

Abstract
Alkaline phosphatase (ALP) responsive polymers, which can unzip from head to tail are reported. Hydrophilic and hydrophobic modification of polymer was carried out for polyelectrolyte complex and amphiphilic nanoassembly formations, respectively, which offered distinct enzyme-triggered disassembly kinetics.
Dysfunctional enzymes are the primary culprits in many pathological conditions.1,2 Therefore, developing enzyme responsive nanomaterials that exhibit programmable and functional response are attractive in applications in many areas including therapeutic delivery, diagnostics and sensing.3–7 To this end, several platforms involving polymeric8,9, oligomeric10 and dendritic11,12 assemblies have been developed, in which a desired response is achieved through substrate-specific activity of enzymes. A major limitation with these amphiphilic self-assembling systems is their slow response to enzymatic treatment, compared to their small molecule substrate counterparts. This is primarily because the substrate functionalities are often buried in the hydrophobic core of these self-assembled structures, thus limiting accessibility for enzymes.13–16
Responsive depolymerization has attracted recent focus, because it offers a convenient pathway for signal amplification.17–20 In these cases, a single stimulus induced activation event at a polymer chain terminus leads to the unzipping of polymer chain from head to tail.18,21,22 Utilizing a specific enzyme as an input signal to trigger the disassembly of nanoparticles formulated using depolymerizable polymers is a promising, yet relatively less explored, approach to address the slow kinetics of enzyme-triggerable materials.23 Specifically, we were interested in utilizing the depolymerization pathway as the mechanism to transduce an enzymatic reaction on the hydrophilic side of the polymer to the rest of the assembly. We envisioned an alkaline phosphatase (ALP) triggerable polymer, represented by the poly(benzyl carbamate) P0 and its hydrophilic and hydrophobic analogues P1 and P2 respectively (Scheme 1A). The phosphate terminus in these polymers are substrate functionalities for ALP, which undergo a cleavage reaction to set-off the depolymerization cascade, because of the revealed phenolic functionality. When these polymers are processed to form nanoparticles, we envisaged that the depolymerization process would cause an enzyme-induced disassembly and a molecular release of the non-covalently bound guest molecules (Scheme 1B).
Scheme 1:
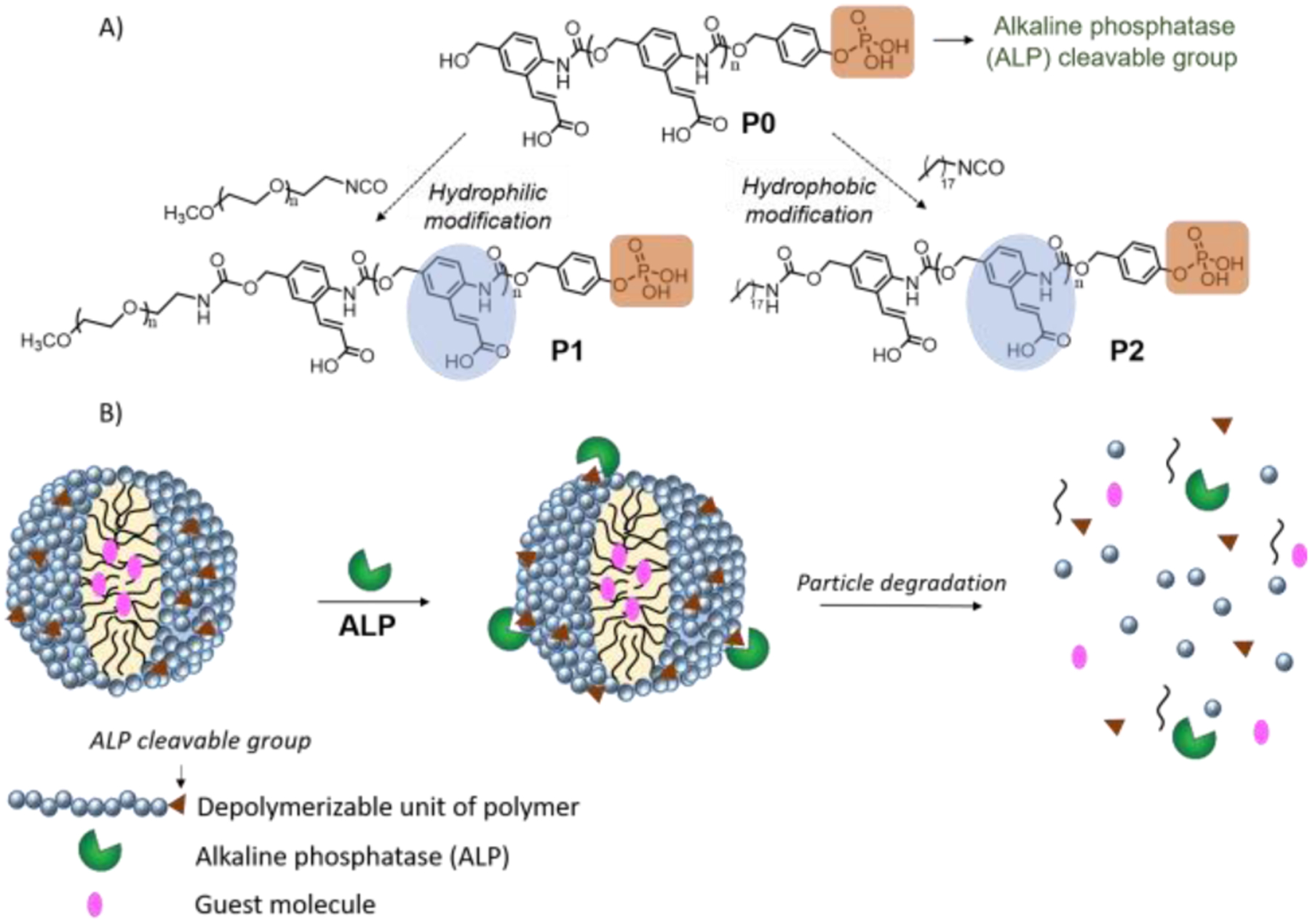
(A) Structure of ALP triggerable polymer, P0; its hydrophilic and hydrophobic modification into P1 and P2; (B) Proposed schematic of particle formulation using P1 and P2 and its triggered disassembly in response to ALP.
Poly(benzyl carbamate) backbone was synthesized using a previously reported procedure24 and its chain terminus was capped using a derivative of benzyl ether protected phosphate trigger. Deprotection of benzyl ether groups from the trigger, and t-butyl ester groups from the side chain of polymer was carried out to synthesize ALP-responsive P0 (see SI). Average number of repeating units in the polymer was ~12. Further, the terminal alcohol group from P0 was used for the hydrophilic and hydrophobic modification using poly(ethylene glycol) isocyanate (Mn ~5000) and octadecylisocyanate to achieve P1 and P2 respectively (see SI for synthetic scheme and details).
Prior to nanoparticle formulation, the enzyme triggered depolymerization process was studied using the non-assembling polymer P0 in bicarbonate buffer (250 mM, pH=8.5). Briefly, the phosphate group is cleaved by ALP to generate a phenolic chain terminus, which triggers the chain unzipping process to afford monomer-like products, including the amino-cinnamic acid derivative R1 (Figure 1A and S1). The distinct UV-absorption features of R1 at 348 nm served as the characterization handle to monitor the chain unzipping process (Figure 1B). First, the initial step of phosphate group cleavage was studied using 31P NMR, which showed the formation of phosphoric acid within 10 min after addition of 3 nM ALP to 0.2 mM concentration of P0 (Figure S2). Then, the temporal evolution of the absorbance peak at 348 nm (Figure 1B), corresponding to the formation of R1, was monitored (the small molecule R1 was independently synthesized to confirm the peak at 348 nm, as shown in Figure 1D). While there is a clear increase in the intensity of this peak with time in the ALP-incubated sample, no such peak was observed without the enzyme (Figure 1C). Formation of R1 was also confirmed using 1H NMR (Figure S3). As an additional control, the enzyme ALP itself also did not show any absorbance peak at 348 nm (Figure S4). To further test if the polymer unzipping event is specific only to ALP-triggering, the polymer solution was incubated with a non-specific enzyme, porcine liver esterase. Here, no change was observed in the absorbance profile (Figure S5), confirming that the depolymerization is indeed specifically triggered by ALP. Using the absorbance calibration curve of R1 (Figure S6), we estimated that ~80% of capped polymer undergoes depolymerization in 90 min (Figure 1E).
Figure 1:
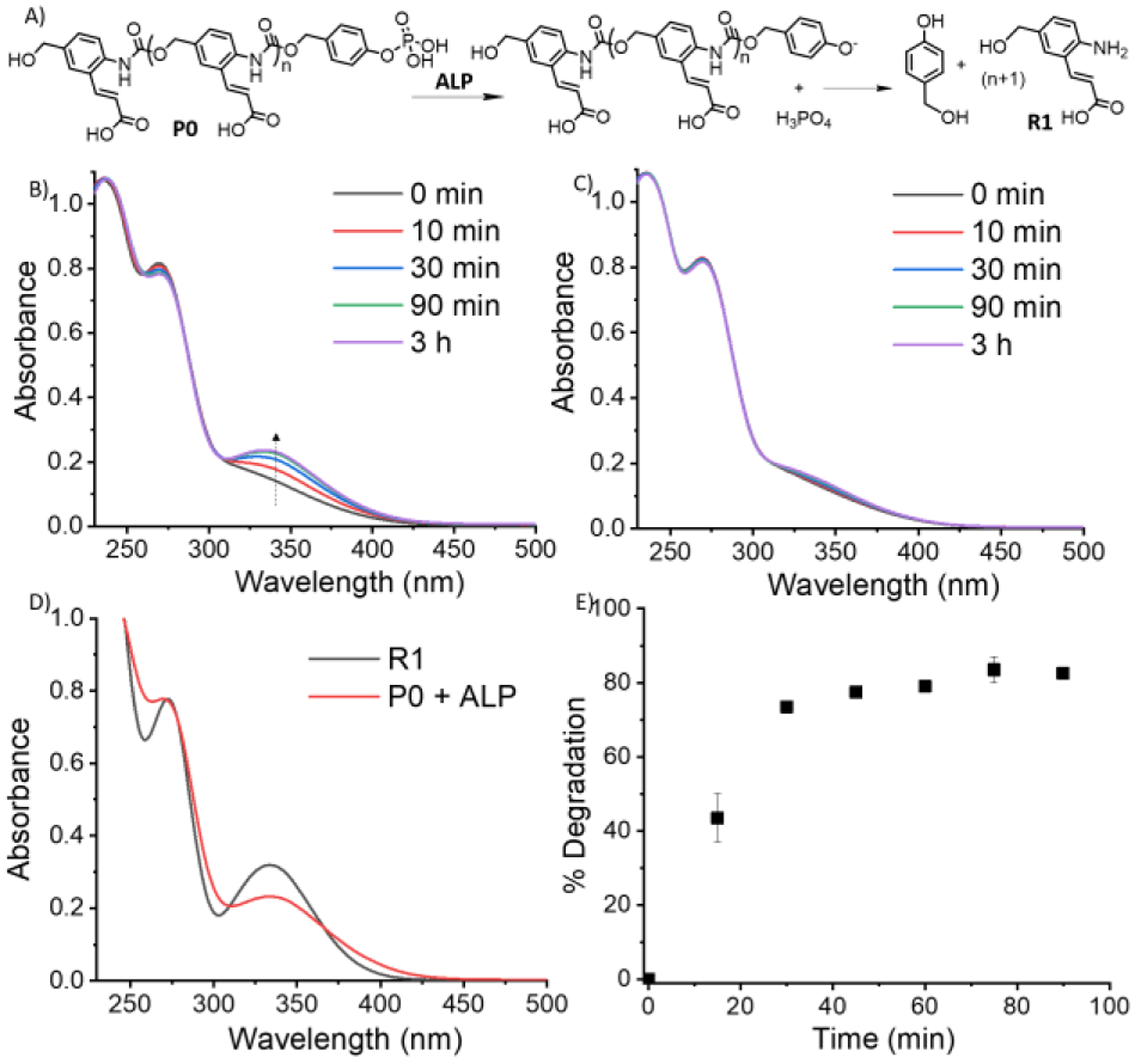
(A) Depolymerization of P0 in presence of ALP to form R1; (B) UV-Vis of P0 solution at different time with ALP incubation; (C) UV-Vis of P0 without ALP incubation; (D) UV-Vis comparison of (P0 + ALP) solution and small molecule reporter, R1; (E) Unzipping % of P0 with time.
Next, the self-assembly behaviour of P0 was studied to test if the enzyme triggered molecular-scale depolymerization behaviour can be translated to nanomaterials with similar kinetics. The aqueous phase solubility of P0 by itself was deemed to be relatively poor. Therefore, we modified P0 with a hydrophilic PEG chain using its terminal alcohol group to react with poly(ethylene glycol)isocyanate. Excess poly(ethylene glycol)isocyanate was removed through multiple washings of the reaction mixture with methanol (Figure S7). The resultant polymer P1 was indeed found to be more water soluble. Carboxylic acid functionalities from the side chain of P1 were then used to form a polyelectrolyte complex with stoichiometric amount of positively charged poly (diallyldimethylammonium chloride) (PDADMAC). Average diameter of the polyelectrolyte complex particles was found to be ~183 nm in dynamic light scattering (DLS) (Figure s9). Particle morphology was studied using transmission electron microscopy (TEM) which revealed spherical solid nanoparticle morphology (Figure S9). The particles were also used to non-covalently encapsulate a fluorescent hydrophobic guest, DiI.
Subsequently, the effect of ALP on the polyelectrolyte complex particles was studied. The nanoparticle disassembly hypothesis here is that the ALP-induced decrease in the polymer chain length would weaken the polyvalent interaction between P1 and PDADMAC. Indeed, the polyelectrolyte nanoparticles revealed the formation of small molecule reporter, R1, with time in the presence of ALP. However, the kinetics was found to be substantially slower, compared to the unzipping of P0 in solution (Figure 2A, 2B, S10). Depolymerization of P0 reached saturation within ~60 min, while the unzipping of P1 in the P1-PDADMAC complex took ~24 h. Also, monitoring the release of the encapsulated hydrophobic dye molecule showed that molecular release from these nanoparticles is negligible over the 24 h time period (Figure 2A). We surmised that the slow response of the complex could be due to the phosphate groups being buried in the polyelectrolyte nanoparticle complex, limiting accessibility to ALP enzyme.23 To test this possibility, time-dependent 31P NMR study of the complex was carried out. NMR peak, corresponding to the phosphate group was not observed initially, likely due to the restricted segmental mobility in the polyelectrolyte complex. However, after incubating the complex particles with ALP, formation of the liberated phosphoric acid was indeed observed, but this appearance was significant only after 20 h (Figure 2C). In contrast to the free polymer P0, the ALP-induced cleavage of the phosphate group itself was substantially slower, supporting the assertion that the availability of phosphate moieties for the enzymes is limited in the polyelectrolyte nanoparticles from P1.
Figure 2:
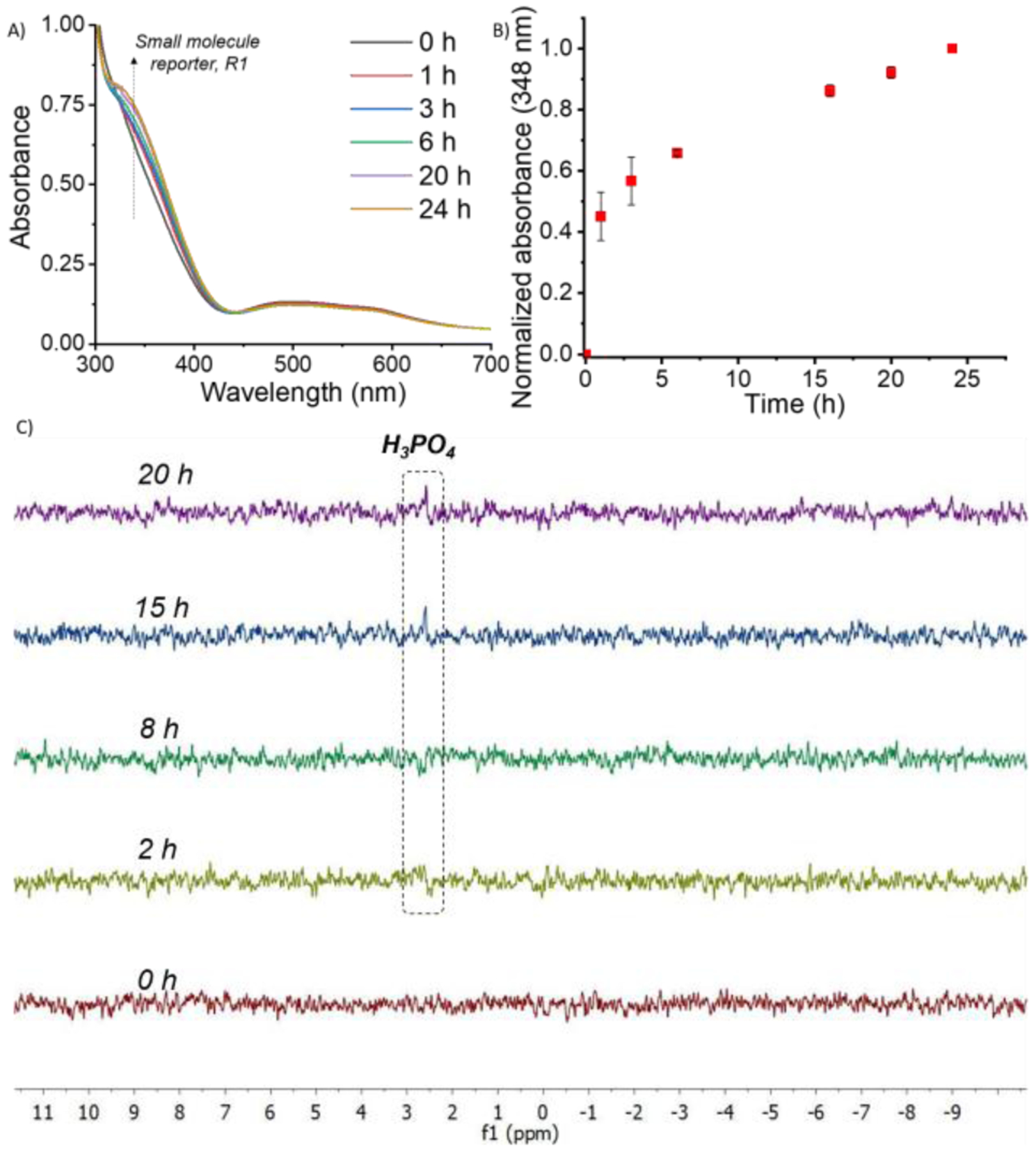
(A) UV-Vis of P1 + PDADMAC complex after ALP incubation; (B) Kinetics of evolution of absorbance at 348 nm after incubation of P1 + PDADMAC complex with ALP; (C) 31P NMR study of P1 + PDADMAC after ALP incubation at different time intervals.
To address this accessibility issue, we targeted a nanoparticle formulation approach that does not involve electrostatic complexation. In addition to P0 being limited in aqueous solubility, we envisaged that it also lacked hydrophilic-lipophilic balance needed to form amphiphilic nanoassemblies. Therefore, a hydrophobic modification of P0 was carried out using octadecyl isocyanate, to form the polymer P2. This polymer is reasonably soluble in volatile organic solvents, such as chloroform. Hence, an oil-in-water emulsion methodology was used for assembly formation.25 Briefly, suspension of P2 and a hydrophobic guest, Nile red, in chloroform (oil phase) was prepared and added to an aqueous solution of poly(vinyl alcohol) co-surfactant (water phase). Introduction of mechanical force using sonication, led to the formation of oil droplets containing polymer and guest molecules, stabilized by poly(vinyl alcohol) at the interface. Evaporation of the organic solvent and subsequent washing of excess surfactant molecules led to the formation of P2-based nanoparticles with encapsulated Nile red.
Characterization of the P2 nanoparticles was done using microscopy imaging techniques and dynamic light scattering. In TEM, spherical morphology with homogeneous distribution of the particles was observed (Figure 3A). Since, we used Nile red as a hydrophobic guest molecule, red fluorescence was observed in fluorescence microscopy image of the particles (Figure 3B). This revealed the confined location of guest molecules in the particle interior. Further, scanning electron microscopy (SEM) was also used to confirm the spherical morphology of nanoparticles (Figure 3C). Average diameter of the particles was found to be ~230 nm, as measured using DLS (Figure 3D).
Figure 3:
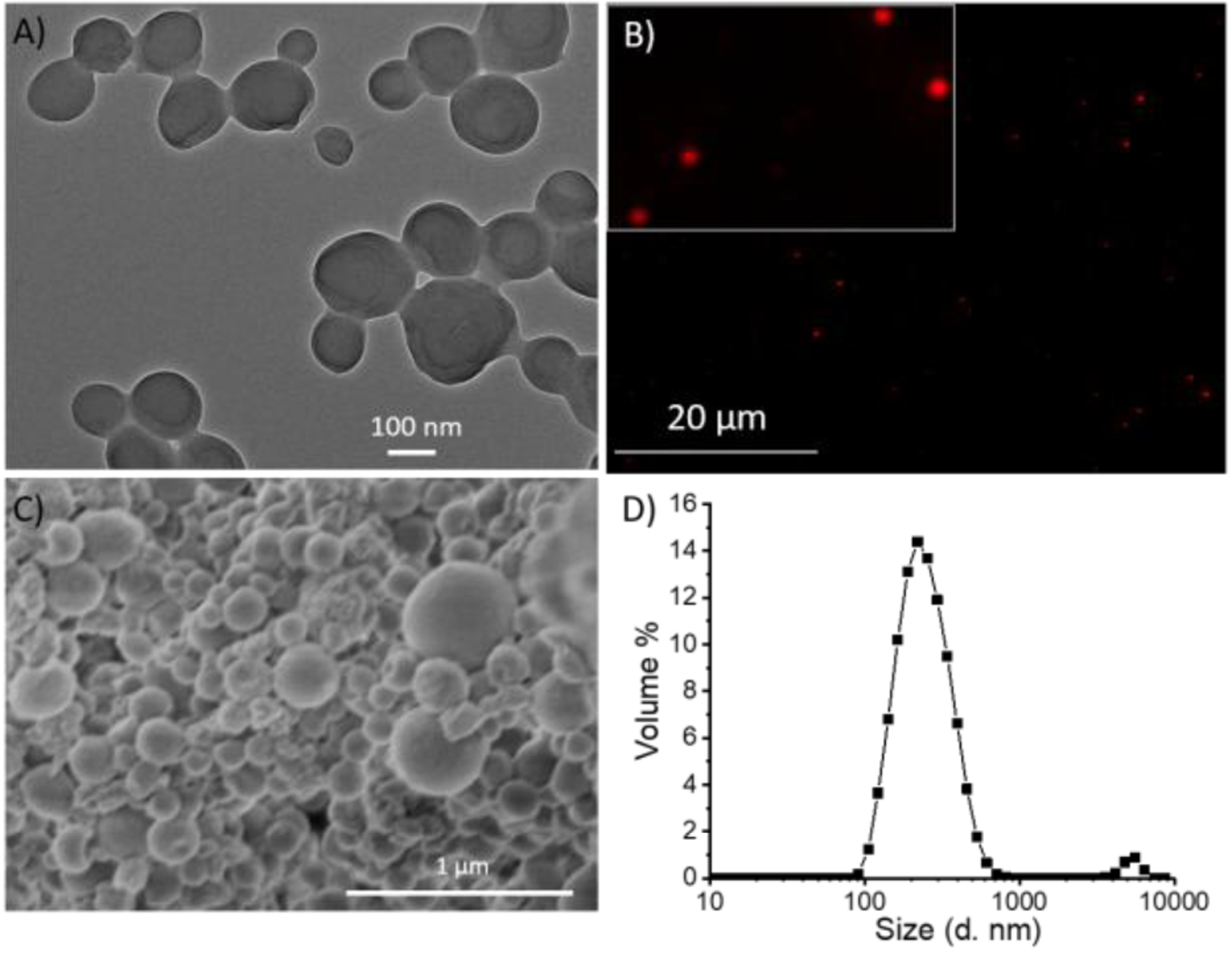
Characterization of emulsion nanoparticles using (A) Transmission electron microscopy (TEM); (B) Fluorescence microscopy; (C) Scanning electron microscopy (SEM); (D) Dynamic light scattering (DLS)
After the successful formation and characterization of nanoparticles using polymer P2, particle disassembly was studied in the presence of ALP. Colloidal suspension of particles was prepared in 2.5 mM bicarbonate buffer (pH=8.5). After incubating the particles with ALP, we observed the formation of small molecule reporter, R1, because of polymer chain unzipping (Figure 4A, 4B, S11). It is noteworthy that significant amount of the unzipping product appeared within just 1h, compared to the formation of the product requiring 24 h in the case of P1-PDADMAC complex. Concurrent with the polymer unzipping, release of encapsulated guest molecules was also observed (Figure 4A, 4C). In the control solution, without ALP, no evolution of the small molecule reporter formation was noted (Figure S12). Additionally, there was no guest release in the absence of enzyme, implying that the particles were stable hosts in the absence of enzyme. After ALP addition, spherical morphology of the particles was also lost, and the resultant particles were ill-defined, as evidenced by TEM studies (Figure 4D). The kinetics of P2 unzipping is in fact comparable to the kinetics of P0, and is much faster than the polyelectrolyte complex-based particles. We attribute this feature to the possibility that the presence of the phosphate moiety at the hydrophilic terminus of the amphiphilic polymer ensures that this moiety is exposed and available for processing by the enzyme in the amphiphilic assembly. This results in a rapid and amplified response leading to nanoparticle disassembly and release of the encapsulated guest molecules.
Figure 4:
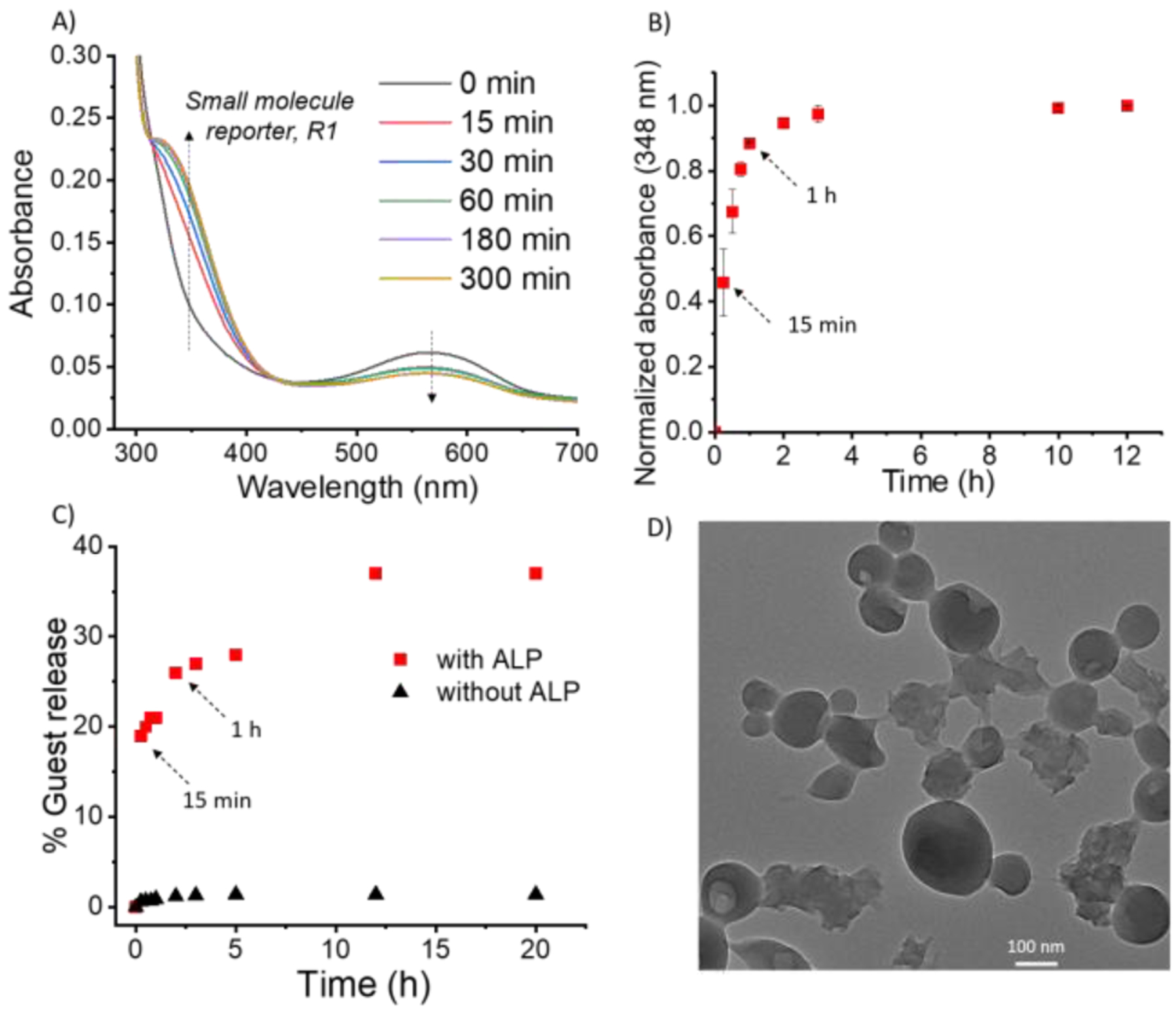
(A) UV-Vis of P2 based emulsion nanoparticles after incubation with ALP; (B) Kinetics of evolution of absorbance at 348 nm after incubating P2 emulsion nanoparticles with ALP; (C) Guest molecule release profile from the particle (red- with ALP; black- without ALP); (D) TEM image of P2 based emulsion nanoparticles after ALP addition.
In summary, using ALP triggerable polymers, structural requirements in nanoparticles for their rapid responses have been evaluated. An ALP-triggerable poly(benzyl carbamate) was modified with hydrophilic or hydrophobic functionalities to polymers that are amenable for polyelectrolyte complexation-based or emulsion-based nanoparticles respectively. The polyelectrolyte nanoparticles were found to be substantially slower in response, compared to the free polymer, while the kinetics of unzipping of the polymer in the amphiphilic assemblies was comparable to that of the free polymer. This molecular scale difference also translated to differences in kinetics of disassembly of the nanomaterials, where the host-guest properties of these materials were compromised by the presence of enzyme. The difference is attributed to the variations in the degree of accessibility of the enzyme-responsive functionalities in the context of their orientations within the nanoparticle. Results outlined in this work are applicable in designing enzyme-triggerable materials for diverse applications such as in controlled release and targeted delivery applications, where the requirements for triggered molecular release are substantially different.
Supplementary Material
Acknowledgements
Support from the U.S. Army Research Office (W911NF-15-1-0568) and the National Institutes of Health (GM-136395) are gratefully acknowledged.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
References
- 1.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA and Chinnaiyan AM, Nature, 2001, 412, 822–826. [DOI] [PubMed] [Google Scholar]
- 2.Sato H, Takino T, Okada Y, Cao J, Shinagawa A, Yamamoto E and Seiki M, Nature, 1994, 370, 61–65. [DOI] [PubMed] [Google Scholar]
- 3.Gao Y, Shi J, Yuan D and Xu B, Nat. Commun, 2012, 3, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Feng Z, Wang H, Wang S, Zhang Q, Zhang X, Rodal AA and Xu B, J. Am. Chem. Soc, 2018, 140, 9566–9573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Liu G, Wang X, Hu J and Liu S, Angew. Chem. Int. Ed, 2016, 55, 1760–1764. [DOI] [PubMed] [Google Scholar]
- 6.Wong C, Stylianopoulos T, Cui J, Martin J, Chauhan VP, Jiang W, Popovíc Z, Jain RK, Bawendi MG and Fukumura D, Proc. Natl. Acad. Sci. U. S. A, 2011, 108, 2426–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kang JH, Asai D, Kim JH, Mori T, Toita R, Tomiyama T, Asami Y, Oishi J, Sato YT, Niidome T, Jun B, Nakashima H and Katayama Y, J. Am. Chem. Soc, 2008, 130, 14906–14907. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez AR, Kramer JR and Deming TJ, Biomacromolecules, 2013, 14, 3610–3614. [DOI] [PubMed] [Google Scholar]
- 9.Rao J and Khan A, J. Am. Chem. Soc, 2013, 135, 14056–14059. [DOI] [PubMed] [Google Scholar]
- 10.Gao J, Wang H, Zhuang J and Thayumanavan S, Chem. Sci, 2019, 10, 3018–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Azagarsamy MA, Sokkalingam P and Thayumanavan S, J. Am. Chem. Soc, 2009, 131, 14184–14185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harnoy AJ, Rosenbaum I, Tirosh E, Ebenstein Y, Shaharabani R, Beck R and Amir RJ, J. Am. Chem. Soc, 2014, 136, 7531–7534. [DOI] [PubMed] [Google Scholar]
- 13.Phillips DJ, Wilde M, Greco F and Gibson MI, Biomacromolecules, 2015, 16, 3256–3264. [DOI] [PubMed] [Google Scholar]
- 14.Ding Y, Kang Y and Zhang X, Chem. Commun, 2015, 51, 996–1003. [DOI] [PubMed] [Google Scholar]
- 15.Blum AP, Kammeyer JK, Yin J, Crystal DT, Rush AM, Gilson MK and Gianneschi NC, J. Am. Chem. Soc, 2014, 136, 15422–15437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Samarajeewa S, Shrestha R, Li Y and Wooley KL, J. Am. Chem. Soc, 2012, 134, 1235–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peterson GI, Larsen MB and Boydston AJ, Macromolecules, 2012, 45, 7317–7328. [Google Scholar]
- 18.Yardley RE, Kenaree AR and Gillies ER, Macromolecules, 2019, 52, 6342–6360. [Google Scholar]
- 19.DiLauro AM, Lewis GG and Phillips ST, Angew. Chem, 2015, 127, 6298–6303. [DOI] [PubMed] [Google Scholar]
- 20.Kumar V, Harris JT, Ribbe A, Franc M, Bae Y, McNeil AJ and Thayumanavan S, ACS Macro Lett, 2020, 9, 377–381. [DOI] [PubMed] [Google Scholar]
- 21.Wang W and Alexander C, Angew. Chem. Int. Ed, 2008, 47, 7804–7806. [DOI] [PubMed] [Google Scholar]
- 22.Esser-Kahn AP, Sottos NR, White SR and Moore JS, J. Am. Chem. Soc, 2010, 132, 10266–10268. [DOI] [PubMed] [Google Scholar]
- 23.Zhuang J, Seçinti H, Zhao B and Thayumanavan S, Angew. Chem. Int. Ed, 2018, 57, 7111–7115. [DOI] [PubMed] [Google Scholar]
- 24.Sagi A, Weinstain R, Karton N and Shabat D, J. Am. Chem. Soc, 2008, 130, 5434–5435. [DOI] [PubMed] [Google Scholar]
- 25.Fomina N, McFearin C, Sermsakdi M, Edigin O and Almutairi A, J. Am. Chem. Soc, 2010, 132, 9540–9542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.