

Abstract
The motor features of Parkinson’s disease (PD) result from the loss of dopaminergic (DA) neurons in the substantia nigra with autophagy dysfunction being closely linked to this disease. A PD-causing familial mutation in VPS35 (D620N) has been reported to inhibit autophagy. In order to identify signaling pathways responsible for this autophagy defect, we performed an unbiased screen using RNA sequencing (RNA-Seq) of wild-type or VPS35 D620N-expressing retinoic acid-differentiated SH-SY5Y cells. We report that VPS35 D620N-expressing cells exhibit transcriptome changes indicative of alterations in extracellular matrix (ECM)-receptor interaction as well as PI3K-AKT signaling, a pathway known to regulate autophagy. Hyaluronan (HA) is a major component of brain ECM and signals via the ECM receptors CD44, a top RNA-Seq hit, and hyaluronan-mediated motility receptor (HMMR) to the autophagy-regulating PI3K-AKT pathway. We find that high (>950 kDa), but not low (15 – 40 kDa), molecular weight HA treatment inhibits autophagy. In addition, VPS35 D620N facilitated enhanced HA-AKT signaling. Transcriptomic assessment and validation of protein levels identified the differential expression of CD44 and HMMR isoforms in VPS35 D620N mutant cells. We report that knockdown of HMMR or CD44 results in upregulated autophagy in cells expressing wild-type VPS35. However, only HMMR knockdown resulted in rescue of autophagy dysfunction by VPS35 D620N indicating a potential pathogenic role for this receptor and hyaluronan signaling in Parkinson’s disease.
Keywords: VPS35, autophagy, extracellular matrix, HMMR, CD44
INTRODUCTION
In the USA, with a prevalence of 1% in people over the age of 60 and an increase to over 4% by the of age 85, Parkinson’s disease is the most common disease of motor system degeneration (de Lau and Breteler, 2006). This disease is clinically characterized by a resting tremor, rigidity, postural instability, and bradykinesia as well as non-motor symptoms that include cognitive dysfunction and constipation. PD’s primary motor features result from a loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SN) region of the midbrain. Specifically, the loss of dopamine produced by this neuronal subset causes dysfunction of the basal ganglia, a cluster of nuclei responsible for coordinating movement. No therapies to date deal with the underlying neuronal loss and thus only symptomatic treatments exist for PD (Jankovic and Stacy, 2007).
Macroautophagy (hereafter referred to as autophagy) is an orderly process of cytoplasmic engulfment by lipid membrane-bound vesicles termed autophagosomes leading to degradation of the encapsulated material (La Spada and Taylor, 2010). Autophagy proceeds in distinct steps that include induction, cargo recognition, autophagosome formation and transport, autophagosome-lysosome fusion, degradation of cargo, and the release of degraded material into the cytoplasm. Following the induction of autophagy, Atg8 (LC3) undergoes cleavage, and a phosphotidylethanolamine (PE) moiety is conjugated (now LC3-II). LC3-II is unique to autophagosomes and is commonly used as a marker for autophagy (He and Klionsky, 2009). Autophagosomes fuse with endosomes and multivesicular bodies that in turn fuse with lysosomes containing lytic enzymes which facilitate the degradation of cargo.
Autophagy is primarily devoted to adapting to nutrient limitation, degrading misfolded proteins, and removing dysfunctional organelles (Choi et al., 2013). Studies of neurodegenerative disease and protein turnover pathways have indicated that the maintenance of protein quality control presents a particular challenge for neurons and other specialized cells of the nervous system (Malgaroli et al., 2006; Taylor et al., 2002). This appears to be of importance to PD pathology as protein aggregates of α-synuclein are believed to be removed by autophagy, at least before fibril formation, and whose presence may signal autophagy dysfunction (Riedel et al., 2010; Tanik et al., 2013). A link between PD and autophagy can also be traced genetically as there are at least seven monogenic causes of PD strongly connected with autophagy regulation and some are directly involved in lysosomal biology (Dehay et al., 2010, 2013). As a result, considerable effort has been made to understand autophagy dysfunction in PD, with the goal of targeting this process for therapy.
In 2011, two independent groups reported the mutation of VPS35 (D620N) as being causal for a familial form of PD (Vilariño-Güell et al., 2011; Zimprich et al., 2011). VPS35 has a well-established and critical role in retromer function. The retromer is a highly conserved complex of proteins and an essential element of the endosomal sorting machinery that directs the recycling of plasma membrane receptors. The retromer is comprised of a cargo recognition trimer containing VPS35-VPS29-VPS26 and a dimer made of an assortment of SNX1, SNX2, SNX5, and SNX6 (McGough and Cullen, 2011). This complex associates with endosomes and facilitates the retrograde transport of transmembrane cargo to the trans-Golgi network, thereby rescuing cargo from degradation by the lysosome.
Interest into the role of VPS35 in mediating autophagy was initially hinted at in work published by Dengjel et al where VPS35 was identified in an autophagosome-interactor screen (Dengjel et al., 2012). Subsequently, Zavodszky et al., presented direct evidence of this by reporting that the PD-causing VPS35 D620N mutation impairs autophagy in HeLa cells through defective WASH complex recruitment to endosomes (Zavodszky et al., 2014). Interestingly, we do not detect an association with the principal WASH complex member WASH1 in retinoic acid (RA)-differentiated SH-SY5Y cells suggesting the existence of a distinct mechanism, at least in these cells (Fig. S1). SH-SY5Y cells are a widely used human neuroblastoma cellular model of PD that, when differentiated using retinoic acid, exhibit similar protein expression and physiological characteristics to dopaminergic neurons (Korecka et al., 2013; Krishna et al., 2014). Conversely, HeLa cells are sourced from a human cervical cancer tumor which may, in part, explain the contrasting findings. As a result, we explored alternative signaling pathways for autophagy disruption by VPS35 D620N. Our current study reveals perturbed hyaluronan-CD44/HMMR signaling in VPS35 mutant cells which may contribute to the associated autophagy defect.
MATERIAL AND METHODS
Tissue culture
SH-SY5Y human neuroblastoma cells (cat# CRL-2266, American Type Culture Collection) were grown in T175 tissue culture flasks and maintained at 37°C with 5% CO2 environment. Cells were cultured in DMEM/F12 50/50 media (cat# 10–090-CV, Corning) with L-glutamine, 15% Fetal Bovine Serum (cat# S11150, Atlanta Biologicals), 1% non-essential amino acids (cat# 25–025-CI, Corning) and 1% penicillin/streptomycin solution (cat# SV30010, Hyclone). Cells were passaged when flasks reached ~80% confluency using 0.25% Trypsin containing 2.2mM EDTA and sodium bicarbonate (cat# 25–053-CI, Corning). Cells at passage numbers 6 thru 13 were used for experiments. For this, cells were plated on multi-well tissue culture plates at the necessary cell concentrations (e.g., 4 million cells per well on a 6-well plate). Prior to experimentation, the cells were treated with 10 μM all-trans retinoic acid (cat# AAA44540–02, CAS# 302-79-4, VWR) for 7 days to induce differentiation into post-mitotic catecholaminergic neuron-like cells. Transgenic cells were also treated 72h prior to experimentation with 2mM sodium butyrate (cat# AAA11079–22, CAS# 156-54-7, VWR) to reactivate transgene expression (Choi et al., 2005). 293TA cells used to generate lentivirus were grown in DMEM media with 1.0 g/L Glucose containing L-glutamine and with sodium pyruvate (cat# VWRL0111–0500, VWR), supplemented with 10% Fetal Bovine Serum, 1% non-essential amino acids and 1% penicillin/streptomycin solution.
Western blotting
Life Technologies Bolt gels and reagents were used for western blotting. Cells were lysed using RIPA lysis buffer (cat# 89901, Thermo Fisher) containing a protease and phosphatase inhibitor cocktail (cat# A32961, Thermo Fisher) and then sonicated. This was then followed by centrifugation at 15000g for 10 minutes and collecting supernatant. Total protein levels were measured and normalized across samples. Polyacrylamide gel electrophoresis was performed on the protein extracts followed by transfer onto a PVDF membrane. Transfers to PVDF membranes were performed using the iBlot2 Dry Transfer device (Life Technologies). Primary antibodies were used to detect LC3 (1:1000, cat# NB100–2220, Novus Biologicals), VPS35 (1:1000, cat# GTX108058, GeneTex), VPS29 (1:500, cat# ab10160, Abcam), VPS26 (1:500, cat# ab23892, Abcam), V5 (1:1000, cat# 13202S, Cell Signaling Technology), CD44 (1:200, cat# TA506726, Origene), phospho-AKT (Ser 473) (1:1000, cat# 4060, Cell Signaling Technology), HMMR (1:1000, cat# GTX54121, Genetex), WASH1 (1:500, cat# NBP1–90464, Novus Biologicals) and β-actin (1:2000, cat# MA1–91399, Thermo Fisher). Secondary antibodies (1:1000, anti-mouse, cat# 7076, Cell Signaling Technology; 1:1000, anti-rabbit, cat# 7074, Cell Signaling Technology) were used for probing PVDF membranes after primary antibody incubation. ECL substrate (cat# 32106, Pierce) or, for more sensitive detection, WesternSure chemiluminescent substrate (cat# 926–95000, Li-cor) was then used to visualize protein bands and images obtained with a ChemiDoc Touch Gel Imager (BioRad). Protein expression was quantified by densitometry using Image J (NIH). Original western blot images for representative figures are provided in the supplemental data.
Lentivirus production and transduction
Life Technologies Virapower lentiviral production kit was used to generate lentivirus. 293TA cells (cat# LT008, Genecopoeia) were transfected with plasmids using Lipofectamine 2000 (cat# 11668019, Life Technologies). Media was replaced the next day. The supernatant media containing active, fully assembled lentiviruses, was collected 48 hours after transfection, filtered through 400 nm pore filters, aliquoted and stored at −80°C until use. For transduction of SH-SY5Y cells, virus containing supernatant was added directly to the culture media. This media was replaced the next day and antibiotic selection (blasticidin or puromycin) for transduced cells was initiated 72 hours following viral supernatant addition. Wild-type human VPS35 CMV promoter-driven overexpression lentiviral plasmid construct was obtained from Addgene (Plasmid #21691) (Scott et al., 2009). Site-directed mutagenesis was performed to create VPS35 D620N in this vector. The mutant was verified by Sanger sequencing. The following lentiviral shRNA constructs were used: scrambled (control) shRNA (cat# SHC016, Sigma-Aldrich), VPS35 shRNA 1 (cat# TRCN0000337085, Sigma-Aldrich), VPS35 shRNA 2 (cat# TRCN0000337019, Sigma-Aldrich), VPS29 shRNA (cat# TRCN0000304146, Sigma Aldrich), HMMR shRNA (cat# LPP-A4000-Lv193–100, Genecopoeia), and CD44 shRNA (cat# LPP-Z0786-Lv193–100, Genecopoeia).
Autophagic flux assay
Bafilomycin A1 (100nM; cat# 200003–360, CAS# 88899-55-2, VWR) was used to block autophagy in experimental cells, four hours prior to lysis. This was then followed by comparison of LC3-II levels normalized to β-Actin between bafilomycin A1 treated and untreated cells obtained from western blot imaging as previously described (Klionsky et al., 2012).
Hyaluronan treatment
Cells were plated at a density of 4 million cells per well on 6 well plates. Cells were then differentiated with RA for 7 days. On the day prior to lysis, cells were treated with high (cat# GLR004, R&D Systems) or low molecular weight (cat# GLR001, R&D Systems) HA in serum-free media and then lysed using RIPA lysis buffer, as described above. For autophagic flux assays, cells were treated with or without HA in the presence or absence of Bafilomycin A1 (100nM) for 4 hours immediately prior to lysis and then autophagic flux was assessed as described above.
Hyaluronan quantification
ELISA measurement of hyaluronan was performed using a Quantikine ELISA kit (cat# DHYAL0, R&D Systems) according to the manufacturer’s instructions. Briefly, cells were plated on 96 well plates at 50,000 cells/well. The cells were then treated with retinoic acid media for seven days. After differentiation, the media was replaced with growth media containing 0.5% FBS and RA. Supernatant was then collected after 48 hours for ELISA analysis. It should be noted that this ELISA system detects all forms of HA (>35kDa). ELISA assay manufacturer-reported validation parameters: intra-assay precision from 3 trials, 20 wells for SD (0.11, 0.26, 0.52) and coefficient of variation (7.2, 5.4, 3.6), inter-assay precision from 3 trials, 20 wells for SD (0.08, 0.23, 0.72) and coefficient of variation (4.8, 4.9, 4.8), minimum detectable dose of hyaluronan ranged from 0.027–0.200 ng/mL, immunoassay is calibrated against Streptococcus pyogenes-derived hyaluronan with a molecular weight range of 75–350 kDa, recovery of hyaluronan spiked through levels throughout the range of the assay was 104%, and linearity for human samples (n=4) for dilutions 1:2, 1:4, 1:8, 1:16 were 99%, 98%, 102%, and 109% of expected, respectively.
Differential gene expression, splicing and gene set enrichment analysis
Transgenic WT and VPS35 D620N-expressing cells grown in 100mm culture plates were RA-differentiated, RNA was extracted using an RNeasy kit (cat # 74104, Qiagen) with DNAse treatment, rRNA depleted, random-primed cDNA made and sequencing subsequently performed using Illumina with HiSeq2000/2500 (>40 million reads/sample). Sequencing and analysis were done by Idaho State University’s Molecular Research Core Facility. Alignment of mRNA-Seq was conducted with a standard protocol as previously described (Fig. S3) (Trapnell et al., 2012). Briefly, raw reads were initially trimmed with FASTX-toolkit to remove adapter sequences, and aligned to a reference genome (human hg19) with TopHat (v2.0.11). To take into account novel splice isoforms, the de novo transcript assembly from the mapped reads was performed using cufflinks (v2.2.1) and the assembled transcripts were then merged into single reference transcripts using cuffmerge.
For differential gene expression analysis, the mapped reads were quantified using cuffdiff and the FPKM (Fragments Per Kilobase of transcript per Million mapped reads) were calculated. Genes were discarded from downstream analysis when the calculated mRNA abundance was lower than FPKM of 0.5, treating them as background noise. The quantified results were further visualized with CummeRbund (Trapnell et al., 2012) or custom scripts in R. To investigate whether the expression level of pre-defined sets of genes correlated to the observed phenotype, the Impact Analysis method was conducted using iPathwayGuide (Advaita) (Draghici et al., 2003; Khatri et al., 2007; Tarca et al., 2009). The number of gene-set permutations was set at 1000, and the most enriched KEGG genesets (Kanehisa et al., 2014) were selected based on Normalized Enrichment Scores (NES).
Gaussia luciferase protein-fragment complementation assay
Plasmids containing transgenic fusion constructs of syn-hGluc (SynLuc), syn-hGLuc1 (S1) and syn-hGLuc2 (S2) were transfected (40ng DNA/well) into transgenic (WT or VPS35 D620N) SH-SY5Y cells in 96 well plates using FuGENE 6 (cat# E2691, Promega) according to manufacturer’s instructions as previously described (Outeiro et al., 2008). Media (conditioned media) was transferred to a new 96 well plate 48 hours later for analysis. The cells were then washed with phosphate buffered saline (cat# 97062–818, VWR) and new media (without phenol red or FBS) was added. Aggregation of alpha-synuclein was determined by assessing complementation of luciferase activity. Luciferase activity was measured within the conditioned media and living cells by automated injection of coelenterazine (40 μM, cat# 55779-48-1, CAS# 55779-48-1, P.J.K.) into each well and detected using a plate reader (Victor X3 microplate reader, Perkin-Elmer) with a 1 second signal integration time.
Quantitative PCR
SH-SY5Y cells were transduced, selected with puromycin, differentiated with RA and treated accordingly. RNA was extracted using a Qiagen RNeasy kit (cat # 74104, Qiagen) and cDNA was made using a kit (cat# 18080051, Thermo Fisher) according to manufacturer instructions. A LightCycler 2.0 (Roche) was used to carry out qPCR reactions in triplicate from three independent experiments using a Fast Evagreen master mix (cat# 31003, Biotium) and the following primer sets: HMMR (cat# HQP100236, Genecopoeia), CD44 (cat# HQP022975, Genecopoeia) and β-Actin (cat# HQP016381, Genecopoeia). Relative expression levels for HMMR and CD44 transcripts were normalized to β-Actin transcript levels for each sample.
Statistical analysis and data management
Statistical analysis was performed using Prism (version 6.0; GraphPad Software). Data between two variables was based on the student’s t-test. Data was considered significantly different at p < 0.05. The datasets generated and/or analyzed during the current study are available from the corresponding author upon request. RNA-Seq data for the described experiments is available in both raw and processed formats at the NCBI Gene Expression Omnibus (GEO) public repository.
RESULTS
VPS35 D620N inhibits autophagy
To determine whether VPS35 D620N represses autophagy in a PD model system, we first engineered a human neuroblastoma cell line (SH-SY5Y) to stably express WT or D620N VPS35 (V5–tagged). This was done by obtaining a VPS35-expressing lentivirus construct from Lynda Chin(Scott et al., 2009) and generating the D620N mutation by site-directed mutagenesis. We next infected low passage SH-SY5Y cells (ATCC) and selected transgene-positive cells using blasticidin. Before conducting experiments, we differentiated SH-SY5Y cultures into postmitotic dopaminergic neuron-like cells using 10uM retinoic acid for 7 days. Western blot revealed that WT and D620N transgenic proteins are expressed equally and at a similar level to endogenous VPS35 (Fig. 1A). In stable cell lines, no endogenous VPS35—only transgenic (shifted) VPS35—was detected, which is likely due to transgenic VPS35 titrating out endogenous VPS35 from the retromer complex. The knockdown of VPS35 results in the knockdown of another core retromer component, VPS29, and vice versa, suggesting that free VPS35 as well as VPS29 are rapidly degraded by the cell (Fig. 2C) (Fuse et al., 2015). Therefore, serendipitously, these cell lines express the transgene at endogenous protein levels, thereby avoiding overexpression artifacts. Upon examination of autophagy in these cells, we found autophagy to be robustly inhibited by D620N, as evidenced by the reduced LC3-II induction following Bafilomycin A1 treatment (Figs. 1B and 1C). We further tested autophagy dysfunction by VPS35 D620N through assessing the clearance of intracellular alpha-synuclein aggregates (Fig. S2) (Outeiro et al., 2008). We report that mutant-expressing cells accumulated significantly higher levels of alpha-synuclein aggregates which supports the notion that VPS35 D620N is a potent inhibitor of autophagy.
Figure 1. VPS35 D620N inhibits autophagy.
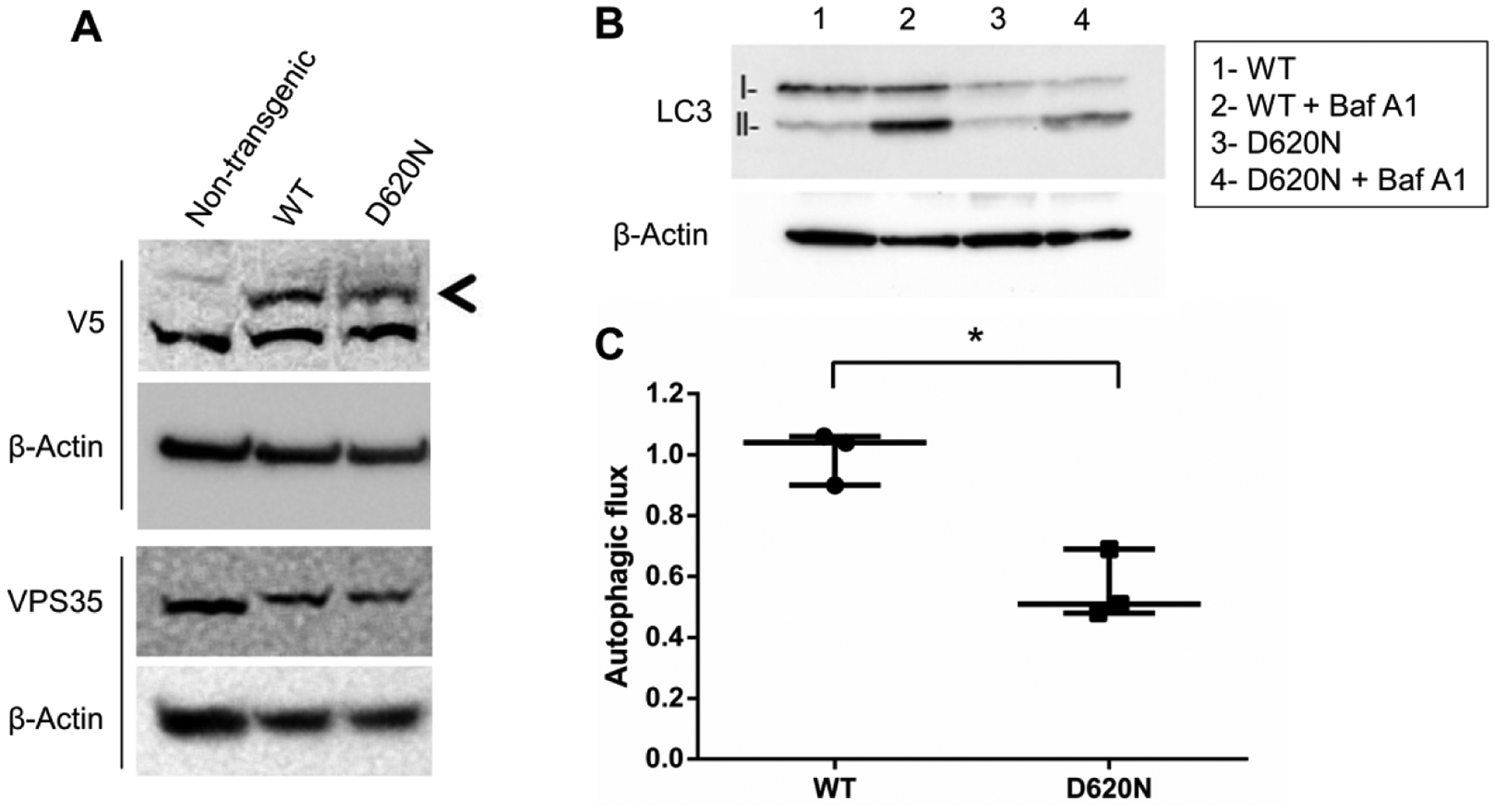
(A) Transgenic and endogenous VPS35 were evaluated by western blot. RA-differentiated SH-SY5Y cells stably expressing transgenic VPS35 (WT or D620N) were treated with or without bafilomycin A1. (B) Western blot of LC3-II was performed and (C) autophagic flux was quantified. Total protein loading was confirmed with β-Actin expression obtained from the same membrane. Results from three independent experiments are displayed (*p<0.05 compared to indicated group, student’s t-test; error bars = SEM).
Figure 2. VPS35 knockdown inhibits autophagy.
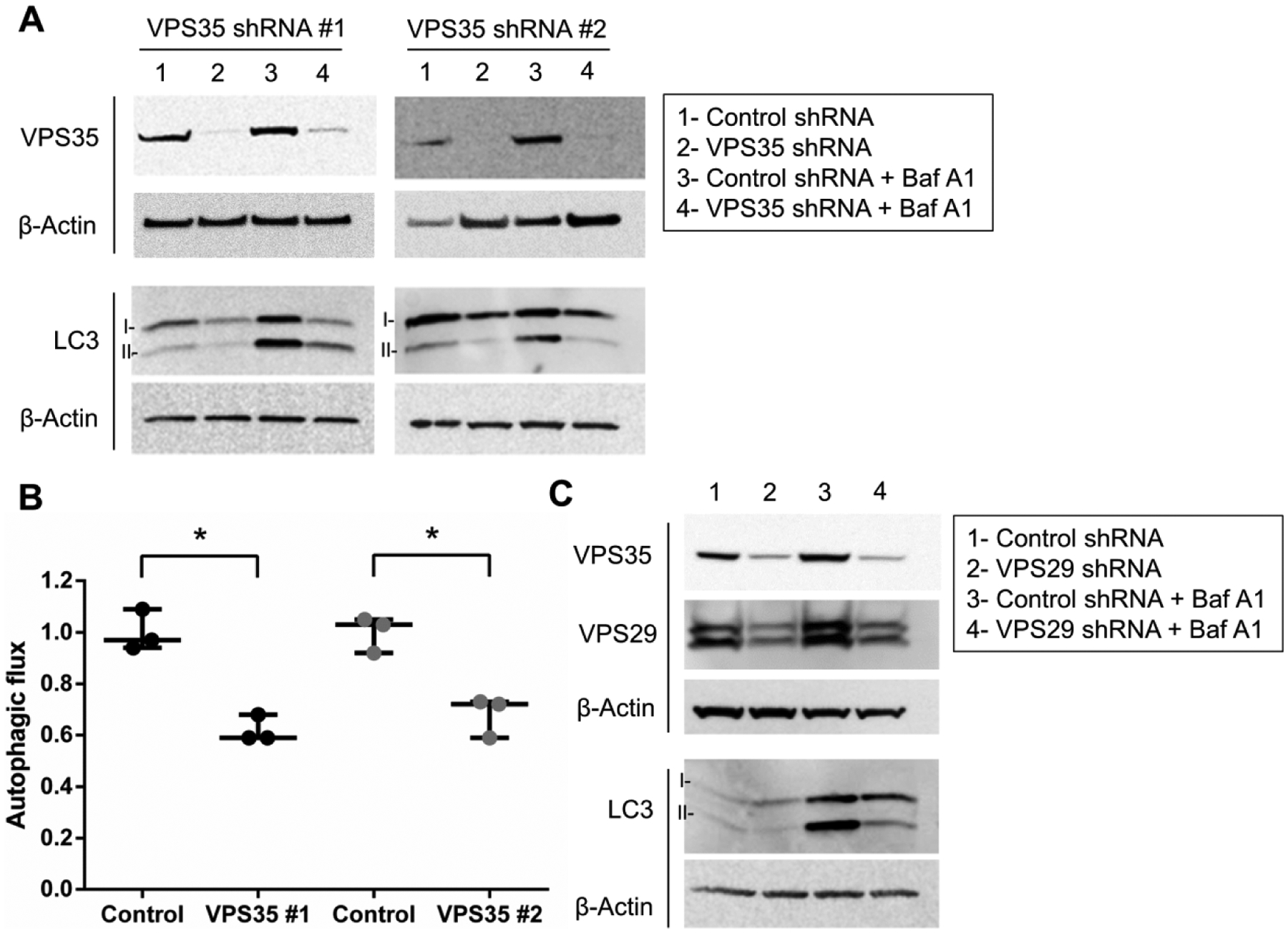
(A) VPS35 expression was knocked down in differentiated SH-SY5Y cells neurons by two distinct lentivirus-delivered shRNA vectors. Following selection with puromycin, transduced cells were treated in the presence or absence of bafilomycin A1. Western blot of LC3-II was performed and (B) autophagic flux quantified. Results from three independent experiments are shown (*p<0.05 versus indicated group, student’s t-test; error bars = SEM). (C) Knockdown of VPS29 by shRNA using a lentiviral vector was also performed. Representative image groupings were obtained from single western blot membranes probed with multiple antibodies as indicated. Total loading for all western blot membranes was assessed using β-Actin.
VPS35 knockdown inhibits autophagy, phenocopying the VPS35 D620N mutation.
To determine if the D620N mutation elicited autophagy blockade through a loss of function in differentiated SH-SY5Y cells, we knocked down VPS35 expression using two distinct shRNA constructs delivered by lentivirus and assessed autophagy (Fig. 2A and 2B). We found that VPS35 knockdown resulted in repression of autophagy consistent with VPS35 D620N being a loss-of-function mutation. As reported by other groups, we found that knockdown of other core retromer components such as VPS29 by shRNA resulted in concomitant knockdown of VPS35 (Fig. 2C) (Fuse et al., 2015). Therefore, it was not surprising to observe that VPS29 knockdown also suppressed autophagy. Collectively, these data suggested that VPS35 D620N is a loss-of-function mutation, which ultimately suppresses autophagic activity.
VPS35 D620N misregulates ECM-receptor interaction and PI3K-AKT pathway gene expression
Having established autophagy dysfunction by VPS35 D620N, likely through a loss-of-function, we next sought to determine a precise molecular mechanism for this phenomenon. To this end, we performed RNA sequencing of the transcriptomes of transgenic WT and D620N VPS35-expressing SH-SY5Y cells. Gene ontology was performed (1.5 fold change cutoff) using iPathways (Advaita) and KEGG annotated pathways were assigned to gene expression changes (Luo et al., 2009; Mootha et al., 2003; Subramanian et al., 2005). Top pathway hits were ECM-receptor interaction and PI3K-AKT signaling which suggests that ECM signaling is perturbed by VPS35 D620N (Table 1). Examination of specific transcripts altered for ECM-receptor interaction and PI3K-AKT KEGG annotated pathways reveals that CD44 mRNA expression is misregulated in VPS35 D620N cells (Fig. 3). The major structural component of brain ECM is hyaluronan (HA), which not only serves as the main anchoring point for brain cells, but is also an important facilitator of intracellular signaling. CD44 and HMMR are primary receptors for HA that signal to AKT upon binding (Lin et al., 2007; Onodera et al., 2015; Trapasso and Allegra, 2012).
Table 1. ECM-receptor interaction and AKT pathway perturbation by VPS35 D620N.
RNA-Seq was performed on WT and VPS35 D620N-expressing SH-SY5Y cells. The top 5 pathways affected by VPS35 D620N expression and relevant significance values are listed.
| Pathway name | Pathway ID | p-value | p-value (FDR) | p-value (Bonferroni) |
|---|---|---|---|---|
| Pathways in cancer | 05200 | 9.878e-8 | 2.683e-5 | 2.914e-5 |
| Systemic lupus erythematosus | 05322 | 1.819e-7 | 2.683e-5 | 5.367e-5 |
| ECM-receptor interaction | 04512 | 2.294e-6 | 2.256e-4 | 6.768e-4 |
| PI3K-AKT signaling pathway | 04151 | 3.124e-6 | 2.304e-4 | 9.215e-4 |
| Focal adhesion | 04510 | 9.627e-6 | 5.680e-4 | 0.003 |
Figure 3. Transcriptome alteration of ECM-receptor and PI3K-AKT pathways by VPS35 D620N.
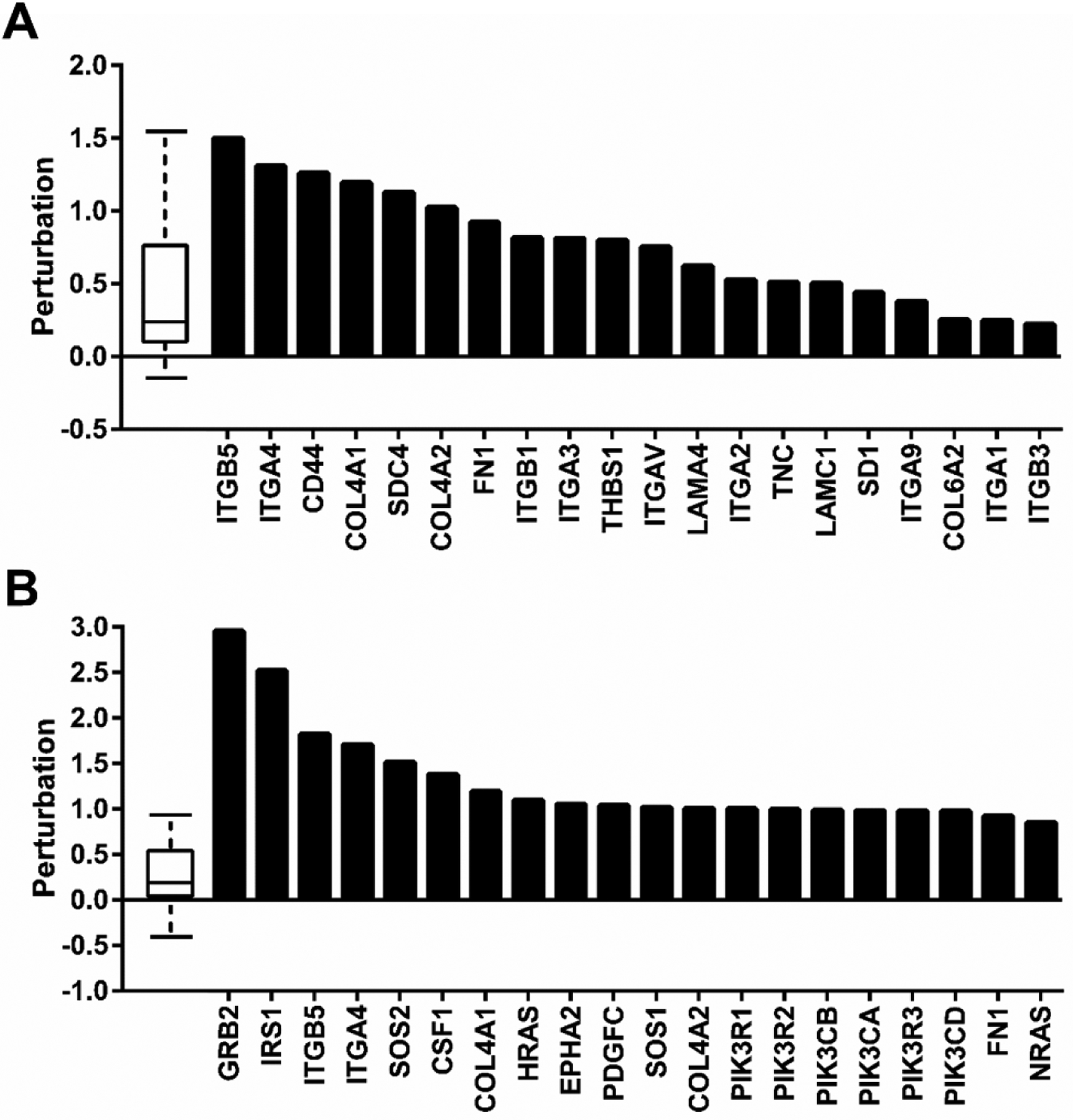
Specific transcript expression changes (LogFoldChange) for (A) ECM-receptor interaction and (B) PI3K-AKT signaling KEGG pathways from RNA-Seq data are shown. The distribution of all gene perturbations in the pathway are represented in the box plot where the first, median and third quartile are indicated.
Hyaluronan inhibits autophagy in differentiated SH-SY5Y cells
Our findings from RNA-Seq data suggest that HA-CD44/HMMR-AKT signaling is perturbed in the VPS35 mutant cell line. These results pose the novel question of whether the HA-AKT signaling axis controls autophagic response. To test this directly, we added HA at concentrations reported to elicit biological effects in human monocytes and fibroblasts to differentiated SH-SY5Y cells(Maharjan et al., 2011). We chose to treat cells with either high (HMW) or low molecular weight (LMW) HA since the polymer size can direct divergent bioactivities (Maharjan et al., 2011). We observed a dose-dependent repression of autophagy following treatment with HMW but not LMW HA (Fig. 4). The finding that HA modulates autophagy may indicate that the composition of ECM is an important factor in the regulation of this key process.
Figure 4. HA inhibits autophagy.
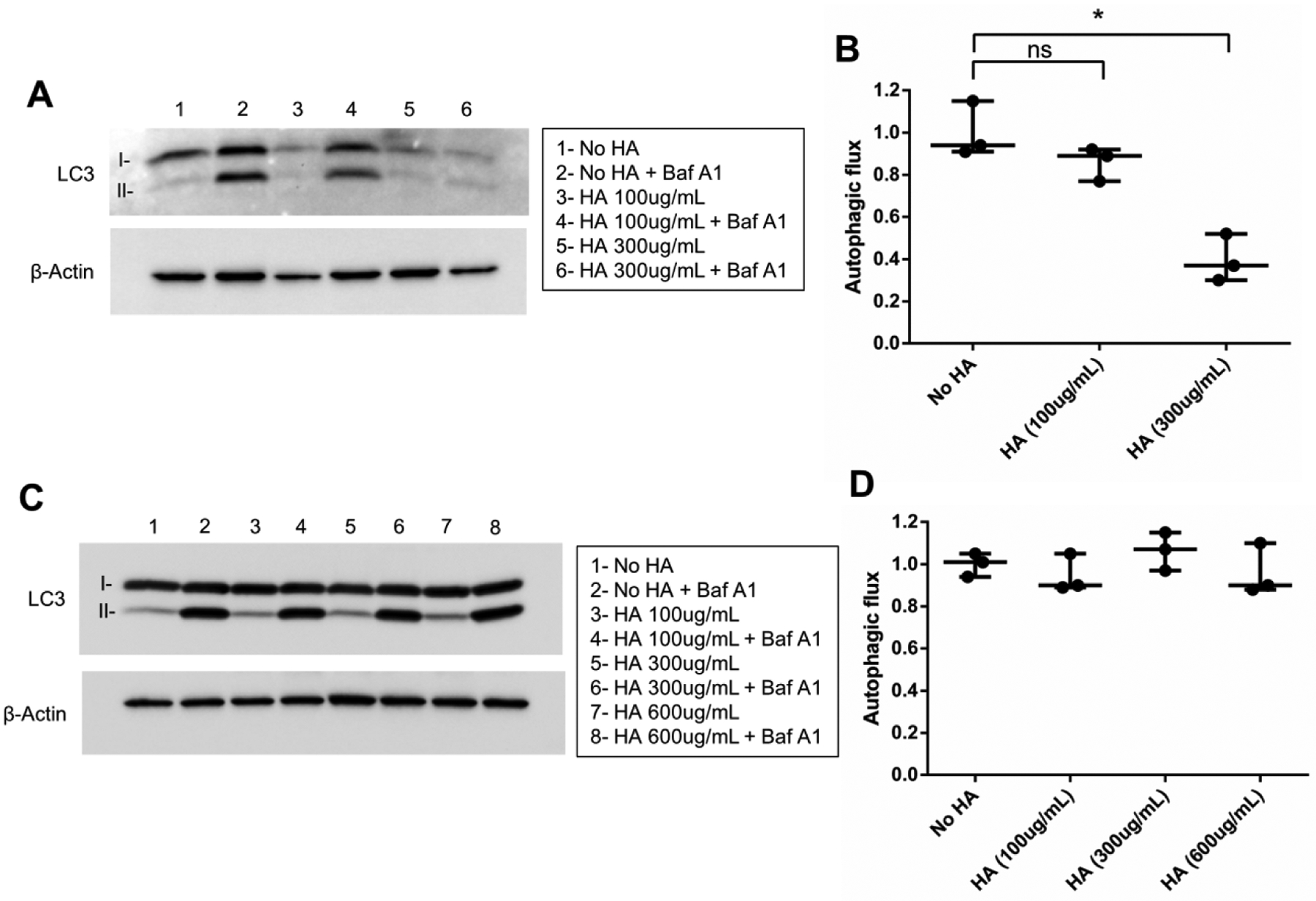
(A) RA-differentiated SH-SY5Y cells were treated with high molecular weight (>950 kDa) HA in serum-free media for 24 hours. The next day, cells were treated with or without bafilomycin A1 and autophagic flux was assessed by LC3-II/β-Actin western blot and (B) quantified from three experiments. (C) Similarly, low molecular weight (15 – 40 kDa) HA treatment was performed and autophagic response was evaluated by LC3-II/β-Actin western blot and (D) represented graphically (*p<0.05 versus indicated group, student’s t-test; error bars = SEM).
Altered hyaluronan-AKT signaling by VPS35 D620N
We next examined whether VPS35 D620N-expressing cells exhibited heightened sensitivity to HA. Addition of HMW HA, following overnight serum deprivation, resulted in elevated AKT activation in mutant cells (Fig. 5A). Conversely, LMW HA failed to induce AKT activation in either WT or mutant cells (Fig. 5B). This suggests that VPS35 D620N facilitates enhanced HMW HA signaling. These results are also consistent with our findings that HMW and not LMW HA repress autophagy via canonical HA-AKT signaling. Interestingly, we observe a trend of mutant cells exhibiting lower levels of steady state p-AKT signaling after serum deprivation that did not rise to statistical significance when compared to wild-type cells. No trending difference in WT or VPS35 D620N-expressing cells was observed in the presence of 15% FBS-containing media (data not shown).
Figure 5. Elevated HA signaling by VPS35 D620N.
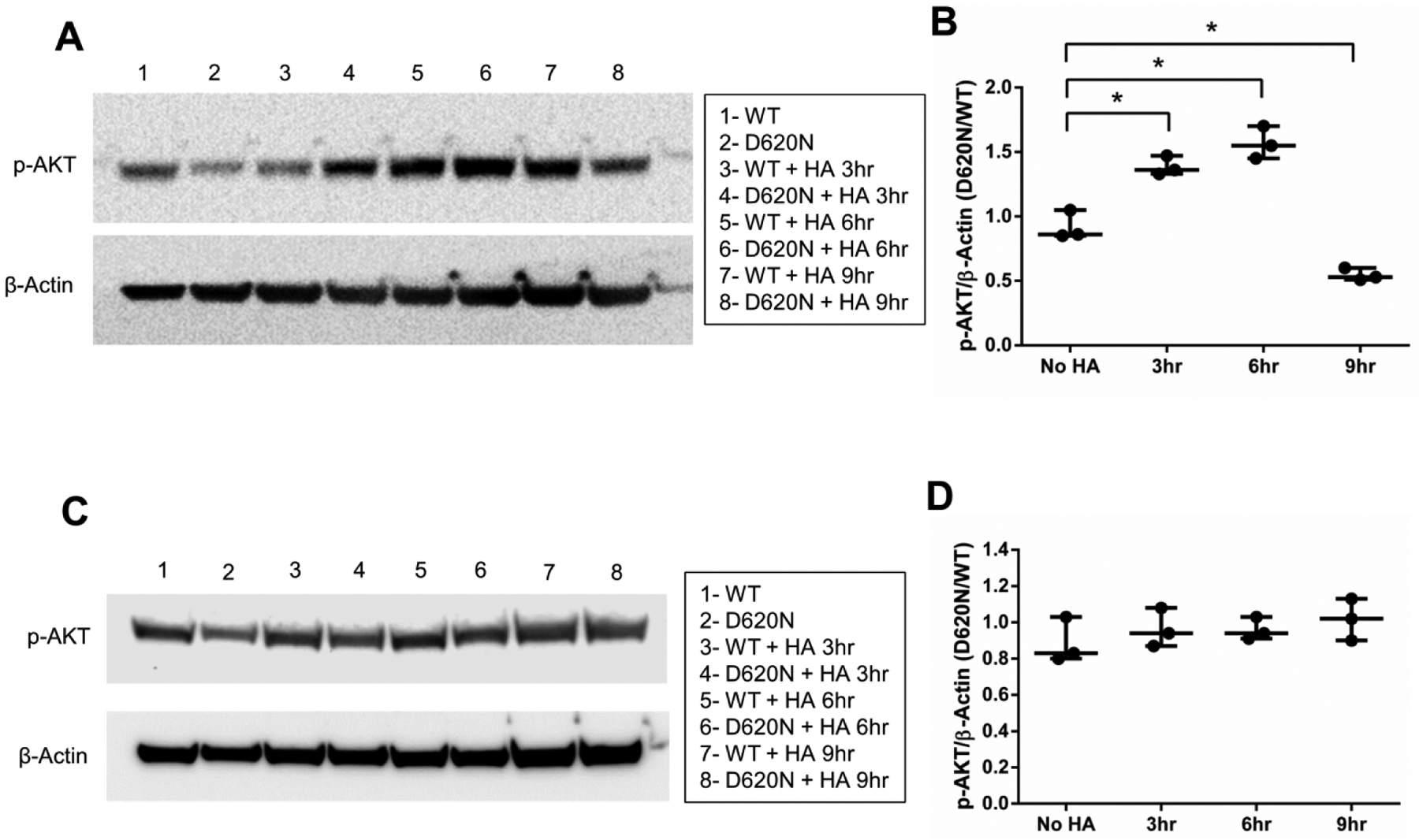
Differentiated SH-SY5Y cells expressing wild-type (WT) or D620N VPS35 were serum-deprived overnight. (A) The following day, high molecular weight (>950 kDa) HA (300 μg/mL) or (C) low molecular weight (15 – 40 kDa) HA (300 μg/mL) was added in serum-free media for the indicated times and western blotting was then performed (p-AKT = Ser473). (B, D) Results are presented as a ratio of D620N/WT for p-AKT/β-Actin by western blot band densitometry for each time point shown (*p<0.05 versus indicated group, student’s t-test; Error bars = SEM).
AKT pathway activation by VPS35 D620N is independent of hyaluronan production
To determine whether HA production contributes to heightened AKT signaling in VPS35-D620N cells, we measured HA produced during a 48 hour period (Fig. 6), but found no detectable difference in wild-type and mutant cells. We also assessed the protein levels for the hyaluronan synthase (HAS) enzymes responsible for HA synthesis. HAS3 was not differentially expressed while we were unable to detect HAS1 and HAS2 in SH-SY5Y cells (Fig. 7A). Taken together, these findings suggest that enhanced HA production is not responsible for the observed change in AKT signaling for VPS35 D620N-expressing cells.
Figure 6. VPS35 D620N does not affect HA production.
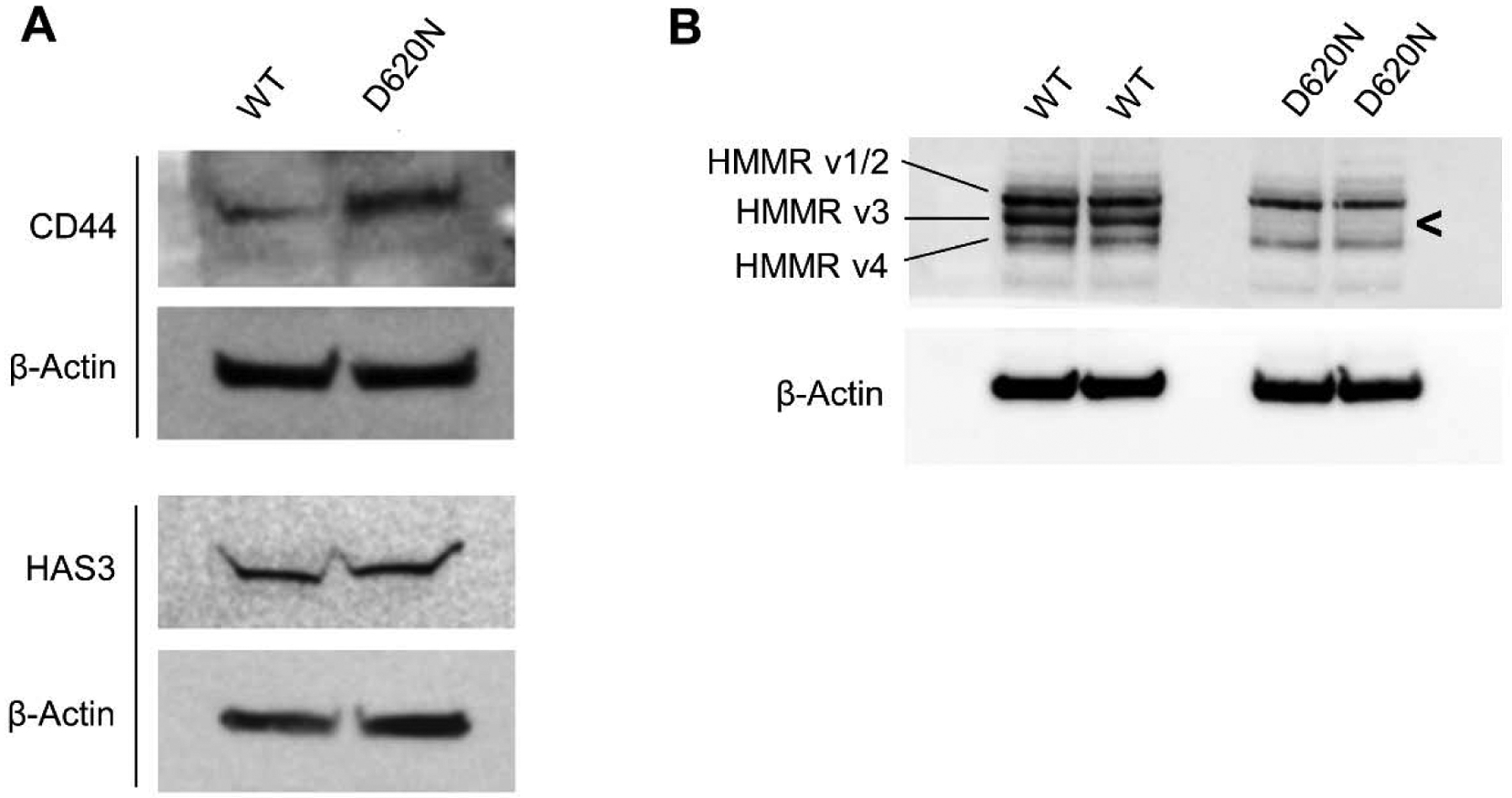
ELISA detection of HA (all forms, >35kDa) production in cell culture supernatant (over 48h in 0.5% FBS-containing media) from differentiated wild-type and mutant SH-SY5Y cells (results from three independent experiments; student’s t-test; p>0.05; error bars = SEM).
Figure 7. VPS35 D620N alters CD44 and HMMR protein expression.
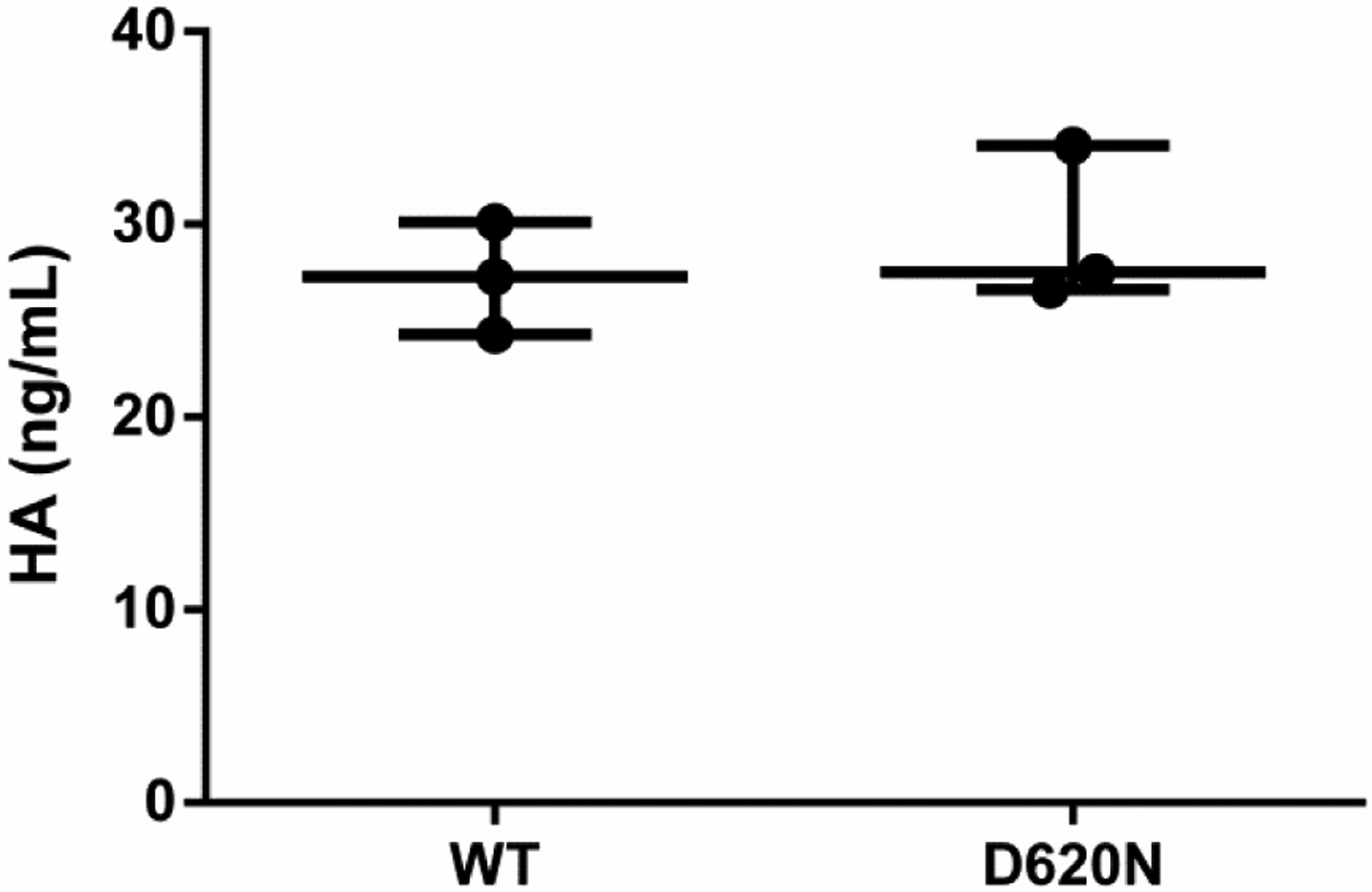
Western blots of differentiated SH-SY5Y cells expressing wild-type (WT) or D620N VPS35. (A) Visualization of CD44 protein required 80μg total lysate and ECL substrate with enhanced sensitivity (WesternSure). (B) HMMR protein detection was performed using 10μg of total lysate and standard ECL reagent.
Misregulated CD44 and HMMR expression by VPS35 D620N
Our RNA Seq analysis revealed that ECM-receptor interaction pathways were a top hit for VPS35 D620N-expressing cells (Table 1). From this dataset we also found that CD44 transcript expression increased significantly in VPS35 D620N-expressing cells. Consistent with CD44 pathway misregulation, assessment of protein expression revealed heightened CD44 expression in the mutant cells (Fig. 7A). Interestingly, only a single truncated isoform of CD44 protein was observed that correlated to CD44v4. Identity of CD44v4 was confirmed by analysis of the RNA Seq data. What’s more, CD44v4 was very lowly expressed at both the protein level, requiring 60–80μg of total lysate and a sensitive ECL reagent (WesternSure) to detect, and at the RNA level, nearing the limit of accurate detection in our RNA-Seq dataset (Table 2). These factors led us to speculate that CD44 likely provides only a minor contribution to HA signaling in SH-SY5Y cells. We then examined the protein levels of HMMR, another primary receptor for HA, and found robust expression of multiple isoforms. However, HMMR isoform 3 (HMMRv3) protein was strikingly absent from VPS35 D620N-expressing cells (Fig. 7B). The identities of all four NCBI annotated isoforms were confirmed at the transcript level by RNA-Seq data. Collectively, these data suggest that the HMMRv3 may contribute to the enhanced activation of HA-AKT signaling and autophagy inhibition by VPS35 D620N mutation, with minor contributions from CD44 misregulation.
Table 2. VPS35 D620N affects CD44 and HMMR RNA splice variant expression.
SpliceR program analysis was performed on the WT and VPS35 D620N RNA-Seq dataset. Relative expression profiles are presented for all NCBI annotated splice variants as a function of FPKM for (A)CD44 and (B)HMMR. Significance for differential expression of WT versus D620N is shown.
| Isoform | Ref. Seq. | Rel. Exp. WT | Rel. Exp. D620N | WT/D620N Ratio | p-value |
|---|---|---|---|---|---|
| V1 | NM_000610 | 0.0000 | 0.0000 | INF | 1 |
| V2 | NM_001001389 | 0.0000 | 0.0142 | 0 | 1 |
| V3 | NM_001001390 | 0.0000 | 0.0000 | INF | 1 |
| V4 | NM_001001391 | 0.3387 | 1.0256 | 0.3303 | 0.0452 |
| V5 | NM_001001392 | 0.1853 | 0.0000 | INF | 1 |
| V6 | NM_001202555 | 0.0000 | 0.0256 | 0 | 1 |
| V7 | NM_001202556 | 0.0000 | 0.0004 | 0.0954 | 1 |
| V8 | NM_001202557 | 0.0444 | 0.0000 | INF | 1 |
| Isoform | Ref. Seq. | Rel. Exp. WT | Rel. Exp. D620N | WT/D620N Ratio | p-value |
|---|---|---|---|---|---|
| V1 | NM_001142556.1 | 5.1525 | 5.5884 | 0.9220 | 0.6159 |
| V2 | NM_012484.2 | 0.9834 | 0.3711 | 2.6500 | 0.3170 |
| V3 | NM_012485.2 | 13.5848 | 13.0535 | 1.0407 | 0.6639 |
| V4 | NM_001142557.1 | 1.4806 | 0.6353 | 2.3306 | 0.2983 |
HMMR contributes to autophagy dysfunction by VPS35 D620N
We next sought to determine if CD44 and HMMR regulated autophagic response. Scrambled as well as shRNA targeted to CD44 or HMMR were delivered to SH-SY5Y cells by lentivirus and pure populations of transduced cells were selected using puromycin. Successful knockdown was confirmed by qPCR (Fig. 8A). Interestingly, knockdown of HMMR resulted in a significant reduction in CD44 transcript expression and vice-versa. This suggests a shared transcription regulatory mechanism. Autophagic flux was then assessed following shRNA-targeted knockdown in differentiated SH-SY5Y cells. Knockdown of either HMMR or CD44 resulted in an increase in autophagic flux in wild-type VPS35-expressing cells (Fig 8B and 8C) while HMMR, and not CD44, knockdown rescued the VPS35 D620N autophagy defect (Fig 8D and 8E).
Figure 8. HMMR mediates autophagy repression by VPS35 D620N.
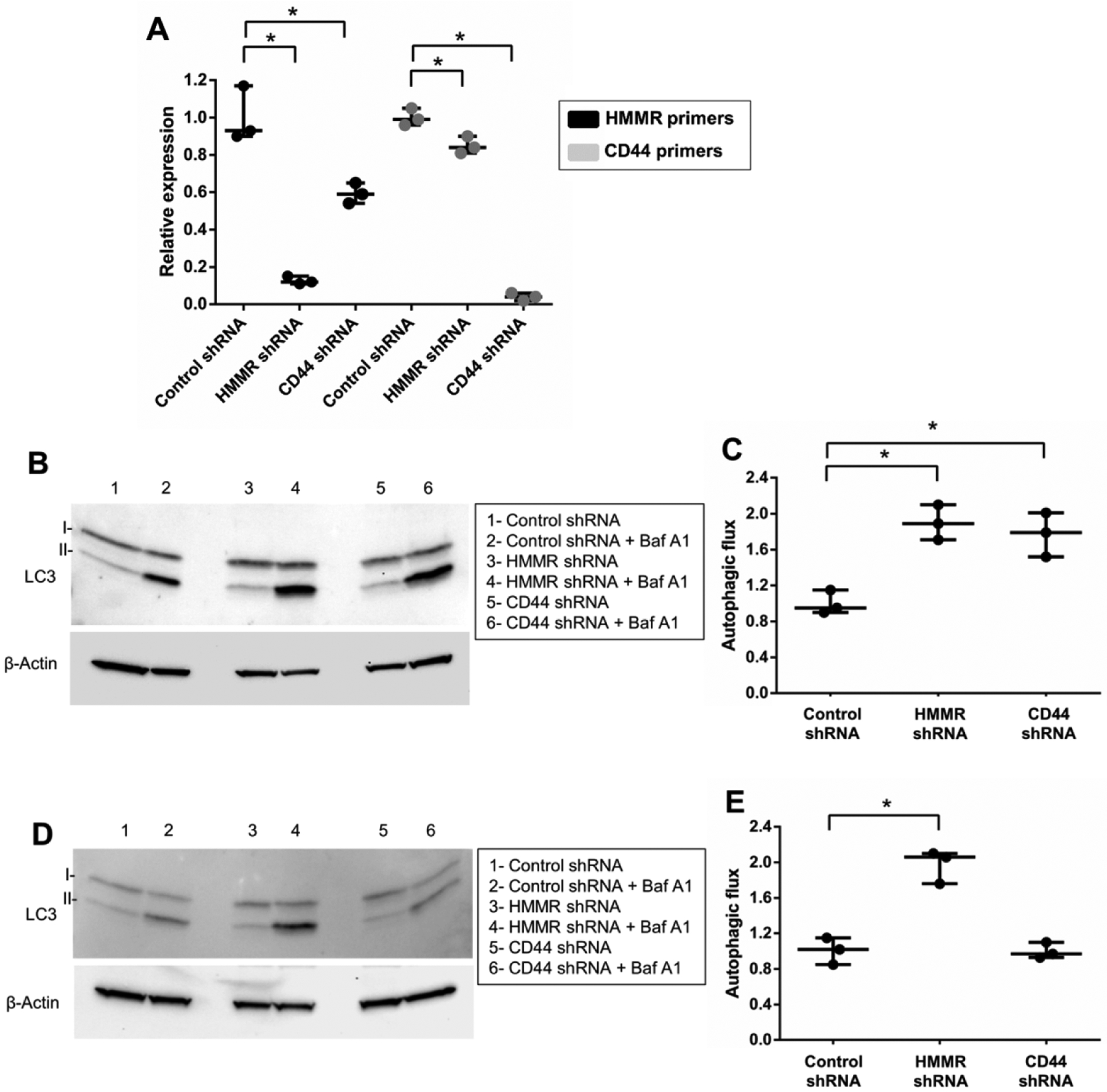
(A) Control, CD44 or HMMR expression was inhibited in differentiated SH-SY5Y cells neurons by lentivirus-delivered shRNA vectors. Transduced cells were selected with puromycin. Knockdown was validated by qPCR using the indicated primer sets and normalized to β-Actin transcript expression levels for each reaction. (B) Autophagic flux was examined in wild-type VPS35-expressing cells by western blot and quantified (C) following shRNA-mediated knockdown, treatment with or without bafilomycin A1 (Baf A1) and normalization of measurements to VPS35 WT shRNA control. (D) Autophagic flux was also assessed by western blot and quantified (E) in VPS35 D620N-expressing SH-SY5Y cells following shRNA transduction, Baf A1 treatment and normalization of measurements to VPS35 D620N shRNA control. Results from three independent experiments are shown (*p<0.05 versus indicated group, student’s t-test; error bars = SEM). Representative image groupings were obtained from single western blot membranes probed with multiple antibodies as indicated. β-Actin antibody was used as a loading control.
DISCUSSION
The study of monogenic causes of PD has yielded valuable insights into the pathogenesis of this disease. The identification of VPS35 D620N as a causal familial factor has allowed for the molecular dissection of signaling pathways that could be important therapeutic points for this and other forms of PD. It is well-established that VPS35 D620N inhibits autophagy and hinders retromer function (Follett et al., 2014; McGough et al., 2014; Zavodszky et al., 2014). Accordingly, research focus in the field has been placed upon canonical retromer interactors and pathways. However, this approach might be limiting as unexpected pathways could play significant roles in VPS35 D620N pathology. Therefore, in an effort to assay all potentially relevant signaling pathways we created isogenic transgenic SH-SY5Y cell lines (isolated from single clones) expressing VPS35 or VPS35 D620N and performed RNA Seq transcriptome analysis. Retinoic acid-differentiated SH-SY5Y cells were chosen due to their post-mitotic, dopaminergic neuron-like properties and suitability for the creation of stably-expressing transgenic cell lines (Brown et al., 1994; Sonnier et al., 2003).
Identification of perturbed HA-AKT signaling as a contributor to VPS35 D620N-mediated autophagy dysfunction may offer a therapeutic target for PD as well as broaden the understanding of autophagy regulation. We report that HMW HA serves as a negative regulator of autophagy. Given the high concentration of HMW HA in the mammalian brain, the physiological consequences of this finding could be significant. In the scope of neurological disease, defective autophagy is linked to a wide variety of maladies (Levine and Kroemer, 2008). If HMW HA and autophagic activity are coupled then the concentration and quality of HMW HA might guide the path of neurodegenerative disease particularly as individuals age and are less adept at maintaining ECM. In the context of PD, recognition of HA-AKT pathway involvement in the misregulation of autophagy in catecholaminergic cells could provide a novel target for treatment.
Our finding that VPS35 D620N-expressing cells have heightened response to HMW but not LMW HA indicates that HA-AKT signaling is not initiated by a traditional ligand-receptor mechanism. This finding would imply that the responsible HA receptor(s) require scaffolding to a large ECM structure for activation. Previous work has indeed shown that cytoarchitecture is important for HA signaling (Neame et al., 1995). Maharjan and colleagues have specifically explored the signaling divergences between HMW and LMW HA(Maharjan et al., 2011). During remodeling, such as following injury, HMW HA is broken down to LMW by matrix metalloproteinases (MMP). This group reported that environments can direct monocyte differentiation and activation based upon HA polymer size. It is therefore possible that brain HA composition could also serve important physiological functions.
CD44 and HMMR are the most established HA receptors. These proteins have been extensively studied in the context of cancer metastasis as being refined sensors for detecting the state of ECM by an invading cancer cell (Savani et al., 2001). Upon identifying defective HA-AKT signaling we examined CD44 and HMMR expression. We report that SH-SY5Y cells express only a single isoform (V4) of CD44 (CD44v4) protein, albeit at very low levels, and that this isoform is found in higher amounts in mutant versus control cells. We also found that CD44v4 protein and mRNA levels correlate closely. However, the extremely low level of CD44 expression coupled with the observation that the CD44 protein expression is likely controlled largely by transcriptional mechanisms suggest that CD44 plays a minor role in VPS35 D620N dysfunction. In contrast, HMMR protein is abundantly expressed yet isoform V3 (HMMRv3) is strikingly absent from VPS35 D620N-expressing cells. Interestingly, HMMRv3 transcript levels are indistinguishable for the wild-type and mutant cells indicating post-splicing regulation of expression and potentially a direct role for VPS35 through retromer activity. A role for both CD44 and HMMR on autophagy is indicated by the enhanced autophagic flux observed following CD44 or HMMR knockdown. However, only HMMR knockdown resulted in rescue of repressed autophagy by VPS35 D620N suggesting that HMMR is an important contributor for the autophagy defect. We can extrapolate, using the observed WT versus mutant autophagic flux inhibition data presented in Figure 1 (48% reduction for VPS35 D620N cells), that the rescue of HMMR knockdown on VPS35 D620N autophagy inhibition is restored to a level comparable to that of control WT VPS35 cells (HMMR knockdown exhibits 93% autophagic flux of WT VPS35 cells). A possible explanation for the improvement in autophagic flux in WT VPS35 cells following knockdown of either HMMR or CD44 is that HMMR signaling is a strong driver of autophagy inhibition. Knockdown of CD44 results in a partial knockdown of HMMR (Fig. 8A) likely through shared transcriptional regulatory mechanisms. This partial knockdown of HMMR might facilitate increased autophagic flux in a wild-type background but is not sufficient to rescue the enhanced HMMR signaling observed in VPS35 D620N cells.
In summary, we report that VPS35 D620N suppresses autophagy in RA-differentiated SH-SY5Y cells in a manner consistent with a loss of function mutation. Using an unbiased RNA Seq screen we identified misregulated ECM-receptor and PI3K-AKT signaling pathways in the mutant cells. Subsequent investigation revealed enhanced HMW HA-AKT signaling and an expression defect in a single isoform of HMMR (HMMRv3) in mutant VPS35-expressing cells. Knockdown of HMMR, and not CD44, rescued the VPS35 D620N autophagy defect. These results provide a compelling rationale to further investigate VPS35 function as it relates to HA-HMMR-AKT signaling and autophagy in PD.
Supplementary Material
Figure S1. WASH1-VPS35 interaction is not detected in SH-SY5Y cells. Non-transgenic (SH-SY5Y), WT-VPS35, and VPS35-D620N-expressing SH-SY5Y cells were RA-differentiated and then WASH1-VPS35 interaction was assessed with co-immunoprecipitation (IP) by pulling transgenic VPS35 down with a V5 antibody. The blot was then subjected to western blotting with the indicated antibodies (> denotes the VPS35 band; * signifies a non-specific band found in all samples). Whole cell lysates (WCL) were also included. Successful VPS35 IP was verified by co-immunoprecipitation of known interactors VPS29, VPS26, and TBC1D5. Each panel shown represents a reprobing of the same western blot membrane. As reported by other groups, we do not observe differential interaction of retromer members (VPS29 and 26A) by VPS35 D620N.
Figure S2. VPS35 D620N enhances intracellular alpha-synuclein aggregation. WT or VPS35 D20N-expressing SH-SY5Y cells were transfected with S1 and S2 plasmids containing full-length alpha-synuclein fused to N-or C-terminal parts of human Gaussia luciferase. Upon aggregation of alpha-synuclein, complementary portions of luciferase produced a functional enzyme that was detected by adding luciferase substrate coelenterazine. Full length luciferase (Luc) and alpha-synuclein conjugated to full-length luciferase (Syn-Luc) were also examined as basal expression controls. Top panel: luciferase activity within cells. Bottom panel: luciferase activity in the culture media (secreted by cells). VPS35 D620N-expressing cells exhibited significantly elevated alpha-synuclein aggregation compared to WT cells (student’s t-test; **P>0.05; Error bars = SEM).
Figure S3. RNA-Seq data assessment. (A) Alignment statistics for sequencing reads. (B) mRNA abundance distribution (density plot) for all genes quantified from RNA-seq analysis. Each replicate was plotted individually and overlaid to show there is no distribution bias. FPKM ≥ 0.5 was arbitrarily set as the cut-off value for downstream analysis. (C) A scatter plot comparing wild-type (wt) and VPS35-D620N (mut) samples. Each dot represents the average expression level (FPKM) of individual genes calculated from three biological replicates. The vast majority of genes were expressed similarly in wt (VPS35) and mut (VPS35-D620N) cells, but genes that were differentially expressed are remotely located from the diagonal dashed-line. (D) A dendrogram of all quantified mRNA from RNA-seq analysis. Clustering among biological replicates suggested that the D620N mutation causes differential gene expression. (E) RNA concentration and purity for RNA Seq analysis is shown.
Figure S4. RNA-Seq pathway gene expression plots. All the differentially expressed genes (WT vs D620N cells) in (A) ECM-receptor interaction and (B) PI3K-AKT KEGG pathways displayed by log fold change (FC) expression change. The downregulated genes are shown in blue, and the upregulated ones are shown in red. The box and whisker plot on the left summarizes the distribution of all the differentially expressed genes in this gene set. The box represents the 1st quartile, the median and the 3rd quartile, while the outliers are represented by circles. The PI3K-AKT pathway results are split into two panels to enhance readability. Consequently, the lower panel in (B) has a box and whisker plot from the original plot attached to the expression panel using image editing software. Data represented using Advaita Corporation iPathways Guide software.
Figure S5. VPS35 D620N does not affect total AKT expression. Cells were serum-deprived overnight and then treated with 300ug/mL of hyaluronan (HA) for 6 hours or without (UN). Western blotting was performed for representative samples from WT and VPS35 D620N cells probed with antibodies against total AKT as well as β-Actin.
HIGHLIGHTS.
VPS35 D620N inhibits autophagy in a loss-of-function manner
RNA Seq identifies extracellular matrix-receptor and PI3K-AKT pathways affected by VPS35 D620N
High molecular weight hyaluronan inhibits autophagy
VPS35 D620N enhances high molecular weight hyaluronan signaling
HMMR knockdown rescues autophagy repression by VPS35 D620N
FUNDING SOURCES
This work was supported by the Institutional Development Awards (IDeA) from the National Institute of General Medical Sciences COBRE program (grants P20GM103408 and P20GM109095) and the National Institute of Neurological Disorders and Stroke (grant R15NS096702) of the National Institutes of Health. Additional support was provided by The Biomolecular Research Center at Boise State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
RESEARCH DATA
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon request. RNA-Seq data for the described experiments is available in both raw and processed formats at the NCBI Gene Expression Omnibus (GEO) public repository.
Conflict of Interest:
None declared.
REFERENCES
- Brown NA, Kemp JA, and Seabrook GR (1994). Block of human voltage-sensitive Na+ currents in differentiated SH-SY5Y cells by lifarizine. Br. J. Pharmacol 113, 600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi AMK, Ryter SW, and Levine B (2013). Autophagy in human health and disease. N. Engl. J. Med 368, 1845–1846. [DOI] [PubMed] [Google Scholar]
- Choi KH, Basma H, Singh J, and Cheng P-W (2005). Activation of CMV promoter-controlled glycosyltransferase and beta -galactosidase glycogenes by butyrate, tricostatin A, and 5-aza-2’-deoxycytidine. Glycoconj. J 22, 63–69. [DOI] [PubMed] [Google Scholar]
- Dehay B, Bové J, Rodríguez-Muela N, Perier C, Recasens A, Boya P, and Vila M (2010). Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci 30, 12535–12544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, Martinez-Vicente M, Caldwell GA, Caldwell KA, Yue Z, Cookson MR, Klein C, Vila M, and Bezard E (2013). Lysosomal impairment in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc 28, 725–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dengjel J, Høyer-Hansen M, Nielsen MO, Eisenberg T, Harder LM, Schandorff S, Farkas T, Kirkegaard T, Becker AC, Schroeder S, et al. (2012). Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol. Cell. Proteomics MCP 11, M111.014035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draghici S, Khatri P, Bhavsar P, Shah A, Krawetz SA, and Tainsky MA (2003). Onto-Tools, the toolkit of the modern biologist: Onto-Express, Onto-Compare, Onto-Design and Onto-Translate. Nucleic Acids Res. 31, 3775–3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett J, Norwood SJ, Hamilton NA, Mohan M, Kovtun O, Tay S, Zhe Y, Wood SA, Mellick GD, Silburn PA, et al. (2014). The Vps35 D620N mutation linked to Parkinson’s disease disrupts the cargo sorting function of retromer. Traffic Cph. Den 15, 230–244. [DOI] [PubMed] [Google Scholar]
- Fuse A, Furuya N, Kakuta S, Inose A, Sato M, Koike M, Saiki S, and Hattori N (2015). VPS29-VPS35 intermediate of retromer is stable and may be involved in the retromer complex assembly process. FEBS Lett. 589, 1430–1436. [DOI] [PubMed] [Google Scholar]
- He C, and Klionsky DJ (2009). Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet 43, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic J, and Stacy M (2007). Medical management of levodopa-associated motor complications in patients with Parkinson’s disease. CNS Drugs 21, 677–692. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, and Tanabe M (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P, Draghici S, Tarca AL, Hassan SS, and Romero R (2007). A System Biology Approach for the Steady-State Analysis of Gene Signaling Networks In Progress in Pattern Recognition, Image Analysis and Applications, (Springer, Berlin, Heidelberg: ), pp. 32–41. [Google Scholar]
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. (2012). Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korecka JA, van Kesteren RE, Blaas E, Spitzer SO, Kamstra JH, Smit AB, Swaab DF, Verhaagen J, and Bossers K (2013). Phenotypic characterization of retinoic acid differentiated SH-SY5Y cells by transcriptional profiling. PloS One 8, e63862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna A, Biryukov M, Trefois C, Antony PMA, Hussong R, Lin J, Heinäniemi M, Glusman G, Köglsberger S, Boyd O, et al. (2014). Systems genomics evaluation of the SH-SY5Y neuroblastoma cell line as a model for Parkinson’s disease. BMC Genomics 15, 1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, and Taylor JP (2010). Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet 11, 247–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau LML, and Breteler MMB (2006). Epidemiology of Parkinson’s disease. Lancet Neurol. 5, 525–535. [DOI] [PubMed] [Google Scholar]
- Levine B, and Kroemer G (2008). Autophagy in the Pathogenesis of Disease. Cell 132, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S-L, Chang D, and Ying S-Y (2007). Hyaluronan stimulates transformation of androgen-independent prostate cancer. Carcinogenesis 28, 310–320. [DOI] [PubMed] [Google Scholar]
- Luo W, Friedman MS, Shedden K, Hankenson KD, and Woolf PJ (2009). GAGE: generally applicable gene set enrichment for pathway analysis. BMC Bioinformatics 10, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maharjan AS, Pilling D, and Gomer RH (2011). High and low molecular weight hyaluronic acid differentially regulate human fibrocyte differentiation. PloS One 6, e26078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malgaroli A, Vallar L, and Zimarino V (2006). Protein homeostasis in neurons and its pathological alterations. Curr. Opin. Neurobiol 16, 270–274. [DOI] [PubMed] [Google Scholar]
- McGough IJ, and Cullen PJ (2011). Recent advances in retromer biology. Traffic Cph. Den 12, 963–971. [DOI] [PubMed] [Google Scholar]
- McGough IJ, Steinberg F, Jia D, Barbuti PA, McMillan KJ, Heesom KJ, Whone AL, Caldwell MA, Billadeau DD, Rosen MK, et al. (2014). Retromer binding to FAM21 and the WASH complex is perturbed by the Parkinson disease-linked VPS35(D620N) mutation. Curr. Biol. CB 24, 1670–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstråle M, Laurila E, et al. (2003). PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet 34, 267–273. [DOI] [PubMed] [Google Scholar]
- Neame SJ, Uff CR, Sheikh H, Wheatley SC, and Isacke CM (1995). CD44 exhibits a cell type dependent interaction with triton X-100 insoluble, lipid rich, plasma membrane domains. J. Cell Sci 108 (Pt 9), 3127–3135. [DOI] [PubMed] [Google Scholar]
- Onodera Y, Teramura T, Takehara T, and Fukuda K (2015). Hyaluronic acid regulates a key redox control factor Nrf2 via phosphorylation of Akt in bovine articular chondrocytes. FEBS Open Bio 5, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outeiro TF, Putcha P, Tetzlaff JE, Spoelgen R, Koker M, Carvalho F, Hyman BT, and McLean PJ (2008). Formation of toxic oligomeric alpha-synuclein species in living cells. PloS One 3, e1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel M, Goldbaum O, Schwarz L, Schmitt S, and Richter-Landsberg C (2010). 17-AAG induces cytoplasmic alpha-synuclein aggregate clearance by induction of autophagy. PloS One 5, e8753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savani RC, Cao G, Pooler PM, Zaman A, Zhou Z, and DeLisser HM (2001). Differential involvement of the hyaluronan (HA) receptors CD44 and receptor for HA-mediated motility in endothelial cell function and angiogenesis. J. Biol. Chem 276, 36770–36778. [DOI] [PubMed] [Google Scholar]
- Scott KL, Kabbarah O, Liang M-C, Ivanova E, Anagnostou V, Wu J, Dhakal S, Wu M, Chen S, Feinberg T, et al. (2009). GOLPH3 modulates mTOR signalling and rapamycin sensitivity in cancer. Nature 459, 1085–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnier H, Kolomytkin O, and Marino A (2003). Action potentials from human neuroblastoma cells in magnetic fields. Neurosci. Lett 337, 163–166. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanik SA, Schultheiss CE, Volpicelli-Daley LA, Brunden KR, and Lee VMY (2013). Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J. Biol. Chem 288, 15194–15210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarca AL, Draghici S, Khatri P, Hassan SS, Mittal P, Kim J-S, Kim CJ, Kusanovic JP, and Romero R (2009). A novel signaling pathway impact analysis. Bioinforma. Oxf. Engl 25, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JP, Hardy J, and Fischbeck KH (2002). Toxic proteins in neurodegenerative disease. Science 296, 1991–1995. [DOI] [PubMed] [Google Scholar]
- Trapasso S, and Allegra E (2012). Role of CD44 as a marker of cancer stem cells in head and neck cancer. Biol. Targets Ther 6, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, and Pachter L (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc 7, 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilariño-Güell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, Soto-Ortolaza AI, Cobb SA, Wilhoite GJ, Bacon JA, et al. (2011). VPS35 mutations in Parkinson disease. Am. J. Hum. Genet 89, 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavodszky E, Seaman MNJ, Moreau K, Jimenez-Sanchez M, Breusegem SY, Harbour ME, and Rubinsztein DC (2014). Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun 5, 3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, et al. (2011). A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet 89, 168–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. WASH1-VPS35 interaction is not detected in SH-SY5Y cells. Non-transgenic (SH-SY5Y), WT-VPS35, and VPS35-D620N-expressing SH-SY5Y cells were RA-differentiated and then WASH1-VPS35 interaction was assessed with co-immunoprecipitation (IP) by pulling transgenic VPS35 down with a V5 antibody. The blot was then subjected to western blotting with the indicated antibodies (> denotes the VPS35 band; * signifies a non-specific band found in all samples). Whole cell lysates (WCL) were also included. Successful VPS35 IP was verified by co-immunoprecipitation of known interactors VPS29, VPS26, and TBC1D5. Each panel shown represents a reprobing of the same western blot membrane. As reported by other groups, we do not observe differential interaction of retromer members (VPS29 and 26A) by VPS35 D620N.
Figure S2. VPS35 D620N enhances intracellular alpha-synuclein aggregation. WT or VPS35 D20N-expressing SH-SY5Y cells were transfected with S1 and S2 plasmids containing full-length alpha-synuclein fused to N-or C-terminal parts of human Gaussia luciferase. Upon aggregation of alpha-synuclein, complementary portions of luciferase produced a functional enzyme that was detected by adding luciferase substrate coelenterazine. Full length luciferase (Luc) and alpha-synuclein conjugated to full-length luciferase (Syn-Luc) were also examined as basal expression controls. Top panel: luciferase activity within cells. Bottom panel: luciferase activity in the culture media (secreted by cells). VPS35 D620N-expressing cells exhibited significantly elevated alpha-synuclein aggregation compared to WT cells (student’s t-test; **P>0.05; Error bars = SEM).
Figure S3. RNA-Seq data assessment. (A) Alignment statistics for sequencing reads. (B) mRNA abundance distribution (density plot) for all genes quantified from RNA-seq analysis. Each replicate was plotted individually and overlaid to show there is no distribution bias. FPKM ≥ 0.5 was arbitrarily set as the cut-off value for downstream analysis. (C) A scatter plot comparing wild-type (wt) and VPS35-D620N (mut) samples. Each dot represents the average expression level (FPKM) of individual genes calculated from three biological replicates. The vast majority of genes were expressed similarly in wt (VPS35) and mut (VPS35-D620N) cells, but genes that were differentially expressed are remotely located from the diagonal dashed-line. (D) A dendrogram of all quantified mRNA from RNA-seq analysis. Clustering among biological replicates suggested that the D620N mutation causes differential gene expression. (E) RNA concentration and purity for RNA Seq analysis is shown.
Figure S4. RNA-Seq pathway gene expression plots. All the differentially expressed genes (WT vs D620N cells) in (A) ECM-receptor interaction and (B) PI3K-AKT KEGG pathways displayed by log fold change (FC) expression change. The downregulated genes are shown in blue, and the upregulated ones are shown in red. The box and whisker plot on the left summarizes the distribution of all the differentially expressed genes in this gene set. The box represents the 1st quartile, the median and the 3rd quartile, while the outliers are represented by circles. The PI3K-AKT pathway results are split into two panels to enhance readability. Consequently, the lower panel in (B) has a box and whisker plot from the original plot attached to the expression panel using image editing software. Data represented using Advaita Corporation iPathways Guide software.
Figure S5. VPS35 D620N does not affect total AKT expression. Cells were serum-deprived overnight and then treated with 300ug/mL of hyaluronan (HA) for 6 hours or without (UN). Western blotting was performed for representative samples from WT and VPS35 D620N cells probed with antibodies against total AKT as well as β-Actin.