

Abstract
Heterobimetallic Mn/Fe proteins represent a new cofactor paradigm in bioinorganic chemistry and pose countless outstanding questions. The assembly of the active site defies common chemical convention by contradicting the Irving-Williams series, while the scope of reactivity remains unexplored. In this work, the assembly and C-H bond activation process in the Mn/Fe R2-like ligand-binding oxidase (R2lox) protein is investigated using a suite of biophysical techniques, including time-resolved optical spectroscopy, global kinetic modeling, X-ray crystallography, electron paramagnetic resonance spectroscopy, protein electrochemistry, and mass spectrometry. Selective metal binding is found to be under thermodynamic control, with the binding sites within the apo-protein exhibiting greater MnII affinity than FeII affinity. The comprehensive analysis of structure and reactivity of wild-type R2lox and targeted primary and secondary sphere mutants indicate that the efficiency of C-H bond activation directly correlates with the Mn/Fe cofactor reduction potentials and is inversely related to divalent metal binding affinity. These findings suggest the R2lox active site is precisely tuned for achieving both selective heterobimetallic binding and high levels of reactivity and offer a mechanism to examine the means by which proteins achieve appropriate metal incorporation.
Graphical Abstract
INTRODUCTION
Heterometallic protein cofactors are responsible for performing some of the most challenging chemical transformations found in nature, including water oxidation,1 oxygen reduction,2 hydrogen production,3 and nitrogen fixation.4 Biological oxidation reactions have typically been attributed to Fe- and Cu-containing cofactors, though the recent discovery of a new class of proteins indicates that Mn/Fe cofactors may perform similar chemistry.5–8 One such class is represented by the R2-like ligand-binding oxidase (R2lox) proteins (Figure 1). First identified a decade ago in Mycobacterium tuberculosis (Mt) as one of the 10 most upregulated proteins in virulent strains of Mt relative to vaccine strains,9 R2lox was initially annotated as a bimetallic ribonucleotide reductase (RNR) based on sequence homology. Upon cloning and expression of the protein, however, R2lox was found to lack RNR activity. Further characterization revealed a fatty acid coordinated directly to the metal center via a long, hydrophobic channel, another trait that is absent in RNR proteins. Instead, R2lox spontaneously assembles a heterobimetallic Mn/Fe cofactor both in vitro and when heterologously expressed in vivo,6 despite each metal being ligated by five oxygen atoms and one histidine-derived nitrogen atom in nearly identical geometries. However, the oxygenic ligands on site B in the reduced state derive solely from carboxylate groups, while site A has one open coordination site that is filled with a water ligand. This cofactor activates oxygen to generate a tyrosine-valine cross-link within its scaffold, representing the first known example of a Mn/Fe cofactor capable of performing two-electron oxidation reactions.6,8,10,11 Such two-electron chemistry is often seen in the highly valuable diiron oxidases, such as soluble methane monooxygenase (sMMO)12–16 and arylamine oxygenase,17–24 which catalyze the activation of inert bonds for methane oxidation and antibiotic synthesis, respectively. Therefore, as a representative member of the potentially equally valuable heterobimetallic oxidases, the study of R2lox—assembly, reactivity, and mechanism—is of particular interest.
Figure 1.
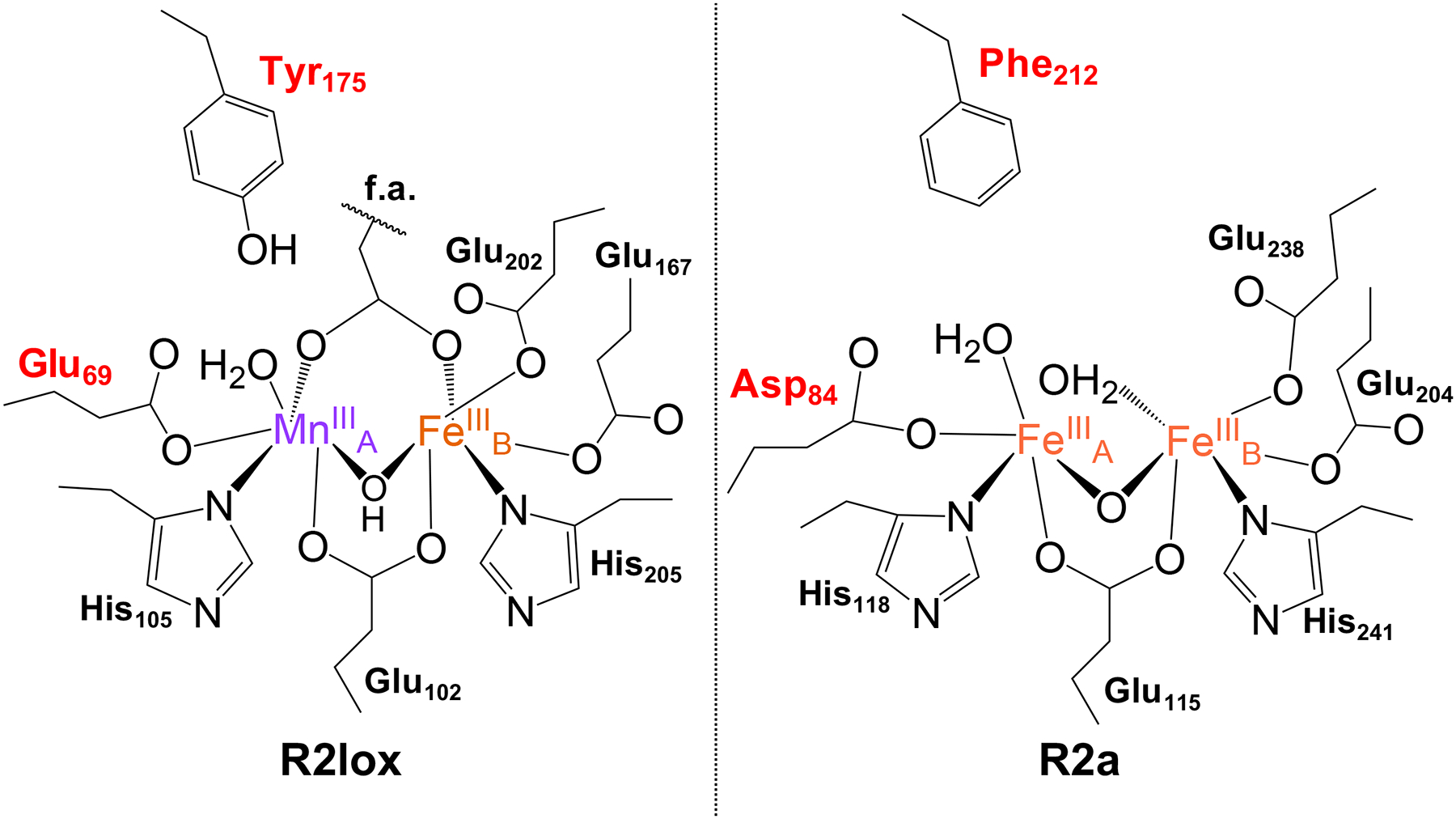
Comparison of the chemical structures of the active sites of aerobically reconstituted R2lox (left, GkR2loxI numbering) and class Ia RNR R2 (right, Ec numbering) proteins. Residues of interest for this study are indicated in red. f.a. = fatty acid ligand.
Heterobimetallic Mn/Fe cofactor assembly in R2lox presents a unique challenge, as the system exhibits behavior that runs contrary to the Irving-Williams (IW) series.25 Over 50 years ago, Irving and Williams formulated a series of empirical rules governing divalent metal binding, where KMn < KFe < KCo < KNi < KCu > KZn, using a number of diverse chelating ligands. Subsequent investigations have shown that this ordering arises from a combination of ligand covalency and ionic bonding, fundamental properties intimately tied to both the metal and ligand identities.26 Proteins, with their carefully constructed three-dimensional scaffolds, are suggested to be able to overcome the metal-binding preferences seen in the small-molecule realm, though the physical processes that enable this discrimination are unclear. Specifically, the underlying mechanism by which a protein such as R2lox selectively binds MnII over FeII in one site and the question of whether metal binding is under thermodynamic or kinetic control remain under investigation.27 Only slight differences are observed between the primary and secondary coordination environments of R2lox and the class Ic Mn/Fe RNR proteins (R2c) or canonical diiron bacterial multicomponent monooxygenase enzymes (BMMs).5,6,10,28,29 However, R2lox exhibits traits seen exclusively in each, i.e., heterobimetallic metalation and C-H bond oxidation, respectively, illustrating the importance of slight changes to the primary and secondary coordination environment in tuning active site properties. Therefore, examining structural differences around the metal binding site “A” across R2lox, RNRs, and the BMMs can provide a platform for investigating molecular control over cofactor assembly, as these distinctions are likely important for modulating cofactor reactivity.
Crystallographic studies suggest that the “B” site of R2lox (Figure 1) appears to obey the IW series; crystals obtained from R2lox metalated in solution using approximately 1:2:1 apo-R2lox:Mn:Fe30 as well as crystallized apo-R2lox metalated by soaking with high concentrations of metals (5 mM)10 have both shown almost exclusive occupation of the B site by FeII. On the other hand, site A shows an equal distribution of FeII and MnII in apo-R2lox crystals soaked with metals under anaerobic conditions,10 while crystals obtained aerobically, whether from metal soaking10 or crystallization of already metalated protein,30 indicate a selectivity for Mn over Fe in site A. Both observations are in direct contrast to predictions following the IW series. It has been proposed that the heterobimetallic cofactor forms as a result of two compounding interactions, which we investigate in this work.10,30–33 The first suggests that MnII cannot bind productively to site A if site B is unoccupied, where productive binding refers to the eventual formation of the “resting state” species, a MnIII(μ-OH)FeIII cofactor. This ordered binding of the two metals was proposed by noting that only the B site is crystallographically resolved in structures of apo-R2lox.10 The second level of control can either be thermodynamic, in which MnII binding to site A (KMnA) is favored over FeII binding to site B (KFeB), in spite of the nearly identical metal binding sites and in opposition to the IW series, or kinetic, where O2 binding to a Mn/Fe site in R2lox is faster than O2 binding to an Fe/Fe site. We note that in our hands, Fe/Fe R2lox cofactor formation is observed to be faster than Mn/Fe cofactor formation (Figure S1); thus, kinetics alone cannot explain the preferential assembly of a heterobimetallic core (Figure S2).
While previous efforts have investigated both the identity and quantification of the metalation state in R2lox, these data have all been collected on the already assembled, resting state of R2lox.10,30,33,34 In this work, data obtained using time-resolved methodologies provide direct insight into the effects of changing metal concentrations on Mn/Fe cofactor assembly, reporting on steps prior to O2 activation. These data provide a foundation upon which to experimentally investigate metal binding affinities and reactivity in R2lox, as well as the role of specific residues in the primary and secondary coordination spheres in modulating these properties, using time-resolved optical experiments, global kinetic modeling, structural characterization, electron paramagnetic resonance (EPR) spectroscopy, protein electrochemistry, and mass spectrometry. Gaining an understanding of the metal binding affinities of each site in R2lox and identifying influential structural elements help to clarify the mechanism underlying the observed selectivity. An inverse correlation between metal binding affinity and reactivity is observed, highlighting the importance of balancing these factors within naturally occurring and engineered protein systems. Resolving the molecular determinants underlying proper metalation has important implications for understanding metal selection in vivo for R2lox and related heterobimetallic proteins.35–39 Furthermore, this knowledge may provide insight into how and why related proteins select for specific cofactors in vivo, an important consideration in understanding how organisms may evolve finely tuned metalloproteins that can respond to dynamic metal availability and withstand conditions of environmental duress.
RESULTS
Prior work from our lab on the Mn/Fe R2lox cofactor assembly mechanism identified two temporally distinct intermediates with different optical profiles at 390 and 620 nm. The feature at 620 nm appears first (I1) and was postulated to derive from a putative Mn/Fe peroxo species, based on comparison to literature absorption profiles for Fe/Fe and Mn/Mn peroxo species; unfortunately, low absolute absorption values suggest this species only accumulates to single μM concentrations.27 The feature at 390 nm resembles the optical profile of the R2c MnIV/FeIV state.27,40 Based on this similarity and the observation that it forms after the postulated peroxo species at 620 nm, the 390 nm species was tentatively assigned to be a high-valent, Mn/Fe intermediate (I2). The lack of a solvent kinetic isotope effect was used to suggest that I2 was responsible for C-H bond activation to initiate cross-link formation. We also identified two new EPR-active species that were initially mapped to the two optical intermediates. This assignment has since been revisited in the context of additional R2lox mutants,41 and we are actively pursuing pulsed EPR studies for structural characterization. The MnIII(μ-OH)FeIII resting state of R2lox exhibits a featureless absorption spectrum from ~300 to 400 nm, characteristic of metal-(hydr)oxo charge-transfer transitions. The appearance of this final species is monitored at 320 nm, with the kinetics of formation clearly lagging behind the formation and decay of both optically resolved I1 and I2 intermediates.
Mn/Fe R2lox Assembly Is Differentially Impacted by Increasing Concentrations of MnII and FeII.
To investigate metal binding, formation of the wild-type (WT) Mn/Fe R2lox resting state was monitored using stopped-flow (SF) absorption spectroscopy at 320 nm as a function of increasing metal concentration. For each set of competition experiments, the amount of either MnII (Figure 2B) or FeII (Figure 2B, inset) added to apo-R2lox was varied, while the other metal was held at a single concentration. To mitigate Fe/Fe R2lox formation and selectively investigate Mn/Fe R2lox assembly, MnII competition experiments were performed using sub-stoichiometric levels of FeII (0.75 equiv), while the corresponding FeII competition experiments utilized an abbreviated FeII concentration series and slightly elevated MnII levels (2 equiv).30 In MnII competition experiments with 100 μM WT R2lox, the absorbance is seen to increase at earlier times when MnII is increased from 50 to 200 μM. However, at [MnII] ≥ 400 μM, the appearance of absorbance features at 320 nm is delayed, with the peak occurring at later times and with lower maximum intensity (Figure S3A).
Figure 2.
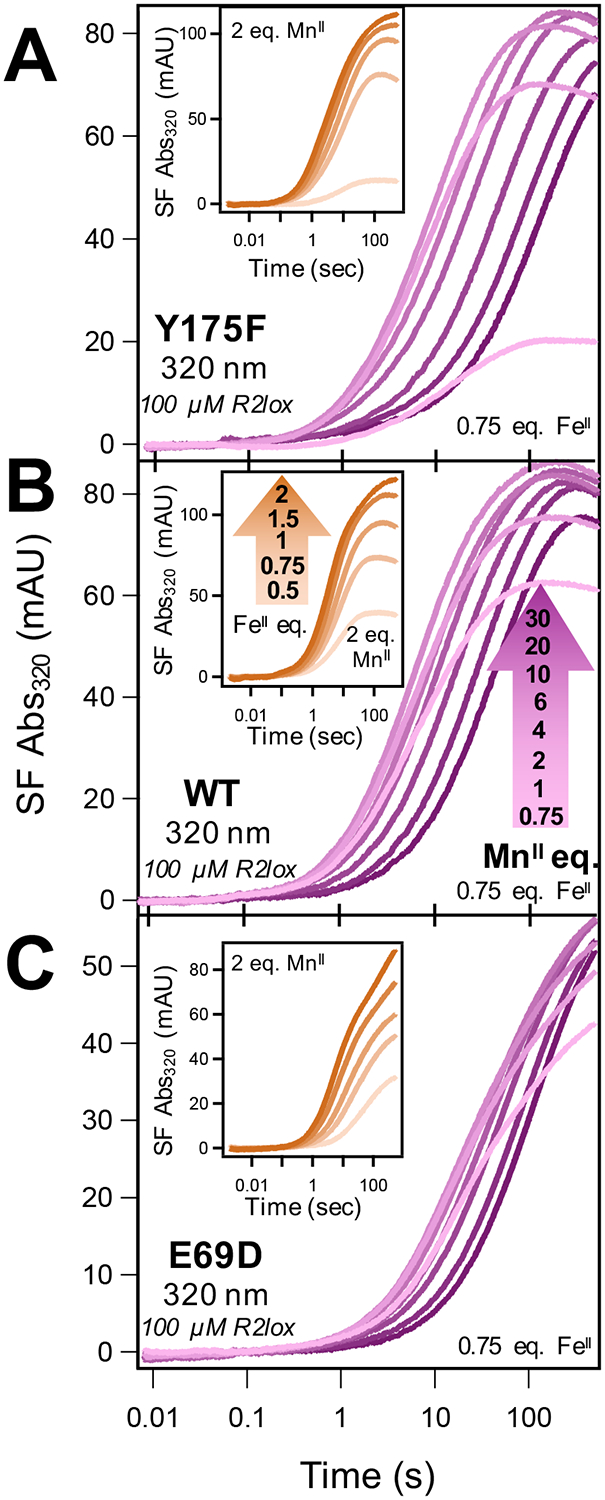
Stopped-flow (SF) absorption kinetic traces at 320 nm of Mn/Fe cofactor assembly in (A) Y175F, (B) WT, and (C) E69D R2lox as a function of metal concentration. FeII was held at 0.75 equiv of apo-R2lox and formation of the resting state monitored at 320 nm with increasing [MnII]. (Insets) Corresponding SF kinetic traces for FeII competition experiments.
In contrast, FeII competition experiments performed in a similar manner did not cause either of these effects. With MnII maintained at 2 equiv (200 μM), formation of the resting state measured at 320 nm increases monotonically with greater FeII concentrations (Figure 2B, inset), and no delay in peak formation is observed (Figure S4A). At super-stoichiometric FeII levels, the rates of assembly appear to increase slightly (Figure S4A). However, these metal ratios have previously been shown to lead to increased formation of the contaminating Fe/Fe cofactor,30 which also has pronounced absorbance at 320 nm and faster overall rates of assembly relative to Mn/Fe R2lox (Figure S1). Thus, the increased absorbance and rates at high FeII concentrations are likely to derive from a mixture of Mn/Fe and Fe/Fe R2lox cofactor formation. Complementary competition experiments were also performed using rapid-mixing optical (UV-vis) spectroscopy to obtain time-resolved measurements across all wavelengths, with the trends observed in the SF experiments mirrored in the UV-vis data (Figures S5 and S6). While we cannot completely discount the adventitious binding of MnII, no evidence for structural changes due to allosteric binding are observed in the EPR spectra, which do not change significantly between 1 and 2 equiv or more of MnII (vide infra).
To further investigate the differential responses in metal competition experiments, and the slowed cofactor assembly observed with increasing [MnII], key residues predicted to influence cofactor assembly and metal binding affinity were targeted for mutation. Specifically, the primary sphere E69 residue that directly coordinates the metal in site A was mutated to an aspartate side chain (E69D), and Y175 in the secondary sphere of site A was replaced with a phenylalanine residue (Y175F) (Figure 1). Time-resolved metal competition experiments monitoring resting state formation, analogous to those described above for WT, were performed on both Y175F and E69D R2lox. This allows for direct comparison between the two mutants and WT R2lox, with noticeable differences in the response to increasing metal concentrations (Figure 2). The decrease in the rates of cofactor formation with increasing MnII concentrations is more pronounced for Y175F than for WT, with traces at [MnII] ≥ 1 mM lacking a clear absorbance maximum at 320 nm (Figure 2A). This is readily observed in the complementary UV-vis competition experiments, where the formation of the resting state at 320 nm is substantially slower for [MnII] ≥ 1 mM (Figure S5B). While a slight overall decrease in absorbance at higher MnII levels also appears in the Y175F SF data, this change is less apparent in the UV-vis data.
Conversely, E69D displays only a minor decrease in the rate of cofactor assembly at high MnII concentrations. However, cofactor formation appears to be slowed overall in comparison to WT, with peak absorbance at 320 nm not resolved on the SF time scale, even at low MnII levels (Figure 2C). This is in agreement with the UV-vis data, which show slowed formation of the resting state across all MnII concentrations for E69D (Figure S5C). Using the UV-vis data to examine overall cofactor formation, it also appears that slight inhibition of cofactor assembly is observed in E69D MnII competition data, but to a lesser degree than in WT.
As in WT R2lox, FeII competition experiments with both Y175F and E69D mutants reveal no apparent inhibition in either SF (Figure 2, insets) or UV-vis (Figure S5E,F) data, although the temporal profiles of the E69D traces at 320 nm are noticeably altered from those seen in WT and Y175F R2lox.
Establishing a Quantitative Metric for Analyzing Metal Response in R2lox.
To quantitatively visualize and compare the response toward MnII and FeII across the WT and the Y175F and E69D mutants, a numerical metric was established by identifying the time point at which the SF absorbance at a given wavelength reached half of its maximum value (t(A1/2), Figure S7). Plotting these values as a function of MnII or FeII concentration results in effective “inhibition curves” for each mutant (Figure 3). Using this analysis, the t(A1/2) values for WT R2lox are found to be approximately constant at 6 s from 75 to 400 μM MnII, which is also the point at which changes are observed in the raw absorbance traces. At higher [MnII], the t(A1/2) increases steadily from 6 to 30 s, indicative of slowed assembly. The MnII inhibition curve for Y175F resembles that of WT at [MnII] below 400 μM; however, above this concentration, the t(A1/2) values increase substantially more than WT, to a maximum of 82 s, in agreement with the experimental data on Y175F that show a more pronounced response to MnII. Similarly, the E69D t(A1/2) values are unchanged at concentrations lower than 400 μM, though the absolute values are higher. As MnII levels are increased beyond 400 μM, the E69D t(A1/2) values increase approximately 3-fold to a maximum of 75 s. When compared to the initial t(A1/2) values at and below 400 μM MnII, this 3-fold increase in E69D is less than that seen for WT or Y175F. However, the absolute change in t(A1/2) values from beginning to end of the E69D MnII inhibition curve is greater than that of WT, resulting in a slightly greater slope than in WT.
Figure 3.
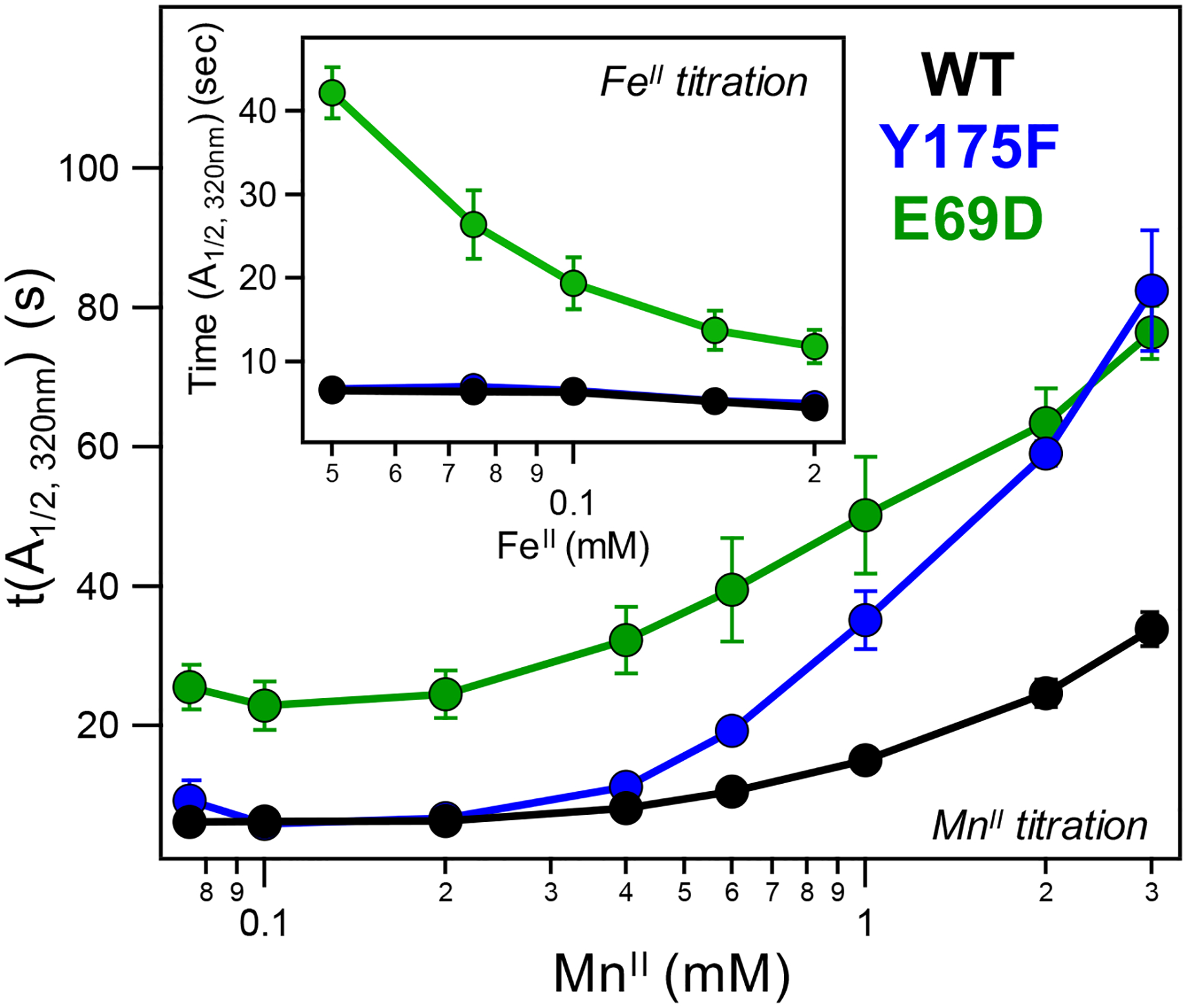
MnII inhibition curves generated from SF competition data at 320 nm for WT (black), Y175F (blue), and E69D (green) R2lox. (Inset) Corresponding FeII inhibition curves of all three mutants at 320 nm (coloring as in main figure).
Applying the same t(A1/2) analysis to the corresponding FeII competition data produces FeII inhibition curves in stark contrast to the MnII inhibition curves (Figure 3, inset). Increased FeII concentrations have minimal effect on the t(A1/2) values of both WT and Y175F, which remain relatively steady across the experiment, in agreement with the similar, monotonic response to increasing [FeII] observed in the raw SF data. Conversely, increased FeII levels have a more pronounced effect on the E69D t(A1/2) values, as also observed in the raw FeII competition data, with greater concentrations of FeII resulting in faster assembly rates and correspondingly smaller values for t(A1/2).
Semi-quantitative Modeling of Metal Binding in WT R2lox.
To identify reaction schemes for MnII and FeII binding to R2lox that are consistent with the experimental data, the metal competition experiments and inhibition curves were considered in conjunction with global kinetic modeling using the program, KinTek.42,43 The model incorporates the full assembly process, including O2 activation and downstream cross-link formation, as described previously.27 Because the UV region used as a marker for the resting state absorption (320 nm) likely also contains contributions from other high-valent species formed earlier in the reaction mechanism, the experimental data collected at that wavelength do not provide a clean representation of only the resting state species, artificially appearing earlier than the final resting state signals develop in EPR.27 Time-resolved absorption traces were also collected at 390 nm to monitor one of the postulated high-valent Mn/Fe intermediates, I2. To reduce ambiguity from potential spectral overlap between intermediate species and the resting state in the near-UV, the 390 nm data were instead chosen for comparison to the modeled data for all considered assembly schemes.27 As with the data at 320 nm, SF competition experiments performed at 390 nm show similar inhibitory effects with increasing MnII (Figure 4A and Figure S8A). This inhibition is also readily apparent in the t(A1/2) values obtained from the experimental data (Figures 4E,F, and Figure S8C), which increase for increasing MnII concentrations. FeII competition experiments did not induce an inhibitory effect on intermediate formation either in the absorbance traces (Figure S7A, inset) or the t(A1/2) values (Figure 4F and Figure S8C, inset). These findings are replicated in the UV-vis kinetic traces, obtained from analysis of data at 390 nm (Figure S8A,C), with good agreement seen between SF and UV-vis data (Figures S9 and S10). We note that the apparent decay of I2 suffers from similar issues as the resting state formation and other high-valent species forming downstream of I2, including the resting state itself, may also have some absorbance at 390 nm. In this case, the experimental absorbance at 390 nm does not decay completely over time and may appear artificially slower than for a single, modeled species (Figure S9).
Figure 4.
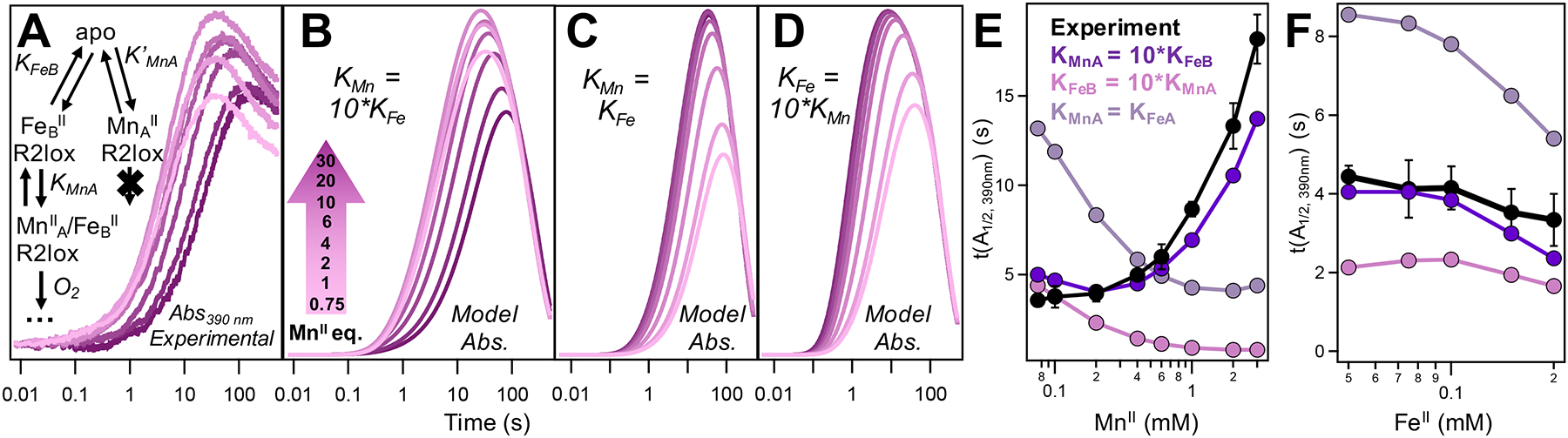
Experimental (A) and simulated (B–D) SF absorption data monitoring I2 formation in WT R2lox at 390 nm as a function of [MnII]. [FeII] held at 75 μM (0.75 equiv), kFe,off = k′Mn,off = kMn,off = 101 s−1 and kFe,on, k′Mn,on, and kMn,on = 104 or 105 M−1 s−1. (A, inset) Scheme for competitive, ordered metal binding used for simulations in (B–D) with K′MnA = KMnA. (E, F) MnII and FeII inhibition curves for experimental data shown in (A) and Figure S8A, inset, respectively, with inhibition curves from data simulated using three thermodynamic regimes overlaid. MnII and FeII concentrations shown in log scale.
Metal binding in R2lox may proceed via multiple, distinct pathways depending on which site binds first. Moreover, the order of site binding may determine whether productive heterobimetallic cofactor formation can occur at all. To address this, the following metal binding schemes were considered, and kinetic models were developed for each: (i) Competitive and ordered, wherein either metal can bind to the respective site first, but only if FeII binds first in site B can productive O2 activation occur. Thus, if MnII binds to site A first, it must then dissociate to allow FeII binding in site B (Figure 4A, inset). (ii) Non-competitive and ordered, with either FeII binding first in site B or MnII binding first in site A, followed by the second metal binding in its respective site and subsequent O2 activation (Figure S11A,B, insets). (iii) Competitive and non-ordered, with either metal able to bind first and support O2 activation (Figure S11K, inset). (iv) Competitive, site non-selective, and non-ordered, in which either metal can bind first and MnII is able to bind in site B, but ultimately only the MnIIA/FeIIB configuration is productive (Figure S11P, inset). Additionally, in (iv), MnII binding in site B prevents any metal from binding to site A. For each metal binding scenario described above, the relative binding affinities for MnII and FeII in sites A and B, respectively, were assessed through consideration of three regimes: KMnA > KFeB, KMnA < KFeB, and KMnA = KFeB (Figure 4 and Figure S9), with the individual off rates for each metal maintained at 10 s−1 and the kon rates set as either 104 or 105 M−1 s−1.
Previous data10,11,30,34 and current work (Figure S12A) using EPR have shown minimal amounts of MnIIA/FeIIB dimers in WT R2lox under solution phase conditions; accordingly, MnII dimer formation was not included in these assembly models. We note that, although R2lox can bind an Fe/Fe cofactor, Fe/Fe assembly in R2lox is observed to be faster than Mn/Fe assembly (Figure S1). Thus, observation of the slower assembly pathway favors the Mn/Fe route. Additionally, no clear spectroscopic handles have been identified for assembly intermediates or resting state in the Fe/Fe pathway. While determining the Fe/Fe R2lox assembly mechanism represents ongoing work in our laboratory, the integer spin states (the majority being ST = 0) of Fe/Fe R2lox intermediates have made EPR studies difficult, and Mössbauer analysis of the Fe/Fe assembly represents a non-trivial feat in terms of instrument time and cost. For these reasons and in the absence of any noticeable inhibitory effect on cofactor assembly during FeII competition experiments, the binding of FeII in site A following FeII binding in site B was not considered.
While trends can be compared between modeled and experimental data, in order to more rigorously draw comparisons between these (and all subsequent) models and the experimental data, MnII and FeII inhibition curves were generated from each modeled dataset showcasing the three different regimes of relative binding constants. These modeled inhibition curves can be directly compared to the experimental inhibition curves (Figure 4E, F, and Figure S11E,J,O,T). In all cases, the MnII and FeII binding affinities for each site were simulated to be independent of the presence of another metal, i.e., the MnII binding affinity of site A without an FeII ion in site B (K′MnA) is the same as the MnII binding affinity of site A with an FeII already present in site B (KMnA). To assess the validity of this and other assumptions made in these models, a number of additional simulations were run, including those considering reversible oxygen binding, variation in kon,O2 under irreversible binding conditions, changes to relative and absolute metal binding affinities, positive and negative cooperativity, and variation in individual kon and koff rates for both metals or just MnIIA (Figures S13–S18).
Although a transient population of MnII binding in site B to slow down productive cofactor formation cannot be ruled out, addition of a non-productive, MnIIB binding step (Figure S11P–T) failed to replicate the experimental trends and MnII inhibition curves generated from experimental data. When examining the downstream effects of O2 binding, neither the inclusion of various levels of reversible O2 binding (Figure S13) nor changes to the rate of irreversible O2 binding (Figure S14) resulted in a modeled MnII inhibition curve that was in better agreement with the experimental data than that obtained initially using irreversible O2 binding (k4 = 1 × 106 M−1 s−1). In situations where cooperative binding was assessed (i.e., K′MnA ≠ KMnA), only when at least one of the MnII binding affinities, K′MnA or KMnA, was greater than KFeB, and the other was equal to KFeB, did the modeled data produce a MnII inhibition curve with any significant inhibition present at higher [MnII] (Figure S16). This was also true in specific cases where the kon and koff rates for the two metals were varied in conjunction with assessing cooperativity, specifically, for k′Mn,off = kMn,off = 100 s−1 and kFe,off = 10 s−1 (Figures S17 and S18). We note that, regardless of the MnII koff rates, for K′MnA > KMnA > KFeB, the modeled inhibition is more pronounced and the absolute t(A1/2) values are greater than those are observed in the experimental data. Conversely, for KMnA > K′MnA > KFeB, the modeled inhibition curves closely resemble the experimental data. In all cases, the competitive and ordered scheme for metal binding (Figure 4A, inset) with KMnA = K′MnA = 104 M−1 and KFeB = 103 M−1 gives rise to t(A1/2) values most closely resembling the experimental data.
Using the KinTek software, attempts were made to directly fit the various models to the experimental I2 data. This proved to be intractable without applying scaling factors to reconcile differences in the modeled vs experimental intensity (concentration) profiles (Figure S19). Using these scaling factors, the profiles of the modeled data could be fit iteratively. This process bounded some of the fundamental rate constants and provided a starting point for heuristic fitting of the data. Although the competitive and non-ordered model also appears to fit using the scaled data, the optimized parameters require an inhibitory MnII binding step with KMnA > KFeB (Figure S19). Thus, the global fits are consistent with the findings obtained through semiquantitative comparison of the modeled and experimental inhibition curves.
Site A Mutations Influence Metal Binding Affinities.
The assembly intermediates observed using time-resolved optical spectroscopy in Y175F R2lox are similar to those observed for WT (Figures S20 and S21). Therefore, the SF metal competition experiments were also performed monitoring 390 nm, and t(A1/2) values were extracted from both MnII and FeII Y175F competition experiments for comparison with WT and modeled data (Figure 5A,B and Figure S8C). For the remaining analyses, the I2 species was used when modeling the Y175F t(A1/2) values. No visible assembly intermediates are readily apparent in the optical spectra of the E69D mutant using either rapid-mixing UV-vis (Figure S20C) or SF spectroscopy at relevant probe wavelengths (Figure S22), potentially due to different relative rates of formation or decay of any transient species, though the optical spectrum of the resting state closely resembles that of WT. While characterizing the precise mechanism for O2 activation following metal binding and identifying potential intermediates in this mutant is beyond the scope of this work, the impact of the E69D mutation on the metal binding affinities can still be projected using the same initial assembly mechanism. In this case, the resting state species was used to obtain modeled t(A1/2) values that can be compared to the experimental E69D data measured at 320 nm (Figure 5C,D). The general trends and differences seen between WT and Y175F t(A1/2) values are reproduced whether the values are extracted from 390 or 320 nm data (Figure 3 and Figure S7C), indicating that increasing metal concentration similarly affects the rate and amount of formation of both species. WT resting state inhibition curves at 320 nm are shown in comparison to those of E69D (Figure 5C, D). The overall greater absolute t(A1/2) values obtained from all modeled resting state data compared to those from the experimental data at 320 nm are consistent with the presence of additional, transient species absorbing at that wavelength (vide supra).
Figure 5.
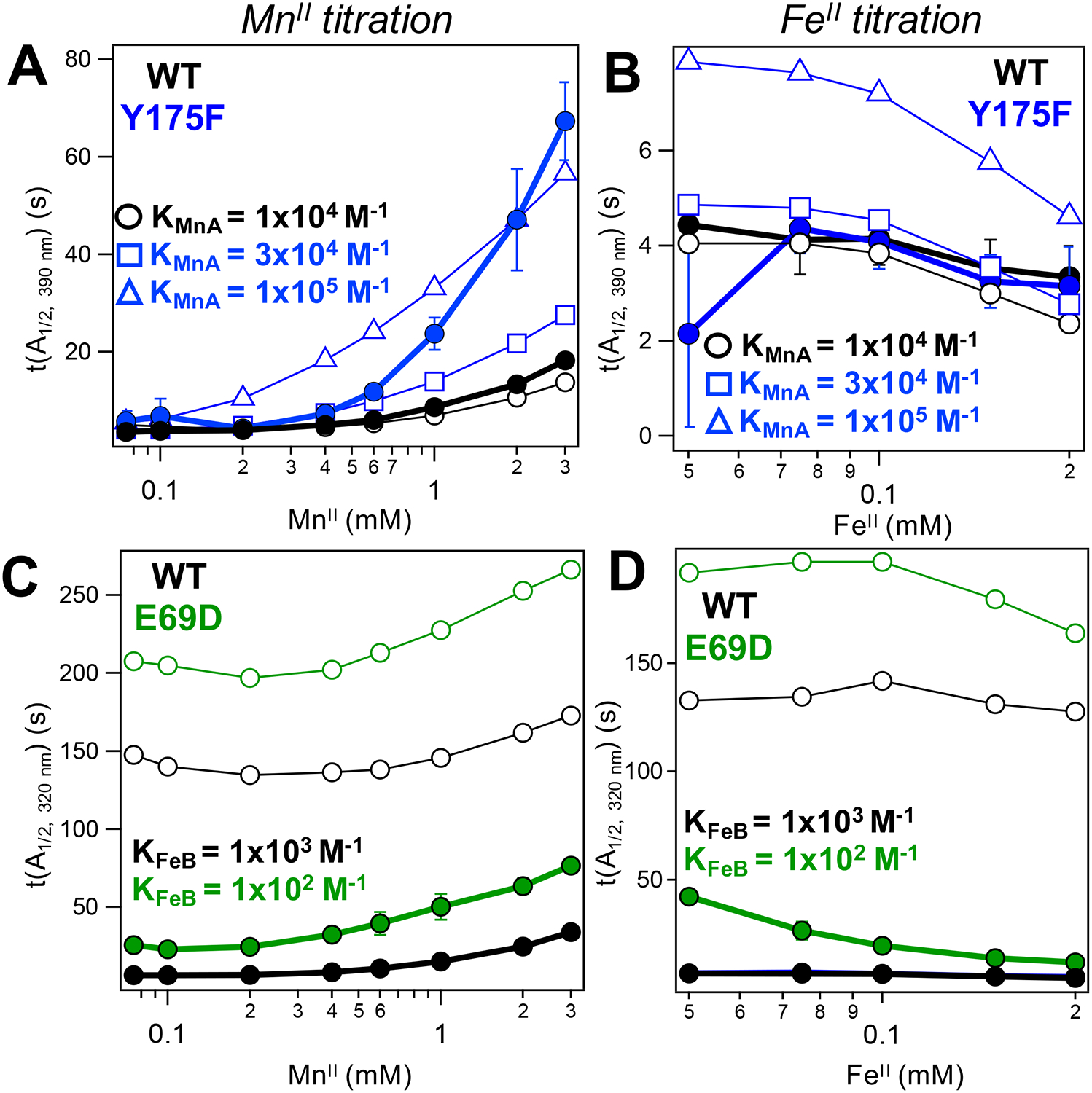
MnII (A, C) and FeII (B, D) inhibition curves for WT (black), Y175F (blue), and E69D (green). Curves generated from SF data (solid symbols) or kinetic modeling (open symbols) for either the I2 (top) or the resting state (bottom) species. Metal binding affinities used to model mutant data are indicated, with koff = 10 s−1 and kon adjusted accordingly; remaining rates were held the same as in the WT competitive/ordered model. For Y175F, modeled inhibition curves generated for KMnA = 3 × 104 M−1 (squares) and 1 × 105 M−1 (triangles) are shown to bound the experimental data. MnII and FeII concentrations shown in log scale.
The MnII competition data for both Y175F and E69D show inhibitory behavior, albeit to a different degree than WT for each mutant (Figure 2, Figure 3, and Figure S8). Using the same competitive/ordered binding model, an effort was made to reproduce the differences observed in the experimental t(A1/2) values of the mutants compared with WT. While Y175F has a slightly higher tendency to assemble MnIIA/FeIIB dimers than WT and E69D, as seen in EPR spectra of Mn-only loaded samples (Figure S12), this dimer formation remains a minor contribution (<10%)30 and was not considered in the kinetic models.
The metal inhibition curves serve as a point of comparison between the experimental and modeled data to assess the effects of each mutation on the metal binding thermodynamics in R2lox. Interestingly, despite both perturbations occurring around the A site, distinct effects were seen for each mutant. The decreased sensitivity to MnII and overall slowed rates of assembly for the E69D mutant were reproduced only when a binding affinity of 102 M−1 for FeIIB was used, which is 10-fold lower than that used in the WT model (Figure 5C and Figure S21). This value also reproduced the E69D FeII inhibition curves (Figure 5D), which show higher rates of assembly with greater [FeII] than seen in WT. In order to reproduce the MnII inhibition curve for the Y175F mutant, which shows a steeper slope at high MnII concentrations, a greater MnIIA binding affinity than in WT R2lox (104 M−1) must be invoked. Values between 3 × 104 and 1 × 105 M−1 give rise to MnII and FeII inhibition curves that offer reasonable bounds to the experimental data while keeping other assembly rate constants fixed (Figure 5A,B and Figure S24).
An attempt was also made to replicate the altered t(A1/2) values and inhibition trends of the mutants solely by perturbing rates downstream of O2 binding within the competitive/ordered model, considering that the mutations may change the O2 activation process or cofactor reactivity relative to WT. However, manipulating the rates of I2 formation or decay alone (Figure S25) or in conjunction with modified binding affinities for FeII (Figure S23) or MnII (Figure S24) were not sufficient to reproduce the changes in the E69D or Y175F metal inhibition curves.
EPR Spectroscopy and X-ray Crystallography of R2lox Mutants Reveal Subtle Changes in Active Site Electronic and Geometric Structures.
The observed changes in assembly intermediates and modeled metal binding affinities in both Y175F and E69D prompt the question of how these mutations affect the resting state structure relative to WT R2lox. To address this issue, X-band EPR spectroscopy was used to resolve electronic structure changes at the Mn/Fe center, while X-ray crystallography was used to probe global protein secondary and tertiary structure.
EPR spectroscopy is a sensitive probe of the electronic structure of a metallocofactor. This is particularly true in the case of spin-coupled systems, where the projected spin Hamiltonian parameters are highly dependent on the properties of both metal centers and the exchange coupling between them. A change in the number or protonation state of the ligands to either metal would be expected to significantly alter this coupling, resulting in a substantial change to the projected g-,55 Mn, and 57Fe hyperfine tensors.
The EPR spectra of the final, resting state species of Y175F and E69D following aerobic incubation with metals were collected and compared to WT R2lox (Figure 6). Data were collected at 5 K to minimize interference from residual MnII, and the resulting spectra were simulated to obtain projected spin Hamiltonian parameters (Table S1). All variants show similar spectral breadth, peak splitting, and additional coupling in samples prepared with 57Fe, indicating that neither mutant resting state species fundamentally deviates from a spin-coupled MnIII(μ-OH)FeIII system.11 However, a subtle but meaningful reduction in Y175F spectral breadth is readily apparent on the lower field edge of the absorption profile, corresponding to the unique axis along the Mn-aqua bond. This is manifested in the Az values for both55 Mn and 57Fe, which display reduced anisotropy relative to those of WT R2lox.
Figure 6.
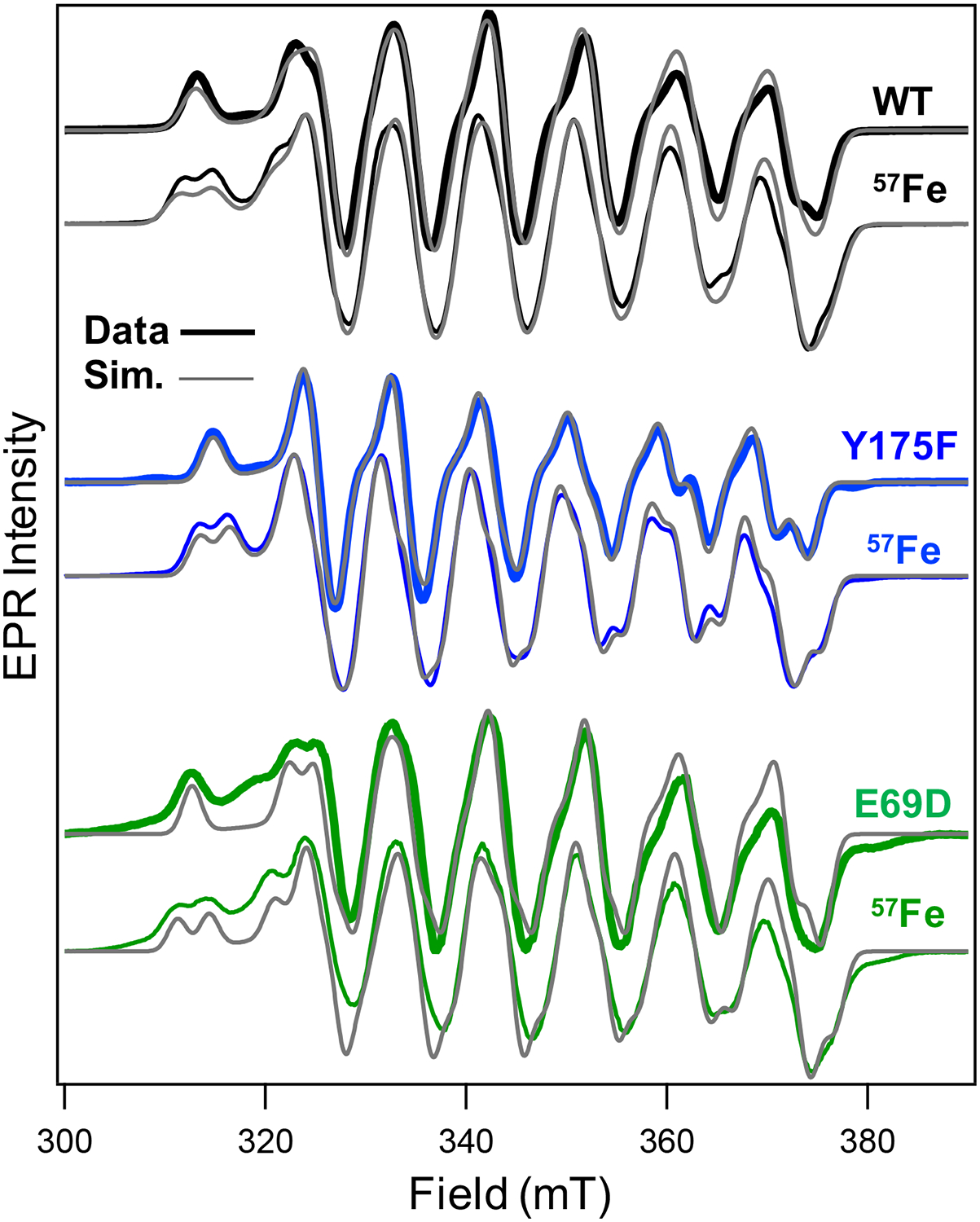
CW X-band (T = 5 K; Pμw = 20 mW) EPR spectra (colored lines) overlaid with spin Hamiltonian simulations (gray) of WT, Y175F, and E69D R2lox resting states prepared with NAFe (thick lines) and 57Fe (thin lines).
X-ray crystal structures were obtained for the E69D and Y175F mutants in the unactivated, reduced state and the oxidized resting state by soaking apo-R2lox crystals with MnII and FeII under anoxic or aerobic conditions, respectively (Figure 7). Similar overall secondary and tertiary geometries are preserved in the R2lox mutants when compared with WT, though the active sites show subtle changes in each mutant state, highlighting the relative plasticity of the protein side chains. The E69D mutation affects the structure of R2lox to a greater extent in the reduced state than in the oxidized state (Figure 7B). In one dimer subunit, the reduced active site resembles the structure of WT, with the mutation causing the metal ions and other ligands as well as Y175 to shift toward the shorter aspartate side chain. In the other subunit, however, the active site appears to be only partially metal-bound, and site A is largely disordered, similar to the unmetalated state of the WT protein (Figure S26).10
Figure 7.
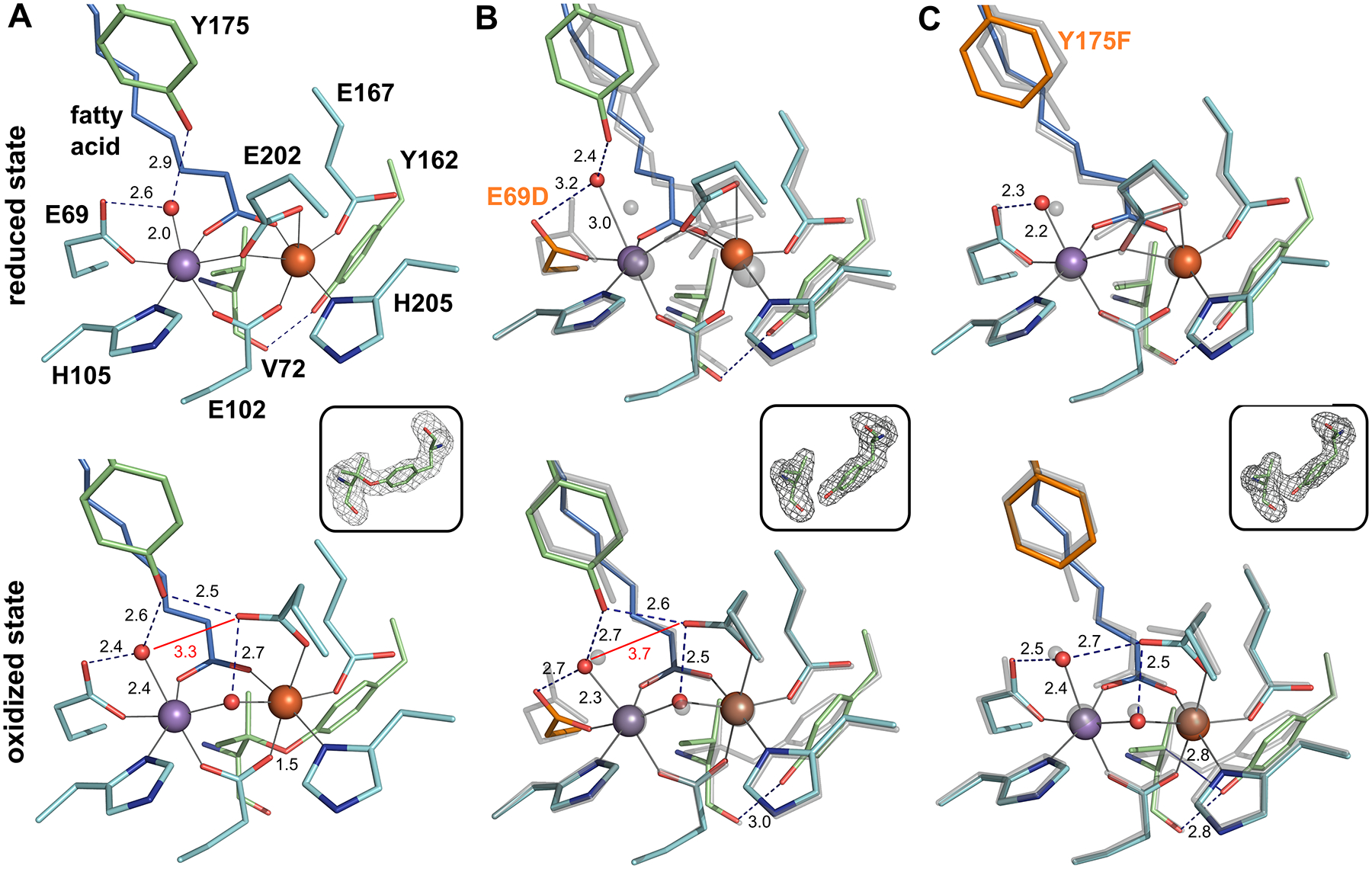
Active site structures of R2lox variants in the unactivated, reduced (top) and O2-oxidized resting state (bottom). All structures are shown in roughly the same orientation, with site A on the left. The E69D and Y175F variants in (B) and (C) are shown in color superimposed with the WT structure in the same redox state in transparent gray. Mutated residues are highlighted in orange. Metal-ligand bonds are indicated by gray lines, hydrogen bonds by dashed blue lines. Selected distances are shown in Å. The insets show mFo – DFc refined omit electron density contoured at 3.0 σ for residues 72 and 162 in oxidized state crystals. (A) WT R2lox (reduced state, PDB ID 4HR4; oxidized state, PDB ID 4HR0): an ether cross-link is formed between the Cβ of V72 and the hydroxyl oxygen of Y162 in the oxidized state. (B) E69D R2lox (reduced state, PDB ID 6QK0; oxidized state, PDB ID 6QJV): the ether cross-link is not observed. (C) Y175F R2lox (reduced state, PDB ID 6QK2; oxidized state, PDB ID 6QK1): the V72-Y162 ether cross-link is partially formed, as indicated by showing both the hydrogen bond between the carbonyl oxygen of V72 and the hydroxyl group of Y162, as well as an unbroken blue line for the ether bond.
In contrast, in the presence of oxygen, both protomers of E69D R2lox react to metalation in the same way, and the structure of oxidized E69D R2lox is remarkably similar to that of WT. Only the water ligand to MnA and Y175 are shifted toward D69 to retain the hydrogen bonding pattern, leading to a larger distortion of the site A geometry compared to WT, though the metal ions are in the same relative positions. The V72-Y162 ether cross-link is not formed to such an extent in E69D R2lox to be crystallographically resolved, despite indications that oxygen activation has occurred, as evidenced by the movement of the bridging E202 residue to a terminal position and the appearance of an oxygenic bridging ligand between the metals.
The Y175F mutation also has only minor effects on the active site X-ray structure (Figure 7C). In the reduced state, the site A water ligand is slightly shifted toward E69, and the MnA-water bond is slightly longer than in WT (although this latter difference is just within the coordinate error). In the oxidized state, the hydrogen-bonding network adapts to the absence of the Y175 hydroxyl group, with E202 shifted slightly so that the water ligand is held in place by hydrogen bonds to E69 and E202 instead of E69 and Y175. The V72-Y162 ether cross-link is also only partially formed in oxidized Y175F R2lox, although, as with E69D, evidence for oxygen activation is present.
Site A Mutations Affect Reactivity of R2lox.
Although the in vivo function of R2lox remains unknown, the formation of a tyrosine-valine ether cross-link within the protein is highly specific and directed, making it an appropriate proxy for monitoring the reactivity of each scaffold toward C-H bond activation.44 The cross-link has been shown to be generated during the O2 activation process, circumventing the canonical radical rebound pathway often observed in the diiron oxidases, which would instead hydroxylate the valine residue.45,46 Importantly, cross-link formation reflects the two-electron oxidation of a C-H bond. To investigate whether metal binding affinity is correlated with efficacy of C-H activation, cross-link formation was quantified using mass spectrometry (Figure 8A) and compared to the Mn/Fe metalation efficiency (Figure 8B) and cofactor reduction potentials (Figure 9) in the E69D and Y175F mutants relative to WT.
Figure 8.
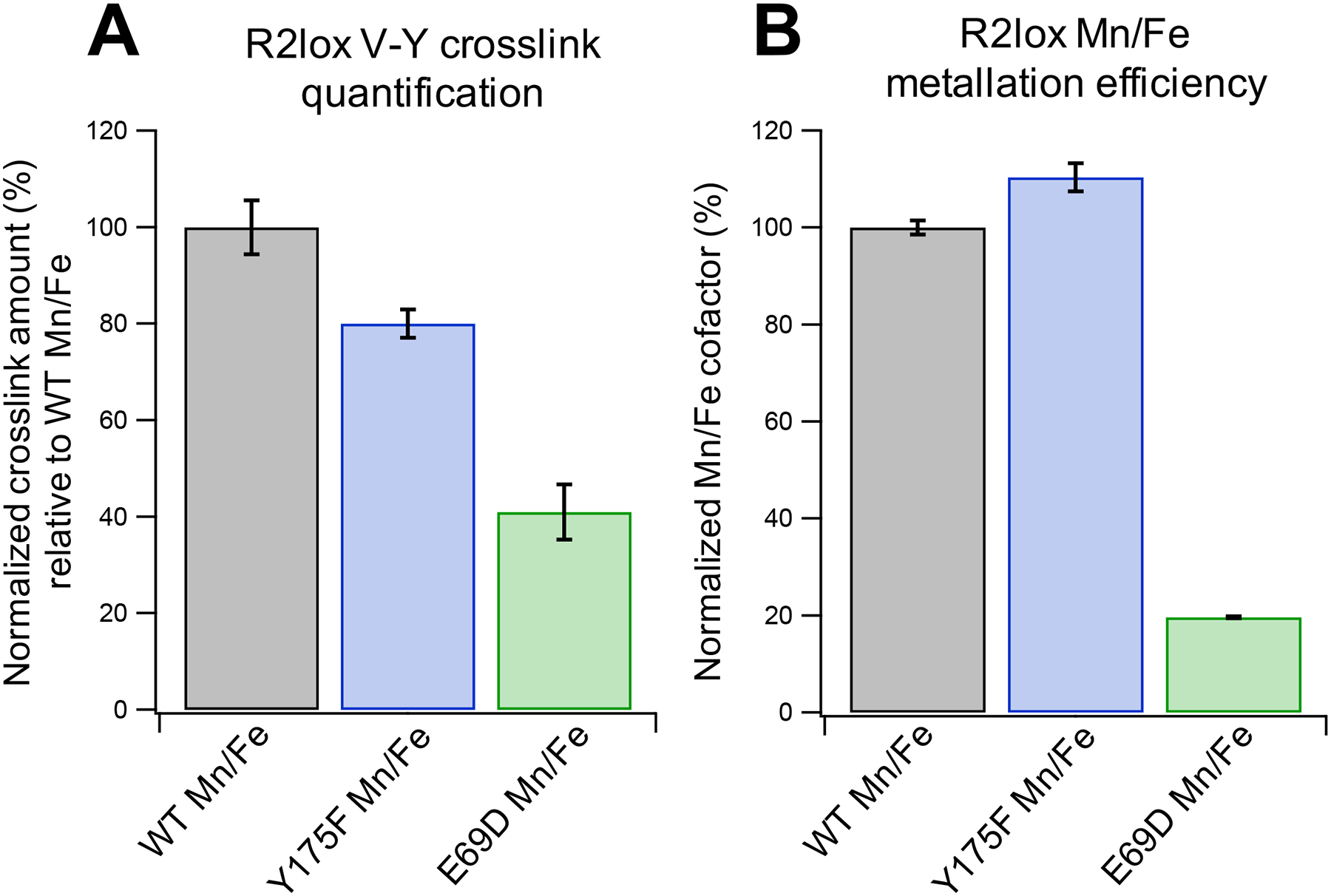
(A) LC-MS analysis of cross-link formation in Mn/Fe WT R2lox compared to Y175F and E69D mutants. Values are from the integrated chromatographic peak areas of the cross-linked peptide AVTLDAVIRAATVYNMIVE, normalized to the R2lox protein area of the respective LC-MS run. Cross-link levels were normalized to average WT values. The error bars represent standard error of mean (SEM) across the 9 runs of each sample type (3 biochemical replicates × 3 technical replicates). (B) MnIII/FeIII cofactor formed in R2lox as determined by EPR spin quantification. Samples were prepared identically to those used in (A). Error bars shown represent SEM across 3 biochemical replicates.
Figure 9.
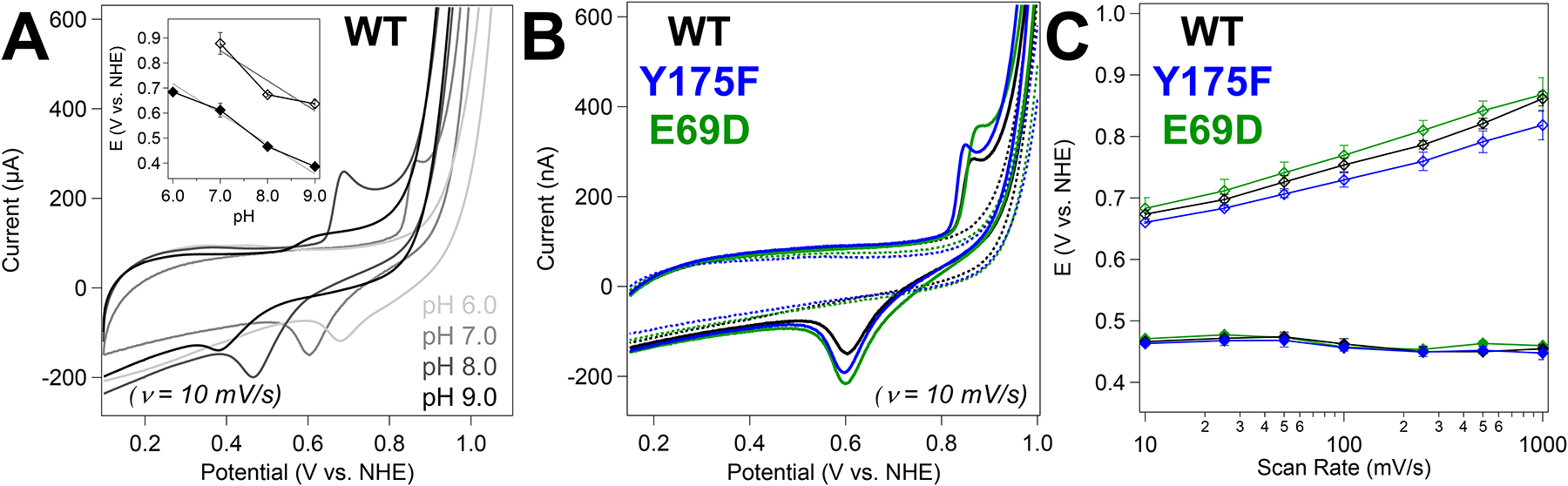
(A) Cyclic voltammograms of WT Mn/Fe R2lox from pH 6 to pH 9 (as indicated). (Inset) Anodic (open symbols) and cathodic (closed symbols) peak potentials for WT Mn/Fe R2lox as a function of pH. Dotted lines indicate a slope of −120 mV/pH unit. (B) Cyclic voltammograms of Mn/Fe (solid) and apo (dashed) R2lox at pH 7.0. (C) Anodic (open symbols) and cathodic (closed symbols) peak potentials as a function of scan rate. All samples contained 100 μM WT (black), Y175F (blue), or E69D (green) R2lox protein with 100 mM NaCl in 100 mM HEPES buffer, pH 8.0. Points and error bars reflect the average and standard deviation of n ≥ 3 trials, respectively. Samples measured with a glassy carbon working electrode.
Following aerobic cofactor assembly and removal of excess unbound metal ions, Glu-C digested protein samples were analyzed by mass spectrometry to measure the amount of cross-linked peptide present in each sample, relative to WT Mn/Fe R2lox (Figure 8A). The amount of cross-link formed in each mutant was less than that seen in the WT scaffold for the Mn/Fe cofactor, with Y175F and E69D cross-link levels at ~80% and ~40% of those of WT, respectively. To investigate if these results could be attributed to lower absolute levels of Mn/Fe cofactor in the R2lox mutants, EPR spin quantification was performed on samples that were prepared in an identical fashion to the mass spectrometry samples (Figure 8B). Despite lower cross-link levels, Y175F appears to form the Mn/Fe cofactor with slightly greater efficiency than WT. Even more striking, the efficiency of Mn/Fe cofactor formation in E69D is only ~20% of that seen in WT R2lox, despite cross-link levels that are found to be ~40% those of WT. To ensure this excess cross-link formation relative to WT is not due to the reactivity of residual Fe/Fe cofactors in the E69D mutant, mass spectrometry was also performed on control samples metalated with only FeII (Figure S27). However, the amount of cross-link formed in Fe/Fe E69D is negligible, at ~6% that of WT Mn/Fe R2lox.
In addition to Mn/Fe metalation efficiency, another metric that is relevant when considering cross-link formation is the reduction potential of the Mn/Fe cofactor across R2lox variants. To directly probe the thermodynamics associated with a high-valent Mn/Fe species in each scaffold, protein electrochemistry was performed (Figure 9). Protein electrochemistry is a highly valuable tool for characterizing reduction potentials and catalysis in electron transport proteins and redox enzymes.47,48 This technique is underutilized in the study of members of the ferritin-like superfamily, and has never been applied to a heterobimetallic Mn/Fe protein.49 Owing to its small size and cambialistic nature, R2lox represents an ideal testbed to develop this technique for interrogating this new class of proteins. While a full analysis of the electrochemical properties of R2lox is beyond the scope of this work and will follow in a subsequent publication, the reduction potential is directly related to the propensity of a species to carry out oxidation reactions. Initial data collected on WT Mn/Fe R2lox at pH 7.0 show well-resolved anodic and cathodic peaks at approximately +878 and +612 mV vs NHE, respectively (Figure 9B). We note the feature at high potentials in all three R2lox variants as well as apo-R2lox likely stems from background water oxidation signals due to the HEPES buffer (Figure S28).50 Experiments measured at varying pH show that the electrochemical process is coupled to deprotonation; cyclic voltammograms recorded from pH 6 to 9 show a pronounced shift of both anodic and cathodic signals of approximately −120 mV/pH unit, indicative of a two-proton, one-electron transfer process (Figure 9A, inset).51 At pH 6, an anodic feature could not be detected under the large oxidative background currents observed at this pH (Figure S28). When compared to the WT data, cyclic voltammograms of Y175F and E69D at pH 7.0 show anodic peaks at +866 and +924 mV vs NHE, respectively, with corresponding cathodic signals observed at +596 and +611 mV (Figure 9B), consistent with a modified resting state reduction potential in the R2lox mutants. The anodic peak potential increases markedly with scan rates in all cases, while the cathodic peak potentials are unaffected (Figure 9C and Figure S29), suggesting that the oxidation process is kinetically coupled to a slow chemical step, such as deprotonation. The invariant cathodic peak positions indicate reduction is either not necessarily coupled to reprotonation associated or the latter process occurs at much higher rates.48
DISCUSSION
Previous work in our laboratory centered around the observation of transient intermediates in heterobimetallic Mn/Fe cofactor assembly formed upon addition of MnII, FeII, and O2 to the WT apo-R2lox protein. It was noted that obtaining metal binding constants from anaerobic experiments was difficult due to poor association of both MnII and FeII to the apo-R2lox scaffold prior to the O2 binding step and the absence of distinct visible optical or EPR signatures for the MnII/FeII R2lox state.27 However, by taking advantage of the heterobimetallic preference of the R2lox cofactor,10,30 information regarding the relative metal binding constants for MnII and FeII can be extracted by monitoring aerobic cofactor assembly rates as the metal concentrations are varied.
Interrogation of Metal Binding in WT R2lox Indicates Thermodynamic Control over Heterobimetallic Selectivity.
Data from metal competition experiments performed on WT R2lox show a differential response to increasing [MnII] and [FeII], suggesting distinct metal binding affinities for each site or for each metal in a given site. Furthermore, the apparent inhibition of cofactor assembly observed during MnII competition experiments but not the corresponding FeII experiments has implications for the necessary order of metal binding; however, these observations necessitated a more rigorous assessment. Although the presence of multiple reactants, i.e., MnII, FeII, apo-R2lox, and O2, make an exact determination of assembly rates difficult, the development of metal inhibition curves by plotting t(A1/2) values as a function of metal concentration provides a quantitative representation of the overall sensitivity of cofactor assembly to increasing levels of MnII and FeII. Indeed, the constructed FeII inhibition curve for WT R2lox remains relatively flat while that corresponding to MnII is clearly curved, supporting a marked influence on cofactor assembly with increasing [MnII] but not [FeII].
With the effects of increasing metal concentration on cofactor assembly more easily visualized through the development of the t(A1/2) metric, quantitative kinetic modeling can be used to investigate the potential role of metal binding affinities in mediating the observed effects. We note that the entire assembly process was used in constructing the kinetic model, with oxygen activation representing an irreversible step in cofactor maturation. This has the effect of trapping the metals that are in the active site upon O2 binding. Consequently, the states following oxidation are not reversibly generated and directly report upon the initial divalent metal binding process, which is reversible, allowing the extraction of thermodynamic information through the modeling of kinetic data. Despite the diversity of models considered, only the competitive/ordered model of metal binding produced results in agreement with the experimental WT data. This competitive and ordered binding effectively results in the presence of a “cul-de-sac” for the divalent metals, where either can move in and out of their respective binding sites, but if MnII is bound in site A without the presence of FeII in site B there is no “open road” leading to the Mn/Fe cofactor, i.e., no path to productive cofactor maturation (Figure 10). Steric hindrance is proposed to play a role in this ordered binding, as premature coordination of a metal to site A may block metal ion access to site B. This hypothesis is supported by the lack of evidence for MnIIA/MnIIB dimer formation and solution-phase EPR experiments showing MnII binds selectively to site A; however, there are likely other viable explanations.10 The results presented here provide the first direct evidence for this binding model, which had previously been postulated based only on the apparent A site disorder in X-ray structures of apo-R2lox. Moreover, it was determined that the MnII binding affinity for site A must be at least an order of magnitude greater than that of FeII (K′MnA = KMnA = 1 × 104 M−1 vs KFeB = 1 × 103 M−1) to reproduce both the observed inhibitory effects and the absolute t(A1/2) values of both the MnII and FeII inhibition curves. While some degree of cooperativity regarding metal binding in sites A and B may play a role in the observed inhibition, the modeled inhibition curves generated using conditions of KMnA > K′MnA > KFeB do not replicate the experimental inhibition curves better than those generated using KMnA = K′MnA > KFeB. Thus, the observed inhibitory effects of excess MnII in WT R2lox assembly can be directly attributed to the higher binding affinity of MnII to site A, which impedes rapid divalent metal exchange and obstructs formation of the FeIIB-R2lox species. This result explicitly opposes the IW series and indicates that selective metal binding in R2lox is a thermodynamically controlled process.
Figure 10.
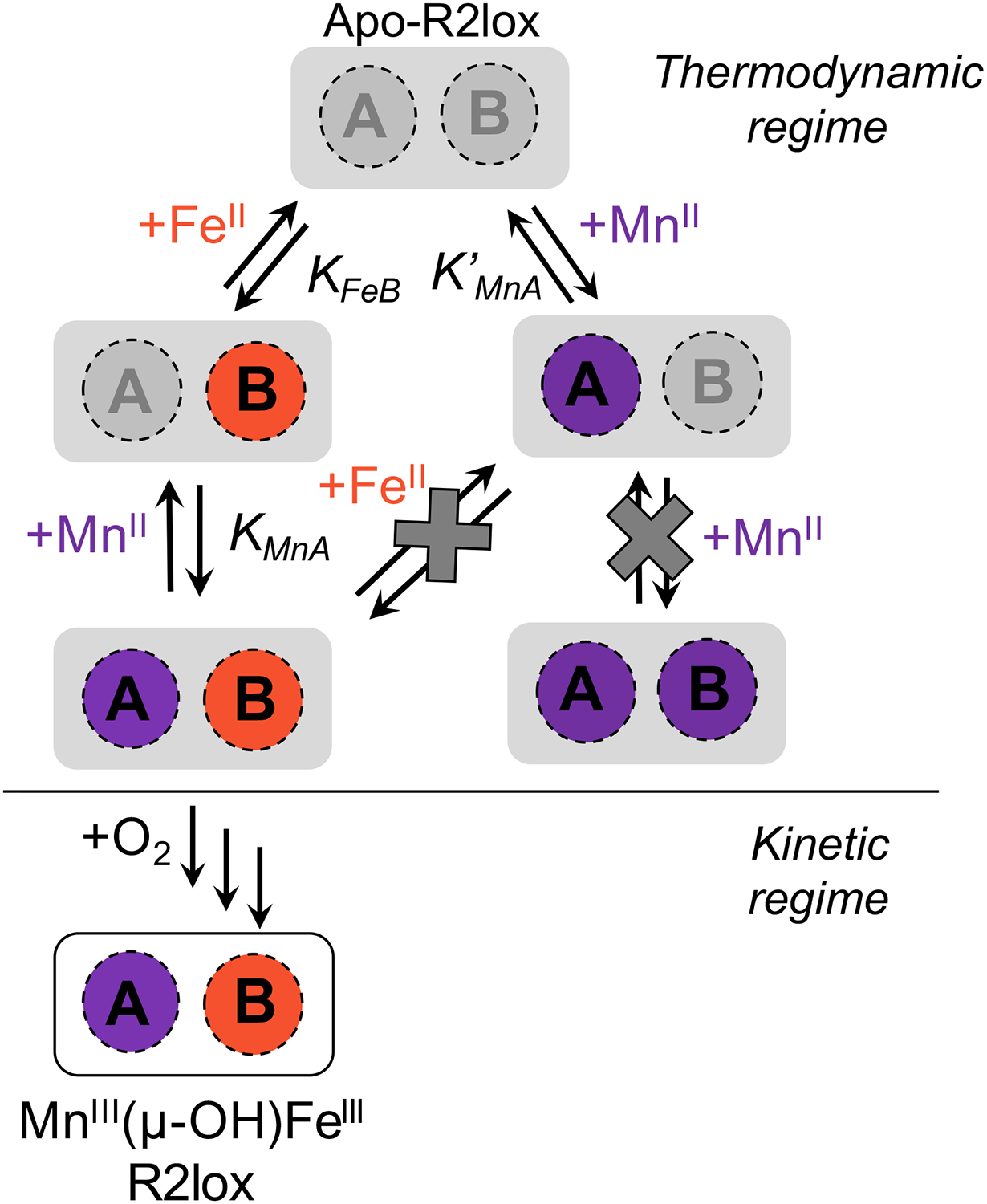
Cartoon schematic for metal binding to WT apo-R2lox when both MnII and FeII are added. The regimes where thermodynamic or kinetic control are thought to dominate are labeled.
The E69D and Y175F mutations reconstruct structural features of RNRs and BMMs to induce distinct changes in metal binding affinity. The bimetallic class I RNRs carry out one-electron radical chemistry to initiate ribonucleotide reduction, using a stable oxidizing equivalent that is either stored directly on the metal center as a high-valent cofactor or in the form of a nearby tyrosine radical.40,52–57 Previous computational work has postulated that a high-valent Mn/Fe cofactor is more stable than an Fe/Fe cofactor; therefore, residues involved in selecting for a high-valent cofactor are of interest for metal selection as well.58 The site A glutamate residue, E69, is conserved in R2lox, class Ic Mn/Fe RNRs, BMMs, and the class Id Mn/Mn RNRs (Figure 1).8,36,52 This residue is instead an aspartate ligand in the class Ia and Ib RNRs, which are Fe/Fe and Mn/Mn proteins, respectively, that utilize a nearby tyrosine residue to hold their stable oxidizing equivalent.8,36,52,59,60 A correlation can then be drawn between using a longer N-terminal coordinating residue and the storage of oxidizing equivalents in the form of a high-valent cofactor (E + MIV/MIII) rather than a nearby side chain (D + MIII/MIII−Y•).8,31 Conversely, a tyrosine residue (Y175) is present in R2lox that is hydrogen-bonded to the MnA water ligand and lines the ligand-binding cavity, suggestive of a potential role for Y175 in substrate binding or as a proton transfer partner during catalysis.61 While this residue is conserved as a phenylalanine in the RNRs, it is not strictly conserved in the BMMs, with an arginine present in sMMO instead.8,62 Taken together, these residues reflect salient distinctions between R2lox, RNRs, and the BMMs and thus serve as targets for exploring molecular influences on metal binding affinity.
Metal competition experiments performed on the two R2lox mutants, E69D and Y175F, show an altered response in both the raw absorbance data and the inhibition curves when compared to those of WT. Through quantitative modeling, these differences can be attributed to modulation of the MnIIA and FeIIB binding affinities in Y175F and E69D, respectively. Specifically, the Y175F mutation results in a MnIIA binding affinity greater than that of WT, while the E69D mutant exhibits a decrease in FeIIB binding affinity. The significantly slowed assembly observed in the E69D absorption profiles would be expected if indeed the FeIIB affinity were weaker, as it would take longer to install the FeII ion in site B, which remains necessary for productive cofactor assembly.
By examining the electronic and geometric structural information on these mutants, the molecular components that are likely responsible for these perturbations in binding affinity can be identified. Small changes in the g-,55 Mn, and 57Fe hyperfine tensors between WT and Y175F likely reflect perturbations to the active site geometry. In particular, the Y175F resting state EPR spectrum exhibits a decrease in the magnitude of hyperfine coupling along the unique axis (Table S1). In the resting state, the unique Jahn-Teller axis of the MnIII center was previously determined to lie along the Mn-aqua bond, the same ligand that is hydrogen bonded to the phenol group of Y175 in WT R2lox.11 It is possible that this hydrogen bonding network pulls the water ligand out of the octahedron, resulting in a more distorted geometry. This would better accommodate the Jahn-Teller distortion needed to stabilize the MnIII species of the resting state but may also disfavor initial binding of MnII. Indeed, in the WT R2lox crystal structures, the Mn-aqua distance increases from 2.0 to 2.4 Å upon oxidation of the metal cofactor.34 The Y175F mutation removes this hydrogen bonding partner to offer an initially more symmetric geometry. This increased symmetry results in a binding site more amenable to accommodating a MnII ion, which does not require Jahn-Teller distortion, giving rise to the increased MnII binding affinity for site A in Y175F compared with WT. The reduced hyperfine anisotropy in the Y175F mutant is consistent with this hypothesis and indicates that eliminating this hydrogen bond thus alleviates some of the distortion at the MnIII site. Furthermore, the subtle increase in MnII-aqua bond distance and movement of the water molecule toward the E69 residue observed in the reduced Y175F crystal structure indicates that the hydrogen bond to Y175 does indeed keep the aqua ligand in place in WT R2lox. These findings align with the kinetic modeling results, indicating greater MnII binding affinity in the case of a more symmetric environment around site A for the Y175F mutant.
In the E69D mutant, the ligand is truncated by one methylene group, which may increase the accessibility of the active site and enable more rapid exchange.34 Higher exchange rates would be expected to increase the metal “off” rate, resulting in a lower overall affinity. The perturbation seen in the reduced crystal structure of E69D, i.e., the shift in metal positions, particularly FeB, in the fully metalated subunit is consistent with a decrease in overall affinity of FeII binding to the B site. Simulations that included this variation were also consistent with the experimental trends, including increased t(A1/2) values and more shallow inhibition curves (Figure S17). A weaker FeII binding affinity would also explain the much slower assembly of E69D compared with WT and Y175F, as FeIIB binding becomes rate-limiting. While the aerobic E69D structure exhibits some modifications from WT, and the V72-Y162 ether cross-link is not crystallographically resolved, E69D R2lox does appear to have activated oxygen. The mass spectrometry data also provide clear evidence that the cross-link is formed under solution-phase conditions, albeit in lower quantities than WT.30 In the diiron R2a proteins, the N-terminal ligand is instead an aspartate, and the opposite mutation, i.e., D84E (E. coli R2a numbering), has been postulated to have a kinetically stabilizing effect, allowing observation of a bridging, μ−1,2-peroxo intermediate.63,64 Such a state is implicated as an intermediate in oxygen activation, and changes to the stability or accessibility of this higher valent species may impact cross-link formation. Mass spectrometry and spin quantitation data are consistent with this hypothesis. An altered reaction mechanism to produce the resting cofactor state of E69D is further supported by the SF and time-resolved UV-vis data, which appear noticeably different for E69D than either WT or Y175F. Conversely, the resting state E69D mutant displays almost identical EPR spectral features and spin Hamiltonian parameters as WT R2lox. Therefore, despite differences in relative metalation affinities and the O2 reduction process, the portion of E69D that does bind a Mn/Fe cofactor and supports O2 activation appears to do so without excessive change to the resting state electronic structure.
We note that differences in O2 binding ability between R2lox variants could also contribute in some way to the observed differences in inhibition trends. However, given that the changes between the mutants become substantially more pronounced as the MnII concentration increases, and O2 activation represents an irreversible process (vide supra), thermodynamic effects likely dominate the observed inhibition of assembly. Taken together, the crystallographic and EPR data demonstrate that the R2lox active site displays remarkable plasticity in the face of drastic alterations of the first and second ligand spheres, retaining the overall cofactor geometry by slight adjustments of the metal ligands.
Heterobimetallic Selectivity and Cofactor Reactivity Are Balanced in the WT R2lox Scaffold.
By examining changes in cross-link formation and reduction potentials in the R2lox mutants compared with WT levels, an assessment of how metalation specificity relates to redox activity can be made. Spin quantification data suggest that the low levels of cross-link observed in E69D, when compared with WT levels, can only partially be attributed to the overall lower efficiency of metalation in E69D compared with WT. While mass spectrometry indicates that the E69D mutant produces ~40% of the cross-linked peptide relative to that seen in WT R2lox, metalation occurs at a significantly lower extent in the E69D mutant, with only ~20% of the Mn/Fe cofactors formed relative to WT. We also note that even in WT Mn/Fe R2lox, assembly as measured by EPR is not quantitative, and investigation of other mutants of R2lox have shown that cross-link formation is not required to generate the EPR spectrum associated with the MnIII(μ-OH)FeIII state (Figure S30).33,65 We consider it likely that, as in the R2a proteins, a high-valent species in R2lox may be able to abstract electrons from FeII in solution,66 which would contribute to the measured EPR signal without necessitating cross-link formation. Thus, the 2-fold increase in cross-link seen in the E69D mutant relative to WT may be attributed to a higher degree of mechanistic coupling between Mn/Fe cofactor formation and oxidation of V72. On the other hand, Y175F forms a greater fraction of the MnIII(μ-OH)FeIII cofactor relative to WT, consistent with a higher MnII binding affinity, though the amount of cross-link formed is lower. This may be due to weaker correlation between cofactor assembly and cross-link formation in this mutant (Table S2), indicating greater potential for branched activation pathways.
The reactivity of WT R2lox with respect to the E69D and Y175F mutants can be better understood upon considering the electrochemistry data. The higher reduction potential of E69D may correlate with greater capacity for C-H bond activation, resulting in higher amounts of cross-link expected, while the lower reduction potential of Y175F relative to WT implies a less potent cofactor, with the lower potentials for the anodic peak at higher scan rates also suggesting a more facile oxidation process within this mutant. The differences in reactivity can likely be traced to subtle structural changes around the active site, reflected primarily in the hydrogen-bonding network around the MnA site (vide supra). Metal-bound water ligands have been proposed to play an important role in the homolytic cleavage of O2 in related protein active sites, e.g., in the diiron hydroxylase T4moHD complex, where hydrogen-bonding of the site A terminal water to a nearby glutamate facilitates the delivery of a proton for O-O bond homolysis.67 A similar role may be ascribed to the MnA-bound water in R2lox, which can propagate changes to the electronic structure of the entire cofactor.
CONCLUSIONS
The contrasting behavior between metal binding affinity and cross-link formation in the Y175F and E69D mutants with respect to WT R2lox highlight the many factors that must be optimized within a naturally occurring, heterobimetallic enzyme. The metal competition experiments in conjunction with global kinetic modeling and structural and spectroscopic analysis represent the first use of time-resolved methods to investigate questions of metal selectivity in R2lox. The resulting data indicate that R2lox follows a competitive and ordered scheme for metal binding. Simply put, MnII binds to site A in the protein with greater affinity than FeII does to site B, indicating assembly of the heterobimetallic active site to be under thermodynamic control. This occurs despite each site having identical coordinating atoms and very similar geometries, further indicating that fine control over metal binding affinity must be achieved. The subtle changes in hydrogen bonding networks between the mutants indicate the critical role that hydrogen bonding plays in controlling metalation, suggesting guidelines for design of synthetic compounds with metalation preferences that oppose the IW series.
The use of mutants has further provided a glimpse into the role that structural elements play in tuning not only metal binding affinity but also the interplay between affinity and reactivity. On one hand, a mutant displaying greater affinity for a Mn/Fe active site, likely owing to a more symmetric coordination environment for MnII, exhibits lower efficacy toward C-H bond activation and a correspondingly decreased reduction potential. Conversely, a mutant with weaker metal binding affinity is more effective for cross-link formation and, accordingly, displays an increased reduction potential. The WT coordination environment thus can be said to offer a compromise between metal binding and reactivity, coregulating the two properties to achieve a “Goldilocks” state. In summation, this work highlights the acute fine-tuning of metal-containing active sites in nature and begins to provide a molecular framework to understand the means by which proteins can encode for selective binding of metals that oppose conventional inorganic trends
Implications for In Vivo Metalation of R2lox.
These observations provide a glimpse into the intricate structural effects that contribute to naturally occurring metal selectivity. This work also offers the first evidence of how a biological system is able to defy the Irving-Williams series and allow MnII to bind to a protein with greater affinity than FeII within a nearly identical coordination environment, owing to careful control over the metal binding environments and hydrogen bonding network within the active site. While determining the relevant in vivo metallome of individual organisms is difficult, the free MnII concentrations within the cell are likely at the μM level, similar to the conditions used for solution-phase metalation experiments in this work.68,69 A higher MnII binding affinity may be necessary within a cellular environment to preferentially assemble a heterobimetallic over a homo-bimetallic cofactor. In the R2lox protein scaffold, the former is more effective for C-H bond activation and thus is thought to be the physiologically relevant state; moreover, the decreased Fe requirement of a Mn/Fe protein relative to a diiron site preserves this often-limiting nutrient for use in other processes. Ultimately, the insight gained from these findings will advance our understanding of how microorganisms use such finely tuned metalloproteins and can promote development of these systems toward accomplishing novel functions and reactivity.
EXPERIMENTAL SECTION
Expression and Purification of GkR2loxI.
Geobacillus kaustophilus (Gk) wild-type (WT) R2loxI (accession number WP_011232245.1) was heterologously expressed and purified in the metal-free (apo) form as previously described.27 Point mutations were introduced into the plasmid by site-directed mutagenesis using the QuikChange Lightning kit (Agilent, Santa Clara, CA) and verified by DNA sequencing (Genewiz, South Plainfield, NJ). All mutants were purified in the metal-free form according to the same protocols as WT R2lox and protein purity was checked via denaturing gel electrophoresis (Figure S29). Adjusted extinction coefficients were used for determination of Y175F concentrations based on a decrease in the ε280 value by 1490 M−1 cm−1, yielding ε280,apo = 46 267 M−1 cm−1 and ε280,holo = 49 073 M−1 cm−1.
Stopped-Flow (SF) and UV-Vis Metal Reconstitutions.
Apo-R2lox aliquots were thawed and diluted into 100 mM HEPES (Goldbio) buffer, pH 7.0, containing 50 mM NaCl (Fisher Scientific), subsequently referred to as “Buffer E”. Reconstitution of the metallocofactor was achieved via 1:1 mechanical mixing in the SF experiments, combining an aqueous metal solution of MnII and FeII with apo-R2lox to a final concentration of 100 μM protein. In the UV-vis experiments, the metallocofactor was reconstituted by rapid hand-mixing of a 25–50 μL aliquot of the aqueous metal solution into 700–725 μL of apo-R2lox to a final concentration of 100 μM protein. Metal stocks were made by dissolving MnCl2·4H2O (Amresco) and (NH4)2Fe(SO4)2·6H2O (Sigma-Aldrich) separately in deionized water to a concentration of 0.1–0.2 M, with further dilutions performed as necessary; fresh stocks were made each day. All solutions were air-saturated unless otherwise noted.
Single-wavelength SF experiments were performed in an Applied Photophysics (Surrey, U.K.) SX-20 apparatus with a dead time of 1 ms; the temperature was held at approximately 22 °C via a recirculating water bath. Kinetic data points were recorded in logarithmic time spacing over 500 s, with a total reaction volume of 125–135 μL per shot. Apo-R2lox and metal solutions were loaded into the instrument with 3 mL plastic syringes (Becton-Dickenson, Franklin Lakes, NJ). Metal syringes were loaded in order of increasing metal concentration, with the line flushed thoroughly with new sample prior to data collection. Because the I1 species previously reported at 620 nm has only low absolute absorbance values (<5 mAU) in the SF experiments, the subtle effects observed at other wavelengths (e.g., 320 and 390 nm) were unresolvable. Therefore, data were analyzed only at 320 and 390 nm.
UV-vis absorption spectra from 900 to 240 nm were recorded on a Shimazdu UV-2600 spectrophotometer using a fast scan speed, giving approximately one scan per minute; metal addition via rapid hand-mixing resulted in a dead time of approximately 1 min. The volume of metal solution used for mixing in the UV-vis competition experiments (25–50 μL) remained constant, with the concentrations of MnII or FeII in the aliquot varied. UV-vis difference absorption spectra were generated by subtracting the t = 60 min trace from earlier traces in time-resolved data sets.
Determination of Time at the Absorbance Half-Maximum (t(A1/2)).
SF data used to generate inhibition curves were the result of 2–4 independent competition experiments, with 1–3 technical repeats performed for each metal concentration and averaged together when possible. The standard deviation of each point was determined using data from no less than 2 competition experiments and contributions from at least 4 individual traces. Each competition data set was analyzed either by hand or through the use of an in-house Matlab (version R2018B) script by normalizing data traces and identifying the half-maximum absorbance across all traces. The time at which each data trace crosses this half-maximum absorbance line was then recorded (Figure S7). Inhibition curves were made by plotting these time points versus the MnII or FeII concentration. To account for situations where maximum absorbance did not occur on SF time scales, e.g., E69D SF traces, a representative UV-vis data set was scaled and overlaid with the SF data so that the kinetics of formation matched with those in the SF experiment. The t(A1/2) was then determined using the UV-vis absorbance maximum. These UV-vis overlays were also performed for WT data sets, even when clear maximum absorbances for WT were observed on the SF time scale, to ensure the validity of this analysis procedure. Overall, use of UV-vis data to determine the maximum absorbance resulted in longer times than those estimated from SF data alone; however, MnII inhibition trends remained the same regardless of which analysis procedure was performed (Figure S32A). To retain consistency across all mutants, the inhibition curves generated using only the SF data were selected for the primary analysis. The same analysis was performed including the UV-vis competition data overlaid with the SF data, with the model run to 3600 s. Identical trends were observed using this approach, albeit with the modeled inhibition curves all yielding higher t(A1/2) values (Figure S32B); therefore, we continue to use the model collected out to 500 s for comparison with the SF-only data.
Kinetic Modeling.
Kinetic modeling was performed using the KinTek software program (version 7.2.180216). The model used in this work is altered slightly from the KinTek model for Mn/Fe WT R2lox assembly previously developed.27 This model includes both divalent metal binding steps, loss of fatty acid coordination, oxygen binding, oxygen reduction to form high-valent intermediate species, intermediate decay with cross-link formation, and ultimate water loss with rebinding of the fatty acid to give the observed resting state species. Shown below are the equations representing the competitive/ordered model of metal binding, where either MnII or FeII may first bind in sites A or B, respectively, and productive cofactor maturation occurring only when binding FeIIB first and then MnIIA (eqs 1–4). Other scenarios for metal binding were also considered (Figure S11, insets) with the indicated adjustments made to the model shown below.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
| (7) |
| (8) |
| (9) |
| (10) |
When referencing the competitive/ordered binding model in this work, the rate constants used are described below unless otherwise stated. In anaerobically prepared crystals of reduced WT R2lox, the fatty acid (FA) ligand can be modeled in two conformations, either coordinating to both metals in a bidentate fashion with the E167 residue as a monodentate ligand to FeB, or unbound to the metals with E167 as a bidentate ligand to FeB; however, upon oxidation, the resting state crystal structures show the fatty acid definitively bound to the metals.34 This dynamic character is suggested to be a model for the transient structure that allows for O2 binding. Therefore, the rearrangement step required after O2 binding in our initial model is instead now modeled as an equilibrium of the fatty acid in a bound or unbound state to the metal center (eq 4). We had previously modeled the rearrangement step as very slow (0.005 s−1) but not reversible, as it occurred following O2 binding. In the new model, the ligand binding state is an equilibrium process (Keq = 0.1), as it occurs prior to O2 binding; however, it is also modeled as a relatively slow process compared to metal and O2 binding, with rates for ligand unbinding and binding of k3 = 10 s−1 and k−3 = 1 M−1 s−1, respectively.27
Prior rate constants were obtained from fitting modeled data to experimentally obtained kinetic traces for both I1 and I2; however, in the absence of additional information, we previously assigned both metal binding steps, as well as O2 binding, as diffusion-limited, with a binding affinity of 103 M−1 for both metals. In light of the extensive KinTek modeling we present here, it does not appear that the equilibrium constants for MnII and FeII are equal to one another (vide supra), while the rate of O2 binding appears to have an upper bound of k4 = 1 × 106 M−1 s−1 (Figure S14). Considering these adjustments, it is expected that the time scales of all downstream species will also be affected. While directly fitting any of the binding models to the experimental data was not possible without the use of scaling factors, described further below, it was possible to use the direct fitting routines in KinTek to bound the rates of formation and decay of the I2 species in a manner that reproduced the appropriate inhibitory trends. On the basis of this, formation of I2 from I1 is now modeled to be more rapid, with k5 = 10 s−1 rather than 0.12 s−1, and its decay is modeled to be slower, with k6 = 0.005 s−1 compared to 0.052 s−1.27 In our previous model, the rate of resting state formation from the penultimate intermediate species was determined by comparison to EPR kinetic data; however, the determination of absolute concentrations was precluded by overlapping signals from multiple species, including residual and unbound MnII, the EPR-active resting state, and multiple ST = 1/2 intermediate(s). Because of this, only an upper bound (0.12 s−1) was estimated.
For the adjusted model given in this work (vide supra), the resting state is modeled to form as the result of the fatty acid ligand rebinding the metal ions. This step was also simulated using an upper bound of the rate constant, which was found to be k9 = 10 M−1 s−1. Identifying useful spectroscopic handles for the reaction intermediates that lie between the decay of I2 and the formation of the final resting state represents ongoing work in our lab; the rates for these “cross-link intermediates” were maintained at k7 = k8 = 10 s−1, which were the rates assigned to the corresponding species in previous work.27
For the purposes of these simplified models, O2 binding is assumed to occur at the same rate for WT, Y175F, and E69D. Additional experiments exploring the effects of variable O2 concentration on assembly represent ongoing work in our lab. This research will provide complementary information on whether differential O2 binding rates contribute to distinct processes in Mn/Fe cofactor formation.
MnII-Only Reconstitutions of Apo-R2lox.
Treatment of apo-R2lox with MnII in the absence of FeII was performed in a similar manner as with both metals present. A solution of apo-R2lox was prepared from frozen stocks via dilution with Buffer E and divided into eight aliquots, 25–36 μL of an aqueous MnII solution were then added to each, and the solutions were hand-mixed with a pipet to give a final protein concentration of 300–400 μM. The concentrations of MnII present in each sample were adjusted by increasing the concentration of the added MnII solution rather than the volume. After mixing, samples were allowed to incubate in the dark for 1–2 h. Each sample was then passed through a HiTrap Desalting column (GE Healthcare, Chicago, IL) that had been equilibrated in Buffer D (25 mM HEPES, 50 mM NaCl, pH 7). Sample concentrations were determined immediately following desalting, and samples were frozen in quartz EPR tubes (Wilmad Lab-Glass 727-SQ-250M).
Preparation of Mn/Fe R2lox EPR Samples.
Solutions of concentrated (~1.0–1.6) mM apo-R2lox were prepared via dilution of protein stocks into O2-saturated Buffer E. Initial metal stocks of 0.1 M were prepared as described above; in samples prepared with 57Fe, frozen stocks of previously prepared27 10 mM 57FeII were used instead. These metal stocks were then diluted into O2-saturated, deionized water. Solutions of apo-R2lox and metals were hand-mixed in a 1:1 ratio, giving final concentrations of ~0.5–0.8 mM, with WT and Y175F containing 20% glycerol. Samples were prepared using equal amounts of sub-stoichiometric metals, with MnII and FeII final concentration set at either 0.5 or 0.75 equiv of apo-R2lox. The mixed solutions were stored at room temperature in the dark and allowed to react for at least 90 min prior to transferring into EPR tubes and freezing in liquid nitrogen. The E69D resting state sample reconstituted using 57Fe was desalted and reconcentrated prior to freezing due to additional MnII signal. Samples were loaded into quartz EPR tubes (either Wilmad Lab-Glass 727-SQ-250 M or Wilmad Lab-Glass 706-PQ-9.50).
EPR Spectroscopy.
CW X-band EPR spectra were collected between 5 and 10 K at either the Ohio Advanced EPR Facility (Miami University) using a Bruker EMX instrument equipped with an Oxford Instruments continuous flow helium cryostat and temperature controller (ESR 900), or in the Analytical Spectroscopy Lab (Ohio State University) using a Bruker EMXPlus instrument equipped with a Cold Edge cryogen-free helium cryostat and recirculation system and an Oxford Instruments MercuryITC temperature controller. All spectra were obtained using a microwave power of 20 mW and a modulation frequency and amplitude of 100 kHz and 10 G, respectively. Spin quantitation was carried out under non-saturating conditions using a copper(II) azurin standard of known concentration, also measured the same day under non-saturating conditions. Spin quantitation numbers of Y175F and E69D were normalized to WT values (37% ± 1.4% of the total possible amount of Mn/Fe cofactor formed).30 Spin quantitation errors are reported as SEM. Spectra were corrected for baseline cavity signals using a spline subtraction in Igor Pro (Wavemetrics, Lake Oswego, OR) data analysis software. For MnII-only loaded samples, the data were normalized for concentration prior to baseline correction. EPR spectral simulations were carried out using the EasySpin (version 5.2.25) toolbox within Matlab.70 Baseline and MnII contributions were subtracted from the E69D NAFe spectrum prior to simulation.
Crystallization and Data Collection.
E69D and Y175F R2lox were crystallized in metal-free form by vapor diffusion in hanging drops at 295 K and yielded well-ordered single diamond-shaped crystals, like the wild-type protein, in essentially the same condition as the wild-type (27.5–30% (w/v) PEG 1500, 100 mM HEPES-Na pH 7.4–7.7). To reconstitute the oxidized resting state Mn/Fe cofactor, crystals of metal-free protein were removed from their drop and soaked in mother liquor additionally containing 5 mM each MnCl2 and (NH4)2Fe(SO4)2 for 1–2 h under aerobic conditions and then briefly washed in 40% (w/v) PEG 1500, 100 mM HEPES-Na (at the pH of the mother liquor) before flash-cooling in liquid nitrogen. To obtain the non-activated reduced Mn/Fe cofactor, apo-protein crystals were soaked in 1 mL of 40% (w/v) PEG 1500, 100 mM HEPES-Na (at the pH of the mother liquor), 5 mM (NH4)2Fe(SO4)2, 5 mM MnCl2, 0.5% (w/v) sodium dithionite, 0.5 mM phenosafranin, and 0.05% (v/v) Tween 20 for 1–2 h and flash-cooled directly without washing.10 Soaking solutions were freshly prepared immediately before use, using freshly dissolved (NH4)2Fe(SO4)2 and dithionite to ensure that the Fe remained in the ferrous state, and that O2 was effectively removed from soaking solutions used to obtain reduced states, with phenosafranin serving as a redox indicator. Data were collected at 100 K at beamline X06SA at the Swiss Light Source (SLS, Villigen, Switzerland).
Structure Determination, Model Building, and Refinement.
Data were processed with XDS.71 Structures of R2lox variants were solved using the structure of the WT protein in the same redox state10 not containing any ligands as a starting model. Crystals of the oxidized states of E69D and Y175F R2lox were obtained in the same space group as the WT, I222, with one molecule in the asymmetric unit, and these structures were consequently solved by Fourier synthesis. Reduced state E69D R2lox crystals were in space group C2, and reduced state Y175F R2lox crystals were in space group P21212, both with two molecules in the asymmetric unit (see Table S3). These structures were solved by molecular replacement using Phaser in Phenix.72,73 Refinement was carried out with phenix.refine72,74 and iterated with rebuilding in Coot.75 Refinement generally included bulk solvent corrections, individual atomic coordinate and isotropic B factor refinement, and occupancy refinement for alternate conformations and metal ions bound on the protein surface, but not the active site metal ions. Metal-ligand bond lengths were restrained. Solvent molecules were added with phenix.refine and manually. Hydrogens were added to the models in the later stages of refinement. The following exceptions were made to this general protocol in the case of reduced state E69D R2lox: The occupancy of the active site metal ions was refined in one of the two chains in the asymmetric unit, and torsion angle NCS restraints were used. Structures were validated using MolProbity.76 Data and refinement statistics are given in Tables S3 and S4. All figures were prepared with PyMOL (version 1.8.6.2, Schrodinger, LLC).
Mass Spectrometry and Electrochemistry Sample Preparation.
For mass spectrometry and electrochemistry sample preparation, samples were protected from light as much as possible.65 For each variant, three replicates of 100 μM apo-R2lox in Buffer E were incubated with 2 equiv of MnII and 1 equiv of FeII for 2–6 h at room temperature; apo-R2lox samples were incubated with equivalent volumes of metal-free deionized water. Excess metal ions were removed by passing protein through a HiTrap Desalting column equilibrated in Buffer D. The reconstituted protein was concentrated to 150–500 μM, and mass spectrometry samples were frozen for further analysis. Using the same protocol, another set of samples was prepared and frozen in EPR tubes (Wilmad Lab-Glass 727-SQ-250M) for use in determining Mn/Fe metalation efficiency for each variant. The total concentration of Mn/Fe cofactor, determined via EPR spin-quantification, was compared to the total protein concentration of the sample, which was determined prior to freezing. Due to differences in ionization efficiencies across different peptide fragments, it is not possible to correlate absolute concentrations across the M/S and EPR methods.
Mass Spectrometry Analysis.
The denatured protein samples were prepared for and analyzed by LC-MS similarly to Griese et al.44 From each sample, 80 μg of protein were subjected to proteolytic digestion by Glu-C (Promega, enzyme:substrate ratio 1:40) in 50 mM phosphate buffer, pH 7.6, using the SP3 sample preparation method.77 From each replicate, 300 μL of digested sample was collected and acidified by addition of 100 μL 10% formic acid (FA) prior to LC-MS.
The auto sampler of a HPLC 1200 system (Agilent Technologies) injected 1 μL (approximately 200 ng of peptides) into a C18 guard desalting column (Zorbax 300SB-C18, 5 × 0.3 mm, 5 μm bead size, Agilent). Then, a 15 cm long C18 picofrit column (100 μm internal diameter, 5 μm bead size, Nikkyo Technos Co., Tokyo, Japan) installed onto the nano electrospray ionization source was used. Solvent A was 97% water, 3% acetonitrile (ACN), 0.1% FA; solvent B was 5% water, 95% ACN, 0.1% FA. At a constant flow of 0.4 μL min−1, a linear gradient went from 2% B up to 40% B in 45 min, followed by a steep increase to 100% B in 5 min, plateau at 100% B for 5 min, and subsequent re-equilibration with 2% B. Online LC-MS was performed using a LTQ Orbitrap Velos Pro mass spectrometer (Thermo Scientific). FTMS master scans (AGC target of 1e6) were acquired with a resolution of 30 000 and were followed by data-dependent MS/MS (AGC target of 1e5) at a resolution of 7500. In data-dependent MS/MS, the top two ions from the master scan were selected first for collision induced dissociation (at 35% energy) and afterward for higher energy collision dissociation (at 30% energy). Precursors were isolated with a 2 m/z window. Dynamic exclusion was used with 2 repeat counts and 30 s duration. Each sample was analyzed in technical triplicates.
To assist in peptide identification, searches of all MS/MS spectra by Sequest were performed using the software platform Proteome Discoverer (v1.4, Thermo Scientific) against the R2lox primary sequence. A precursor mass tolerance of 10 ppm and product mass tolerance of 0.02 Da were used. Oxidation of methionine was considered as variable modification. The proteolysis enzyme used was Glu-C, with up to 2 missed cleavages allowed. PD1.4 can calculate protein areas based on the MS1 peak area integration of the three most abundant identified peptides. The MS1 peak area in each LCMS run of m/z 689.038, z = 3 (which corresponds to the cross-linked peptide AVIRAATVYNMIVEAVTLD), normalized to the protein area of the same run, was used as surrogate marker for the amount of cross-link in the samples.
Electrochemical Data Collection and Analysis.
Cyclic voltammetry electrochemistry experiments were conducted using a CHI 760E potentiostat (CH Instruments). Experiments were performed under anaerobic conditions within a Vigor Technologies glovebox (O2 < 1 ppm). A three-electrode system was employed using a 3 mm glassy carbon working electrode (CH Instruments), a platinum wire counter electrode, and a Ag/AgCl (sat. KCl) reference electrode. Prior to each experiment, the glassy carbon working electrode was polished using 1.0 μm alumina powder, extensively rinsed with deionized water, and then polished with 0.05 μm alumina powder (CH Instruments). The electrode was again extensively rinsed with deionized water before sonicating for 3 min. Samples were prepared immediately prior to experimentation. R2lox stock solutions were diluted to a final concentration of 100 μM using a buffer of 100 mM HEPES with 100 mM NaCl, pH 7.0.
Potential values were reported against NHE by the addition of +198 mV to the experimentally determined value. All analysis was performed using IGOR Pro 8. The cyclic voltammogram (CV) of the corresponding apo-R2lox control was subtracted from the CV of the metalated sample. To resolve the anodic and cathodic potentials for the Mn/Fe samples, the derivatives of the apo-subtracted CVs were used. The cathodic peak position was defined as the point where the derivative crosses zero. The peak potential for the anodic process was defined as the halfway point between the peak and trough of the derivative due to large background slopes at higher potentials. This analysis procedure is illustrated in greater detail in the Supporting Information (Figure S33).
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Högbom lab and the staff at beamline X06SA/SLS for assistance with X-ray data collection, and members of the Shafaat lab for helpful discussions. This work was supported by the National Institutes of Health R35 GM-128852 grant to H.S.S., a Pelotonia Graduate Fellowship from The Ohio State University to E.C.K., a Presidential Fellowship from The Ohio State University Graduate School to C.R.S., the European Community’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 283570 (for BioStruct-X), the Swedish Research Council (2017-04018), the European Research Council (HIGH-GEAR 724394), and the Knut and Alice Wallenberg Foundation (Wallenberg Academy Fellows (2017.0275)) to M.H., and the Swedish Research Council (2016-03770) to J.J.G.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c00333.
Detailed experimental methods, raw optical spectroscopy data, supplemental kinetic models, EPR spectra, electrochemical workup, and supplemental structure and crystallographic statistics, including Figures S1–S33 and Tables S1–S4 (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c00333
The authors declare no competing financial interest.
Contributor Information
Effie C. Kisgeropoulos, The Ohio State Biochemistry Program, The Ohio State University, Columbus, Ohio 43210, United States;.
Julia J. Griese, Department of Biochemistry and Biophysics, Stockholm University, SE-106 91 Stockholm, Sweden; Department of Cell and Molecular Biology, Uppsala University, SE-751 24 Uppsala, Sweden;.
Zachary R. Smith, Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, United States
Rui M. M. Branca, Department of Oncology-Pathology, Science for Life Laboratory, Karolinska Institutet, SE-171 21 Solna, Sweden
Camille R. Schneider, The Ohio State Biochemistry Program, The Ohio State University, Columbus, Ohio 43210, United States
Martin Högbom, Department of Biochemistry and Biophysics, Stockholm University, SE-106 91 Stockholm, Sweden;.
Hannah S. Shafaat, The Ohio State Biochemistry Program and Department of Chemistry and Biochemistry, The Ohio State University, Columbus, Ohio 43210, United States;.
REFERENCES
- (1).Cox N; Pantazis DA; Neese F; Lubitz W Biological Water Oxidation. Acc. Chem. Res 2013, 46 (7), 1588–1596. [DOI] [PubMed] [Google Scholar]
- (2).Adam SM; Wijeratne GB; Rogler PJ; Diaz DE; Quist DA; Liu JJ; Karlin KD Synthetic Fe/Cu Complexes: Toward Understanding Heme-Copper Oxidase Structure and Function. Chem. Rev 2018, 118 (22), 10840–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lubitz W; Ogata H; Rüdiger O; Reijerse E Hydrogenases. Chem. Rev 2014, 114 (8), 4081–4148. [DOI] [PubMed] [Google Scholar]
- (4).Einsle O; Tezcan FA; Andrade SLA; Schmid B; Yoshida M; Howard JB; Rees DC Nitrogenase MoFe-Protein at 1.16 A Resolution: A Central Ligand in the FeMo-Cofactor. Science 2002, 297 (5587), 1696–1700. [DOI] [PubMed] [Google Scholar]
- (5).Jiang W; Yun D; Saleh L; Barr EW; Xing G; Hoffart LM; Maslak M-A; Krebs C; Bollinger JM A Manganese(IV)/Iron(III) Cofactor in Chlamydia Trachomatis Ribonucleotide Reductase. Science 2007, 316 (5828), 1188–1191. [DOI] [PubMed] [Google Scholar]
- (6).Andersson CS; Högbom M A Mycobacterium Tuberculosis Ligand-Binding Mn/Fe Protein Reveals a New Cofactor in a Remodeled R2-Protein Scaffold. Proc. Natl. Acad. Sci. U. S. A 2009, 106 (14), 5633–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Högbom M Metal Use in Ribonucleotide Reductase R2, Di-Iron, Di-Manganese and Heterodinuclear—an Intricate Bioinorganic Workaround to Use Different Metals for the Same Reaction. Metallomics 2011, 3 (2), 110–120. [DOI] [PubMed] [Google Scholar]
- (8).Högbom M The Manganese/Iron-Carboxylate Proteins: What Is What, Where Are They, and What Can the Sequences Tell Us? JBIC, J. Biol. Inorg. Chem 2010, 15 (3), 339–349. [DOI] [PubMed] [Google Scholar]
- (9).Schmidt F; Donahoe S; Hagens K; Mattow J; Schaible UE; Kaufmann SHE; Aebersold R; Jungblut PR Complementary Analysis of the Mycobacterium Tuberculosis Proteome by Two-Dimensional Electrophoresis and Isotope-Coded Affinity Tag Technology. Mol. Cell. Proteomics 2004, 3 (1), 24–42. [DOI] [PubMed] [Google Scholar]
- (10).Griese JJ; Roos K; Cox N; Shafaat HS; Branca RMM; Lehtiö J; Gräslund A; Lubitz W; Siegbahn PEM; Högbom M Direct Observation of Structurally Encoded Metal Discrimination and Ether Bond Formation in a Heterodinuclear Metalloprotein. Proc. Natl. Acad. Sci. U. S. A 2013, 110 (43), 17189–17194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Shafaat HS; Griese JJ; Pantazis DA; Roos K; Andersson CS; Popović-Bijelić A; Gräslund A; Siegbahn PEM; Neese F; Lubitz W; Högbom M; Cox N Electronic Structural Flexibility of Heterobimetallic Mn/Fe Cofactors: R2lox and R2c Proteins. J. Am. Chem. Soc 2014, 136 (38), 13399–13409. [DOI] [PubMed] [Google Scholar]
- (12).Lee SK; Nesheim JC; Lipscomb JD Transient Intermediates of the Methane Monooxygenase Catalytic Cycle. J. Biol. Chem 1993, 268 (29), 21569–21577. [PubMed] [Google Scholar]
- (13).Lee SK; Fox BG; Froland WA; Lipscomb JD; Münck E A Transient Intermediate of the Methane Monooxygenase Catalytic Cycle Containing an FeIVFeIV Cluster. J. Am. Chem. Soc 1993, 115 (14), 6450–6451. [Google Scholar]
- (14).Liu KE; Wang D; Huynh BH; Edmondson DE; Salifoglou A; Lippard SJ Spectroscopic Detection of Intermediates in the Reaction of Dioxygen with the Reduced Methane Monooxygenase/Hydroxylase from Methylococcus Capsulatus (Bath). J. Am. Chem. Soc 1994, 116 (16), 7465–7466. [Google Scholar]
- (15).Brazeau BJ; Lipscomb JD Kinetics and Activation Thermodynamics of Methane Monooxygenase Compound Q Formation and Reaction with Substrates. Biochemistry 2000, 39 (44), 13503–13515. [DOI] [PubMed] [Google Scholar]
- (16).Tinberg CE; Lippard SJ Dioxygen Activation in Soluble Methane Monooxygenase. Acc. Chem. Res 2011, 44 (4), 280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Korboukh VK; Li N; Barr EW; Bollinger JM Jr.; Krebs C A Long-Lived, Substrate-Hydroxylating Peroxodiiron(III/III) Intermediate in the Amine Oxygenase, AurF, from Streptomyces Thioluteus. J. Am. Chem. Soc 2009, 131 (38), 13608–13609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Winkler R; Hertweck C Sequential Enzymatic Oxidation of Aminoarenes to Nitroarenes via Hydroxylamines. Angew. Chem., Int. Ed 2005, 44 (26), 4083–4087. [DOI] [PubMed] [Google Scholar]
- (19).Simurdiak M; Lee J; Zhao H A New Class of Arylamine Oxygenases: Evidence That p-Aminobenzoate N-Oxygenase (AurF) Is a Di-Iron Enzyme and Further Mechanistic Studies. ChemBioChem 2006, 7 (8), 1169–1172. [DOI] [PubMed] [Google Scholar]
- (20).Park K; Li N; Kwak Y; Srnec M; Bell CB; Liu LV; Wong SD; Yoda Y; Kitao S; Seto M; Hu M; Zhao J; Krebs C; Bollinger JM Jr.; Solomon EI Peroxide Activation for Electrophilic Reactivity by the Binuclear Non-Heme Iron Enzyme AurF. J. Am. Chem. Soc 2017, 139 (20), 7062–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lu H; Chanco E; Zhao H CmlI Is an N-Oxygenase in the Biosynthesis of Chloramphenicol. Tetrahedron 2012, 68 (37), 7651–7654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Makris TM; Vu VV; Meier KK; Komor AJ; Rivard BS; Münck E; Que L; Lipscomb JD An Unusual Peroxo Intermediate of the Arylamine Oxygenase of the Chloramphenicol Biosynthetic Pathway. J. Am. Chem. Soc 2015, 137 (4), 1608–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Jasniewski AJ; Komor AJ; Lipscomb JD; Que L Unprecedented (μ−1,1-Peroxo)Diferric Structure for the Ambiphilic Orange Peroxo Intermediate of the Nonheme N-Oxygenase CmlI. J. Am. Chem. Soc 2017, 139 (30), 10472–10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Wang C; Chen H Convergent Theoretical Prediction of Reactive Oxidant Structures in Diiron Arylamine Oxygenases AurF and CmlI: Peroxo or Hydroperoxo? J. Am. Chem. Soc 2017, 139 (37), 13038–13046. [DOI] [PubMed] [Google Scholar]
- (25).Irving H; Williams RJP The Stability of Transition-Metal Complexes. J. Chem. Soc 1953, 3192–3210. [Google Scholar]
- (26).Gorelsky SI; Basumallick L; Vura-Weis J; Sarangi R; Hodgson KO; Hedman B; Fujisawa K; Solomon EI Spectroscopic and DFT Investigation of [M{HB(3,5-IPr2pz)3}-(SC6F5)] (M = Mn, Fe, Co, Ni, Cu, and Zn) Model Complexes: Periodic Trends in Metal-Thiolate Bonding. Inorg. Chem 2005, 44 (14), 4947–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Miller EK; Trivelas NE; Maugeri PT; Blaesi EJ; Shafaat HS Time-Resolved Investigations of Heterobimetallic Cofactor Assembly in R2lox Reveal Distinct Mn/Fe Intermediates. Biochemistry 2017, 56 (26), 3369–3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Högbom M; Stenmark P; Voevodskaya N; McClarty G; Gräslund A; Nordlund P The Radical Site in Chlamydial Ribonucleotide Reductase Defines a New R2 Subclass. Science 2004, 305 (5681), 245–248. [DOI] [PubMed] [Google Scholar]
- (29).Sazinsky MH; Lippard SJ Correlating Structure with Function in Bacterial Multicomponent Monooxygenases and Related Diiron Proteins. Acc. Chem. Res 2006, 39 (8), 558–566. [DOI] [PubMed] [Google Scholar]
- (30).Kutin Y; Srinivas V; Fritz M; Kositzki R; Shafaat HS; Birrell J; Bill E; Haumann M; Lubitz W; Högbom M; Griese JJ; Cox N Divergent Assembly Mechanisms of the Manganese/Iron Cofactors in R2lox and R2c Proteins. J. Inorg. Biochem 2016, 162, 164–177. [DOI] [PubMed] [Google Scholar]
- (31).Griese JJ; Srinivas V; Högbom M Assembly of Nonheme Mn/Fe Active Sites in Heterodinuclear Metalloproteins. JBIC, J. Biol. Inorg. Chem 2014, 19 (6), 759–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kositzki R; Mebs S; Marx J; Griese JJ; Schuth N; Högbom M; Schünemann V; Haumann M Protonation State of MnFe and FeFe Cofactors in a Ligand-Binding Oxidase Revealed by X-Ray Absorption, Emission, and Vibrational Spectroscopy and QM/MM Calculations. Inorg. Chem 2016, 55 (19), 9869–9885. [DOI] [PubMed] [Google Scholar]
- (33).Griese JJ; Kositzki R; Haumann M; Högbom M Assembly of a Heterodinuclear Mn/Fe Cofactor Is Coupled to Tyrosine–Valine Ether Cross-Link Formation in the R2-like Ligand-Binding Oxidase. JBIC, J. Biol. Inorg. Chem 2019, 24 (2), 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Griese JJ; Kositzki R; Schrapers P; Branca RMM; Nordström A; Lehtiö J; Haumann M; Högbom M Structural Basis for Oxygen Activation at a Heterodinuclear Manganese/Iron Cofactor. J. Biol. Chem 2015, 290 (42), 25254–25272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cotruvo JA Jr.; Stubbe J Metallation and Mismetallation of Iron and Manganese Proteins in Vitro and in Vivo: The Class I Ribonucleotide Reductases as a Case Study. Met. Integr. Biometal Sci 2012, 4 (10), 1020–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Dassama LMK; Krebs C; Bollinger JM Jr.; Rosenzweig AC; Boal AK Structural Basis for Assembly of the MnIV/FeIII Cofactor in the Class Ic Ribonucleotide Reductase from Chlamydia Trachomatis. Biochemistry 2013, 52 (37), 6424–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Carboni M; Latour J-M Enzymes with an Heterodinuclear Iron–Manganese Active Site: Curiosity or Necessity? Coord. Chem. Rev 2011, 255 (1–2), 186–202. [Google Scholar]
- (38).Ardini M; Fiorillo A; Fittipaldi M; Stefanini S; Gatteschi D; Ilari A; Chiancone E Kineococcus Radiotolerans Dps Forms a Heteronuclear Mn–Fe Ferroxidase Center That May Explain the Mn-Dependent Protection against Oxidative Stress. Biochim. Biophys. Acta, Gen. Subj 2013, 1830 (6), 3745–3755. [DOI] [PubMed] [Google Scholar]
- (39).Mitić N; Noble CJ; Gahan LR; Hanson GR; Schenk G Metal-Ion Mutagenesis: Conversion of a Purple Acid Phosphatase from Sweet Potato to a Neutral Phosphatase with the Formation of an Unprecedented Catalytically Competent MnIIMnII Active Site. J. Am. Chem. Soc 2009, 131 (23), 8173–8179. [DOI] [PubMed] [Google Scholar]
- (40).Jiang W; Hoffart LM; Krebs C; Bollinger JM Jr. A Manganese(IV)/Iron(IV) Intermediate in Assembly of the Manganese(IV)/Iron(III) Cofactor of Chlamydia Trachomatis Ribonucleotide Reductase. Biochemistry 2007, 46 (30), 8709–8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Kutin Y; Kositzki R; Branca RMM; Srinivas V; Lundin D; Haumann M; Högbom M; Cox N; Griese JJ Chemical Flexibility of Heterobimetallic Mn/Fe Cofactors: R2lox and R2c Proteins. J. Biol. Chem 2019, 294 (48), 18372–18386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Johnson KA F itting Enzyme Kinetic Data with KinTek Global Kinetic Explorer In Computer Methods Part B; Johnson ML, Brand L, Eds.; Methods in Enzymology 467; Elsevier, 2009; pp 601–626. [DOI] [PubMed] [Google Scholar]
- (43).Johnson KA; Simpson ZB; Blom T FitSpace Explorer: An Algorithm to Evaluate Multidimensional Parameter Space in Fitting Kinetic Data. Anal. Biochem 2009, 387 (1), 30–41. [DOI] [PubMed] [Google Scholar]
- (44).Griese JJ; Branca RMM; Srinivas V; Högbom M Ether Cross-Link Formation in the R2-like Ligand-Binding Oxidase. JBIC, J. Biol. Inorg. Chem 2018, 23 (6), 879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Huang X; Groves JT Beyond Ferryl-Mediated Hydroxylation: 40 Years of the Rebound Mechanism and C–H Activation. JBIC, J. Biol. Inorg. Chem 2017, 22 (2–3), 185–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Lippard SJ Hydroxylation of C–H Bonds at Carboxylate-Bridged Diiron Centres. Philos. Trans. R. Soc., A 2005, 363 (1829), 861–877. [DOI] [PubMed] [Google Scholar]
- (47).Armstrong FA; Hill HAO; Oliver BN; Walton NJ Direct Electrochemistry of Redox Proteins at Pyrolytic Graphite Electrodes. J. Am. Chem. Soc 1984, 106 (4), 921–923. [Google Scholar]
- (48).Armstrong FA; Hirst J Reversibility and Efficiency in Electrocatalytic Energy Conversion and Lessons from Enzymes. Proc. Natl. Acad. Sci. U. S. A 2011, 108 (34), 14049–14054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Bernhardt PV; Schenk G; Wilson GJ Direct Electrochemistry of Porcine Purple Acid Phosphatase (Uteroferrin). Biochemistry 2004, 43 (32), 10387–10392. [DOI] [PubMed] [Google Scholar]
- (50).Schneider CR; Lewis LC; Shafaat HS The Good, the Neutral, and the Positive: Buffer Identity Impacts CO2 Reduction Activity by Nickel(II) Cyclam. Dalton Trans. 2019, 48 (42), 15810–15821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Warren JJ; Tronic TA; Mayer JM Thermochemistry of Proton-Coupled Electron Transfer Reagents and Its Implications. Chem. Rev 2010, 110 (12), 6961–7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Rose HR; Ghosh MK; Maggiolo AO; Pollock CJ; Blaesi EJ; Hajj V; Wei Y; Rajakovich LJ; Chang W; Han Y; Hajj M; Krebs C; Silakov A; Pandelia M-E; Bollinger JM Jr.; Boal AK Structural Basis for Superoxide Activation of Flavobacterium Johnsoniae Class I Ribonucleotide Reductase and for Radical Initiation by Its Dimanganese Cofactor. Biochemistry 2018, 57 (18), 2679–2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Stubbe J Di-Iron-Tyrosyl Radical Ribonucleotide Reductases. Curr. Opin. Chem. Biol 2003, 7 (2), 183–188. [DOI] [PubMed] [Google Scholar]
- (54).Nordlund P; Reichard P Ribonucleotide Reductases. Annu. Rev. Biochem 2006, 75 (1), 681–706. [DOI] [PubMed] [Google Scholar]
- (55).Cotruvo JA; Stubbe J An Active Dimanganese(III)-Tyrosyl Radical Cofactor in Escherichia Coli Class Ib Ribonucleotide Reductase. Biochemistry 2010, 49 (6), 1297–1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Cotruvo JA; Stich TA; Britt RD; Stubbe J Mechanism of Assembly of the Dimanganese-Tyrosyl Radical Cofactor of Class Ib Ribonucleotide Reductase: Enzymatic Generation of Superoxide Is Required for Tyrosine Oxidation via a Mn(III)Mn(IV) Intermediate. J. Am. Chem. Soc 2013, 135 (10), 4027–4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Bollinger JM Jr.; Jiang W; Green MT; Krebs C The Manganese(IV)/Iron(III) Cofactor of Chlamydia Trachomatis Ribonucleotide Reductase: Structure, Assembly, Radical Initiation, and Evolution. Curr. Opin. Struct. Biol 2008, 18 (6), 650–657. [DOI] [PubMed] [Google Scholar]
- (58).Roos K; Siegbahn PEM A Comparison of Two-Electron Chemistry Performed by the Manganese and Iron Heterodimer and Homodimers. JBIC, J. Biol. Inorg. Chem 2012, 17 (3), 363–373. [DOI] [PubMed] [Google Scholar]
- (59).Boal AK; Cotruvo JA; Stubbe J; Rosenzweig AC Structural Basis for Activation of Class Ib Ribonucleotide Reductase. Science 2010, 329 (5998), 1526–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Högbom M; Galander M; Andersson M; Kolberg M; Hofbauer W; Lassmann G; Nordlund P; Lendzian F Displacement of the Tyrosyl Radical Cofactor in Ribonucleotide Reductase Obtained by Single-Crystal High-Field EPR and 1.4-Å X-Ray Data. Proc. Natl. Acad. Sci. U. S. A 2003, 100 (6), 3209–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Martinie RJ; Blaesi EJ; Krebs C; Bollinger JM Jr.; Silakov A; Pollock CJ Evidence for a Di-μ-Oxo Diamond Core in the Mn(IV)/Fe(IV) Activation Intermediate of Ribonucleotide Reductase from Chlamydia Trachomatis. J. Am. Chem. Soc 2017, 139 (5), 1950–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Rosenzweig AC; Brandstetter H; Whittington DA; Nordlund P; Lippard SJ; Frederick CA Crystal Structures of the Methane Monooxygenase Hydroxylase from Methylococcus Capsulatus (Bath): Implications for Substrate Gating and Component Interactions. Proteins: Struct., Funct., Genet 1997, 29 (2), 141–152. [PubMed] [Google Scholar]
- (63).Moënne-Loccoz P; Baldwin J; Ley BA; Loehr TM; Bollinger JM Jr. O2 Activation by Non-Heme Diiron Proteins: Identification of a Symmetric μ−1,2-Peroxide in a Mutant of Ribonucleotide Reductase. Biochemistry 1998, 37 (42), 14659–14663. [DOI] [PubMed] [Google Scholar]
- (64).Bollinger JM Jr.; Krebs C; Vicol A; Chen S; Ley BA; Edmondson DE; Huynh BH Engineering the Diiron Site of Escherichia Coli Ribonucleotide Reductase Protein R2 to Accumulate an Intermediate Similar to H peroxo, the Putative Peroxodiiron(III) Complex from the Methane Monooxygenase Catalytic Cycle. J. Am. Chem. Soc 1998, 120 (5), 1094–1095. [Google Scholar]
- (65).Maugeri PT; Griese JJ; Branca RM; Miller EK; Smith ZR; Eirich J; Högbom M; Shafaat HS Driving Protein Conformational Changes with Light: Photoinduced Structural Rearrangement in a Heterobimetallic Oxidase. J. Am. Chem. Soc 2018, 140 (4), 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Bollinger JM Jr.; Tong WH; Ravi N; Huynh BH; Edmonson DE; Stubbe J Mechanism of Assembly of the Tyrosyl Radical-Diiron (III) Cofactor of E. Coli Ribonucleotide Reductase. 2. Kinetics of the Excess Fe2+ Reaction by Optical, EPR, and Mössbauer Spectroscopies. J. Am. Chem. Soc 1994, 116 (18), 8015–8023. [Google Scholar]
- (67).Acheson JF; Bailey LJ; Brunold TC; Fox BG In-Crystal Reaction Cycle of a Toluene-Bound Diiron Hydroxylase. Nature 2017, 544 (7649), 191–195. [DOI] [PubMed] [Google Scholar]
- (68).Dudev T; Lim C Competition among Metal Ions for Protein Binding Sites: Determinants of Metal Ion Selectivity in Proteins. Chem. Rev 2014, 114 (1), 538–556. [DOI] [PubMed] [Google Scholar]
- (69).Osman D; Martini MA; Foster AW; Chen J; Scott AJP; Morton RJ; Steed JW; Lurie-Luke E; Huggins TG; Lawrence AD; Deery E; Warren MJ; Chivers PT; Robinson NJ Bacterial Sensors Define Intracellular Free Energies for Correct Enzyme Metalation. Nat. Chem. Biol 2019, 15 (3), 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Stoll S; Schweiger A EasySpin, a Comprehensive Software Package for Spectral Simulation and Analysis in EPR. J. Magn. Reson 2006, 178 (1), 42–55. [DOI] [PubMed] [Google Scholar]
- (71).Kabsch W XDS. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (2), 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: A Comprehensive Python-Based System for Macromolecular Structure Solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (2), 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser Crystallographic Software. J. Appl. Crystallogr 2007, 40 (4), 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Afonine PV; Grosse-Kunstleve RW; Echols N; Headd JJ; Moriarty NW; Mustyakimov M; Terwilliger TC; Urzhumtsev A; Zwart PH; Adams PD Towards Automated Crystallographic Structure Refinement with Phenix. Acta Crystallogr., Sect. D: Biol. Crystallogr 2012, 68 (4), 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and Development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Chen VB; Arendall WB; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: All-Atom Structure Validation for Macromolecular Crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66 (1), 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Hughes CS; Foehr S; Garfield DA; Furlong EE; Steinmetz LM; Krijgsveld J Ultrasensitive Proteome Analysis Using Paramagnetic Bead Technology. Mol. Syst. Biol 2014, 10 (10), 757. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.