

Abstract
Asymmetric, radical C–H functionalizations are rare, yet powerful tools for solving modern synthetic challenges. Specifically, the enantio- and regio-selective C–H amination of alcohols to access medicinally valuable, chiral β-amino alcohols remains elusive. To solve this challenge, a radical relay chaperone strategy was designed, wherein an alcohol is transiently converted to an imidate radical that undergoes intramolecular H-atom transfer (HAT). This regioselective HAT was also rendered enantioselective by harnessing energy transfer catalysis to mediate selective radical generation and interception by a chiral copper catalyst. The successful development of this multi-catalytic, asymmetric, radical C–H amination enables broad access to chiral β-amino alcohols from a variety of alcohols containing alkyl, allyl, benzyl, and propargyl C–H bonds. Mechanistic experiments reveal triplet energy sensitization of a Cu-bound radical precursor facilitates catalyst-mediated HAT stereoselectivity – enabling the synthesis of several important classes of chiral β-amines by enantioselective, radical C–H amination.
Graphical Abstract
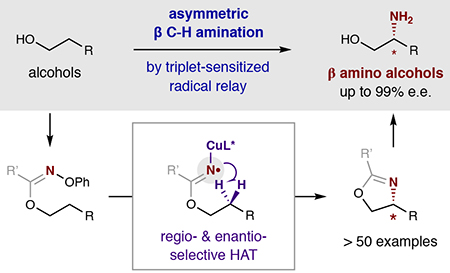
Five of the six most commonly employed chemical reactions in the discovery of new medicines entail the construction of a C–N bond1. To supplement these classical methods for amination of preformed C–X bonds, modern synthetic efforts have focused on designing strategies for direct C–H amination2,3. To this end, aminations of aryl4–6 and alkyl7–9 C–H bonds have been developed. In the latter cases, stereochemical control of sp3 C–H amination affords an added challenge, with rare solutions entailing nitrenes and heavy metals, porphyrins, or enzymes10–15. Nonetheless, the synthesis of β-amino alcohols by enantio- and regio-selective β-C–H amination remains an unsolved problem. Here we report a new radical relay chaperone strategy, wherein an alcohol is transiently converted to an imidate radical16 that enables intramolecular H-atom transfer (HAT).17 This regioselective 1,5-HAT18,19 has now also been rendered enantioselective – a rare example for C–H amination – by harnessing energy transfer catalysis to mediate selective radical generation and interception by a chiral copper catalyst20–22.
To selectively prepare chiral β-amino alcohols – a privileged motif in nature, medicine, and catalysis – we sought to develop an enantio- and regio-selective, radical C–H amination of alcohols, as shown in Fig. 1a. This strategy complements current methods to access vicinal amino alcohols that rely on the chiral pool (e.g. amino acids), chiral auxiliaries (e.g. imines), or multi-step conversion of chiral diols23–25. However, we were cognizant of the dearth of methods for stereoselective, radical C–H amination. In fact, asymmetric radical C–H functionalizations, in general, are rare – with pioneering solutions existing only for alkylation, arylation, and deracemization26–32. Our analysis of this problem led us to propose the challenge lies in the typical generation of radicals by non-selective H-abstraction of weak C–H bonds (e.g. benzylic, α-hetero), leading to radical chain processes that are difficult to render stereoselective – especially in the case of C–H amination33. We proposed that if these carbon radicals are instead mildly generated by intramolecular HAT from N-centered radicals, then catalytic systems could be engineered to exercise complete regio- and stereo-selective control over radical C–H amination.
Fig. 1: Design of a selective, radical C–H amination for the synthesis of β-amino alcohols.
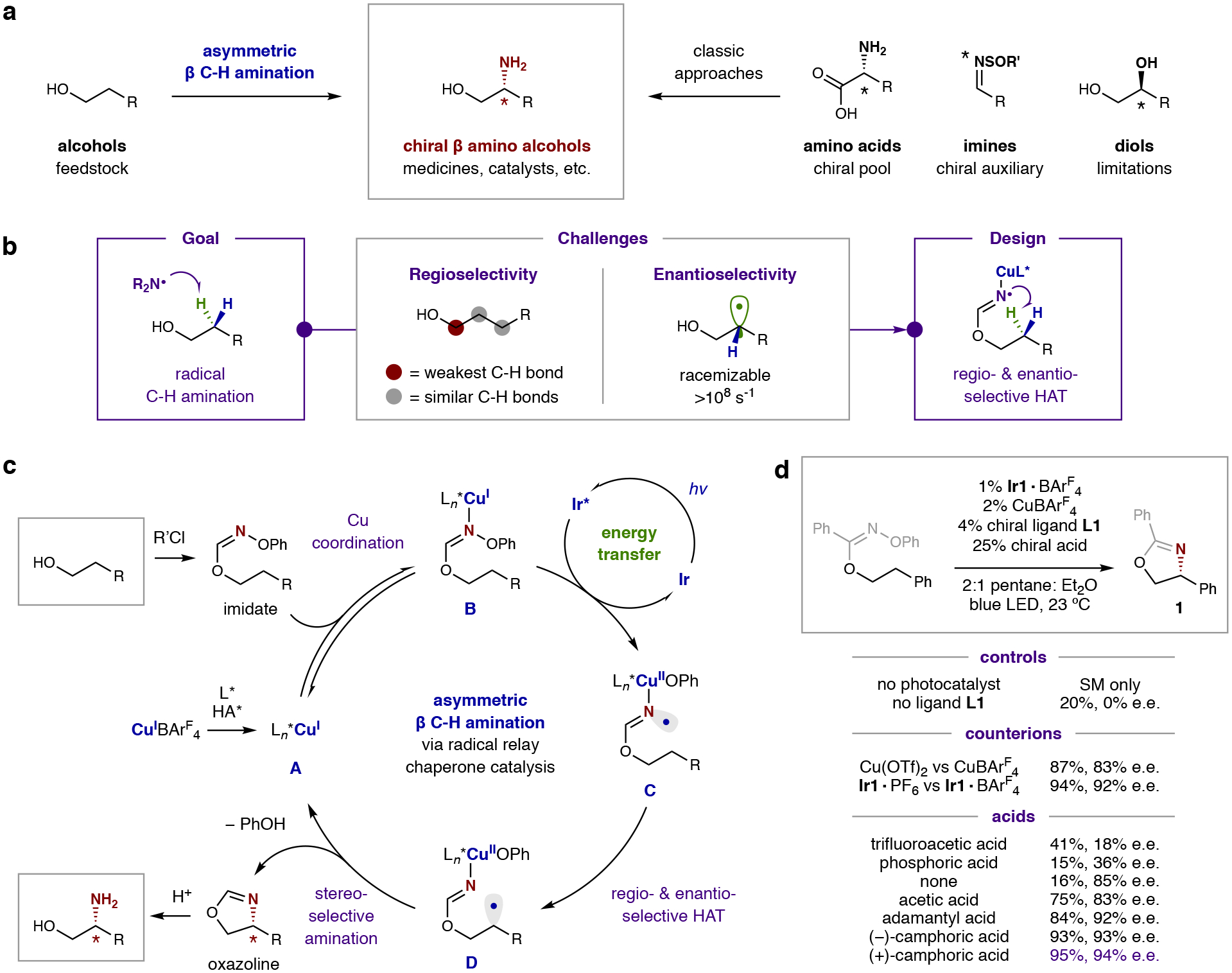
a, Radical C–H amination of cheap and abundant alcohols enables access to the privileged β-amino alcohol architecture – complementing classic approaches that are limited by availability of chiral pool precursors (e.g. amino acids), stoichiometric chiral auxiliaries (e.g. imines), or multi-step manipulation of chiral reagents (e.g. diols). b, Dual challenges for a radical approach include selective formation and trapping of a transient radical intermediate. Specifically, β-regioselectivity is disfavored compared to abstraction of the weaker α C–H bond by an N-centered radical. Moreover, enantioselectivity is both challenging to control and retain in this process, as it may rapidly erode by radical epimerization. c, Proposed mechanism: Alcohol addition to an imidoyl chloride chaperone furnishes an oxime imidate, which binds to a chiral Cu catalyst. This complex then forms an N-centered radical via selective, triplet sensitization by an excited Ir photocatalyst. Regio- and enantio-selective HAT, followed by stereoselective amination, affords a chiral oxazoline. Hydrolysis yields the enantioenriched β-amino alcohol. d, Development of this asymmetric β-C–H amination required a multi-catalytic strategy, wherein a photocatalyst selectively excites a chiral Cu catalyst complex when it is bound to an imidate-activated alcohol. Mechanistic probes illustrate the necessity of each component in ensuring the efficiency and selectivity of this radical C–H amination.
Results and discussion
Design plan.
In designing a HAT-mediated synthesis of chiral β-amino alcohols via radical C–H amination, we noted several challenges, as outlined in Fig. 1b. First, the α-C–H of an alcohol is weaker than the β- and γ-methylene C–H bonds (BDE: 90 vs 98 kcal/mol)34. We have shown a radical relay chaperone strategy, wherein an alcohol is temporarily converted to an imidate radical, facilitates selective β-radical formation via 1,5-HAT to overcome this thermodynamic bias16. Yet, this approach has only been rendered racemic because of its reliance on non-selective, iodine atom transfer to generate and trap the requisite radicals. Alternatively, we envisioned a chiral catalyst could mediate asymmetric C–H functionalization via an enantioselective HAT – only if radical generation is dependent on the presence of a chiral catalyst, such as through a multi-catalytic, triplet sensitization mechanism. Moreover, the low barrier for inversion of alkyl radicals (0.5 kcal/mol; >108 s−1)35 necessitates that this β-radical also be rapidly and stereoselectively trapped by the chiral catalyst. In this manner, and in contrast to concerted, nitrene insertion mechanisms, we envisioned a radical-mediated approach could enable stereoselectivity by exercising catalyst control over two elementary steps: (i) H-atom abstraction, and (ii) alkyl radical trapping. We anticipated catalytic stereocontrol over both these radical generating and terminating events would provide highly enantioenriched β-amino alcohols.
Proposed mechanism.
The chemo-, regio-, and enantio-selective radical C–H amination described herein is mediated by multiple catalysts working in concert to enable this valuable, asymmetric transformation via an energy transfer mechanism, as illustrated in Fig. 1c. In this strategy, an alcohol is converted to an oxime imidate by combination with a bench-stable, imidoyl chloride chaperone, prepared in two steps from H2N-OPh36. This radical precursor is then coordinated by copper catalyst A bearing a chiral bisoxazoline ligand, as well as a chiral carboxylate, to generate Cu-imidate complex B. Upon excitation with visible light, an Ir photocatalyst may then sensitize this organocopper complex via an energy transfer mechanism37, which generates N-centered radical C by triplet sensitization of the oxime N–O bond to enable a net photoinduced oxidative addition. The resulting chiral Cu-bound imidate radical C can undergo enantio- and regio-selective HAT to afford chiral β-radical D. Amination may then occur with further stereocontrol by either: (i) diastereoselective metalation of the alkyl radical38 and reductive elimination of the ensuing alkyl Cu(III) intermediate39, or (ii) diastereoselective, coupling of the radical within a Cu(II) complex40. Both pathways would afford enantioenriched oxazoline, along with PhOH and regenerated Cu catalyst A. Finally, acidic hydrolysis furnishes the chiral β-amino alcohol from this multi-catalytic, radical relay mechanism.
Reaction development.
The execution of our design for asymmetric β-C–H amination, shown in Fig. 1d, requires a multi-catalytic approach that includes chiral copper and acid co-catalysts, working in concert with an energy transfer photocatalyst, to selectively initiate and trap an intramolecular HAT mechanism. Specifically, the benzoic acid-derived oxime imidate of 2-phenyl ethanol was converted to chiral oxazoline 1 (95% yield, 94% e.e.; enantiomeric excess) by combination of 1% Ir1 photocatalyst (Ir{[dF(CF3)ppy]2dtbbpy}BArF4), 2% CuBArF4, 4% bisoxazoline ligand L1, and 25% camphoric acid in a 2:1 mixture of pentane:Et2O, with blue LED irradiation for 1 hr. Although asymmetric, α-amido amination has been shown by direct photoexcitation of Cu carbazolides40, neither Cu acetate A, nor Cu imidate B, significantly absorb visible light (λabs > 400 nm). Therefore, an external triplet sensitizer (Ir1) was used to mediate this reaction with blue LED irradiation. Control experiments confirm this photosensitizer is required (only starting material observed without Ir1), and other non-redox active, triplet sensitizers increase reaction efficiency under UV irradiation (e.g. naphthalene, xanthone; see SI section VIII). Additionally, chiral ligand L1 impacts both reaction efficiency and selectivity (20% yield, 0% e.e., in its absence), and the BArF4 counterion improves stereocontrol (83–94% e.e.)41. Finally, the acid co-catalyst plays a pivotal role in controlling efficiency and selectivity. For example, acetic acid significantly improves yield relative to the absence of acid or use of strong acids, whose anions are poorly coordinating (75% vs 16% yield). Notably, bulkier acids (e.g. adamantyl, camphoric) afford optimal efficiency (84–95% yield) and stereocontrol (92–94% e.e.). Together, these data indicate a bulky, albeit not necessarily chiral, acid is required – likely to stabilize the higher oxidation state Cu intermediates.
Synthetic scope.
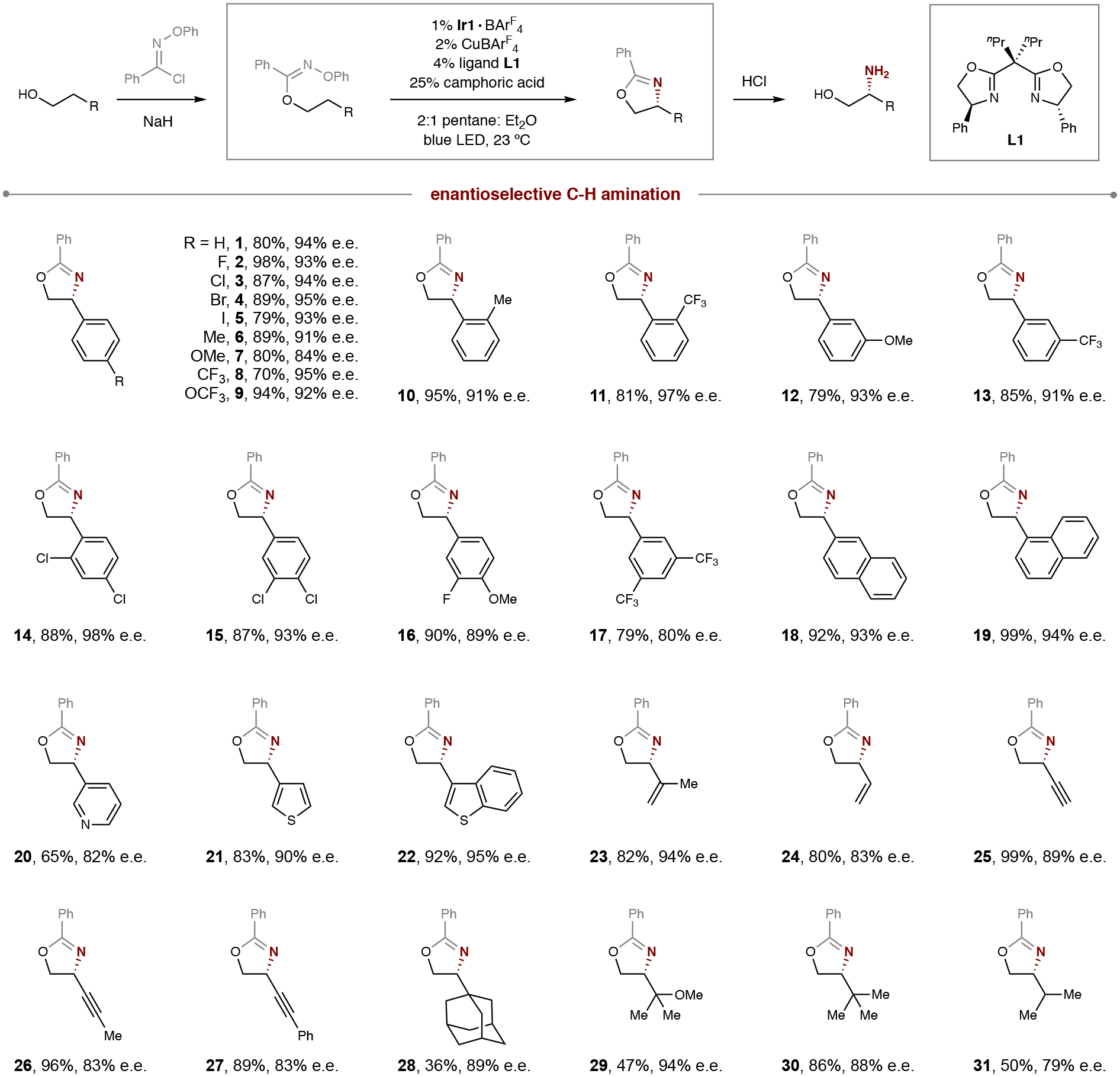
To probe the synthetic utility of this enantioselective β-C–H amination, we subjected a range of sterically and electronically diverse alcohols to this catalytic, radical relay sequence. As illustrated in Table 1, the HAT mechanism is robust and furnishes β-amino variants of a wide range of para-substituted 2-aryl ethanols (1-9) – spanning Hammett σp constants of −0.3 (OMe) to +0.5 (CF3) – with little effect on efficiency or selectivity (70–98%; up to 95% e.e.). Intrigued by the potential medicinal value of these β-amino alcohols (e.g. 7 is an isomeric analog of the neurotransmitter, octopamine, wherein the N and O atoms of the β-amino alcohol are inverted), we then explored various ortho and meta substitutions (10-13) as well as di-substituted arenes (14-17), which were also smoothly aminated. Similarly, alcohols containing polyaromatics (18-19) and heteroarenes (20-22) afford β-C–H amination with high enantioselectivity.
Table 1.
Synthetic scope of alcohols in enantioselective, radical C–H amination.
|
Conditions: 1% Ir{[dF(CF3)ppy]2dtbbpy}BArF4, 2% CuBArF4, 4% ligand L1, 25% camphoric acid, pentane: Et2O (2:1), blue LED irradiation, room temperature, 1 hour. See General Procedure 3 in SI section II for full experimental details. Isolated yield and enantiomeric excess (e.e.) indicated below each entry.
Having established robust reactivity for benzylic C–H amination, we next probed this strategy for alcohols with stronger allylic, propargylic, and aliphatic C–H bonds. In the allylic case, a more sterically encumbered 1,1-disubstituted alkene affords greater enantioselectivity than a simple vinyl substituent (23, 94% e.e. vs 24, 83% e.e.), although both are similarly efficient (80–82% yield). To our surprise, the smaller sp-hybridized substituent of propargylic alcohols also affords β-C–H amination with high selectivity (25-27, 83–89% e.e.). The observation that the smallest ethynyl substituent affords greatest efficiency and selectivity among the trio suggests these alkynes may interact with the Cu catalyst in an additional manner. With the hopes of removing all such possible coordination from the substrate, we then probed a series of aliphatic alcohols bearing β-methylene units. Notably, simple 2-alkyl ethanols with large β-substituents afford enantioselectivities as high as the 2-aryl ethanols (28-30, up to 94% e.e.), suggesting the role of the aryl group is merely a sterically repulsive, catalyst-recognition element. When the β-substituent size is decreased (e.g. to an iPr group), stereoselectivity drops (31, 79% e.e.), yet complete β-C–H regioselectivity remains intact as the kinetically favored 1,5-HAT mechanism outcompetes 1,6-HAT of the weaker γ-tertiary C–H (Δ 2 kcal/mol)34.
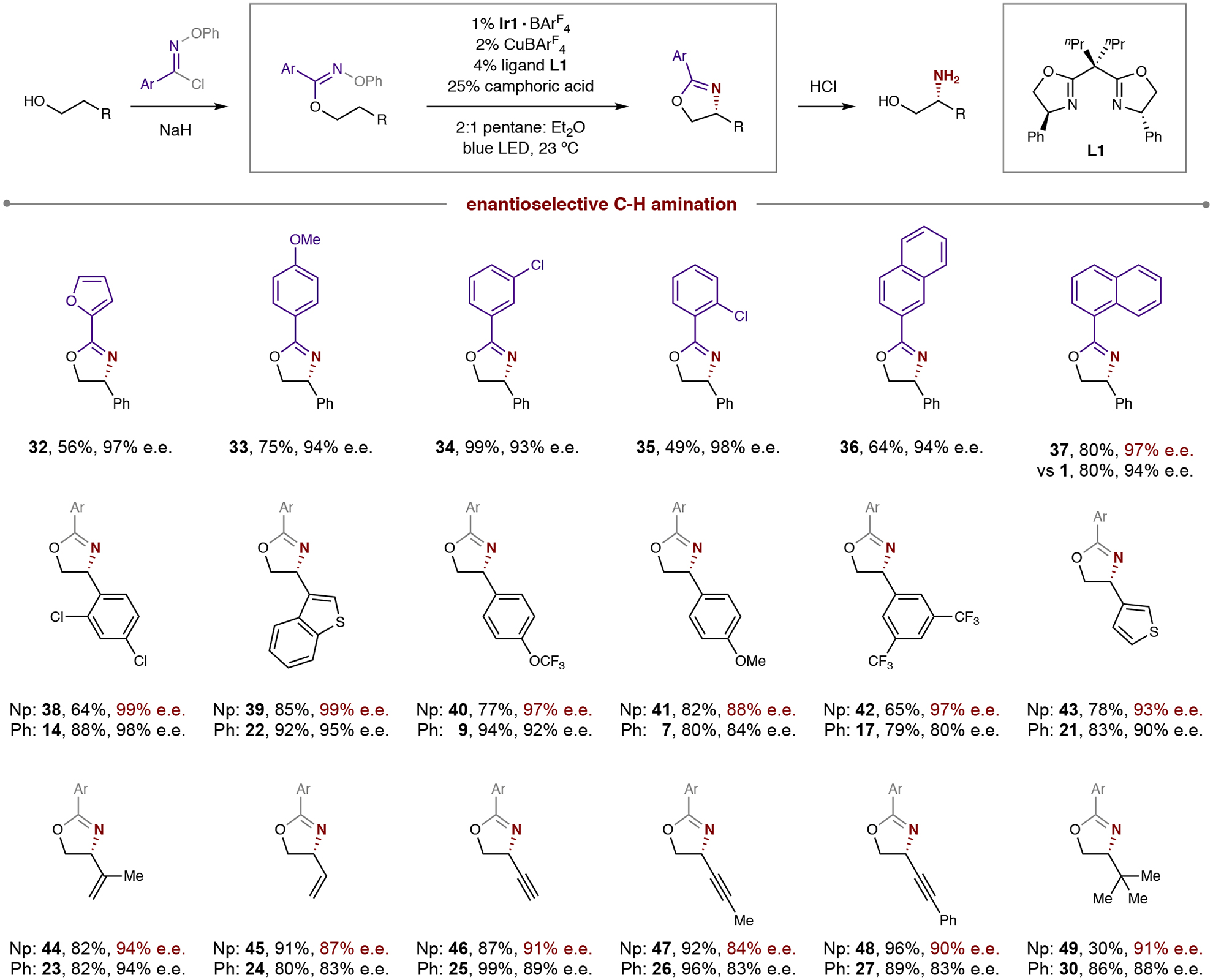
Having investigated the alcohol scope exclusively using simple benzimidate chaperones (derived from benzoyl chloride), we were interested in exploring the synthetic robustness of this β-C–H amination in accessing other chiral oxazolines. To this end, a variety of aryl imidates were prepared and subjected to the multi-catalytic sequence, as shown in Table 2. Interestingly, a smaller 3-furanyl substituent affords unexpectedly high selectivity (32, 97% e.e.). Other arenes with stereoelectronically diverse substitution also afford highly enantioenriched oxazolines (33-35, 93–98% e.e.), albeit with varying efficiency (49–99%). To our delight, we observed a 1-naphthyl substituent (37) affords similar efficiency to the Ph-imidate (80% in both cases), as well as boosted stereoselectivity (97 vs 94% e.e.; 1). To further investigate this effect, we probed the naphthyl (Np) imidates of a variety of alcohols from Table 1. In several cases, we observed near complete enantiocontrol (38-39, 99% e.e.), with the improvement in enantioselectivities ranging from modest (38, Δ 1% e.e.) to large (42, Δ 17% e.e.). This effect was recapitulated for the vinyl, alkynyl, and alkyl substituted alcohols (44-49), which are boosted to >90% e.e. in many cases.
Table 2.
Synthetic scope and effect of chaperone in enantioselective, radical C–H amination.
|
Conditions: 1% Ir{[dF(CF3)ppy]2dtbbpy}BArF4, 2% CuBArF4, 4% ligand L1, 25% camphoric acid, pentane:Et2O (2:1), blue LED irradiation, room temperature, 1 hour. See General Procedure 3 in SI section II for full experimental details. Isolated yield and enantiomeric excess (e.e.) indicated below each entry. Np affords improved enantioselectivity in many cases. Abbreviations: Ar, aryl, Np, 1-naphthyl.
Mechanistic investigations.
The rationale for the proposed mechanism illustrated in Fig. 1b is provided by a combination of data from cyclic voltammetry, as well as computational, photo-quenching, isotopic labeling, competition, desymmetrization, and radical clock experiments, as shown in Fig. 2. Probing the role of the photocatalyst in radical initiation (Fig. 2a), we noted that a more strongly reducing Ir complex Ir2 (–1.5 V; see SI, section VIII, for details) is less efficient than a less reducing catalyst Ir1 (–1.4 V) (14% vs 80% yield), which is surprising, given the oxime imidate reduction potential is ‒1.6 V (all potentials vs SCE)36. Yet, the more efficient complex (Ir1) has a significantly higher triplet energy (62 vs 49 kcal/mol) and longer-lived excited state (2300 vs 557 ns)42, which suggests energy transfer is more likely operative than an electron transfer pathway. Significant catalyst counterion effects (BArF4 > PF6 > OTf) – on both yield and enantioselectivity – were also observed, likely due to increased triplet energies or lifetimes, which may facilitate more efficient energy transfer41. Concerning the proposed mechanism of radical formation, photoinduced oxidative addition into the N–O bond of the oxime imidate is likely a result of its bond-weakening. For example, upon catalytic triplet sensitization, the N–O bond of the triplet state is significantly weaker than the ground state (–18 vs 35 kcal/mol), facilitating N–O scission (see SI section IX for computational details).
Fig. 2: Mechanistic experiments.
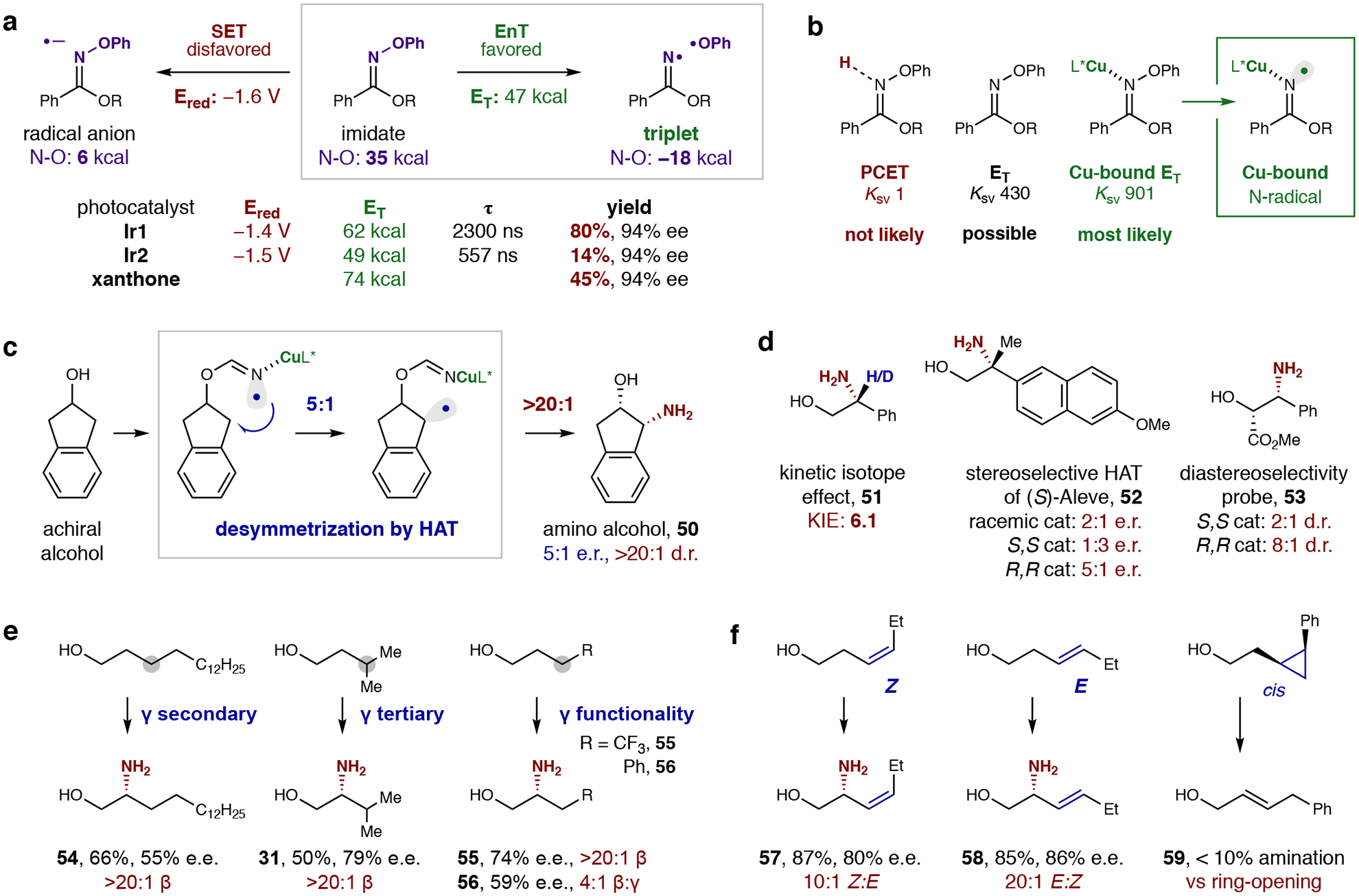
a, An energy transfer (EnT) pathway (vs single-electron transfer: SET) is supported by CV and DFT data. Specifically, a less reducing photocatalyst (Ir1 vs Ir2) provides greater efficiency due to a higher triplet energy (ET) and longer-lived excited state (τ). Triplet sensitizer, xanthone, also affords reactivity – indicating the imidate triplet, which is computed to have a significantly weakened N–O bond, is the source of N-centered radical. b, Photo-quenching experiments illustrate the favorability of Cu catalyst coordination during the triplet sensitization and subsequent N-radical generation. c, Desymmetrization of a secondary alcohol demonstrates the ability of this Cu-bound N-radical to discriminate among H-atoms by enantioselective HAT (5:1 e.r.). High diastereoselectivity (>20:1 d.r.) illuminates the role of Cu radical trapping in further upgrading the high enantioselectivity observed for primary alcohols. d, A significant KIE indicates HAT is the product-determining step. Stereoselectivity of the HAT was further probed with two chiral alcohols. Selectivity and efficiency are highly catalyst-dependent for both (S)-Aleve, which contains a β-stereocenter, as well as an enantioenriched secondary alcohol. e, A highly regioselective 1,5-HAT (>20:1 β) outcompetes 1,6-HAT even when much weaker γ-C–H bonds are present. f, Radical clocks indicate radical trapping by Cu is more rapid than allyl radical isomerization, but slower than cyclopropyl ring-opening.
The interaction of the excited Ir catalyst with the imidate was further investigated through Stern-Volmer quenching experiments (Fig. 2b). Since an acid co-catalyst was pivotal in obtaining high efficiency and selectivity (cf. Fig. 1d), a PCET mechanism was first considered18. However, addition of acid to the imidate decreases quenching (KSV 901 vs 430), discounting the likely involvement of the Brønsted acid in radical generation. On the other hand, addition of the copper catalyst increases quenching (KSV 901 vs 430), suggesting the Cu-coordinated imidate is the likely acceptor of triplet energy from the excited Ir photocatalyst. This Cu-bound energy transfer mechanism also explains the proximity of chiral information at the outset of radical generation to overcome racemic mechanisms, involving either energy transfer43 or Cu catalysis44, as well as to mediate a stereoselective HAT pathway.
To test the hypothesis that a Cu-bound N-centered radical mediates enantioselective HAT, we subjected an achiral secondary alcohol to the C–H amination (Fig. 2c). Given the likely irreversibility of the HAT, the observed desymmetrization (50; 5:1 e.r.; enantiomeric ratio) indicates the coordinated chiral Cu is involved in promoting enantioselective HAT. The subsequent diastereoselectivity (>20:1 d.r.; diastereomeric ratio) illustrates the Cu catalyst’s additional role in stereoselectively trapping this intermediate. The higher enantioselectivity observed for primary alcohols versus this secondary alcohol (c.f. Tables 1–2) can be considered a result of these additive effects.
Further insight into the key HAT mechanism (Fig. 2d) was provided by a strong, primary KIE observed in the intramolecular H/D competition (51, KIE: 6.1), which indicates HAT is the product-determining step. To further test the likelihood of a stereoselective HAT, an enantiopure β-chiral alcohol derived from nonsteroidal anti-inflammatory drug, (S)-Aleve, was converted to β-amine 52. A pronounced catalyst match/mismatch scenario was observed wherein S,S catalyst affords 1:3 e.r., while R,R catalyst affords 5:1 e.r., and racemic catalyst affords 2:1 e.r. – all with varying levels of efficiency (R,R > rac > S,S) indicating stereocontrol of both HAT and subsequent alkyl radical trapping. Likewise, subjecting an enantioenriched secondary alcohol as a diastereoselectivity probe, 53, affords between 2:1 d.r. (S,S catalyst) to 8:1 d.r. (R,R catalyst).
To probe regioselectivity of the intramolecular HAT, several classes of alcohols were subjected to this amination (Fig. 2e). Notably, n-hexadecanol affords a single β-aminated regioisomer (54) resulting from exclusive 1,5-HAT. Additionally, α-amination is not observed in any case, indicating 1,5-HAT is highly favored over 1,4-HAT despite an 8 kcal/mol weaker α-oxy C-H bond. To elucidate 1,5-vs 1,6-HAT regioselectivity, sterically and electronically diverse γ substituents – including those with weaker γ-C–H bonds – were investigated. However, β-selectivity (via 1,5-HAT) was exclusively observed (>20:1) for both γ-tertiary (31) and γ-CF3 substituents (55). Even in the case of a 10 kcal/mol weaker γ-benzyl C–H bond, 4:1 β:γ regioselective amination is observed (56).
Since stereoselective C–H amination by HAT is much rarer than nitrene insertion, we wished to investigate whether these metal-bound nitrogen radicals behave more like radicals or nitrenes. If the former case is operative, we envisioned a radical pathway could allow for catalyst stereocontrol over two steps (both HAT and alkyl radical trapping) rather than a single, C–H insertion step as in nitrene reactivity. Thus, a homoallylic alcohol was subjected to the amination (Fig. 2f). Interestingly, this probe exclusively affords C–H amination (57-58, >85% yield, >80% e.e.) without any aziridination, as sometimes observed in nitrene mechanisms10,11,45. Interestingly, a Z-alkene is tolerated, and only slightly isomerized (57, 10:1 Z, from 20:1 Z starting material) despite a low inversion barrier for the allyl radical intermediate (15.7 kcal/mol)46, indicating radical trapping by the Cu catalyst is rapid. As a control, an E-alkene completely retains its configuration (58, 20:1 E). As further evidence of a radical-mediated HAT mechanism, a β-cyclopropyl ring predominantly undergoes ring-opening or reduction (59), rather than C–H amination – unlike with Rh-nitrenes11. Combining these observations, we conclude alkyl radical trapping by the Cu catalyst likely occurs at a rate between 108 s–1 (tBu radical isomerization)35 and 1011 s–1 (cyclopropyl ring-opening)47, which is rapid enough to retain (and enhance) chiral memory via diastereoselective catalyst coordination38,48. The large KIE (6.1) also contrasts with those observed for concerted C–H insertion by an Ir-nitrene (KIE: 1.5)9. Lastly, unlike nitrene-based methods that require azides or heterocyclic nitrene precursors10–15, this Cu-catalyzed radical relay strategy has the synthetic benefit of enabling C–H amination of ubiquitous alcohols via their readily available imidates.
Diversification of chiral products.
Finally, the synthetic utility of this enantioselective β-C–H amination for accessing medicinally relevant motifs is illustrated in Fig. 3 via derivatization of oxazoline 1. For example, subjecting 1 to HCl at 100 °C affords free β-amino alcohol 60, while hydrolysis with HCl at 23 °C affords β-benzamide 61. This amide may then be oxidized by RuCl3/NaIO4 to α amino acid 62. Conversely, instead of an acidic work-up, double reduction by DIBAL at 0 °C yields Bn-protected β-amine 63, and C–O displacement by silyl halides (Me3Si–X) afford vicinal benzamide halides (64-66). Importantly, in each of these oxazoline derivatizations, the stereocenter was retained with greater than 98% enantiospecificity (e.s.). Lastly, applying this method to streamlined access of privileged catalyst architectures, oxazoline may be selectively deprotonated with nBuLi, then quenched with Ph2PCl, to afford phosphinooxazoline (PHOX) ligand49. Alternatively, the bis-imidate 67 of a meta-diol was converted to inverted bisoxazoline 68 (66% yield, 98% e.e.; 15:1 d.r.) – providing rapid access to a new C2 symmetric ligand class.
Fig. 3. Synthetic applications.
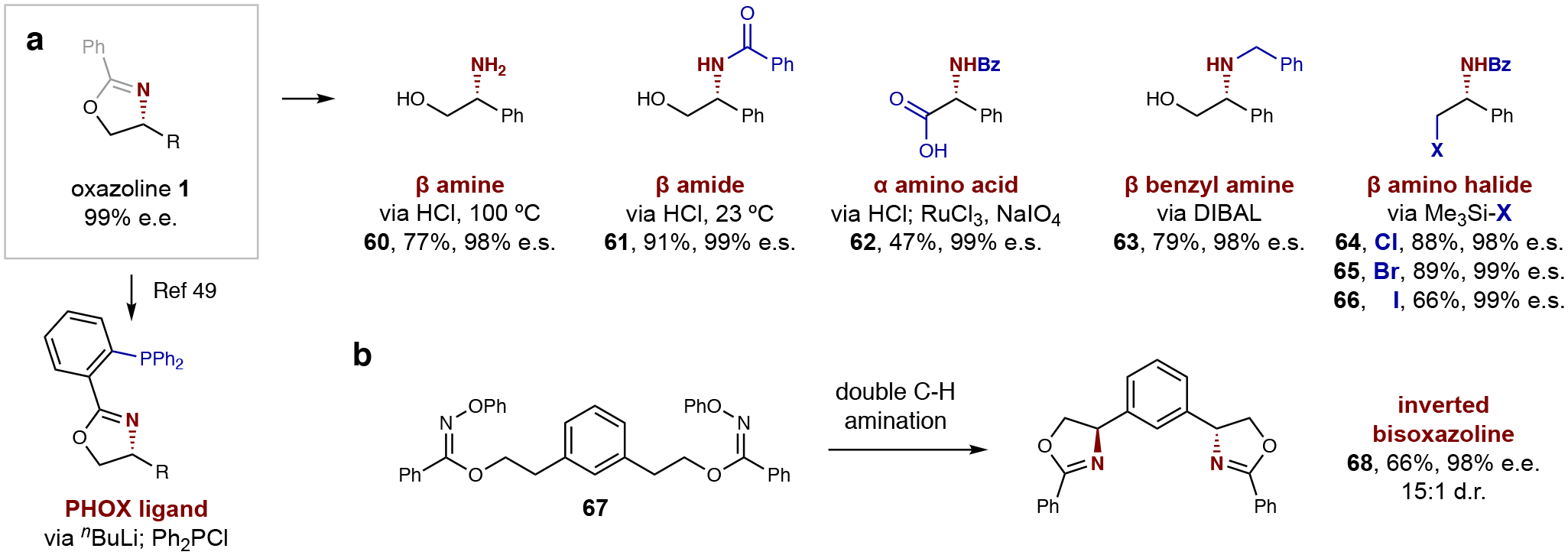
a, The enantioenriched oxazoline intermediate is rapidly converted to a family of valuable, chiral products, including amino acids and β-amines, by acidic hydrolysis, oxidation, reduction, or halide substitution. Reagents, yield, and enantiospecificity (e.s.) provided below each example, and full details in SI section VII. Alternatively, directed lithiation and phosphinylation may afford access to PHOX ligands49. b, Double C–H amination of a diol affords a new class of chiral catalysts.
Conclusion
In summary, we expect this enantio- and regio-selective, radical β-C–H amination will streamline the synthesis of important molecules with the chiral, vicinal amino alcohol motif. Notably, this approach bypasses the use of chiral auxiliaries (imines) and chiral pool precursors (amino acids), and enables the enantioselective synthesis of several new β-amino alcohols. Moreover, this radical relay chaperone strategy will likely also enable development of other classes of stereoselective C–H functionalization.
Methods
Enantioselective C–H amination.
An oven-dried vial was charged with a stir bar, imidate (0.1 mmol), and (+)-camphoric acid (25 mol%). Catalyst solutions of Ir1 (1 mol%), as well as CuBArF4 (2 mol%) and L1 (4 mol%) in Et2O (see below for details) were added in a glove box. After dilution with pentane (4 mL), the vial was irradiated with a 455 nm blue LED for 1 hr while cooling by fan. Upon completion, the reaction mixture was purified by column chromatography to afford the enantioenriched oxazoline. Further hydrolysis with HCl affords the enantioenriched amino alcohol.
Preparation of catalyst solutions.
L1·CuBArF4.
A flame-dried flask was charged with a stir bar, CuBArF4 (0.04 mmol), and L1 (0.08 mmol) in a glove box. Et2O (20 mL) was added and stirred at 23 °C for 1 hr to afford a 0.002 M solution (1 mL was used in each experiment).
Ir1.
A flame-dried flask was charged with Ir{[dF(CF3)ppy]2dtbbpy}BArF4 photocatalyst (0.02 mmol) in a glove box. Et2O (20 mL) was added to afford a 0.001 M solution (1 mL was used in each experiment).
Synthesis of imidate starting materials.
A flame-dried flask was sequentially charged with a stir bar, alcohol (2 mmol), THF (20 mL), and NaH (1.5 equiv.). After stirring for 1 hr, imidoyl chloride (1.1 equiv.) was added and the resulting solution was further stirred. Upon complete consumption of alcohol, the reaction mixture was purified by column chromatography to afford the corresponding oxime imidate in 80–99% yields.
Supplementary Material
Acknowledgments
Financial support was provided by the National Institutes of Health (NIH R35 GM119812) and National Science Foundation (NSF CAREER 1654656). L.M.S. is supported by an NSF graduate fellowship. Calculations were performed using resources at the Ohio Supercomputer Center.
Footnotes
Data availability. The data that support the findings of this study are available within the article and its Supplementary Information files.
Competing interests
The authors declare no competing interests.
References:
- 1.Roughley SD & Jordan AM The Medicinal Chemist’s Toolbox: An Analysis of Reactions Used in the Pursuit of Drug Candidates. J. Med. Chem 54, 3451–3479 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Park Y, Kim Y & Chang S Transition Metal-Catalyzed C-H Amination: Scope, Mechanism, and Applications. Chem. Rev 117, 9247–9301 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Davies HML & Manning JR Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 451, 417–424 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Romero NA, Margrey KA, Tay NE & Nicewicz DA Site-selective arene C-H amination via photoredox catalysis. Science 349, 1326–1330 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Paudyal MP et al. Dirhodium-catalyzed C-H arene amination using hydroxylamines. Science 353, 1144–1147 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ruffoni A et al. Practical and regioselective amination of arenes using alkyl amines. Nat. Chem 11, 426–433 (2019). [DOI] [PubMed] [Google Scholar]
- 7.McNally A, Haffemayer B, Collins BSL & Gaunt MJ Palladium-catalysed C-H activation of aliphatic amines to give strained nitrogen heterocycles. Nature 510, 129–33 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Sharma A & Hartwig JF Metal-catalysed azidation of tertiary C-H bonds suitable for late-stage functionalization. Nature 517, 600–604 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hong SY et al. Selective formation of γ-lactams via C-H amidation enabled by tailored iridium catalysts. Science 359, 1016–1021 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Reddy RP & Davies HML Dirhodium tetracarboxylates derived from adamantylglycine as chiral catalysts for enantioselective C-H aminations. Org. Lett 8, 5013–5016 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Zalatan DN & Du Bois J A chiral rhodium carboxamidate catalyst for enantioselective C-H amination. J. Am. Chem. Soc 130, 9220–9221 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milczek E, Boudet N & Blakey S Enantioselective C-H amination using cationic ruthenium(II)-pybox catalysts. Angew. Chem. Int. Ed 47, 6825–6828 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Park Y & Chang S Asymmetric formation of γ-lactams via C–H amidation enabled by chiral hydrogen-bond-donor catalysts. Nat. Catal 2, 219–227 (2019). [Google Scholar]
- 14.Hu Y et al. Enantioselective Radical Construction of 5-Membered Cyclic Sulfonamides by Metalloradical C–H Amination. J. Am. Chem. Soc 141, 18160–18169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Y, Cho I, Qi X, Liu P & Arnold FH An enzymatic platform for the asymmetric amination of primary, secondary and tertiary C(sp3)–H bonds. Nat. Chem 11, 987–993 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wappes EA, Nakafuku KM & Nagib DA Directed β C–H Amination of Alcohols via Radical Relay Chaperones. J. Am. Chem. Soc 139, 10204–10207 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stateman LM, Nakafuku KM & Nagib DA Remote C-H Functionalization via Selective Hydrogen Atom Transfer. Synthesis 50, 1569–1586 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi GJ, Zhu Q, Miller DC, Gu CJ & Knowles RR Catalytic alkylation of remote C-H bonds enabled by proton-coupled electron transfer. Nature 539, 268–271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chu JCK & Rovis T Amide-directed photoredox-catalysed C-C bond formation at unactivated sp 3 C-H bonds. Nature 539, 272–275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Twilton J et al. The merger of transition metal and photocatalysis. Nat. Rev. Chem 1, 52 (2017). [Google Scholar]
- 21.Wang F, Chen P & Liu G Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res 51, 2036–2046 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Hossain A, Bhattacharyya A & Reiser O Copper’s rapid ascent in visible-light photoredox catalysis. Science 364, eaav9713 (2019). [DOI] [PubMed] [Google Scholar]
- 23.Ager DJ, Prakash I & Schaad DR 1,2-Amino alcohols and their heterocyclic derivatives as chiral auxiliaries in asymmetric synthesis. Chem. Rev 96, 835–875 (1996). [DOI] [PubMed] [Google Scholar]
- 24.Robak MT, Herbage MA & Ellman JA Synthesis and applications of tert-butanesulfinamide. Chem. Rev 110, 3600–3740 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Chang HT & Sharpless KB A practical route to enantiopure 1,2-aminoalcohols. Tetrahedron Lett. 37, 3219–3222 (1996). [Google Scholar]
- 26.Zhang W et al. Enantioselective cyanation of benzylic C-H bonds via copper-catalyzed radical relay. Science 353, 1014–1018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy JJ, Bastida D, Paria S, Fagnoni M & Melchiorre P Asymmetric catalytic formation of quaternary carbons by iminium ion trapping of radicals. Nature 532, 218–222 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Wang C, Harms K & Meggers E Catalytic Asymmetric Csp3–H Functionalization under Photoredox Conditions by Radical Translocation and Stereocontrolled Alkene Addition. Angew. Chem. Int. Ed 55, 13495–13498 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Proctor RSJ, Davis HJ & Phipps RJ Catalytic enantioselective Minisci-type addition to heteroarenes. Science 360, 419–422 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Wu L, Chen P & Liu G Enantioselective Arylation of Benzylic C–H Bonds by Copper-Catalyzed Radical Relay. Angew. Chem. Int. Ed 58, 6425–6429 (2019). [DOI] [PubMed] [Google Scholar]
- 31.Shin NY, Ryss JM, Zhang X, Miller SJ & Knowles RR Light-driven deracemization enabled by excited-state electron transfer. Science 366, 364–369 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J et al. Site-specific allylic C–H bond functionalization with a copper-bound N-centred radical. Nature 574, 516–521 (2019). [DOI] [PubMed] [Google Scholar]
- 33.Ni Z et al. Highly regioselective copper-catalyzed benzylic C-H amination by N-fluorobenzenesulfonimide. Angew. Chem. Int. Ed 51, 1244–1247 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Luo YR Comprehensive handbook of chemical bond energies. Comprehensive Handbook of Chemical Bond Energies (CRC Press, 2007). doi: 10.1201/9781420007282 [DOI] [Google Scholar]
- 35.Griller D, Ingold KU, Krusic PJ & Fischer H Configuration of the tert-butyl radical. J. Am. Chem. Soc 100, 6750–6752 (1978). [Google Scholar]
- 36.Nakafuku KM, Fosu SC & Nagib DA Catalytic Alkene Difunctionalization via Imidate Radicals. J. Am. Chem. Soc 140, 11202–11205 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Welin ER, Le C, Arias-Rotondo DM, McCusker JK & MacMillan DWC Photosensitized, energy transfer-mediated organometallic catalysis through electronically excited nickel(II). Science 355, 380–385 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gutierrez O, Tellis JC, Primer DN, Molander GA & Kozlowski MC Nickel-catalyzed cross-coupling of photoredox-generated radicals: Uncovering a general manifold for stereoconvergence in nickel-catalyzed cross-couplings. J. Am. Chem. Soc 137, 4896–4899 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kochi JK Electron-transfer mechanisms for organometallic intermediates in catalytic reactions. Acc. Chem. Res 7, 351–360 (1974). [Google Scholar]
- 40.Kainz QM et al. Asymmetric copper-catalyzed C-N cross-couplings induced by visible light. Science 351, 681–684 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farney EP et al. Discovery and Elucidation of Counteranion Dependence in Photoredox Catalysis. J. Am. Chem. Soc 141, 6385–6391 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strieth-Kalthoff F, James MJ, Teders M, Pitzer L & Glorius F Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev 47, 7190–7202 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Blum TR, Miller ZD, Bates DM, Guzei IA & Yoon TP Enantioselective photochemistry through Lewis acid-catalyzed triplet energy transfer. Science 354, 1391–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y-F, Chen H, Zhu X & Chiba S Copper-Catalyzed Aerobic Aliphatic C–H Oxygenation Directed by an Amidine Moiety. J. Am. Chem. Soc 134, 11980–11983 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Alderson JM, Corbin JR & Schomaker JM Tunable, Chemo- and Site-Selective Nitrene Transfer Reactions through the Rational Design of Silver(I) Catalysts. Acc. Chem. Res 50, 2147–2158 (2017). [DOI] [PubMed] [Google Scholar]
- 46.Korth HG, Trill H & Sustmann R [1–2H]Allyl Radical: Barrier to Rotation and Allyl Delocalization Energy. J. Am. Chem. Soc 103, 4483–4489 (1981). [Google Scholar]
- 47.Newcomb M, Johnson CC, Manek MB & Varick TR Picosecond radical kinetics. Ring openings of phenyl-substituted cyclopropylcarbinyl radicals. J. Am. Chem. Soc 114, 10915–10921 (1992). [Google Scholar]
- 48.Gloor CS, Dénès F & Renaud P Memory of chirality in reactions involving monoradicals. Free Radic. Res 50, S102–S111 (2016). [DOI] [PubMed] [Google Scholar]
- 49.Wüstenberg B & Pfaltz A Homogeneous hydrogenation of tri- and tetrasubstituted olefins: Comparison of iridium-phospinooxazoline [Ir-PHOX] complexes and crabtree catalysts with hexafluorophosphate (PF6) and tetrakis[3,5-bis(trifluoromethyl) phenyl]borate (BArF) as counterions. Adv. Synth. Catal 350, 174–178 (2008). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.