homogeneized samples were centrifuged at 3,000 x g at 4°C to reduce foam and then rotated for 10 min at 4°C. Samples were sonicated at 30% power for 10 sec on and 10 sec off on ice for 1 min with a Virtus Virsonic sonicator followed by rotation for 2 hr at 4°C. Whole cell lysates were centrifuged at 14,000 x g for 20 min at 4°C and supernatant containing protein was transferred to a fresh pre-chilled 1.5 ml microcentrifuge tube. When necessary, samples were concentrated by filtration through Amicon 3K filters for 30 minutes at 4°C, followed by inversion for collection into a fresh pre-chilled microcentrifuge tube. Protein was quantitated as described for cell lysates by Bio-Rad microassay protein assay and the whole cell lysates were stored at −20°C until further use.
2.5. Preparation of RNA from human tissue for RT-qPCR
Tissue (20 mg) was dissected as described above in section 2.3 and RNA was prepared using the Qiagen DNA/RNA/miRNA Universal kit according to manufacturer’s protocol. Purified RNA was quantitated by Nanodrop and stored at −80°C until use.
2.6. ARGO Assay to quantify MTH1-specific 8-oxodGTPase enzymatic activity
The ARGO assay is based on the ability of MTH1 to enzymatically cleave a previously described chimeric two-headed dinucleotide probe combining 8-oxo-dGTP and ATP into 8-oxo-dGMP and ATP, the latter which can be measured by standard luciferase assay. ATP-depleted protein lysates prepared as described in section 2.3 were diluted to 1 μg/μl in ice-cold hypotonic buffer and 1 μg lysate was assayed in triplicate in a 20 μl reaction volume containing 1 x NEB PNK buffer (New England Biolabs), 40 μM ARGO probe, 1 mM Na3V04 and either 1% DMSO or 20 μM S-crizotinib (ApexBio), an MTH1 inhibitor [26]. Each sample was assayed both with and without S-crizotinib to determine MTH1 specific activity. Samples were incubated at 30°C for 1 hour to allow the endogenous MTH1 to cleave the ARGO probe and then placed on ice. Recombinant MTH1 (Abcam ab99390) was used to generate a standard curve and a reaction containing all reagents except lysate or recombinant MTH1 was used to determine background level of the probe. Both the standard curve and background reactions contained an equivalent amount of DMSO in the place of S-crizotinib. The resultant cleaved ATP was measured by assaying 5 μl of each reaction in a 100 μl reaction containing Firefly Luciferase and luciferin using the Invitrogen ATP Determination Kit according to manufacturer’s protocol in an opaque white flat-bottom 96-well microtiter plate. Reactions and substrate were mixed quickly on ice and then read at 1 min intervals for a total of 15 minutes at 570 nM on a Molecular Devices Spectramax L luminometer with an integration time of 0.5 sec, MaxRange sensitivity and a 5 sec shake before the first read. The peak value of unknowns, standards and background fell between 6–9 minutes dependent on the time it took to add the luciferase reagents to the reaction before starting the instrument read. Background was subtracted from all unknown and MTH1 standard readings and the time of data collection for unknowns was determined by when the recombinant MTH1 standard reactions peaked. In general, all peak values for unknowns fell within a minute of the peak of the MTH1 standards. Specific MTH1 activity was calculated by subtracting the peak value of the reaction containing S-crizotinib from the peak value of the reaction without S-crizotinib. Linear regression of the peak values for the MTH1 standards was used to determine the equation of the line and to calculate the amount of functional MTH1 activity present in tissue lysates. Data is presented in pg MTH1-specific activity/1 μg lysate. All data presented are derived from at least three independent experiments each performed in triplicate.
2.7. Western Blot assay of MTH1 in colon tissue RIPA protein lysates
Whole cell lysates from cells (30 ug) or from colon tumor and matched normal tissues (150 μg) prepared in RIPA buffer as described above in section 2.4 were resolved along with Bio-Rad Precision Plus Protein WesternC prestained marker on a NuPAGE (Invitrogen) 4–12% 1.5 mm x 10 well Bis-Tris gel using MOPS buffer for 50 min at 200 V. Protein was transferred to PVDF for 1 hr at 30 V. Membranes were blocked with 10% w/v nonfat milk powder in 1 x PBS containing 0.1% Tween-20 (PBST buffer) for 15 min at room temperature after which they were cut according to molecular weight and incubated overnight at 4°C with 1° antibody, either a 1:5000 dilution of anti-alpha-actinin (Millipore MAB1682) as an internal loading control or with a 1:1000 dilution of anti-MTH1 (Novus NB100–109). All antibody dilutions were made in 1 x PBST buffer containing 5% w/v nonfat milk powder. Membranes were washed 3 × 15 sec, followed by 2 × 10 min in 1 x PBST buffer after which they were incubated for 1 hr at RT with a 1:10,000 dilution of 2° antibody (Horseradish peroxide [HRP]-labeled-Goat anti-mouse IgG for alpha-actinin; HRP-labeled-Donkey anti-rabbit IgG for MTH1). Bio-Rad Precision Protein™ StrepTactin-HRP conjugate was added at 1:10,000 dilution to visualize the WesternC protein markers. After washing as described above, protein-antibody complexes were detected using WesternBright™ ECL (Advansta) and imaged on a Bio-Rad ChemiDoc MP imager. Quantitation of bands was determined using Bio-Rad Image Lab software. All samples were run at least twice on Western blot with a representative blot shown.
2.8. cDNA synthesis and RT-qPCR
RNA (1 μg) from tumor and matched normal tissues prepared as described in section 2.4 was reverse transcribed into cDNA using the Qiagen Quantitect RT kit according to manufacturer’s protocol. RT-qPCR was performed in triplicate in a 5 μl reaction using Maxima® Probe/ROX qPCR Master Mix (Fermentas) with 0.05 μg of cDNA and MTH1 specific primers (MTH1 Forward: CTCAGCGAGTTCTCCTGG; MTH1 Reverse: GGAGTGGAAACCAGTAGCTGTC) or GAPDH specific primers (GAPDH Forward: AGCCACATCGCTCAGACAC; GAPDH Reverse: GCCCAATACGACCAAATCC) as an internal control. Samples were run on an ABI QuantStudio 12K Flex instrument using 1 cycle at 50°C for 2 min, 95°C for 10 min followed by 40 cycles of 95°C for 15 sec, 60°C for 1 min and a melt curve of 95°C for 15 sec, 60°C for 1 min, 95°C for 15 sec. Data from 3 separate experiments were analyzed using the delta Ct method and the mean from these experiments is reported.
2.9. Statistical analysis
All results were confirmed in a least 3 independent experiments and data is presented as mean plus standard deviation except for Western blot which was run a minimum of 2 times with representative blots presented. When two groups were compared, Student’s t-test was applied except in the comparison of Western blot to ARGO assay, in which case a Pearson’s Correlation test was used.
3. Results
3.1. The ARGO assay measures MTH1 functional activity in cells and tissue
MTH1 hydrolyzes 8-oxodGTP by cleaving between the α and ß phosphates to release pyrophosphate. This knowledge was used to design the ARGO probe, a specific reporter to measure MTH1 functional activity, by appending adenosine monophosphate to the terminal phosphate group of 8-oxodGTP (Fig. 1) [25]. Upon cleavage by MTH1, the leaving group in this reaction is ATP which can be readily measured by commercially available ATP-determination kits using luciferase and luciferin. The specificity of the ARGO assay arises from a combination of the probe structure and the assay itself, which includes measures to remove background from other sources including endogenous ATP and enzymes other than MTH1 that might accept the probe as a substrate. Specifically, endogenous ATP that could react with luciferase and luciferin, is removed from the cells by repeated centrifugation through filtration units with a 3 kDa molecular weight cut-off that allows ATP to pass through while retaining larger proteins including MTH1. In addition, to avoid interference from non-MTH1 background in cells and tissues, the assay was carefully designed to measure signal distinct from background by using an MTH1-specific inhibitor (S-crizotinib). As demonstrated by a direct-binding isothermal titration calorimetry assay and pyrophosphate release assay, S-crizotinib is a low nanomolar inhibitor of MTH1[26]. The assay measures the difference in ARGO-generated signal with and without a saturating concentration of inhibitor, and this difference amounts to the MTH1-specific signal. To further validate the specificity of the ARGO assay for measuring MTH1 activity, we obtained two commercially available MTH1 KO cell lines and used the ARGO assay to measure MTH1-specific activity as compared to a parental line. Western blot was performed to confirm that there was no MTH1 protein expression in the KO cell lines as compared to the parental line with recombinant MTH1 protein used as a positive control and α-actinin used as an internal control (Fig. 2A). In Fig. 2B, the ARGO assay was performed in the parental cell line and the two MTH1 KO cell lines in the absence (Total signal) and presence (Other signal) of 20 uM S-crizotinib and the differences in ARGO-generated signal were calculated (MTH1-specific activity). Using the equation derived from linear regression of a recombinant MTH1 standard curve, the pg amount of MTH1-specific activity per 1 μg of protein lysate was determined. There was an approximate 3-fold difference in signal with and without inhibitor in the parental cell line indicating the presence of a significant amount of MTH1-specific activity in these cells. Contrarily, there was no significant difference in ARGO-generated signal with and without S-crizotinib in either of the MTH1 KO cell lines showing that these cell lines were indeed void of MTH1-specific activity. This provides evidence that the design of the ARGO assay can distinguish MTH1-specific activity from other background sources present in cells or tissues that can cleave the ARGO probe and that this assay can be used to determine quantitative measurements of MTH1 activity.
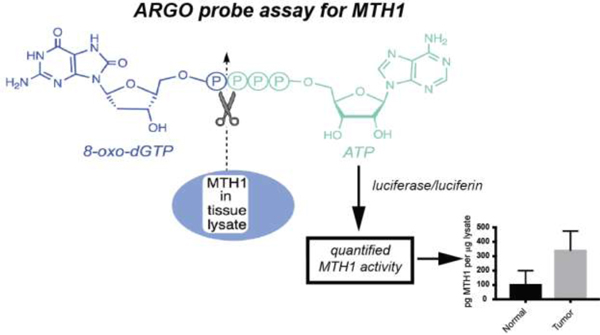
Fig. 1. The ARGO probe assay allows quantification of functional MTH1 enzyme activity in tissue lysates.

The chimeric ARGO probe combines 8-oxodGTP (shown in blue) with ATP (shown in green). MTH1 repair enzyme from tissue lysates cleaves the probe, releasing ATP, which can then trigger luminescence signals via firefly luciferase and luciferin. MTH1-specific activity is determined by assaying lysates with and without the MTH1 inhibitor S-crizotinib and subtracting the activity that remains in the presence of inhibitor from the total 8-oxodGTPase activity. Activity is quantified in pg per μg lysate by running a standard curve with recombinant MTH1 enzyme.
Fig. 2. The ARGO assay measures MTH1-specific 8-oxodGTPase activity.

A. Western blot of MTH1in Hap1 parental cells and two MTH1 KO cell lines demonstrates that there is no MTH1 protein expression in the KO cell lines. Recombinant MTH1 and α-actinin were used as positive and internal controls, respectively. B. ARGO assay showing Total signal (minus S-crizotinib), Other signal (plus S-crizotinib) and MTH1-specific 8-oxodGTPase activity (‘Total’– ‘Other’). No significant differences between ‘Total signal’ and ‘Other signal’ in the MTH1 KO cell lines indicate that in the absence of MTH1 expression, no MTH1-specific 8-oxodGTPase activity is detected by the ARGO assay. Green arrows highlight that there is no significant MTH1-specific 8-oxodGTPase activity in the MTH1 KO cell lines (p-values of .532 and .292 respectively, when compared by two-tailed Student’s T-test to zero).
3.2. MTH1 activity is higher in cancers of the colon, lung and pancreas compared to matched normal tissue
Although previous studies have shown that MTH1 is often increased in a variety of cancer cells compared to normal cells, the methods of measuring this increase have been indirect metrics such as mRNA or protein levels or pyrophosphate release assays which suffer from low sensitivity and selectivity as pyrophosphate formation is not specific to this enzyme pathway. With the development of the ARGO assay, we have the ability to measure functional enzyme activity of MTH1 in tissues. Flash-frozen CRC, NSCLC and PDAC tissues were obtained along with paired normal tissues taken from areas adjacent to the tumor and the ARGO assay was used to determine the pg amount of MTH-specific 8-oxodGTPase activity in 1 μg of protein lysate for both normal and tumor tissue within each pair (Fig. 3).
Fig. 3. MTH1-specific activity is higher in cancer than normal tissue in A) colon, B) lung and C) pancreas.

MTH1-specific activity was quantified by ARGO assay and is expressed as pg per μg lysate as calculated using linear regression of a standard curve of recombinant human MTH1 with R2 ≥ 0.9900. Pairs are identified by colored symbols as indicated in the legend with connecting lines to show the pair-wise change in MTH1. The fold increase in MTH1 activity based on group means for normal and tumor are listed above each graph along with the associated p-values. Individual pairs also showed a significant (p< .05) increase in MTH1 activity in tumor except for one pair in each tissue type that had equal amounts of MTH1 in normal and tumor tissue (indicated with fuchsia triangles). For pancreas, normal samples 6, 90 and 95 had undetectable MTH1 activity and are graphed at zero on the y-axis for the purpose of this figure. Each tissue pair was tested in triplicate for a minimum of 3 independent experiments from which the mean of MTH1 quantity was determined. Student’s t-test was used to determine significance.
For colon tissue (Fig. 3A), 9/10 (90%) pairs had more functional MTH1-specific activity in tumor tissue compared to matched normal tissue. Taken as a whole, this group had 5.5-fold higher MTH1-specific activity in tumor vs normal tissue (T/N) (p= .0009) with individual pairs ranging from no change for pair 13 (0.93-fold; p= .501; fuchsia triangle) to a high of 13.7-fold (p= .018) for pair 4 (pink circle). Actual amounts of MTH1 ranged from 29 to 307 pg/μg tissue lysate in normal colon and 184 to 569 pg/μg tissue lysate for tumor tissue. Interestingly, the normal tissue from pair 13 which had a T/N ratio of 0.93 had the highest amount of MTH1 among the normal samples which was more in line with the range seen for the tumor tissues and therefore, explains why there was no significant increase seen in its matched tumor tissue. This might indicate that an underlying issue such as high ROS is inducing higher levels of MTH1 in this normal tissue.
Similar results were obtained for lung (Fig. 3B) and pancreas (Fig. 3C) with 10/11 (91%) and 7/8 (87.5%) pairs respectively having more MTH1-specific activity in tumor compared to normal. Lung had a considerably higher overall fold T/N of MTH1-specific activity of 8.2 (p= .0033) compared to colon, while pancreas had 4.8-fold T/N (p=.0017). For lung tissue, the fold T/N for individual pairs ranged from no change for pair 4 (0.77-fold; p= .582; fuschia triangle) to 22.1-fold for pair 13 (p= .043; green circle). Amounts of MTH1 activity in normal lung tissue ranged from 10.1 to 46.1 pg/μg tissue lysate while the range in lung tumor was from 28.1 to 579.6 pg/μg tissue lysate. As in colon, the normal tissue from Pair 4 that did not show an increase in MTH1-specific activity in tumor had one of the highest amounts of MTH1 activity (36.68 pg/μg tissue lysate) compared to the other normal tissues. Only the normal tissue from pair 11 (blue hexagon) had a higher amount of MTH1-specific activity.
In PDAC, three tumor samples had matched normal in which no MTH1-specific activity was detectable [pairs 6 (blue square), 90 (green triangle) and 95 (red circle)] and therefore, a fold T/N could not be calculated for these pairs. For the remaining PDAC pairs, T/N MTH1-specific activity ranged from unchanged for pair 94 (0.99-fold; p= .977; fuchsia triangle) to 35.0-fold for pair 91 (p= .01; purple diamond). Pair 91 normal tissue had very low amounts of MTH1-specific activity (mean= 0.73 pg/μg lysate), thus accounting for this high fold difference with tumor tissue. MTH1-specific activity ranged from 0.73 to 35.17 pg/μg tissue lysate in the pancreatic normal samples and from 25.13 to 93.82 pg/μg tissue lysate in the PDAC samples.
3.3. Decreased tumor percentage could lead to an underestimation of MTH1-specific activity in tumor samples
Because we did not have access to histological slides for the NSCLC and PDAC tumor samples to determine tumor percentage, we conducted a proof-of-principle experiment to see what effect a decreased tumor percentage would have on measuring MTH1-specific activity. We performed ARGO assays on two matched normal/tumor sample pairs each from colon, NSCLC and PDAC in which we progressively added in more of the normal lysate while proportionally decreasing the amount of tumor lysate, keeping the total lysate at 1 ug per assay (Fig 4). Ratios were 100%, 75%, 50%, 25% and 0% tumor lysate with 0%, 25%, 50%, 75% and 100% normal lysate, respectively. In each case, diluting tumor lysate with normal lysate lead to a decrease in the amount of MTH1-specific activity measured. This suggests that in tumor samples with lower tumor percentage, the ARGO assay would likely underestimate the amount of MTH1-specific activity, further supporting that there is higher MTH1 enzymatic activity in these tumor cells than in normal cells.
Fig. 4. Decreasing tumor percentage results in an underestimation of MTH1-specific activity.

MTH1-specific activity was measured by ARGO assay in tumor samples diluted with increasing amounts of normal lysate from 0–100%. Amounts of MTH1-specific activity were normalized to the 100% tumor only sample.
3.4. ARGO assay is a more reliable method to determine functional MTH1 activity compared to mRNA and protein expression
Previous studies have used mRNA or protein expression to determine the level of MTH1 in cells and tissues [26–33], but it is unclear whether these are accurate surrogate measures of MTH1 functional activity. Having used the newly-developed ARGO assay to measure enzymatic MTH1 function in normal and tumor tissues, we compared these results to both mRNA and protein expression as measured by RT-qPCR and Western blot in our colon tissue set (Fig. 5A, B). All three methods show that overall there is a significant increase in MTH1 levels in tumor tissue compared to normal tissue with RT-qPCR, Western blot and ARGO assay showing a mean increase of 2.6-fold (p= .01), 6.1-fold (p< .0001) and 5.5-fold (p= .0009), respectively. However, the fold T/N increase for MTH1 mRNA is significantly lower than for MTH1 measured by Western blot (p= .004) or ARGO assay (p= .036). There is no significant difference in the mean fold T/N increase in MTH1 between Western blot and ARGO assay (p= .693). Upon examining the data from the perspective of individual pairs, a few demonstrate good correlation across all three methodologies [Pairs 10 (teal hexagon) and 12 (purple diamond)], but most do not, with RT-qPCR being the method that is least correlative. Pearson correlation comparing RT-qPCR to Western blot or ARGO assay yields R values of −0.2118 and 0.0412, respectively, indicating that there is no linear relationship between MTH1 mRNA expression and MTH1 expression measured by either Western blot or ARGO assay (data not shown). Conversely, a Pearson correlation comparing MTH1 measured by Western blot and ARGO assay gave an R value of 0.8523 with a p value of .0017 indicating that there is a significant positive correlation between these two methods as an R value between 0.7 and 1 is considered a strong correlation (Fig. 5C). Despite this positive correlation, there are some pairs that fall outside of the 95% confidence interval, specifically pairs 2, 7, 11 and 13 with these latter two pairs falling further outside this interval. Pair 13 is particularly notable in that there is clearly more MTH1 protein seen in the tumor tissue compared to normal tissue in the Western blot (Fig. 5B), yet there is no change in the T/N ratio of MTH1 activity by ARGO assay. For pairs 2, 7 and 11, both Western blot and ARGO assay show higher amounts of MTH1 protein or MTH1 activity in tumor, but the amplitude of the increase differs between the two methods. It is also interesting to note that 3 out of 4 (75%) of these pairs that are less correlative have a lower fold increase in MTH1 enzymatic activity measured by ARGO assay compared to the fold increase seen in Western blot. This indicates that Western blot, a method that measures all MTH1 protein expression, whether it is enzymatically active or not, may have a tendency to overestimate MTH1 activity in certain tumor samples and types when used as a surrogate.
Fig. 5. MTH1 enzyme activity measured by ARGO assay correlates with MTH1 protein expression but not with mRNA expression in colon tissue.

A) Comparison of MTH1 mRNA expression by RT-qPCR, protein expression by Western blot and functional MTH1 activity by ARGO assay. Data for each technique is expressed as Fold MTH1 (Tumor/Normal) to allow for comparisons. Pairs are indicated by colored symbols as shown in the legend. RT-qPCR and ARGO assays were tested in triplicate for a minimum of 3 independent experiments. Western blot was repeated a minimum of 2 times for each pair. Group mean of T/N fold and p-value is listed above each technique. An asterix denotes a significant difference for the group means between the indicated techniques (RT-qPCR vs Western blot or ARGO assay). B) Representative Western blot of colon pairs. Not all blots displayed were run on the same gel, but each tissue pair was run at least twice. Alpha-actinin was used as an internal loading control to normalize MTH1. C) Pearson correlation of Western blot and ARGO assay. The R value and p value of the correlation is listed above the graph. Dotted lines denote the 95% CI.
4. Discussion
The role of MTH1 in cancer and whether it is a promising anti-cancer target for small molecule inhibitors remains disputed with often contradictory results confounded by the use of different cell lines, distinct methods of inhibiting MTH1 (small molecule inhibitors, knock-down or KO) and varied methodologies for measuring the effect of these interventions on MTH1 function (reviewed in [24]). To provide a more accurate measure of MTH1 in tumor cells, it is imperative to have a reliable, easy to use and sensitive assay to measure MTH1 functional activity that can be utilized to yield more consistent results across different research groups and from one model system to the next. We have shown herein that the ARGO assay can be used to directly measure 8-oxodGTP cleavage in a set of CRC, NSCLC and PDAC tissues alongside patient-matched normal tissue, thereby giving a readout of functional MTH1 activity in these tissues. We found that there is significantly higher MTH1 activity in tumors of the colon, lung and pancreas compared with patient-matched normal tissue, agreeing for the most part with the conclusion of previous studies in which MTH1 activity was measured by surrogate methods [34] [15]. However, in certain cases, our study goes further. For example, in lung, Speina et al. measured total 8-oxodGTPase activity in lysates from NSCLC and matched normal tissue using HPLC measurements of 8-oxodGMP and found that while 8-oxodGTPase activity is higher in tumor than normal tissues as a group, there is no correlation when examining individual normal/tumor tissue pairs [35]. In addition, their study did not determine MTH1-specific activity, but rather measured total 8-oxodGTPase activity which could include cleavage by enzymes other than MTH1. In contrast, we determined MTH1-specific 8-oxodGTPase activity and found that there are significantly higher levels of functional MTH1 activity in NSCLC compared to normal tissue not only when viewed overall as groups, but also in 11/12 (92%) of the matched patient pairs. In another study in NSCLC, Fujishita et al. used immunohistochemical staining to show that higher MTH1 expression is associated with worse prognosis, but unlike in our study, no comparisons were made to patient-matched normal tissues [21]. Furthermore, studies of MTH1 in PDAC have been limited to cell lines with no comparison to normal pancreatic ductal cells, thus showing only that there is MTH1 mRNA expression in MIAPaCa-2 and Panc-1 cells [33]. Therefore, our data showing that functional MTH1 activity is significantly higher in 7/8 (88%) patient-matched PDAC/normal tissue pairs is a novel finding.
A strength of the ARGO assay is its specificity for measuring MTH1-derived 8-oxodGTPase activity through the use of an MTH1 inhibitor. One possible source of overlapping signal in the ARGO assay would be if the MTH1 inhibitor used here also acted as a potent inhibitor of a background-generating enzyme; one hypothetical candidate is NUDT15 (also known as MTH2). We consider this to be unlikely, as recent structural studies of NUDT15 revealed several differences in the active site relative to MTH1, suggesting that it would be very improbable that MTH1 inhibitors such as S-crizotinib would be able to bind potently [36]. A second NUDIX-family enzyme, NUDT18 (also known as MTH3), uses 8-oxo-dG-diphosphate as its preferred substrate, and has very low activity with 8-oxodGTP [37]. Given this distinctly different substrate preference, it is also unlikely that MTH1 inhibitors would also inhibit this enzyme. In addition, the complete lack of detectable MTH1-specific 8-oxodGTPase activity in the HAP1 MTH1 KO cell lines (Fig. 2) convincingly demonstrates the specificity of the ARGO assay. Thus, the ARGO assay is likely reporting primarily on MTH1 activity in these clinical tissues; however, more work will be needed to rule out contributions from the aforementioned related enzymes and other sources.
Before the development of the ARGO assay, functional assays of MTH1 activity were restricted to low sensitivity or cumbersome assays such as HPLC measurement of nucleotide cleavage of 8-oxodGTP to 8-oxodGMP and colorimetric or luminescent measurement of pyrophosphate that could not reliably be used in native tissues. Because of this, researchers generally have used RT-qPCR or Western blot as an indirect way to measure MTH1 activity in tissues and even cells. Our data shows that although MTH1 mRNA expression is higher in CRC than normal tissue, the fold increase is significantly lower than seen with Western blot or ARGO assay. This poorer correlation between MTH1 mRNA measured by RT-qPCR and either Western blot or ARGO assay, at least in this set of CRC samples, suggests that mRNA expression by itself may not be the most accurate surrogate for MTH1 activity. Furthermore, although there is a strong positive correlation between MTH1 levels detected by Western blot and ARGO assay, some pairs are more poorly correlated. Our data shows that of the 4 pairs that fell outside of the 95% CI of the correlation, 3 of these pairs have a higher fold increase in MTH1 measured by Western blot than that measured by ARGO assay. This suggests that measuring total protein expression as a surrogate for function can in certain cases lead to an overestimation and that the ARGO assay gives a more accurate profile of MTH1 enzyme activity in cancer.
While the ability to measure enzymatically active MTH1 is the defining hallmark of the ARGO assay over methods that do not assess function, this assay also offers other benefits including a low requirement for input material. In our comparison to the ARGO assay in CRC and normal tissue, it was necessary to resolve 150 μg of total cellular lysate on Western blots in order to visualize MTH1 protein in some of the normal tissues in contrast to needing only 1 μg of total cellular lysate for the ARGO assay. Even functional assessment of MTH1 activity by HPLC measurement of 8-oxodGTP cleavage into 8-oxodGMP requires significantly more lysate. For example, a study in NSCLC required 0.5 to 1000 μg of protein per assay and the researchers could only assess 33 out of 56 pairs of matched tumor/normal tissues because they had already exhausted their lysates in other experiments [35]. Patient samples are usually very limited and being able to significantly decrease the amount of lysate needed for measurement means that more varied assessments can be conducted with these samples. Another benefit of the ARGO assay is the short time required as compared with Western blot and RT-qPCR measurements. ARGO assay can be completed in under 2 hours from start to finish, whereas Western blot typically requires several hours to run and transfer the gel and up to an additional day to incubate with antibodies and then detect bands. RT-qPCR, necessitating 3–4 hours to complete reverse transcription and qPCR, is more comparable to the ARGO assay in terms of total run time, but requires many costly reagents including a reverse transcription kit, gene-specific primers and SYBR green or TaqMan reagents. A final benefit of ARGO is that for cell line-based experiments, the ARGO assay is also amenable to high-throughput workflows to enable library screening of compounds or the assessment of many cell perturbation conditions at one time compared to the time-intensive Western blot.
Another interesting aspect of the ARGO assay is that it measures both total and MTH1-specific 8-oxodGTPase activity. Supplemental Fig 2 shows that in colon tissue, there is substantial residual activity in both normal and tumor tissue after the addition of 20 μM S-crizotinib, suggesting that there may be other, as yet unidentified, enzymes present in the tissues that are capable of hydrolyzing the ARGO probe, and by inference, 8-oxodGTP. This is also true for the lung and pancreatic tissues pairs (data not shown). It could be argued that in such cases S-Crizotinib does not completely inhibit MTH1, but during the original characterization of the ARGO probe, the IC50 of S-Crizotinib with recombinant MTH1 enzyme was determined to be 220 nM, and at 20 μM, all MTH1-specific 8-oxodGTPase activity is abolished [25]. Using HPLC to measure 8-oxodGMP in NSCLC as a measurement of MTH1 activity, Speina et al. also found that while 8-oxodGTPase activity increased in tumors, there was no inverse correlation between this activity and levels of 8-oxoGTP in either normal or tumor tissues, as might be expected if MTH1 was solely responsible for maintaining the level of 8-oxoGTP in DNA [35]. In another observation from our data, total 8-oxodGTPase activity is higher in tumor than in normal tissues for all colon pairs, but there is a higher proportion of residual activity in normal tissues that cannot be inhibited by S-crizotinib. This might suggest that MTH1-independent mechanisms of 8-oxodGTPase activity are more prevalent in normal tissues compared to tumor tissues.
In summary, we demonstrate that increased MTH1 activity is a marker of cancer in CRC, NSCLC and PDAC. We have addressed a deficit in the area of reliable MTH1 enzymatic activity measurement with the ARGO assay that offers distinct advantages over other methodologies including ease of use, low input of starting material, and a sensitive measure of functional enzymatic activity versus protein levels. Due to the intense interest in inhibiting MTH1 as an anti-cancer therapy, the adoption of the ARGO assay across the field may help clarify some of the contradictory results reported in different models to date and tease out whether these cell-specific and inhibitor-specific effects are real or due to the use of different methodologies to measure MTH1. Use of the ARGO assay can also be used to decipher whether MTH1 has a role in normal cells in maintaining genomic stability and preventing tumorigenesis.
Supplementary Material
Highlights.
ARGO assay uses a chimeric substrate for luminescent measurement of functional MTH1 activity
MTH1-specific 8-oxodGTPase activity is higher in CRC, NSCLC and PDAC compared to normal tissue
In CRC, MTH1 activity measured by ARGO assay correlates to MTH1 protein level but not mRNA expression
Acknowledgements
We would like to thank Carlos Juan Suarez, Clinical Assistant Professor in the Pathology Department at the Stanford University School of Medicine, for performing the pathological determination of tumor percent for colon tumors.
Funding
This work was supported by the National Institutes of Health/National Cancer Institute grants R01CA217809 (ETK and JMF) and R01CA175086 (PR) and by Sylvester Comprehensive Cancer Center research support funding (PR).
Abbreviations
- ROS
Reactive oxygen species
- MTH1
MutT Homolog 1
- ARGO
ATP-releasing guanine-oxidized
- CRC
Colorectal cancer
- NSCLC
Non-small cell lung cancer
- PDAC
Pancreatic ductal adenocarcinoma
- 8-oxoG
8-oxo-7,8-dihydroguanine
- SSBs
Single-stranded DNA breaks
- OGG1
8-oxoguanine DNA Glycosylase
- KO
Knock-out
- H&E
Haemotoxylin and Eosin
- PBST buffer
PBS containing 0.1% Tween-20
Footnotes
Conflict of Interests
The authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D and Weinberg RA, Hallmarks of cancer: the next generation. Cell, 2011. 144(5): p. 646–74. [DOI] [PubMed] [Google Scholar]
- 2.Loeb LA, Human cancers express mutator phenotypes: origin, consequences and targeting. Nat Rev Cancer, 2011. 11(6): p. 450–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gorrini C, Harris IS, and Mak TW, Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov, 2013. 12(12): p. 931–47. [DOI] [PubMed] [Google Scholar]
- 4.Kryston TB, et al. , Role of oxidative stress and DNA damage in human carcinogenesis. Mutat Res, 2011. 711(1–2): p. 193–201. [DOI] [PubMed] [Google Scholar]
- 5.Panieri E and Santoro MM, ROS homeostasis and metabolism: a dangerous liason in cancer cells. Cell Death Dis, 2016. 7(6): p. e2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rai P and Sobol RW, Mechanisms of MTH1 inhibition-induced DNA strand breaks: The slippery slope from the oxidized nucleotide pool to genotoxic damage. DNA Repair (Amst), 2019. 77: p. 18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Son Y, et al. , Reactive oxygen species in the activation of MAP kinases. Methods Enzymol, 2013. 528: p. 27–48. [DOI] [PubMed] [Google Scholar]
- 8.Taguchi K, Motohashi H, and Yamamoto M, Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Genes Cells, 2011. 16(2): p. 123–40. [DOI] [PubMed] [Google Scholar]
- 9.Moriya M, et al. , Site-specific mutagenesis using a gapped duplex vector: a study of translesion synthesis past 8-oxodeoxyguanosine in E. coli. Mutat Res, 1991. 254(3): p. 281–8. [DOI] [PubMed] [Google Scholar]
- 10.Tubbs A and Nussenzweig A, Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell, 2017. 168(4): p. 644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayakawa H, et al. , Generation and elimination of 8-oxo-7,8-dihydro-2’-deoxyguanosine 5’-triphosphate, a mutagenic substrate for DNA synthesis, in human cells. Biochemistry, 1995. 34(1): p. 89–95. [DOI] [PubMed] [Google Scholar]
- 12.Sakumi K, et al. , Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J Biol Chem, 1993. 268(31): p. 23524–30. [PubMed] [Google Scholar]
- 13.Giribaldi MG, et al. , MTH1 expression is required for effective transformation by oncogenic HRAS. Oncotarget, 2015, 6(13): p. 11519–11529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abbas HHK, et al. , MTH1 deficiency selectively increases non-cytotoxic oxidative DNA damage in lung cancer cells: more bad news than good? BMC Cancer, 2018. 18(1): p. 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kumagae Y, et al. , Overexpression of MTH1 and OGG1 proteins in ulcerative colitis-associated carcinogenesis. Oncol Lett, 2018. 16(2): p. 1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Okamoto K, et al. , Overexpression of human mutT homologue gene messenger RNA in renal-cell carcinoma: evidence of persistent oxidative stress in cancer. Int J Cancer, 1996. 65(4): p. 437–41. [DOI] [PubMed] [Google Scholar]
- 17.Pompsch M, et al. , The presumed MTH1-inhibitor TH588 sensitizes colorectal carcinoma cells to ionizing radiation in hypoxia. BMC Cancer, 2018. 18(1): p. 1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qing X, et al. , Anticancer effect of (S)-crizotinib on osteosarcoma cells by targeting MTH1 and activating reactive oxygen species. Anticancer Drugs, 2018. 29(4): p. 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song WJ, et al. , Expression of Cytoplasmic 8-oxo-Gsn and MTH1 Correlates with Pathological Grading in Human Gastric Cancer. Asian Pac J Cancer Prev, 2015. 16(15): p. 6335–8. [DOI] [PubMed] [Google Scholar]
- 20.Wani G, Milo GE, and D’Ambrosio SM, Enhanced expression of the 8-oxo-7,8-dihydrodeoxyguanosine triphosphatase gene in human breast tumor cells. Cancer Lett, 1998. 125(1–2): p. 123–30. [DOI] [PubMed] [Google Scholar]
- 21.Fujishita T, et al. , Association of MTH1 expression with the tumor malignant potential and poor prognosis in patients with resected lung cancer. Lung Cancer, 2017. 109: p. 52–57. [DOI] [PubMed] [Google Scholar]
- 22.Li J, et al. , MutT-related proteins are novel progression and prognostic markers for colorectal cancer. Oncotarget, 2017. 8(62): p. 105714–105726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MTH1, A Phase I, Study on Tumors Inhibition, First in Human, First in Class. Available from: https://ClinicalTrials.gov/show/NCT03036228.
- 24.Samaranayake GJ, Huynh M, and Rai P, MTH1 as a Chemotherapeutic Target: The Elephant in the Room. Cancers (Basel), 2017. 9(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ji D, et al. , A Chimeric ATP-Linked Nucleotide Enables Luminescence Signaling of Damage Surveillance by MTH1, a Cancer Target. J Am Chem Soc, 2016. 138(29): p. 9005–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huber KV, et al. , Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature, 2014. 508(7495): p. 222–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dai X, et al. , (S)-crizotinib induces apoptosis in human non-small cell lung cancer cells by activating ROS independent of MTH1. J Exp Clin Cancer Res, 2017. 36(1): p. 120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellermann M, et al. , Novel Class of Potent and Cellularly Active Inhibitors Devalidates MTH1 as Broad-Spectrum Cancer Target. ACS Chem Biol, 2017. 12(8): p. 1986–1992. [DOI] [PubMed] [Google Scholar]
- 29.Flick KM, et al. , Grr1-dependent inactivation of Mth1 mediates glucose-induced dissociation of Rgt1 from HXT gene promoters. Mol Biol Cell, 2003. 14(8): p. 3230–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gad H, et al. , MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature, 2014. 508(7495): p. 215–21. [DOI] [PubMed] [Google Scholar]
- 31.Giribaldi MG, et al. , MTH1 expression is required for effective transformation by oncogenic HRAS. Oncotarget, 2015. 6(13): p. 11519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hashiguchi K, et al. , The roles of human MTH1, MTH2 and MTH3 proteins in maintaining genome stability under oxidative stress. Mutat Res, 2018. 808: p. 10–19. [DOI] [PubMed] [Google Scholar]
- 33.Ikejiri F, et al. , TH588, an MTH1 inhibitor, enhances phenethyl isothiocyanate-induced growth inhibition in pancreatic cancer cells. Oncol Lett, 2018. 15(3): p. 3240–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Obtulowicz T, et al. , Oxidative stress and 8-oxoguanine repair are enhanced in colon adenoma and carcinoma patients. Mutagenesis, 2010. 25(5): p. 463–71. [DOI] [PubMed] [Google Scholar]
- 35.Speina E, et al. , Contribution of hMTH1 to the maintenance of 8-oxoguanine levels in lung DNA of non-small-cell lung cancer patients. J Natl Cancer Inst, 2005. 97(5): p. 384–95. [DOI] [PubMed] [Google Scholar]
- 36.Carter M, et al. , Crystal structure, biochemical and cellular activities demonstrate separate functions of MTH1 and MTH2. Nat Commun, 2015. 6: p. 7871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Carreras-Puigvert J, et al. , A comprehensive structural, biochemical and biological profiling of the human NUDIX hydrolase family. Nat Commun, 2017. 8(1): p. 1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.