

Abstract
Clinical trials are a fundamental component of medical research and serve as the main route to obtain evidence of the safety and efficacy of a treatment before its approval. A trial’s ability to provide the intended evidence hinges on appropriate design, from background knowledge and trial rationale to sample size and interim monitoring rules. In this article, we present some general design principles for investigators and their research teams to consider when planning to conduct a trial.
Keywords: principles, clinical trial, design
Introduction.
Clinical trials are a fundamental component of medical research. Before any treatment is approved and offered to patients in the general population, rigorous evidence of its safety and efficacy must be shown. Clinical trials are the main route to obtain this required evidence. In this article, we present some general principles of good clinical trial design, which are often used as the basis to evaluate the quality of the evidence presented in manuscripts reporting trial results. By trial “design,” we include aspects from background knowledge and trial rationale to sample size and interim monitoring rules. Given that mistakes in design can seldom be later rectified, we strongly encourage investigators to consider these guidelines before beginning a study.
The critical component for a successful design is the relationship among the different members of the scientific team. This is important because each person on the team contributes their area of expertise to come up with a feasible study that meets the scientific hypothesis. It is crucial to involve statisticians in the very early stage of the study design instead of waiting to involve them at the time of data analysis. Not only can statisticians help with assessing the design parameters and calculating the sample size needed to address the study aims, they also ensure that the statistical hypotheses appropriately align with the study objectives and that the corresponding statistical analyses are correctly applied. Note that it is very difficult to fix a poorly-designed study once it is implemented.
The design process of a clinical trial is iterative in nature with some of the steps being inherently connected to others, but it can be helpful to divide the process into two phases – conceptual planning and implementation (Figure 1). The conceptual planning phase includes establishing prior knowledge/background, thinking through the rationale for the proposed trial as it relates to the patient population and the intervention under consideration, considering the outcomes of interest and statistical design parameters including stratification factors, and determining trial phase. The implementation phase is where the design parameters necessary to actually run the trial are specified, and consists of performing sample size calculations, defining interim monitoring and stopping rules, and conducting simulation studies to evaluate the operating characteristics of the proposed design. In the remainder of this paper, we frame our guidelines around these phases in the design process of a clinical trial. Throughout the paper, we provide references for the interested reader to find further details and explanations of concepts and terms.
Figure 1:
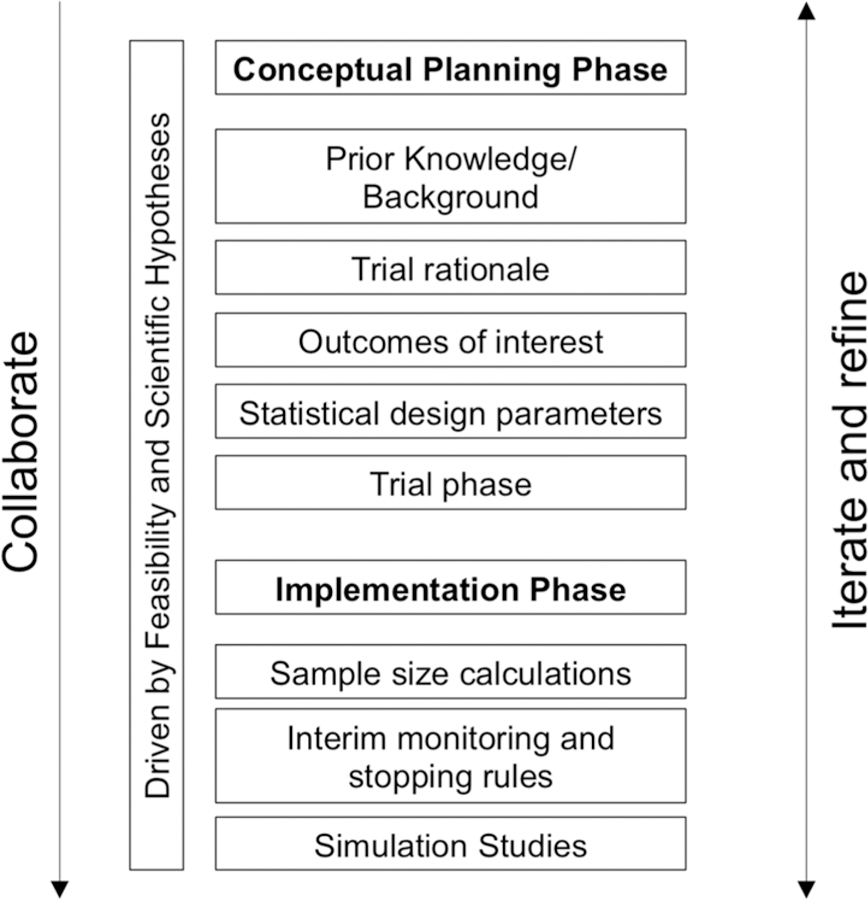
Principles for conceptual planning and implementation stages
Conceptual Phase
In this section, we outline the different areas that need careful attention when considering a clinical trial.
Prior knowledge/background:
A first step in designing a clinical trial is to establish what is known about the disease being studied. Specifically, this includes identifying the current standard of care and reviewing what is already known about the intervention(s) being studied including its safety profile and whether it has been tested in humans.
Trial rationale:
It is important to justify the need for the proposed trial, to identify the population of interest and to determine the disease or biomarker prevalence in this population. When the disease is rare and/or a targeted subgroup is of interest, then specific study designs for these settings may need to be considered; see Le-Rademacher et al. (2018), Gupta et al. (2011), and Mandrekar and Sargent (2009). Similarly, there is extensive work in the literature on study designs for personalized medicine in oncology, see for example Renfro and Mandrekar (2018).
Outcomes of interest:
Once the rationale for a trial has been established, selection of the outcome(s) of interest is essential. Trial outcomes can be either health- or treatment-related. Examples of health outcomes include quality of life, symptoms, adverse events, and patient-reported outcomes. Treatment outcomes include assessing safety or efficacy of the intervention; examples include tumor shrinkage, hematologic outcomes, intermediate or surrogate outcomes, time to event outcomes (e.g. overall survival or progression-free survival), and surgical outcomes. It is common to have one or two primary outcomes, and one or two secondary outcomes. The primary outcome should be directly related to the mechanism of action of the intervention, clinically meaningful, relevant to the patient, clearly defined, and measurable. These principles highlight the importance of the design process being collaborative, not only among clinicians and statisticians, but also including patient advocates, patients and their caregivers. Friedman et al. (2010) and Wu and Sargent (2010) offer more considerations on choosing endpoints.
Statistical design:
Estimating treatment effect is a common goal of many studies. Single-arm designs – wherein all patients receive the same intervention and are generally compared to a historical control group – can provide some information on treatment effect. However, often the single arm of patients and the historical control group do not represent the same populations of interest nor receive treatment under similar trial conditions. As such, single-arm designs are limited in the conclusions they can draw and less desirable than randomized trials. In randomized trials, there are at least 2 treatment groups (or “arms”) to which patients are randomly assigned. The random assignment, or randomization, aims to create groups that are similar with respect to all factors, besides the intervention, that might affect the outcome. This is a key principle of randomized trials that ensures a fair comparison. Randomized trials can additionally incorporate other design components. Common examples include the use of a control arm (i.e. an arm that receives the standard of care) and blinding (i.e. patient and/or clinician do not know the treatment assignment) to reduce bias. Randomization can be balanced where both groups are of equal size or unbalanced where groups are of unequal size. Finally, when confounding factors may be of concern, stratification may be considered as an additional design component. Although randomization aims to reduce confounding by making treatment groups as similar as possible except for the treatment assigned, it is nevertheless possible for the groups to differ with respect to some important factors. Examples of such factors include gender and age, and other factors specific to the study context. To avoid this possibility, identify these potential confounding factors and include stratification as part of the randomization process. Specifically, patients are grouped into strata according to the important factors and then randomized within each stratum. For considerations on other study designs, including adaptive, group sequential and Bayesian designs, see Pallmann et al. (2018), Bhatt and Mehta (2016), Vandemeulebroecke (2008), Lee and Chu (2012), and Berry (2006). For considerations when drafting a statistical analysis plan for clinical trials, see Gamble et al. (2017).
Trial phase:
The traditional development of new therapeutic interventions occurs in phases of trials, from pre-clinical to post-market, and so one must consider the available information about the intervention, the targeted population, etc. to better understand the trial phase for the study under consideration. Early phases of clinical studies include pilot studies, phase I, phase II single arm, and proof of concept. Later phases of clinical studies include randomized phase II, phase II/III, and phase III trials. Phase II trials aim to further understand the safety and efficacy of an intervention to help decide whether or not to proceed to a phase III trial. Phase II and Phase III trials typically have different endpoints; Phase II trials utilize short-term, early endpoints such as response rate or event-free survival rate at a predetermined time point whereas Phase III trials utilize longer-term clinical outcomes such as overall survival (Foster, Le-Rademacher, & Mandrekar, 2019). Given the role of Phase II trials in determining the go/no-go decision to proceed for further testing in large confirmatory Phase III trials, it is crucial to select an appropriate endpoint, particularly in Phase II trials. Phase II endpoints should ideally be a strong surrogate for the Phase III endpoints (Yin et al., 2018).
Implementation Stage
Once the design elements in the conceptual phase have been identified, and there is consensus to move forward with designing a clinical trial, the design elements necessary for actually running the trial need to be specified. This constitutes the implementation phase, the steps for which are outlined below.
Sample size calculation:
The purpose of sample size calculation is to determine the number of patients needed to enroll in the study to provide sufficient information to address the primary objectives. For traditional randomized designs, this depends on three primary factors that the research team must decide together – effect size, (statistical) power, and statistical significance level. Effect size refers to the minimum treatment effect that one hopes to detect in the study. Power refers to the likelihood of detecting an effect when in fact there is an effect of a priori specified size. Significance level refers to the p-value threshold for concluding statistically significant results; it also corresponds to the type I error rate (the chance of concluding an effect when in fact none exists). In general, larger sample sizes are needed to detect a smaller effect size, achieve greater power, and/or reduce the type I error rate. In addition to these factors, sample size calculations for trials should anticipate loss to follow-up and withdrawals, patient non-compliance to treatment, and protocol violations and ineligibility. Sample size calculations should be adjusted (specifically, increased) based on expected rates of these various sources of patient “drop-out.” Finally, examining the population of interest will help determine the expected accrual rate, and in turn, the expected time to accrue the total required number of patients to the trial. Sample size considerations for Bayesian designs depend on additional factors, most notably the prior distribution of the effect size; see Pezeshk (2003).
Interim monitoring and stopping rule:
Clinical trial monitoring is critical to the conduct – especially the ethical conduct – of the trial, and as part of this, it is important to decide the number and timing of interim analyses to be conducted prior to the completion of data collection, and build this as part of the design. Further, it is important to specify parameters for all stopping rules for stopping the trial early. In a Frequentist design, stopping rules are defined in terms of boundaries for safety, efficacy and futility; see Chapter 8 in Ellenberg, Fleming, and DeMets (2002). In a Bayesian design, stopping rules are typically defined in terms of posterior probabilities or predictive probabilities; see Saville et al. (2014).
Simulation studies:
Finally, even with the best trial design, actual trials seldom go as planned as unanticipated scenarios may arise. Therefore in designing the trial, it is helpful to brainstorm as much as possible these unanticipated scenarios and understand their implications using simulation studies. Simulation studies, when designed well with realistic scenarios, are a valuable tool for evaluating different trial designs and scenarios without exposing patients to an ineffective or harmful therapy or incurring the high financial costs associated with running an actual trial. The insights gained from simulation studies can help further guide the design process.
Conclusion
The goal of this paper is to provide an initial guidance to investigators through the design process of a clinical trial. It is not meant to be a strict set of rules to be followed in some prescribed order, rather it is meant to be a set of guidelines to consider in active collaboration with the study team including a statistician. These principles should apply for designing any clinical trial, regardless of who initiates and conducts the study (e.g. research group vs. industry). The involvement of the statistician throughout the entire research cannot be overemphasized. The statistician can aid in each step, from formulating appropriate scientific hypotheses to designing and conducting simulation studies. In addition to being collaborative, the design process is also iterative; it may be that some design elements need to be modified after other design elements are considered. For example, trial phase is typically driven by the level of available evidence on the drug being tested. However, occasionally the choice of trial phase (e.g. Phase II vs. Phase III) may be driven by feasibility to launch a large trial. Ultimately the design must be feasible and appropriate to answer the research question(s) of interest.
This paper is also not meant to provide an extensive review of design principles; for that, we refer the interested reader to the references included in this paper that offer detailed guidelines for designing trials. Further, the International Conference on Harmonisation has drafted two reports on statistical design: International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceutical for Human Use (1998, 2016). The frequently-cited reference by Altman et al. (1983) outlines statistical guidelines for preparing a manuscript for medical journals.
Expanding these principles for novel study designs, including immunotherapy and cellular therapy trials, and cancer care delivery research that spans multiple disciplines and where randomization must be made at the patient, provider, and site levels, could be considered in future work.
ACKNOWLEDGMENTS:
This work is funded in part by 5K12CA090628 and P30CA15083 (Mayo Clinic Comprehensive Cancer Center Grant).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICTS OF INTEREST: The authors declare no potential conflicts of interest.
References
- 1.Le-Rademacher J, Dahlberg S, Lee JJ, Adjei AA, Mandrekar SJ. Biomarker Clinical Trials in Lung Cancer: Design, Logistics, Challenges, and Practical Considerations. J Thorac Oncol 2018. November;13(11):1625–1637. [DOI] [PubMed] [Google Scholar]
- 2.Gupta S, Faughnan ME, Tomlinson GA, Bayoumi AM. A framework for applying unfamiliar trial designs in studies of rare diseases. J Clin Epidemiol 2011;64: 1085–1094. [DOI] [PubMed] [Google Scholar]
- 3.Mandrekar SJ, Sargent DJ. Clinical trial designs for predictive biomarker validation: theoretical considerations and practical challenges. J Clin Oncol 2009;27(24):4027–4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renfro LA, Mandrekar SJ. Definitions and statistical properties of master protocols for personalized medicine in oncology. J of Biopharmaceutical Statistics 2018, 28(2)217–229. [DOI] [PubMed] [Google Scholar]
- 5.Friedman LM, Furberg CD, and DeMets DL. Fundamentals of Clinical Trials, 4th Edition. Springer; 2010 [Google Scholar]
- 6.Wu W, Sargent D. Choice of Endpoints in Cancer Clinical Trials. Oncology Clinical Trials: Successful Design, Conduct, and Analysis (Eds. Kelly and Halabi). demosMEDICAL; NY: 2010. [Google Scholar]
- 7.Pallmann P, Bedding AW, Choodari-Oskooei B, et al. Adaptive designs in clinical trials: why use them, and how to run and report them. BMC Med 2018; 16:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bhatt DL, Mehta CM. Adaptive Designs for Clinical Trials. NEJM (2016) 375:65–74. [DOI] [PubMed] [Google Scholar]
- 9.Vandemeulebroecke M Group Sequential and Adaptive Designs – A Review of Basic Concepts and Points of Discussion. Biometrical Journal (2008) 4:541–557. [DOI] [PubMed] [Google Scholar]
- 10.Lee JJ and Chu CT. Bayesian clinical trials in action. Stat Med 2012; 31:2955–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Berry DA. Bayesian clinical trials. Nat Rev Drug Discov 2006; 5(1)27–36. [DOI] [PubMed] [Google Scholar]
- 12.Gamble C, Krishan A, Stocken D, et al. Guidelines for the Content of Statistical Analysis Plans. JAMA 2017. 2337–2343. [DOI] [PubMed]
- 13.Foster JC, Le-Rademacher J, & Mandrekar SJ (2019). Design Considerations for Phase II In Roychoudhury S, & Lahiri S, Statistical Approaches in Oncology Clinical Development (pp. 97–114). London: CRC Press Taylor and Francis Group. [Google Scholar]
- 14.Yin J, Dahlberg SE, Mandrekar SJ. Evaluation of End Points in Cancer Clinical Trials. J Thorac Oncol 2018. June; 13(6):745–757. [DOI] [PubMed] [Google Scholar]
- 15.Pezeshk H Bayesian techniques for sample size determination in clinical trials: a short review. Statistical Methods in Medical Research 2003; 12: 489–504. [DOI] [PubMed] [Google Scholar]
- 16.Ellenberg SS, Fleming TR, DeMets DL. Data Monitoring Committees in Clinical Trials: A Practical Perspective. John Wiley & Sons; 2002. [Google Scholar]
- 17.Saville BR, Connor JT, Ayers GD, et al. The utility of Bayesian predictive probabilities for interim monitoring of clinical trials. Clinical Trials 2014; 11(4)485–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. (1998). ICH Harmonised Tripartite Guideline: Statistical Principles for Clinical Trials E9. London, England: European Medicines Agency. [Google Scholar]
- 19.International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2016). ICH Harmonised Guideline: Integrated addendum to ICH E6(R1): Guideline for Good Clinical Practice E6(R2). London, England: European Medicines Agency. [Google Scholar]
- 20.Altman DG, Gore SM, Gardner MJ, & Pocock SJ. Statistical guidelines for contributors to medical journals. British Medical Journal. 1983. 286(6376)1489–1493. [DOI] [PMC free article] [PubMed] [Google Scholar]