

Abstract
We are just beginning to understand the diversity of the peripheral T cell compartment, which arises from the specialization of different T cell subsets and the plasticity of individual naive T cells to adopt different fates. Although the progeny of a single T cell can differentiate into many phenotypes following infection, individual T cells are biased towards particular phenotypes. These biases are typically ascribed to random factors that occur during and after antigenic stimulation. However, the T cell compartment does not remain static with age, and shifting immune challenges during ontogeny give rise to T cells with distinct functional properties. Here, we argue that the developmental history of naive T cells creates a ‘hidden layer’ of diversity that persists into adulthood. Insight into this diversity can provide a new perspective on immunity and immunotherapy across the lifespan.
While much is known about T cell development on the individual cell level, we still lack critical information about how the T cell compartment is ‘put together’ as a whole. In general, it is thought that T cells are generated in the thymus, and a steady stream of T cells are exported to the periphery until the compartment is ‘full’1,2. Most of the variation in the peripheral T cell compartment is characterized on the basis of the antigenic experience of the cell. When a naive T cell encounters an antigen and undergoes progressive differentiation, it expresses a different set of surface markers, which can be used to distinguish naive T cells from effector and memory T cells3,4. Over the years, additional markers have been added to this classification scheme to identify new subsets of effector T cells (short-lived effector cells and memory precursor effector cells) and memory T cells (central memory cells, effector memory cells, long-lived effector cells and tissue-resident memory cells) on the basis of their distinct location and functional properties5–9. By contrast, naive T cells are often classified as a single subset of cells (CD44lowCD62Lhi cells in mice and CD45RAhiCD45ROlowCCR7hi cells in humans)10. As a result, the naive T cell compartment is typically viewed as a homogenous pool of cells.
Previous work has also viewed naive T cells as having equal potential to become effector or memory T cells, their fates determined by stochastic events in the host environment following microbial infection11,12. For example, individual T cell precursors from OT-I mice, which express an identical T cell receptor (TCR), display a wide range of effector phenotypes and clonal burst sizes after infection12. On the basis of these findings and other work, it has been proposed that the short-term and long-term fates of naive T cells are simply explained by the amount and type of stimulation they received during infection, which bias processes such as asymmetric cell division and differentiation13–17. Recent data, however, indicate that the differentiation trajectory of naive T cells is also influenced by when they were initially created in the host18–21. Naive T cells that are identical in every way except their developmental origin or age adopt different fates during infection, even when stimulated with equal amounts of an antigen and inflammation22–25. These studies suggest that not all naive T cells are created equal.
The link between T cell function and developmental origin has largely been confined to the field of neonatal immunity, where it has served as a useful explanation for why neonatal T cells behave differently to their adult counterparts. By contrast, studies of adult immune responses have generally not considered the developmental origins of cells, as this variable has not been considered relevant to immune responsiveness in adulthood. However, new studies in mice have shown that neonatal T cells persist into adulthood and retain their cell-intrinsic properties26,27, indicating that the schism between the study of adult and neonatal responses needs to be overcome and that it is important to consider the developmental history of cells in the starting population.
These new studies have prompted us to reconsider our understanding of the structure and function of the naive T cell compartment. Instead of a continuous ‘stream’ of homogeneous cells, the naive T cell compartment appears to be built from a shifting palette of T cells that are produced at defined ages and periods of development27,28. These cells persist, leading to a highly dynamic and heterogeneous pool of cells with different functional properties26,29,30. However, a more comprehensive picture of T cell ontogeny is required to fully understand the diversity of T cell responses at different stages of life.
In this Perspective, we discuss how the production and function of naive T cells adapt to meet the challenges of a changing external environment during development. We explore how the phenotype of naive T cells changes at different ages and how alterations to the formation of the naive T cell compartment might contribute to individual immune variation across the lifespan. Although we largely focus on mechanistic studies in mice, the same developmental processes exist in humans and provide an exciting opportunity for future translational research.
Immune challenges in development
T cells produced at various stages of development are faced with different immune environments and challenges (FIG. 1). During fetal life, T cells emerge into a relatively sterile environment, in which they must maintain tolerance to maternal alloantigens. Given that the fetus expresses MHC molecules inherited from both parents, a primary goal of fetal T cells is to learn how to exist in a semiallogeneic host31. Fetal T cells must also become tolerant of other harmless antigens, such as food antigens that are transferred across the placenta32,33. Thus, in utero, there is less of a need for T cells to protect the host against harmful pathogens and more of a demand to maintain tolerance to harmless antigens.
Fig. 1 |. Immune challenges and solutions during development.
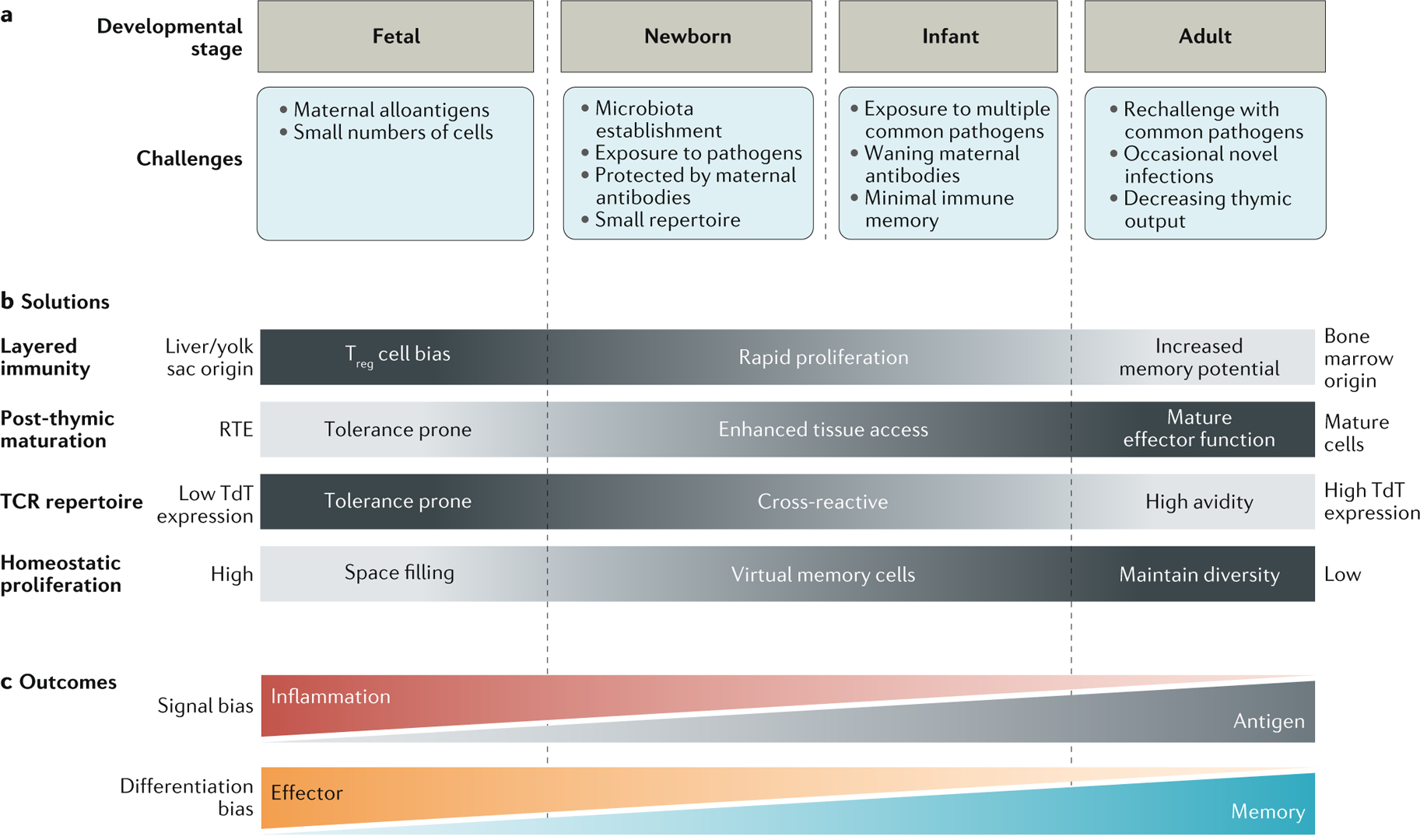
a | Development from fetal life to adulthood requires progressive adaptation to environmental and infectious challenges. Prenatal life is characterized by the need for tolerance and the absence of infectious threats. Once born, an animal must cope with exposure to a diversity of novel antigens, learn to differentiate pathogenic colonization from commensal colonization and develop an effective memory response to common pathogens. b | The host adapts to these various immune challenges by alternating haematopoietic stem cell origins, changing the duration of post-thymic maturation, changing T cell receptor (TCR) diversity and varying the amounts of homeostatic proliferation. c | The interplay of specific challenges and solutions results in age-related biases in signal responsiveness and commitment to effector or memory fates. RTE, recent thymic emigrant; TdT, terminal deoxynucleotidyltransferase; Treg cell, regulatory T cell.
Following birth, the fetus transitions from the more sterile environment of the uterus to the foreign antigen-rich environment of the outside world. During this time, the major goal of neonatal T cells is to learn the complicated task of tolerating commensal organisms, while also offering immune defence against dangerous microorganisms. Making matters more complicated, the total number of T cells is small, and effective T cell diversity is likely smaller still owing to increased homeostatic proliferation of more TCR-restricted neonatal clonotypes34–39. The neonatal T cell pool lacks immunological memory40, placing further constraints on its ability to protect the host against the multitude of pathogens encountered in early life.
With advancing age, the major goal of T cells shifts to mounting appropriate responses against novel infections and protecting the host against reinfection with common pathogens. This goal becomes even more important in adulthood, as the production of new T cells wanes with the involution of the thymus41–43. As a consequence, existing T cells in the periphery must maintain their diversity to be capable of responding to any novel pathogens encountered in old age44. At the same time, adult T cells must contain high-avidity TCRs that can both respond to specific pathogens and limit autoreactive responses against self-antigens.
From fetal life to adulthood, the changing ecological landscape presents an evolving challenge for a properly functioning T cell compartment. An important question is how does the host mediate changes within the peripheral T cell compartment to adapt?
Immune solutions
In this section, we propose that a number of different programmes of T cell production and maintenance are conserved in mice and humans, which enable T cells to meet the rapidly changing demands of their environment (FIG. 1). These developmentally related solutions to environmental challenges are important drivers of functional heterogeneity in the naive T cell compartment.
Layered immune development.
One solution to the problem of needing T cells with different functions at various stages of development is to derive them from separate progenitors. During immune ontogeny, the thymus is first colonized by fetal haematopoietic stem cells (HSCs) and later by adult HSCs33,45–48. Fetal HSCs give rise to fetal T cells that are biased towards becoming regulatory T (Treg) cells after stimulation with allogeneic cells, which help to promote tolerance in utero20,49. Fetal progenitors continue to give rise to T cells in neonatal life, which exhibit distinct epigenomic profiles50 and have an enhanced capacity to proliferate and differentiate during infection21,23–25,51,52, potentially compensating for the less diverse naive T cell pool and absence of memory cells in early life. By contrast, adult-derived T cells respond to infection with slower kinetics, but they exhibit an enhanced capacity to form memory T cells, which protect the organism against reinfections21,23–25,52. The progressive transition from fetal-origin to adult-origin HSCs during early life leads to a spectrum of cell types that are poised to differentiate into cell types (effector or memory) most useful to the host at different stages of life27.
TCR repertoire diversity.
Another way to alter T cell function at various stages of life is to change antigen recognition patterns. In early life, this is largely accomplished by delaying the expression of terminal deoxynucleotidyltransferase (TdT; the enzyme responsible for insertion of random nucleotide additions)53,54. As a result, the TCR repertoire in neonatal mice is less diverse and comprises more germline-encoded clonotypes36,37,55,56. It was initially assumed that restricting TCR repertoire diversity in early life limits pathogenic T cell responses during critical stages of growth and development. However, evidence suggests that even the more limited TCR repertoire in the developing fetus is diverse enough to recognize foreign pathogens57. Moreover, germline-encoded TCRs are more cross-reactive58, providing a mechanism to maximize immune recognition by the small number of cells present in early life. While the delay in TdT expression leads to increased cross-reactivity, it may come at the cost of reduced TCR avidity36,59. Once TdT is active, T cells made later in life show both high avidity and specificity and thus are ideal for forming pathogen-specific memory responses.
Post-thymic maturation.
In addition to the ontogenic changes in T cell production, another solution for the rapidly changing immunological demands is to have T cell function evolve over the lifespan of the individual cell. Indeed, it is well established that CD4+ and CD8+ T cells express different markers and display different functions at various times after thymic egress60. Functional changes that occur during the post-thymic maturation period align well with the needs of the host during early stages of development. For example, the newly minted naive T cells (denoted ‘recent thymic emigrants’ (RTEs)) (BOX 1), which are most abundant in early life, have an enhanced capacity to migrate to peripheral organs and are more readily tolerized in the absence of inflammation61–63.
Box 1 |. Naive T cell subsets.
Different programmes of T cell production and maintenance create naïveT cell subsets that are phenotypically and functionally distinct. Markers used to identify common naive T cell subsets in mice and humans are depicted in the table. Recent thymic emigrants (RTEs) are a well-known subset of naive T cells60. Historically, RTEs were characterized as ‘hypofunctional’, as they are generally less proliferative and exhibit diminished cytokine profiles compared with mature T cells after in vitro stimulation18,114,115. However, recent data indicate that RTEs may not be as defective as previously thought, as they undergo robust expansion and effector cell differentiation in the presence of inflammation19,61
Mature naive (MN) cells and virtual memory (VM) cells are two other subsets of naive T cells71,76,116. MN cells are what we might consider the ‘classical’ naive T cells, whereas VM cells are naive T cells that have undergone extensive homeostatic proliferation in the periphery. Functionally, VM cells respond more quickly to antigenic stimulation than MN cells and have a unique ability to undergo bystander activation in response to innate cytokines (IL-12 and IL-18) alone72,73. Thus, it is not surprising that the VM T cells are the first to respond to both inflammation and antigens during infection72.
Subsets of naive T cells behave differently depending on their stem cell origin, providing an additional layer of diversity to the naive T cell pool. For example, neonatal and adult RTEs respond differently to homeostatic cues and produce different cytokines after stimulation117. Moreover, adult CD8+ RTEs19, but not neonatal CD8+ RTEs24, mount robust recall responses against secondary infection. Similarly, MN and VM cells derived from fetal haematopoietic stem cells adopt different fates after infection to their counterparts derived from adult haematopoietic stem cells 23. These developmental differences were nicely illustrated in a series of dual adoptive experiments, which showed that naive T cells are biased towards terminal differentiation in the following order: neonatal VM cells > neonatal MN cells > adult VM cells > adult MN cells.
| Species | RTEs | Mature naive T cells | Virtual memory T cells | |||
|---|---|---|---|---|---|---|
| Cell type | Phenotype | Cell type | Phenotype | Cell type | Phenotype | |
| Mouse | CD4+and CD8+ | CD24+ CD62L+ | CD4+and CD8+ | CD44− CD62L+Qa2+ | CD4+and CD8+ | CD122+CD44+ CD49d− |
| Human | CD4+ only | CD45RA* CCR7+ PTK7+CD31* | CD4+and CD8+ | CD45RA* CCR7+ | CD8+only | CD45RA*KIR+ and/or NKC2A |
CCR7, CC-chemokine receptor 7; KIR, killer cell immunoglobulin-like receptor; PTK7, protein-tyrosine kinase 7.
Antigen versus inflammation sensitivity.
A major challenge for neonatal cells in early life is differentiating between commensal and pathogenic organisms after birth. That is, there is no easy way for the TCR to differentiate between healthy microbiota-derived peptides and potential pathogen-derived peptides on the basis of peptides alone. Only the ‘danger’ signals associated with pathogens allow any discrimination by the TCR. Thus, it is adaptive for neonatal cells to be more ‘inflammation responsive’27,63–69. For example, human CD8+ T cells in umbilical cord blood preferentially express Toll-like receptor 2 (TLR2) and TLR5, and human neonatal CD4+ T cells are equipped to respond to TLR1 and TLR2 ligands65–67. In adults, RTEs also exhibit increased expression of innate-like receptors (TLRs, complement receptors and natural killer cell receptors), which may help them to discriminate between harmless and pathogenic microorganisms63,68,69.
Homeostatic proliferation.
The differentiation trajectory of naive T cells is also influenced by their previous levels of homeostatic proliferation70–73. The current dogma is that T cells undergo increased rates of homeostatic proliferation in early life because they are exported to peripheral environments that are devoid of other T cells and, therefore, exposed to greater amounts of homeostatic cytokines (IL-7 and IL-15) on a per cell basis35,74,75. However, recent data demonstrate that cells made early in life have an inherent propensity to undergo homeostatic proliferation, probably owing to their HSC origin27. Naive T cells that have undergone extensive homeostatic proliferation upregulate markers and obtain functions that are typically associated with memory T cells induced by a foreign antigen71,76,77 (BOX 1). Thus, in addition to rapidly filling the peripheral pool and providing some naive T cells with additional functional properties, homeostatic proliferation may also serve as a useful stop-gap defence mechanism until more true memory cells can be made. In adulthood, homeostatic proliferation may be reduced to maintain repertoire diversity78.
Structure dictates function
Viewing the naive T cell compartment through the lens of immune development changes our perspective of the T cell response to infection with progressing age. In the past, the focus was on how age alters the cell-intrinsic properties of individual cells. For example, it is generally believed that CD8+ T cells from aged individuals respond less vigorously to infection than younger cells because they have been subjected to years of protein misfolding and oxidative damage79. However, different drivers (HSC origin, post-thymic maturation or homeostatic proliferation) sculpt the peripheral T cell compartment during various windows of development, resulting in dramatic changes to the composition of the naive T cell pool at various stages of life. If we zoom out and consider how the distribution of naive T cell subsets varies over time, we can begin to appreciate how age-related changes in the starting population alter the host response to infection. The traditional ‘neonatal’ and ‘adult’ phenotypes represent just two snapshots in time across a changing immune landscape. In this section, we take into account the developmental biology of the peripheral T cell compartment and offer a different perspective on the ‘neonatal’ and ‘adult’ T cell responses to infection (FIG. 2).
Fig. 2 |. Naive T cell subsets differ in their ability to respond to antigens and inflammation.
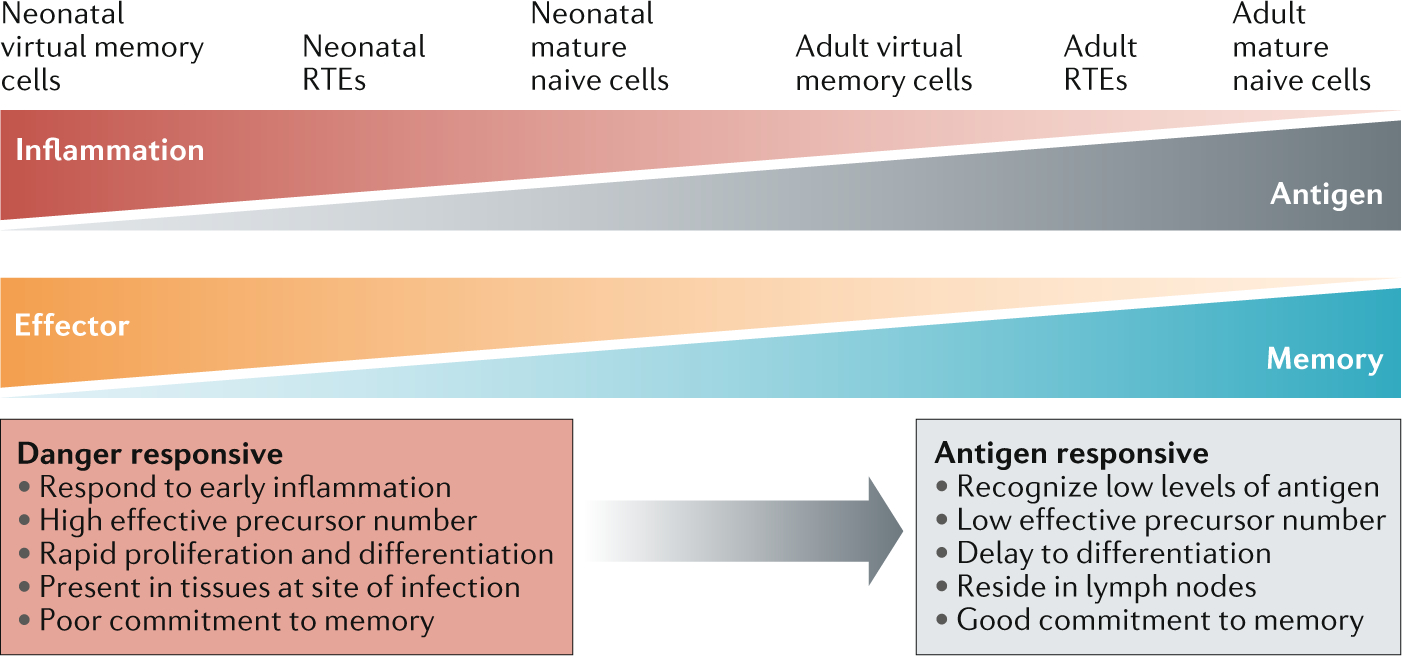
Immune challenges elicit a spectrum of responses from developmentally distinct subsets. Cells made early in life are more responsive to inflammatory signals and undergo rapid proliferation and differentiation. This allows rapid effector responses but is detrimental for the generation of robust memory. By contrast, cell subsets generated in adulthood respond to low levels of an antigen and make strong memory responses but require a significant time to become functional. RTEs, recent thymic emigrants.
The classical picture of T cell responses to novel pathogens assumes a long delay between the recognition of a peptide by a small number of naive antigen-specific precursors and their subsequent proliferation, differentiation and ability to mount an effector response. In the very young, in which nearly all infections are novel, this slow responsiveness represents a major risk to the individual. This risk is mitigated by the unique composition of T cells present in early life. First, there is a larger relative number of virtual memory cells in the neonatal pool, which are sensitive to inflammation and can be activated by innate cytokines alone23,27,64,80. The ability of (non-antigen-specific) neonatal cells to respond to inflammatory stimuli means a larger proportion of T cells can respond to infection in a ‘bystander’ manner81 (but only once the pathogen has caused sufficient tissue damage). Second, the broad cross-reactivity of germline-encoded clonotypes58 may allow a larger proportion of neonatal T cells to recognize a particular epitope. Although the neonatal precursors may have lower avidity for a cognate antigen, this limitation is potentially overcome by the fact that most neonatal T cells are also RTEs and can therefore receive additional (costimulatory) signalling via TLRs65–67. Lastly, the virtual memory cells and RTEs in early life are derived from fetal HSCs, which enable them to divide and differentiate more rapidly than their adult-derived counterparts, and quickly contribute to the response23,27,82,83. The major drawback of the neonatal T cell response is the inability to form high-avidity, long-lived memory responses24,25,36. Although the bias towards effector cell differentiation in early life is often viewed as a defect, we propose that it is actually a useful adaptation that prioritizes immediate survival over future survival.
However, later in life, there is a shift from fighting infection with novel pathogens to fighting recurring exposure to common pathogens. The composition of the naive T cell pool in adulthood is well adapted to meet these demands. Adult-derived naive T cells are produced from a more diverse pool of TCRs and largely have a mature naive phenotype. Thus, they have the potential to be highly sensitive to an antigen and give rise to memory cells, but they tend to exist at low precursor numbers and exhibit slower proliferation rates21,23–25,52,82,83. Fortunately, the transition from neonatal to adult immunity does not involve the complete loss of neonatal cells. Neonatal phenotype cells persist throughout early life even as they are gradually replaced by adult phenotype cells26. Newly arrived adult cells also have an RTE phenotype, are sensitive to danger signals and offer some innate-like functions63,68,69. Thus, in adolescence, we see the presence of rapid early responses by neonatal virtual memory cells and adult-derived RTEs, followed by the development of mature (adult-derived) effector and memory responses. As the host accumulates a pool of memory cells for common pathogens, we see a shift from inflammation responsiveness to antigen-specific memory, which is accompanied by a decrease in the number of neonatal and RTE phenotype cells26. However, even in adult life, we observe that neonatal cells expand early in infection by novel pathogens, before being overtaken by the naive cell-derived adult response27.
Variations in immune development
As discussed already, we propose that the naive T cell pool evolves over the lifespan of the individual. However, the evolution of the peripheral T cell compartment likely differs among individuals, depending on the unique set of conditions encountered during growth and development84–86. In anthropology, this concept is referred to as ‘developmental acclimatization’, and it has been used to explain how physiological systems adapt to certain conditions in early life87. Early life is a critical phase of rapid adaptation of the peripheral T cell compartment to the external environment, and thus stressors acting in this phase can act to alter either the number or the function of cells surviving into adulthood88 (FIG. 3). As different proportions of neonatal, RTE and virtual memory cells alter how an individual responds to inflammation and antigens (self, commensal or foreign), factors that affect immune cell production and survival in early life may have long-term effects on the susceptibility to infectious or autoimmune diseases.
Fig. 3 |. Evolution and adaptation of the T cell compartment with progressing age.
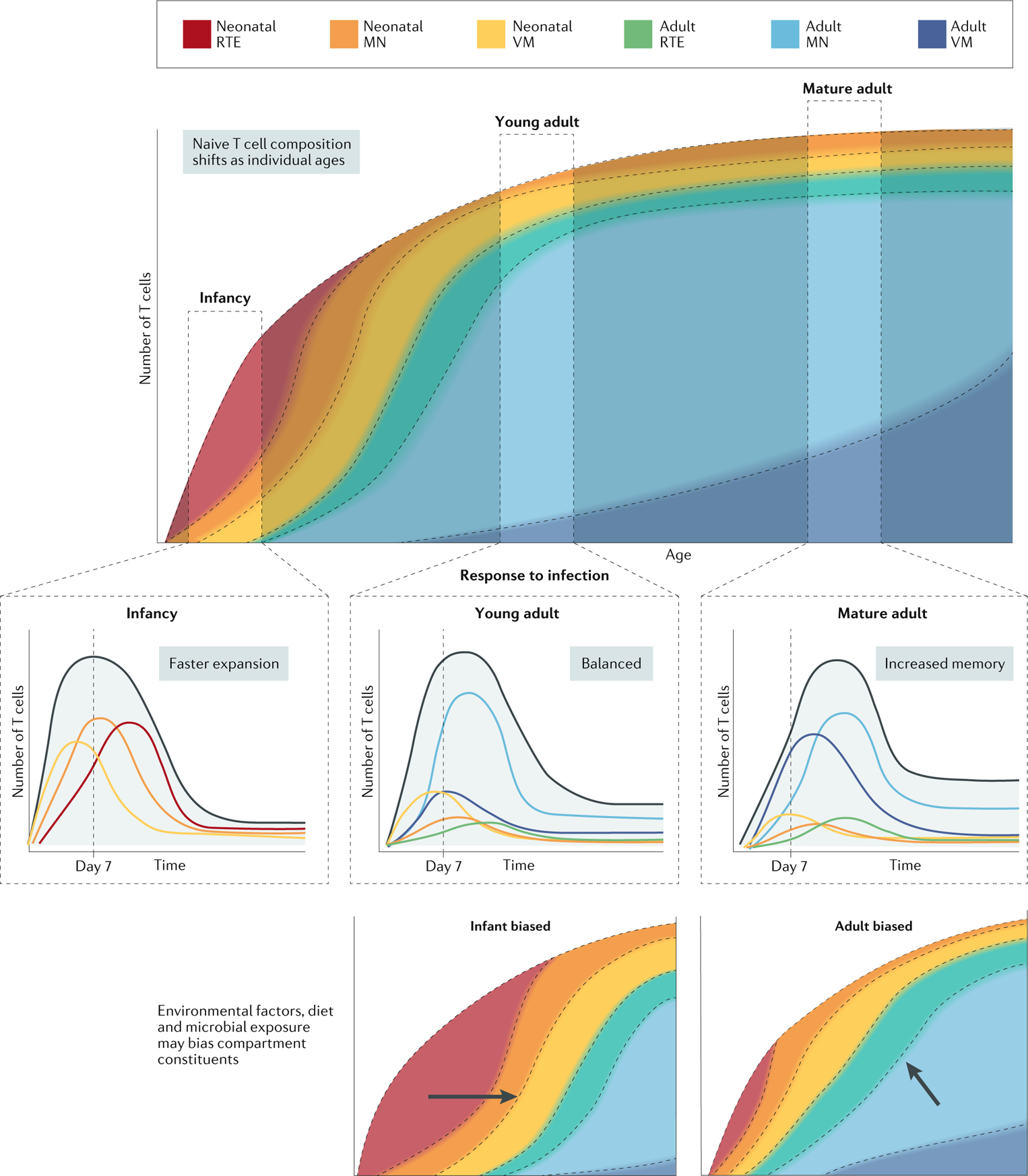
The composition of the pool of naive T cells changes over time owing to changes in stem cell origin, T cell production, post-thymic T cell differentiation and survival. In infancy, the peripheral T cell pool is dominated by neonatal recent thymic emigrants (RTEs), which progressively differentiate to neonatal phenotype mature naive (MN) cells and virtual memory (VM) cells as a result of homeostatic proliferation. In time, these are complemented by the arrival of adult RTEs, which also differentiate over time into MN cells and VM cells, albeit at a slower rate than in infancy. At any given age, the composition of the responding naive T cell pool is unique and leads to changes in how the pool responds to novel infections. Shortly after birth, the response is dominated by fast-esponding, inflammation-sensitive subsets. Through infancy, fast-responding neonatal cells persist to provide protection, while newly arrived adult subsets begin to establish high-avidity immunological memory. In adulthood, the highly antigen-sensitive adult-derived subsets are mostly highly represented, leading to slower responses but enhanced memory formation. Perturbations in normal immune development due to infection, malnutrition or environmental factors can perturb the T cell pool, resulting in it containing higher or lower levels of neonatal immune cells.
One factor that likely alters the layered development of the peripheral T cell compartment is microbial exposure. Indeed, twin studies have shown that persistent infection with cytomegalovirus is a major driver of immune variation in humans84, and population studies suggest that geographical location can profoundly affect immune development89. However, it is important to consider how the timing of exposure could impact the composition of the T cell compartment. Exposure to cytomegalovirus in infancy might lead to an outgrowth of neonatal T cells, while later exposure could favour the expansion of adult T cells90–92. Changing the distribution of subsets in the naive T cell compartment could alter an individual’s inherent ability to form effector and memory T cells, as well as their response to new infections. Other types of pathogen can also alter the architecture of the peripheral T cell compartment and subsequent responsiveness to future infection. For example, mice exposed to helminths (such as Heligmosomoides polygyrus) have a larger population of virtual memory cells and are more resistant to bacterial pathogens93,94. Depending on the timing of infection, infection with H. polygyrus could lead to an outgrowth of neonatal or adult virtual memory cells and change the inflammatory response during early stages of infection.
It is also interesting to speculate how reductions in the overall number of infections affect immune development at the population level95,96. For example, the ‘hygiene hypothesis’ suggests that growing up in the unusually ‘clean’ environment of high-income countries may predispose individuals to allergic and autoimmune reactions97–99. Proponents of the hygiene hypothesis argue that in the absence of routine pathogen exposure, the immune system overreacts to what should be harmless. However, a developmentally driven interpretation might suggest that differential layering of T cells during ontogeny may contribute to these shifts in responsiveness. Factors such as malnutrition or obesity in early life may also have long-term effects on immunity if they perturb the overall structure of the peripheral T cell compartment100–105.
The importance of naive T cell diversity may also be revealed in circumstances in which normal development is interrupted. For example, recovery from bone marrow transplants (or HIV-mediated depletion of CD4+ T cells) is typically measured by replacement of total T cell numbers. However, what might appear as a superficially ‘normal’ repopulation may lack naturally occurring phenotypic diversity, owing to a loss of ontogenic layers. If cells produced at different times are building blocks in the establishment of a fully functional naive peripheral T cell compartment, removal of the original structure (via irradiation or chronic infection) could result in profound changes in how the new T cell compartment responds to infection.
Conclusion
Collectively, the studies discussed herein point towards a new model, in which different developmental histories in the naive T cell pool contribute to diversity in the T cell response to infection. These observations are similar to those made with the ontogeny of B cells106–108, macrophages109,110, innate lymphoid cells111 and γδ T cells112,113: successive waves of phenotypically distinct cells populate the periphery, persist into adulthood and respond to infection with distinct kinetics. There is now ample evidence to suggest that different subsets of naive T cells in adults exhibit unique roles during infection, which are linked to when they were initially created in the host. We believe the evidence warrants a reframing of how we view adult immunity. Rather than focusing solely on events that occur after priming, we need to better understand how the heterogeneity in the starting population of T cells affects both the response to acute infection and the quality of long-term memory responses.
Moving forwards, the challenge will be to generate models and tools to assess the proportions of neonatal versus adult cells, RTEs versus mature cells, and mature naive versus virtual memory phenotype cells that make up the naive pool in mice and humans at various stages of life (BOX 2). It will also be important to understand how these subsets respond to infection in neonatal and adult environments. In mice, we have the ability to use sophisticated fate-mapping mouse models to understand how extrinsic factors (such as diet and microorganisms) perturb the ontogeny of the T cell compartment and, consequently, the host response to infection. Such models may also prove to be useful in mapping out the developmental pathways of different subsets of naive T cells, as well as for developing testable hypotheses for future human studies.
Box 2 |. Ontogeny of T cells in mice and humans.
Much of our present knowledge of T cell ontogeny comes from studies in mice, in which marking and tracking of cells makes it possible to study their origins. The work on immune development in mice provides a framework in which to better understand the similarities and differences in humans (see the figure118–124). Although the underlying mechanisms of T cell development are conserved between species, notable distinctions are worth mentioning. First, the diversification of the repertoire occurs at an earlier stage of development in humans than in mice. Delayed expression of terminal deoxynucleotidyltransferase (TdT) extends the ‘fetal repertoire’ of mice into the first few weeks after birth53,54. Second, the post-thymic maturation period is shorter in mice. This difference in timing reflects the longer lifespan of T cells in humans than in mice2. Third, there is more detailed information on how developmental layering alters the naive T cell compartment in mice than in humans, as more sophisticated models are available to track the fates of T cells in mice. As a result, it is still not entirely clear when the developmental switch from fetal to adult haematopoietic stem cells (HSCs) occurs in humans. Lastly, some evidence suggests that mice rely more on thymic output to maintain the T cell compartment, whereas humans rely more on homeostatic proliferation2. Also, there are subsets of T cells with a memory-like phenotype that are present in newborn humans but not in newborn mice125. However, these studies involved mice housed in specific pathogen-free conditions, so it remains unclear whether these species-specific differences can be attributed to genetic or environmental factors.
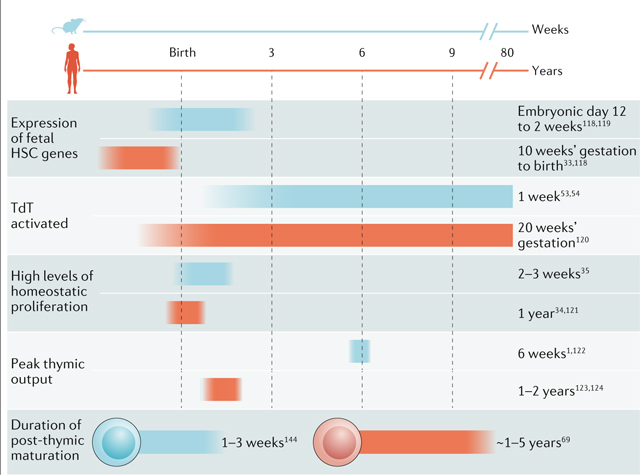
In humans, it will be important to use the newer single-cell technologies (single-cell RNA sequencing and single-cell assay for transposase-accessible chromatin using sequencing) to identify reliable markers for different subsets of naive T cells and to better understand how the peripheral T cell compartment is altered at the individual cell level and the population level at various stages of development. If we can understand changes in the peripheral T cell pool from birth to old age, we may one day be able to predict infection outcomes on the basis of the structure of the naive T cell pool. A better understanding of the ontogeny of the naive T cell compartment may also yield advancements in the development of personalized medicine. For example, it may be possible to personalize the delivery of vaccines to different individuals to improve protective immunity, or to predict effectiveness and adverse outcomes associated with immunomodulatory therapies. Finally, we may find that certain subsets of naive T cells are particularly well suited for specific therapeutic applications, such as cancer immunotherapy.
Footnotes
Competing interests
The authors declare no competing interests.
Peer review information
Nature Reviews Immunology thanks T. Burt and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.van Hoeven V et al. Dynamics of recent thymic emigrants in young adult mice. Front. Immunol 8, 933 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.den Braber I et al. Maintenance of peripheral naive T cells is sustained by thymus output in mice but not humans. Immunity 36, 288–297 (2012). [DOI] [PubMed] [Google Scholar]
- 3.Sallusto F, Lenig D, Forster R, Lipp M & Lanzavecchia A Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401, 708–712 (1999). [DOI] [PubMed] [Google Scholar]
- 4.Mackay CR Homing of naive, memory and effector lymphocytes. Curr. Opin. Immunol 5, 423–427 (1993). [DOI] [PubMed] [Google Scholar]
- 5.Jameson SC & Masopust D Understanding subset diversity in T cell memory. Immunity 48, 214–226 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Omilusik KD & Goldrath AW Remembering to remember: T cell memory maintenance and plasticity. Curr. Opin. Immunol 58, 89–97 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kumar BV, Connors TJ & Farber DL Human T cell development, localization, and function throughout life. Immunity 48, 202–213 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marshall HD et al. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4+ cell properties during viral infection. Immunity 35, 633–646 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerlach C et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity 45, 1270–1284 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van den Broek T, Borghans JAM & van Wijk F The full spectrum of human naive T cells. Nat. Rev. Immunol 18, 363–373 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Stemberger C et al. A single naive CD8+ T cell precursor can develop into diverse effector and memory subsets. Immunity 27, 985–997 (2007). [DOI] [PubMed] [Google Scholar]
- 12.Plumlee CR, Sheridan BS, Cicek BB & Lefrancois L Environmental cues dictate the fate of individual CD8+ T cells responding to infection. Immunity 39, 347–356 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerlach C et al. Heterogeneous differentiation patterns of individual CD8+ T cells. Science 340, 635–639 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Joshi NS et al. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity 27, 281–295 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van Faassen H et al. Reducing the stimulation of CD8+ T cells during infection with intracellular bacteria promotes differentiation primarily into a central (CD62LhighCD44high) subset. J. Immunol 174, 5341–5350 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Masson F, Mount AM, Wilson NS & Belz GT Dendritic cells: driving the differentiation programme of T cells in viral infections. Immunol. Cell Biol 86, 333–342 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Chang JT et al. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science 315, 1687–1691 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Hendricks DW & Fink PJ Recent thymic emigrants are biased against the T-helper type 1 and toward the T-helper type 2 effector lineage. Blood 117, 1239–1249 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Makaroff LE, Hendricks DW, Niec RE & Fink PJ Postthymic maturation influences the CD8 T cell response to antigen. Proc. Natl Acad. Sci. USA 106, 4799–4804 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mold JE et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science 330, 1695–1699 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reynaldi A et al. Modeling the dynamics of neonatal CD8+ T-cell responses. Immunol. Cell Biol 94, 838–848 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tabilas C et al. Cutting edge: elevated glycolytic metabolism limits the formation of memory CD8+ T cells in early life. J. Immunol 203, 2571–2576 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J et al. Fetal and adult progenitors give rise to unique populations of CD8+ T cells. Blood 128, 3073–3082 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Smith NL et al. Rapid proliferation and differentiation impairs the development of memory CD8+ T cells in early life. J. Immunol 193, 177–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zens KD et al. Reduced generation of lung tissue-resident memory T cells during infancy. J. Exp. Med 214, 2915–2932 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reynaldi A et al. Fate mapping reveals the age structure of the peripheral T cell compartment. Proc. Natl Acad. Sci. USA 116, 3974–3981 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith NL et al. Developmental origin governs CD8(+) T cell fate decisions during infection. Cell 174, 117–130.e114 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Yang S, Fujikado N, Kolodin D, Benoist C & Mathis D Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348, 589–594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pogorelyy MV et al. Persisting fetal clonotypes influence the structure and overlap of adult human T cell receptor repertoires. PLoS Comput. Biol 13, e1005572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B et al. Glimpse of natural selection of long-lived T-cell clones in healthy life. Proc. Natl Acad. Sci. USA 113, 9858–9863 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mold JE et al. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science 322, 1562–1565 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burt TD Fetal regulatory T cells and peripheral immune tolerance in utero: implications for development and disease. Am. J. Reprod. Immunol 69, 346–358 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mold JE & McCune JM Immunological tolerance during fetal development: from mouse to man. Adv. Immunol 115, 73–111 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Schonland SO et al. Homeostatic control of T-cell generation in neonates. Blood 102, 1428–1434 (2003). [DOI] [PubMed] [Google Scholar]
- 35.Le Campion A et al. Naive T cells proliferate strongly in neonatal mice in response to self-peptide/self-MHC complexes. Proc. Natl Acad. Sci. USA 99, 4538–4543 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rudd BD et al. Acute neonatal infections ‘lock-in’ a suboptimal CD8+ T cell repertoire with impaired recall responses. PLoS Pathog. 9, e1003572 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rudd BD, Venturi V, Davenport MP & Nikolich-Zugich J Evolution of the antigen-specific CD8+ TCR repertoire across the life span: evidence for clonal homogenization of the old TCR repertoire. J. Immunol 186, 2056–2064 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carey AJ et al. Public clonotypes and convergent recombination characterize the naive CD8+ T-cell receptor repertoire of extremely preterm neonates. Front. Immunol 8, 1859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schelonka RL et al. T cell receptor repertoire diversity and clonal expansion in human neonates. Pediatr. Res 43, 396–402 (1998). [DOI] [PubMed] [Google Scholar]
- 40.D’Arena G et al. Flow cytometric characterization of human umbilical cord blood lymphocytes: immunophenotypic features. Haematologica 83, 197–203 (1998). [PubMed] [Google Scholar]
- 41.Nikolich-Zugich J The twilight of immunity: emerging concepts in aging of the immune system. Nat. Immunol 19, 10–19 (2018). [DOI] [PubMed] [Google Scholar]
- 42.Goronzy JJ & Weyand CM Mechanisms underlying T cell ageing. Nat. Rev. Immunol 19, 573–583 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thome JJ et al. Longterm maintenance of human naive T cells through in situ homeostasis in lymphoid tissue sites. Sci. Immunol 1, eaah6506 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nikolich-Zugich J, Slifka MK & Messaoudi I The many important facets of T-cell repertoire diversity. Nat. Rev. Immunol 4, 123–132 (2004). [DOI] [PubMed] [Google Scholar]
- 45.Jotereau F, Heuze F, Salomon-Vie V & Gascan H Cell kinetics in the fetal mouse thymus: precursor cell input, proliferation, and emigration. J. Immunol 138, 1026–1030 (1987). [PubMed] [Google Scholar]
- 46.Tavian M & Peault B The changing cellular environments of hematopoiesis in human development in utero. Exp. Hematol 33, 1062–1069 (2005). [DOI] [PubMed] [Google Scholar]
- 47.Herzenberg LA & Herzenberg LA Toward a layered immune system. Cell 59, 953–954 (1989). [DOI] [PubMed] [Google Scholar]
- 48.Adkins B Developmental regulation of the intrathymic T cell precursor population. J. Immunol 146, 1387–1393 (1991). [PubMed] [Google Scholar]
- 49.Ng MSF, Roth TL, Mendoza VF, Marson A & Burt TD Helios enhances the preferential differentiation of human fetal CD4(+) naive T cells into regulatory T cells. Sci. Immunol 4, eaav5947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rudd BD Neonatal T cells: a reinterpretation. Annu. Rev. Immunol 38, 229–247 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Connors TJ et al. Developmental regulation of effector and resident memory T cell generation during pediatric viral respiratory tract infection. J. Immunol 201, 432–439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Siefker DT & Adkins B Rapid CD8+ function is critical for protection of neonatal mice from an extracellular bacterial enteropathogen. Front. Pediatr 4, 141 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bogue M, Candeias S, Benoist C & Mathis D A special repertoire of alpha:beta T cells in neonatal mice. EMBO J. 10, 3647–3654 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Feeney AJ Junctional sequences of fetal T cell receptor beta chains have few N regions. J. Exp. Med 174, 115–124 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venturi V et al. The neonatal CD8+ T cell repertoire rapidly diversifies during persistent viral infection. J. Immunol 196, 1604–1616 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carey AJ et al. Rapid evolution of the CD8+ TCR repertoire in neonatal mice. J. Immunol 196, 2602–2613 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rechavi E & Somech R Survival of the fetus: fetal B and T cell receptor repertoire development. Semin. Immunopathol 39, 577–583 (2017). [DOI] [PubMed] [Google Scholar]
- 58.Gavin MA & Bevan MJ Increased peptide promiscuity provides a rationale for the lack of N regions in the neonatal T cell repertoire. Immunity 3, 793–800 (1995). [DOI] [PubMed] [Google Scholar]
- 59.Kedzierska K et al. Terminal deoxynucleotidyltransferase is required for the establishment of private virus-specific CD8+ TCR repertoires and facilitates optimal CTL responses. J. Immunol 181, 2556–2562 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fink PJ The biology of recent thymic emigrants. Annu. Rev. Immunol 31, 31–50 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Friesen TJ, Ji Q & Fink PJ Recent thymic emigrants are tolerized in the absence of inflammation. J. Exp. Med 213, 913–920 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.He Q et al. Thymic development of autoreactive T cells in NOD mice is regulated in an age-dependent manner. J. Immunol 191, 5858–5866 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Das A et al. Adaptive from innate: human IFN-γ+CD4+ T cells can arise directly from CXCL8-producing recent thymic emigrants in babies and adults. J. Immunol 199, 1696–1705 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akue AD, Lee JY & Jameson SC Derivation and maintenance of virtual memory CD8 T cells. J. Immunol 188, 2516–2523 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCarron M & Reen DJ Activated human neonatal CD8+ T cells are subject to immunomodulation by direct TLR2 or TLR5 stimulation. J. Immunol 182, 55–62 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Komai-Koma M, Jones L, Ogg GS, Xu D & Liew FY TLR2 is expressed on activated T cells as a costimulatory receptor. Proc. Natl Acad. Sci. USA 101, 3029–3034 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sinnott BD, Park B, Boer MC, Lewinsohn DA & Lancioni CL Direct TLR-2 costimulation unmasks the proinflammatory potential of neonatal CD4+ T cells. J. Immunol 197, 68–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van den Broek T et al. Neonatal thymectomy reveals differentiation and plasticity within human naive T cells. J. Clin. Invest 126, 1126–1136 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pekalski ML et al. Neonatal and adult recent thymic emigrants produce IL-8 and express complement receptors CR1 and CR2. JCI Insight 2, e93739 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fulton RB et al. The TCR’s sensitivity to self peptide-MHC dictates the ability of naive CD8+ T cells to respond to foreign antigens. Nat. Immunol 16, 107–117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.White JT, Cross EW & Kedl RM Antigen-inexperienced memory CD8+ T cells: where they come from and why we need them. Nat. Rev. Immunol 17, 391–400 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lee JY, Hamilton SE, Akue AD, Hogquist KA & Jameson SC Virtual memory CD8 T cells display unique functional properties. Proc. Natl Acad. Sci. USA 110, 13498–13503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haluszczak C et al. The antigen-specific CD8+ T cell repertoire in unimmunized mice includes memory phenotype cells bearing markers of homeostatic expansion. J. Exp. Med 206, 435–448 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Min B et al. Neonates support lymphopenia-induced proliferation. Immunity 18, 131–140 (2003). [DOI] [PubMed] [Google Scholar]
- 75.Schuler T, Hammerling GJ & Arnold B Cutting edge: IL-7-dependent homeostatic proliferation of CD8+ T cells in neonatal mice allows the generation of long-lived natural memory T cells. J. Immunol 172, 15–19 (2004). [DOI] [PubMed] [Google Scholar]
- 76.Jameson SC, Lee YJ & Hogquist KA Innate memory T cells. Adv. Immunol 126, 173–213 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Goldrath AW, Bogatzki LY & Bevan MJ Naive T cells transiently acquire a memory-like phenotype during homeostasis-driven proliferation. J. Exp. Med 192, 557–564 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mold JE et al. Cell generation dynamics underlying naive T-cell homeostasis in adult humans. PLoS Biol. 17, e3000383 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Miller RA The aging immune system: primer and prospectus. Science 273, 70–74 (1996). [DOI] [PubMed] [Google Scholar]
- 80.Adkins B, Guevara P & Rose S Thymic and extrathymic contributions to T helper cell function in murine neonates. Haematol. Rep 2, 9–13 (2006). [PMC free article] [PubMed] [Google Scholar]
- 81.White JT et al. Virtual memory T cells develop and mediate bystander protective immunity in an IL-15-dependent manner. Nat. Commun 7, 11291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Adkins B Peripheral CD4+ lymphocytes derived from fetal versus adult thymic precursors differ phenotypically and functionally. J. Immunol 171, 5157–5164 (2003). [DOI] [PubMed] [Google Scholar]
- 83.Adkins B, Williamson T, Guevara P & Bu Y Murine neonatal lymphocytes show rapid early cell cycle entry and cell division. J. Immunol 170, 4548–4556 (2003). [DOI] [PubMed] [Google Scholar]
- 84.Brodin P et al. Variation in the human immune system is largely driven by non-heritable influences. Cell 160, 37–47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Liston A, Carr EJ & Linterman MA Shaping variation in the human immune system. Trends Immunol. 37, 637–646 (2016). [DOI] [PubMed] [Google Scholar]
- 86.Carr EJ et al. The cellular composition of the human immune system is shaped by age and cohabitation. Nat. Immunol 17, 461–468 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Frisancho AR Developmental functional adaptation to high altitude: review. Am. J. Hum. Biol 25, 151–168 (2013). [DOI] [PubMed] [Google Scholar]
- 88.Olin A et al. Stereotypic immune system development in newborn children. Cell 174, 1277–1292 e1214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hill DL et al. Immune system development varies according to age, location, and anemia in African children. Sci. Transl Med 12, eaaw9522 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Marchant A et al. Mature CD8+ T lymphocyte response to viral infection during fetal life. J. Clin. Invest 111, 1747–1755 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Karrer U et al. Memory inflation: continuous accumulation of antiviral CD8+ T cells over time. J. Immunol 170, 2022–2029 (2003). [DOI] [PubMed] [Google Scholar]
- 92.Munks MW et al. Four distinct patterns of memory CD8 T cell responses to chronic murine cytomegalovirus infection. J. Immunol 177, 450–458 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Rolot M et al. Helminth-induced IL-4 expands bystander memory CD8+ T cells for early control of viral infection. Nat. Commun 9, 4516 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lin JS et al. Virtual memory CD8 T cells expanded by helminth infection confer broad protection against bacterial infection. Mucosal Immunol. 12, 258–264 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reese TA et al. Sequential infection with common pathogens promotes human-like immune gene expression and altered vaccine response. Cell Host Microbe 19, 713–719 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Beura LK et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 532, 512–516 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zanvit P et al. Antibiotics in neonatal life increase murine susceptibility to experimental psoriasis. Nat. Commun 6, 8424 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kirjavainen PV et al. Farm-like indoor microbiota in non-farm homes protects children from asthma development. Nat. Med 25, 1089–1095 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bach JF The hygiene hypothesis in autoimmunity: the role of pathogens and commensals. Nat. Rev. Immunol 18, 105–120 (2018). [DOI] [PubMed] [Google Scholar]
- 100.Sureshchandra S, Marshall NE & Messaoudi I Impact of pregravid obesity on maternal and fetal immunity: Fertile grounds for reprogramming. J. Leukoc. Biol 106, 1035–1050 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kanneganti TD & Dixit VD Immunological complications of obesity. Nat. Immunol 13, 707–712 (2012). [DOI] [PubMed] [Google Scholar]
- 102.Iyer SS et al. Protein energy malnutrition impairs homeostatic proliferation of memory CD8 T cells. J. Immunol 188, 77–84 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chatraw JH, Wherry EJ, Ahmed R & Kapasi ZF Diminished primary CD8 T cell response to viral infection during protein energy malnutrition in mice is due to changes in microenvironment and low numbers of viral-specific CD8 T cell precursors. J. Nutr 138, 806–812 (2008). [DOI] [PubMed] [Google Scholar]
- 104.van de Pavert SA et al. Maternal retinoids control type 3 innate lymphoid cells and set the offspring immunity. Nature 508, 123–127 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kongsbak M, Levring TB, Geisler C & von Essen MR The vitamin d receptor and T cell function. Front. Immunol 4, 148 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Vanhee S et al. Lin28b controls a neonatal to adult switch in B cell positive selection. Sci. Immunol 4, (2019). [DOI] [PubMed] [Google Scholar]
- 107.Hardy RR & Hayakawa K A developmental switch in B lymphopoiesis. Proc. Natl Acad. Sci. USA 88, 11550–11554 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kantor AB, Stall AM, Adams S, Herzenberg LA & Herzenberg LA Differential development of progenitor activity for three B-cell lineages. Proc. Natl Acad. Sci. USA 89, 3320–3324 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McGrath KE, Frame JM & Palis J Early hematopoiesis and macrophage development. Semin. Immunol 27, 379–387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Perdiguero EG & Geissmann F The development and maintenance of resident macrophages. Nat. Immunol 17, 2–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Schneider C et al. Tissue-resident group 2 innate lymphoid cells differentiate by layered ontogeny and in situ perinatal priming. Immunity 50, 1425–1438 e1425 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tieppo P et al. The human fetal thymus generates invariant effector gammadelta T cells. J. Exp. Med 217, 20190580 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ikuta K et al. A developmental switch in thymic lymphocyte maturation potential occurs at the level of hematopoietic stem cells. Cell 62, 863–874 (1990). [DOI] [PubMed] [Google Scholar]
- 114.Boursalian TE, Golob J, Soper DM, Cooper CJ & Fink PJ Continued maturation of thymic emigrants in the periphery. Nat. Immunol 5, 418–425 (2004). [DOI] [PubMed] [Google Scholar]
- 115.Priyadharshini B, Welsh RM, Greiner DL, Gerstein RM & Brehm MA Maturationdependent licensing of naive T cells for rapid TNF production. PLoS One 5, e15038 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hussain T & Quinn KM Similar but different: virtual memory CD8 T cells as a memory-like cell population. Immunol. Cell Biol 97, 675–684 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Opiela SJ, Koru-Sengul T & Adkins B Murine neonatal recent thymic emigrants are phenotypically and functionally distinct from adult recent thymic emigrants. Blood 113, 5635–5643 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yuan J, Nguyen CK, Liu X, Kanellopoulou C & Muljo SA Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science 335, 1195–1200 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim I, Saunders TL & Morrison SJ Sox17 dependence distinguishes the transcriptional regulation of fetal from adult hematopoietic stem cells. Cell 130, 470–483 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bonati A et al. T-cell receptor beta-chain gene rearrangement and expression during human thymic ontogenesis. Blood 79, 1472–1483 (1992). [PubMed] [Google Scholar]
- 121.Rufer N et al. Telomere fluorescence measurements in granulocytes and T lymphocyte subsets point to a high turnover of hematopoietic stem cells and memory T cells in early childhood. J. Exp. Med 190, 157–167 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hale JS, Boursalian TE, Turk GL & Fink PJ Thymic output in aged mice. Proc. Natl Acad. Sci. USA 103, 8447–8452 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Elder RW et al. Immunologic aging in adults with congenital heart disease: does infant sternotomy matter? Pediatr. Cardiol 36, 1411–1416 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Steinmann GG, Klaus B & Muller-Hermelink HK The involution of the ageing human thymic epithelium is independent of puberty. A morphometric study. Scand. J. Immunol 22, 563–575 (1985). [DOI] [PubMed] [Google Scholar]
- 125.Zhang X et al. CD4 T cells with effector memory phenotype and function develop in the sterile environment of the fetus. Sci. Transl Med 6, 238ra272 (2014). [DOI] [PubMed] [Google Scholar]