

Abstract
Identifying genetic factors underlying neuroanatomical variation has been difficult. Traditional methods have used brain regions from predetermined parcellation schemes as phenotypes for genetic analyses, although these parcellations often do not reflect brain function and/or do not account for covariance between regions. We proposed that network-based phenotypes derived via source-based morphometry (SBM) may provide additional insight into the genetic architecture of neuroanatomy given its data-driven approach and consideration of covariance between voxels. We found that anatomical SBM networks constructed on ~ 20 000 individuals from the UK Biobank were heritable and shared functionally meaningful genetic overlap with each other. We additionally identified 27 unique genetic loci that contributed to one or more SBM networks. Both GWA and genetic correlation results indicated complex patterns of pleiotropy and polygenicity similar to other complex traits. Lastly, we found genetic overlap between a network related to the default mode and schizophrenia, a disorder commonly associated with neuroanatomic alterations.
Keywords: genome-wide association analysis (GWAS), genome-wide complex trait analysis (GCTA), linkage disequilibrium score regression (LDSC), source-based morphometry (SBM), structural magnetic resonance imaging (sMRI)
Introduction
Neuroanatomic variation, as measured in vivo by magnetic resonance imaging (MRI), is associated with individual differences in cognitive ability (Aljondi et al. 2018), emotional processing (Muhlert and Lawrence 2015), and psychiatric symptomatology (Turetsky et al. 1995; Dotson et al. 2009). While the directionality of this association is unknown, it is clear that genetic variation influences both neuroanatomic and behavioral traits (Toga and Thompson 2005; Briley and Tucker-Drob 2017). Furthermore, it is possible that genes influencing neuroanatomy also affect neurocognitive performance and/or risk for mental illness. Thus, a more complete understanding of the genetic control of neuroanatomic variability may yield insight into the genetic architecture of normal and abnormal brain function.
Studies in twins and families (see Jansen et al. 2015) for a comprehensive review), as well as unrelated individuals (Elliott et al. 2018), consistently find that brain volume measures are heritable. Identifying specific genes related to normal variation in brain volume, however, is difficult. Candidate gene studies indicate some genetic effects (e.g., for APOE, COMT and BDNF) (Knickmeyer et al. 2013) but show little convergence across studies (Chételat and Fouquet 2013; Harrisberger et al. 2014), likely due to the complex genetic architecture of neuroanatomy coupled with small sample sizes that are under powered (Strike et al. 2015a). Large-scale genome-wide association (GWA) studies provide an unbiased approach for identifying specific loci associated with neuroanatomy (see Strike et al. 2015a for a list). The most recent and largest GWA analyses from the UK Biobank (Elliott et al. 2018) identified 17 loci associated with volumetric measures across 33 regions, although only 3 of these regions were from cortex (specifically the precentral gyrus and intracalcarine cortices). This result is surprising given the high heritability of cortical regions, although the level of multiple testing correction necessary for this study may have played arole.
Traditionally, both candidate gene and GWA studies have used cortical phenotypes based on predetermined parcellation schemes of the brain, the most common being the Desikan–Killiany atlas (Desikan et al. 2006). These regional parcellations are often constructed based on anatomical rather than functional information, like sulci and gyri, and each region is treated as an independent entity for genetic analysis. There is, however, strong evidence for structural covariance between brain regions (Xu et al. 2009; Segall et al. 2012; Gupta et al. 2014), part of which is genetic in nature (Strike et al. 2015b), indicating that genetic effects could influence subcomponents of regions or multiple regions that are not necessarily adjacent to each other. There is also evidence that structural covariance mirrors patterns seen in functional brain networks (Alexander-Bloch et al. 2013), suggesting that phenotypes that take into account covariance information may provide insight into the functional and behavioral consequences of genetic effects on neuroanatomy.
Perhaps an alternative way to probe genetic influences on neuroanatomy may be to use structural covariance in the brain to create network-based phenotypes that are derived from the data itself. Such phenotypes may provide complementary information on how genetics influence brain anatomy while avoiding issues related to predetermined assumptions and data reduction. Source-based morphometry (SBM) (Xu et al. 2009) is a promising method for constructing such phenotypes. SBM is a multivariate method that uses independent component analysis (ICA) to obtain spatially distinct anatomical networks of regions that covary across a set of individuals. The ICA decomposition is performed on data from voxel-based morphometry (VBM), which considers volumetric measures at the voxel level throughout the brain. As such, this method can pool information across voxels and consider the relationship between them in order to construct anatomical networks of regions in a hypothesis-free manner. SBM networks, in particular, are also of interest because they have been used widely in the neuroimaging literature to understand neuroanatomic variation in psychiatric disease (Xu et al. 2009; Kašpárek et al. 2010; Kubera et al. 2014; Ciarochi et al. 2016; Bergsland et al. 2018; Pappaianni et al. 2018) and the relationship between brain structure and function (Luo et al. 2012), making further quantification of their genetic underpinnings a beneficial endeavor. The goal of this paper was to validate the possibility of using SBM networks as potential phenotypes for genetic analyses by understanding some of their basic genetic contributions. We aimed to do this by 1) establishing their heritability and understanding more about their genetic architecture, primarily if they are polygenic like other complex traits, 2) evaluating their genetic relationship (or pleiotropy) with each other at the global level and at the level of individual loci, and 3) given their frequent use to identify neuroanatomical alterations in disease, understanding their genetic relationship with three psychiatric disorders commonly associated with neuroanatomical disruptions (McIntosh et al. 2006; Holmes et al. 2012; Lee et al. 2016): schizophrenia (SZ), bipolar disorder (BP), and major depressive disorder (MDD). We expected that some genetic influences would be specific to particular SBM networks, while others may be shared across networks, although we anticipated that these would be different from genetic contributions to whole brain gray matter volume (GMV). We also predicted that genetic influences would overlap with psychiatric phenotypes that are genetically related to brain structure.
Materials and Methods
Participants
The UK Biobank (http://www.ukbiobank.ac.uk) is a population-based prospective cohort of ~ 50 000 individuals, ages 40–69 recruited between 2006 and 2010 throughout Great Britain. Data were collected on thousands of variables including those related to physical and mental health, cognitive functioning, lifestyle, demographics, genetics, and imaging. Recruitment procedures for the UK Biobank are described elsewhere (Sudlow et al. 2015). Participants in this study included 17 257 individuals (7870 males, mean age [standard deviation] = 62.5 [7.4]) with available imaging and genetic data as of 19 October 2018 who passed our quality control and filtering procedures for both imaging and genetic data. The UK Biobank received ethical approval from the Research Ethics Committee (REC reference 11/NW/0382). The present analyses were conducted under UK Biobank data application number 4844.
Phenotypic Analysis
Acquisition of MRI Images
Imaging protocols were designed by the UK Biobank Imaging Working Group (www.ukbiobank.ac.uk/expert-working-groups) (see Alfaro-Almagro et al. 2018 for additional details). Imaging datasets were collected at one of three dedicated imaging centers with identical scanners (3T Siemens Skyra [software platform VD13]), which refrained from major software or hardware updates during the study. Each center was equipped with a 32-channel head coil and used the following 3D MPRAGE protocol to quantify brain structure: T1-weighted, TI/TR = 880/2000 ms, sagittal acquisition, resolution = 1.0 × 1.0 × 1.0 mm, FOV = 208 × 256 × 256, in-plane acceleration factor = 2, 4 min 54 s scan duration.
Image Preprocessing
Datasets were part of the first two imaging releases by the UK Biobank. All image preprocessing was performed with FMRIB Software Library v10.0 (FSL) (Jenkinson et al. 2012). Segmented gray matter images, which were part of the structural T1 processing pipeline by the UK Biobank (Alfaro-Almagro et al. 2018), were input into a customized VBM pipeline based on the protocol used in FSL (Ashburner and Friston 2000). A study-specific template was created using an average T1-weighted image (provided by the UK Biobank) from 5000 subjects. To generate the template, brain extraction and tissue segmentation was performed on the average T1-weighted image. The gray matter image from the segmentation was then registered to the avg152T1_gray template available in FSL. Segmented gray matter images from each subject were nonlinearly registered to the study-specific template. Each registered gray matter image was also multiplied by the Jacobian of the warp field as a compensation (or “modulation”) for the contraction/enlargement due to the nonlinear component of the transformation. The resulting gray matter image was then smoothed with a 6 mm Gaussian kernel. Twenty-one thousand three hundred eighty-eight subjects were run through our preprocessing protocol. For quality assurance, the unmodulated registered T1 image for each subject was correlated with the study-specific template. After visual inspection of 80 individuals around the border of our cutoff (r = 0.78), subjects with a correlation less than or equal to the cutoff were eliminated from further analysis (N = 1083), resulting in analyzable imaging data for 20 306 subjects. Lastly, for subjects passing the 0.78 cutoff, we calculated an estimate of GMV. An average gray matter mask (generated automatically as part of the VBM pipeline from our subjects) was applied to each subject’s output image from the preprocessing pipeline. GMV estimates were the average value across voxels included in thismask.
Source-Based Morphometry
SBM was performed on 20 306 subjects using the GIFT toolbox (http://mialab.mrn.org/software/gift/). Segmented gray matter images output from the preprocessing pipeline were flattened into row vectors and stacked to form a subjects-by-voxel matrix in preparation for spatial ICA decomposition (Xu et al. 2009; Calhoun et al. 2001; Gupta et al. 2018). ICA decomposes the gray matter matrix into a mixing matrix (subjects-by-components), representing the relative strength (weight) of each subject on every component, and the source matrix (components by voxels), representing the maximally spatially independent gray matter regions. This results in networks of anatomical regions that covary across the brain and subject-level loading coefficients for each network, which indicate how representative that subject is on that component pattern. ICA was performed with the infomax algorithm with a model order of 25, to stay consistent with previous work and focus on large-scale networks (Xu et al. 2009).
Genetic Analyses
Phenotypes
Phenotypes for all analyses were subject loadings on each of the 25 SBM networks and GMV. For all genetic analyses, we applied an inverse-normal transformation to all phenotypes and used the following covariates—age, age2, sex, age × sex, age2 × sex, genotype array (UK BiLEVE or UK Biobank Axiom)—and 20 genetic principal components calculated by the UK Biobank to account for population stratification.
Genotyping and Imputation
All genetic analyses performed in this study used the imputed genotypes provided by the UK Biobank. Blood sample handling and storage details are in Peakman and Elliot (2008). DNA was extracted at one of the UK Biobank assessment centers and sent to Affymetrix Research Services Laboratory in 96-well plates containing 94 UK Biobank samples. Special attention was paid to ensure that plates or timing of extraction did not correlate systematically with baseline phenotypes (age, sex, ethnic background, or time and location of sample collection). Details of SNP genotyping, extensive QC of genotyped data, and SNP phasing and imputation from the genotype panel are described in detail elsewhere (Bycroft et al. 2017). In short, participants were genotyped on either the UK BiLEVE Axiom Array by Affymetrix1 (807 411 markers) or the closely related Applied Biosystems UK Biobank Axiom Array (825 927 markers). Both arrays were specifically designed for the UK Biobank and have a 95% marker overlap. Genotyping was performed with a GeneTitan® Multi-Channel (MC) Instrument using batches (106 total) of multiple plates (~4700 samples each); this was an optimized genotype calling pipeline that takes advantage of the multiple batch design and is suited for biobank-scale genotyping. The Biobank performed six marker QC tests, four within batch (batch effect, plate effect, departures from Hardy–Weinberg equilibrium, and sex effect) and two across batch (array effect and discordance across controls). If a marker failed one within batch test, it was set to missing for that batch; markers that failed any one of the four within batch tests in every batch or any one of the across batch tests were excluded from the dataset, resulting in 805 426 unique markers (bi-allelic SNPs and indels) from both arrays. For imputation, phasing on the autosomes was carried out using SHAPEIT3 (https://mathgen.stats.ox.ac.uk/genetics_software/shapeit/shapeit.html) with the 1000 Genomes Phase 3 dataset (Consortium 2015) as a reference panel. SNPs were imputed with IMPUTE4 (https://jmarchini.org/software/) for the Haplotype Reference Consortium (McCarthy et al. 2016), UK10K (Consortium UK 2015), and 1000 Genomes Phase 3 reference panels, resulting in 92 693 895 autosomal SNPs, short indels, and large structural variants in 487 409 individuals. SNP (rs) IDs were assigned where possible from the UCSC genome annotation database for the GRCh37 assembly of the human genome; otherwise they are notated as such chr: position_allele1_allele2.
Sample and Variant Filtering
Subjects with genetic data were initially excluded from our analysis if they were of non-British ancestry (N = 78 674), had a mismatch between reported and genotypic sex (N = 378), sex chromosome aneuploidy (N = 652), and/or high rates of heterozygosity or missingness (N = 968), resulting in 80 121 exclusions (some subjects belonged to more than one category of exclusion). These measures were calculated as part of the quality control procedure for genotype data by the UK Biobank and provided as a list to researchers. We then took the intersection between the remaining subjects (N = 407 288) and our subjects who had analyzable imaging data (N = 20 306), resulting in 17 396 subjects. Using IBD estimation in PLINK v1.9 (http://pngu.mgh.harvard.edu/purcell/plink/) (Purcell et al. 2007) and the available genotype data, we eliminated a further 139 individuals for genetic relatedness with other subjects (PI-HAT > 0.2). The final sample size was 17 257 subjects with acceptable imaging and genetic data. Within our final sample, we also filtered out variants in the imputed data based on the following criteria using qctool v2 (https://www.well.ox.ac.uk/~gav/qctool_v2/): MAF < 0.01, Hardy–Weinberg Equilibrium (HWE) P-value ≤ 0.001, missingness > 0.05, INFO < 0.3. The final number of imputed variants was 9 777 841.
Heritability and Genetic Architecture of SBM Networks
Heritability was estimated with the genome-wide complex trait analysis (GCTA) package (Yang et al. 2011). GCTA uses a genomic relatedness matrix (GRM) (a pairwise measure of genetic similarity between individuals) and a genome-based restricted maximum likelihood (GREML) procedure to estimate the total contribution of SNPs to the phenotypic variance of a trait and is termed SNP-based heritability (h2SNP). To further understand the genetic architecture of SBM networks, we aimed to determine whether genetic contributions were preferentially located on a particular chromosome or distributed evenly across the genome like other highly polygenic traits (Yang et al. 2010; Shi et al. 2016). In highly polygenic traits, the chromosome-based h2SNP is roughly proportional to chromosomal size, whereas for less polygenic traits, smaller fractions of the genome can contribute disproportionately to the total h2SNP (Shi et al. 2016). We therefore partitioned the total h2SNP across chromosomes, performed a regression between the fractional of h2SNP of a particular chromosome and its size, and noted whether any chromosomes fell appreciably above the regressionline.
Pleiotropy Between SBM Networks
To determine whether SBM networks shared genetically meaningful information with each other, we adopted two approaches. The first was a global approach, by which we applied bivariate GREML analyses (an extension of the univariate GREML analysis used for calculating h2SNP) to estimate the co-heritability or genetic correlation between each pair of SBM networks (Lee et al. 2012). This is considered a global measure as the correlation involves genetic effects from across the genome, rather than information about a specific variant. In addition to pairwise genetic correlations between SBM networks, we also calculated the genetic correlation between each SBM network and GMV to understand how genetic contributions to SBM networks may be similar or unique to genetic influences on whole brain measures. We additionally applied agglomerative hierarchical clustering (using the corrplot package in R v3.5.1) to the resulting symmetric genetic correlation matrix between each pair of phenotypes and GMV in order to define genetic modules or groups of brain phenotypes that were most similar in their genetic correlation pattern.
Our second approach was more targeted and focused on identifying genomic regions that may influence one or more SBM networks and/or GMV. We performed genome-wide association (GWA) analysis for each phenotype (N = 26, 25 components, and GMV) to see if genetic loci were shared across, or unique to, a particular brain phenotype. Genome-wide tests were performed on the filtered imputed genotypes (N = 9 777 841 variants) with 17 257 individuals using BGENIE v1.3 (Bycroft et al. 2017). Bonferroni correction was used for multiple comparison correction (0.05/[26 × 9 777 841]), resulting in a P < 1.96 × 10−10 threshold for significance.
Pleiotropy Between SBM Networks and Psychiatric Disease
We evaluated the genetic relationship between SBM networks and three psychiatric disorders (SZ, BP, and MDD) using cross-trait LD score regression (LDSC). LDSC is an extension of single-trait LD score regression and tests the genetic correlation between two traits using GWA summary statistics rather than individual-level genotype data (Bulik-Sullivan et al. 2015). We used GWA results from the 25 SBM networks generated in our study and the GWA results from three studies available for download from the Psychiatric Genetics Consortium (PGC), one for each psychiatric disorder of interest. There were multiple datasets per diagnosis available from the PGC; we chose the dataset with the largest number of individuals and the larger number of variants in order to maximize overlap with variants in the UK Biobank. False discovery rate (FDR) (Benjamini and Hochberg 1995) was applied to the LDSC results across all 25 networks within a disorder to account for false positives due to multiple testing. The SZ sample was a meta-analysis across 36 989 cases and 113 075 controls with 9 444 230 variants (Schizophrenia Working Group of the Psychiatric Genomics 2014). The BP sample was part of a larger study evaluating the genetic commonalities/uniqueness between bipolar disorder and schizophrenia and included 20 129 bipolar cases and 54 065 controls with 8 958 989 variants (Consortium 2018). Lastly, the MDD sample was part of a mega-analysis of MDD and included 9240 cases and 9519 controls and 1 235 109 variants (Ripke et al. 2013). All three samples were collected in those with European ancestry, although 6% of the sample for the SZ study was Asian.
Results
SBM Networks
SBM identified 25 components, each representing a spatially distinct covarying anatomical network in the cortex and/or cerebellum (see Fig. 1 and Supplementary Figure 1 for subject loading distributions for each component). Network maps were thresholded at |Z| > 2.5 for visualization purposes, although subject loadings refer to a pattern of voxel-wise coefficients across the whole brain. Each of the 25 networks was assigned to a functional module based on their similarity to functional networks from resting state or task-based fMRI as defined in Abrol et al. (2017): 7 networks were assigned to a cognitive control (CC) module (components 1, 3, 4, 9, 15, 23, and 24), 6 networks to a default mode (DMN) module (components 5, 8, 10, 12, 17, and 20), 5 networks to a sensory module (Sens) module (components 2, 14, 16, 18, and 21), and 7 networks to a cerebellar (Cereb) module (components 6, 7, 11, 13, 19, 22, and 25). Regions with the highest loading voxels for each network along with peak MNI coordinates are listed in Supplementary Table 1. Networks are subsequently referenced with their functional module and component number (e.g., CC1, DMN5, Sens2, and Cereb6).
Figure 1.
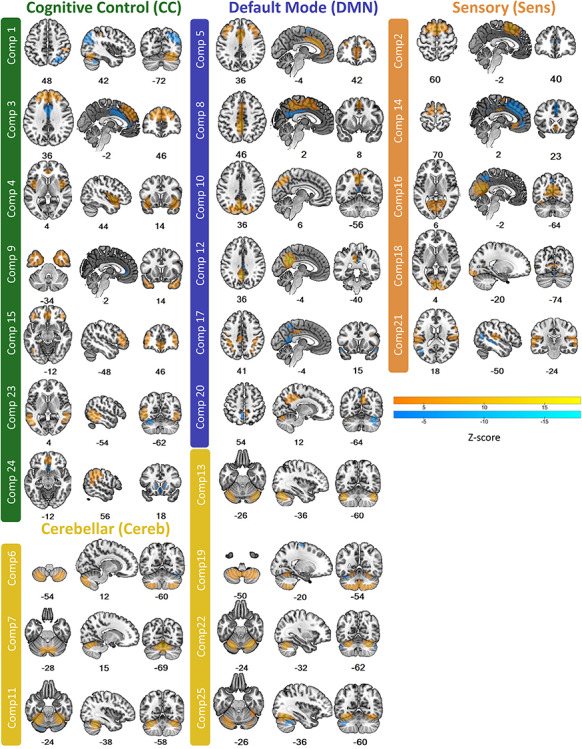
Anatomical brain networks from SBM. Networks are color coded by functional module. Although subject loadings correspond to a whole brain pattern of voxel-wise coefficients, we threshold maps at |Z| > 2.5 to indicate voxels that most highly contribute to the component pattern. Warm colors indicate positive loadings; cool colors indicate negative loadings. Numbers are z, y, and x coordinates in MNI space.
Genetic Contributions to SBM Networks
All brain phenotypes were significantly and moderately heritable with total h2snp estimates ranging from 0.14 to 0.38, with most exceeding 0.30 (see Table 1). Relationships between fractional h2snp and chromosome length were generally positive for all SBM networks and GMV (see Supplementary Figure 2). The level of polygenicity of the phenotype is represented by how close each chromosome is to the regression line. Some networks were highly polygenic (e.g., Cereb22 and Sens18) in that there was a strong relationship between the amount of heritability accounted for and chromosome size (R2 = 0.73 and R2 = 0.72, respectively). For other networks, there were noticeable deviations from the regression line, even though there was still a significant relationship between chromosome size and fractional h2snp. This was particularly true for DMN17 and Sens21, although less extreme cases were present in other SBM networks (e.g., Sens16 andCC9).
Table 1.
SNP-based heritability of SBM networks
| Phenotype | Total h2SNP | P |
|---|---|---|
| CC1 | 0.14 | 6.61 × 10−12 |
| CC3 | 0.14 | 7.78 × 10−12 |
| CC4 | 0.27 | 8.88 × 10−31 |
| CC9 | 0.34 | 5.45 × 10−49 |
| CC15 | 0.32 | 1.77 × 10−50 |
| CC23 | 0.34 | 4.23 × 10−51 |
| CC24 | 0.26 | 8.68 × 10−34 |
| DMN5 | 0.33 | 6.29 × 10−49 |
| DMN8 | 0.25 | 1.25 × 10−29 |
| DMN10 | 0.20 | 3.44 × 10−20 |
| DMN12 | 0.21 | 7.53 × 10−21 |
| DMN17 | 0.33 | 9.09 × 10−50 |
| DMN20 | 0.16 | 5.80 × 10−15 |
| Sens2 | 0.19 | 5.50 × 10−18 |
| Sens14 | 0.22 | 4.57 × 10−24 |
| Sens16 | 0.30 | 1.20 × 10−39 |
| Sens18 | 0.29 | 3.97 × 10−38 |
| Sens21 | 0.28 | 6.84 × 10−42 |
| Cereb6 | 0.38 | 1.43 × 10−63 |
| Cereb7 | 0.32 | 2.60 × 10−46 |
| Cereb11 | 0.30 | 5.60 × 10−39 |
| Cereb13 | 0.32 | 3.02 × 10−46 |
| Cereb19 | 0.23 | 3.58 × 10−24 |
| Cereb22 | 0.33 | 2.86 × 10−48 |
| Cereb25 | 0.26 | 2.41 × 10−32 |
| GMV | 0.35 | 3.20 × 10−55 |
Notes: Values calculated with GCTA. CC, cognitive control; DMN, default mode; Sens, sensory; Cereb, cerebellum; GMV, gray matter volume.
Genetic Overlap Among SBM Networks
Genetic correlations between brain phenotypes were both positive and negative and can be seen in Figure 2a. Hierarchical clustering of genetic correlations across phenotypes identified several clusters or genetic modules (GM) comprised of phenotypes that shared similar genetic correlation patterns (see Fig. 2b). Some of these genetic modules were dominated by phenotypes from the same functional category; this was especially true for GM4 which was almost completely composed of cerebellar networks. Other genetic modules were more varied in their composition of functional categories, particularly GM2 which included a wide range of SBM networks from cortex as well asGMV.
Figure 2.
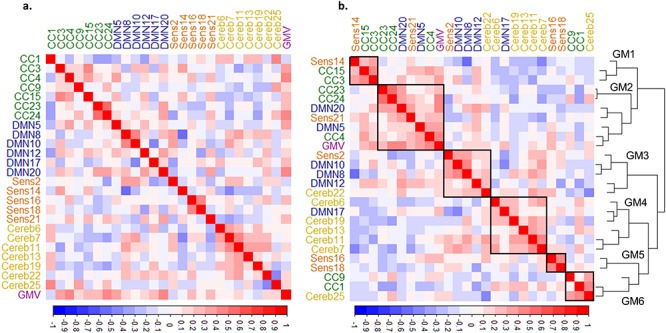
Genetic overlap between SBM networks and GMV. (a) Plot shows genetic correlations between each pair of SBM networks ordered and color coded by functional module. (b) The same as a but ordered by agglomerative hierarchical clustering; lines represent the clustering dendrogram. The clustering algorithm identified six genetic modules (GM) outlined by the black squares and numbered in the dendrogram. CC, cognitive control; DMN, default mode; Sens, sensory; Cereb, cerebellar; GMV, gray matter volume.
In order to identify individual loci that may contribute to the overlap (or non-overlap) in genetic variance found using GCTA, we performed individual GWA analyses on each SBM network and GMV. After multiple comparison correction, 27 LD-independent (Machiela and Chanock 2017) SNPs were significantly associated with one or more of our brain phenotypes (see Table 2). Each SNP was assigned to a locus (defined as a window 250 kb above and below the tagging SNP). The phenotype-wide Manhattan plot (see Fig. 3) shows that loci were scattered across the genome, although there was a large concentration of significant associations on chromosomes 7 and 14 (4 loci each). There were three loci where the same SNP was associated with more than one phenotype. This included locus 1 (rs6658111) for Sens21 and Cereb25, locus 5 (rs13107325) for DMN12 and Cereb19, and locus 16 (rs12146713) for CC4, DMN17, and Cereb25. Additionally, there were phenotypes that were associated with the same locus but were identified by different SNPs that were in LD with each other. These included GMV and DMN17 (locus 2); Cereb6 and Cereb13 (locus 19); DMN12, Sens16, and Sens18 (locus 20); and DMN8, DMN10, and DMN17 (locus 25). Individual Manhattan plots and corresponding quantile–quantile plots for each phenotype are in Supplementary Figure 3.
Table 2.
Significant GWA associations for brain phenotypes
| Phenotype | SNP ID | Chr | Position | Allele 1* | Allele 2 | P-value | β | se | Minor allele | MAF | INFO | Genes within 500 kb locus window | Locus No. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Sens21, Cereb25 | rs6658111 | 1 | 47 980 916 | G | T | 1.1 × 10−13 | 0.07 | 0.01 | G | 0.36 | 0.99 | STIL, CMPK1, FOXE3, FOXD2-AS1, FOXD2, TRABD2B | 1 |
| GMVa | rs1452628 | 1 | 215 139 887 | T | A | 5.4 × 10−15 | 0.06 | 0.01 | T | 0.38 | 1.00 | KCNK2 | 2 |
| DMN17a | rs6540873 | 1 | 215 137 222 | C | A | 6.9 × 10−23 | 0.09 | 0.01 | C | 0.38 | 1.00 | KCNK2 | 2 |
| Cereb13 | rs7589256 | 2 | 217 775 937 | T | C | 1.6 × 10−10 | -0.07 | 0.01 | C | 0.40 | 0.99 | IGFBP2, IGFBP5, TNP1, LOC105373876 | 3 |
| Cereb13 | rs1039906 | 4 | 4 764 026 | T | C | 1.3 × 10−11 | -0.08 | 0.01 | T | 0.26 | 1.00 | STX18, STYX-AS1, LOC101928279, MSX1 | 4 |
| DMN12, Cereb19 | rs13107325 | 4 | 103 188 709 | T | C | 1.2 × 10−11 | 0.14 | 0.02 | T | 0.07 | 1.00 | BANK1, SLC39A8, NFKB1 | 5 |
| Cereb11 | rs58972771 | 5 | 122 498 768 | GCTT | G | 6.1 × 10−11 | 0.09 | 0.01 | GCTT | 0.17 | 0.99 | SNX24, PPIC, PRDM6, CEP120 | 6 |
| DMN17 | rs77560239 | 6 | 126 710 804 | CTGTG | C | 1.3 × 10−11 | -0.06 | 0.01 | CTGTG | 0.43 | 0.97 | CENPW | 7 |
| Sens16 | rs4719573 | 7 | 19 617 100 | C | G | 6.3 × 10−11 | -0.07 | 0.01 | C | 0.34 | 0.99 | TWISTNB, MIR3146, TMEM196 | 8 |
| CC15 | rs6465524 | 7 | 96 188 457 | G | A | 3.9 × 10−12 | 0.08 | 0.01 | G | 0.27 | 1.00 | SLC25A13, SEM1 | 9 |
| GMV | rs55963900 | 7 | 120 977 734 | A | G | 9.8 × 10−12 | 0.06 | 0.01 | A | 0.28 | 1.00 | CPED1, WNT16, FAM3C | 10 |
| DMN5 | rs4128399 | 7 | 151 498 679 | C | T | 7.2 × 10−13 | 0.10 | 0.01 | C | 0.17 | 0.96 | PRKAG2, LOC644090, PRKAG2-AS1, GALNTL5, GALNT11 | 11 |
| Cereb6 | rs72754248 | 9 | 119 061 396 | A | G | 6.8 × 10−15 | 0.16 | 0.02 | A | 0.07 | 1.00 | PAPPA, ASTN2, ASTN2-AS1, ZNF407, ZADH2, TSHZ1, SMIM21 | 12 |
| CC4 | rs556819846 | 10 | 104 646 044 | T | TCCTCCCTC | 6.9 × 10−14 | -0.14 | 0.02 | T | 0.08 | 0.99 | TRIM8, ARL3, SFXN2, WBP1L, CYP17A1, BORCS7, AS3MT, CNNM2, NT5C2 | 13 |
| Cereb11 | rs4937515 | 11 | 130 268 147 | C | G | 1.0 × 10−11 | -0.07 | 0.01 | G | 0.41 | 0.99 | ST14, ZBTB44, ZBTB44-DT, ADAMTS8, ADAMTS15 | 14 |
| CC9 | rs79487293 | 12 | 65 905 126 | T | C | 5.7 × 10−12 | -0.08 | 0.01 | T | 0.33 | 1.00 | MSRB3, LOC105369187, RPSAP52 | 15 |
| CC4, DMN17, Cereb25 | rs12146713 | 12 | 106 476 805 | C | T | 2.1 × 10−11 | -0.11 | 0.02 | C | 0.09 | 1.00 | NUAK1, CKAP4, TCP11L2 | 16 |
(Continued)
Figure 3.
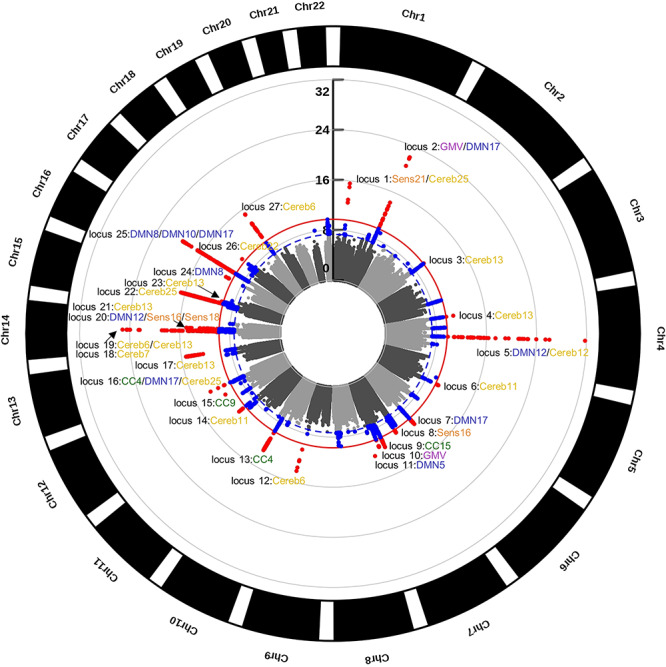
GWA analyses results. Circular Manhattan plot of all GWA analyses (n = 26). Blue dots are variants exceeding the nominal GWA significance threshold (blue dashed line; P < 5.0 × 10−8); red dots are variants exceeding the Bonferroni corrected threshold (solid red line; P < 1.96 × 10−10). Locus peaks are labeled with their respective SBM networks. Some loci were identical for multiple networks, whereas other loci had overlapping windows (particularly in chromosomes 14 and 16). The GWA analyses identified 27 unique loci across 18 SBM networks and GMV. Scale is in log10. CC, cognitive control; DMN, default mode; Sens, sensory; Cereb, cerebellar; GMV, gray matter volume; Chr, chromosome.
To better visualize each locus’ contribution to each SBM network, we plotted the minimum P-value within each locus for each brain phenotype (see Fig. 4 and Supplementary Table 2). While it was clear that there was overlap across all brain phenotypes (mostly mid-dark-shaded green cells), there were loci with more specific contributions to particular phenotypes including locus 3, locus 9, locus 21, and locus 23. Figure 4 also shows that loci significantly associated with a particular SBM network were not necessarily associated with GMV, indicating some distinction in their genetic influences.
Figure 4.
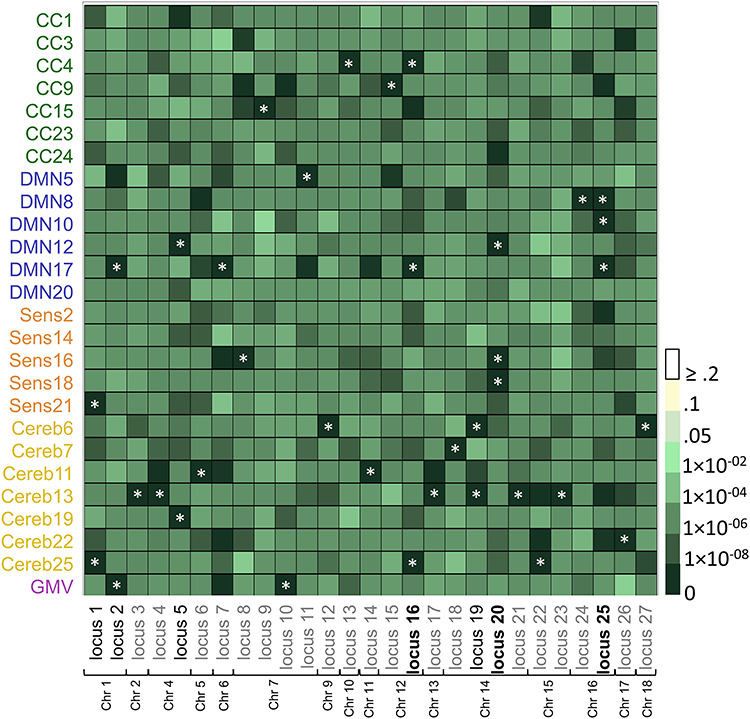
Overlap in genetic loci across SBM components. Plot shows the minimum P-value within each of the 27 unique GWA loci for all 25 SBM networks and GMV. X-axis shows SBM networks and GMV; y-axis shows significant GWA loci (Bonferroni corrected P < 1.96 × 10−10) ordered by chromosome. Asterisks indicate which phenotype the locus was associated with. Loci in black were significantly associated with two phenotypes; loci in black and bolded were significantly associated with three phenotypes. CC, cognitive control; DMN, default mode; Sens, sensory; Cereb, cerebellar; GMV, gray matter volume; Chr, chromosome.
Genetic Overlap Between SBM Networks and Psychiatric Disorders
LDSC analysis between each component and three psychiatric disorders including schizophrenia (SZ), bipolar disorder (BPD), and major depressive disorder (MDD) yielded seven nominally significant associations (see Fig. 5 and Supplementary Table 3): three for SZ, four for BP, and one for MDD. However, only the correlation between DMN10 and SZ (rg = 0.18 (0.06), P = 0.002) survived FDR correction.
Figure 5.
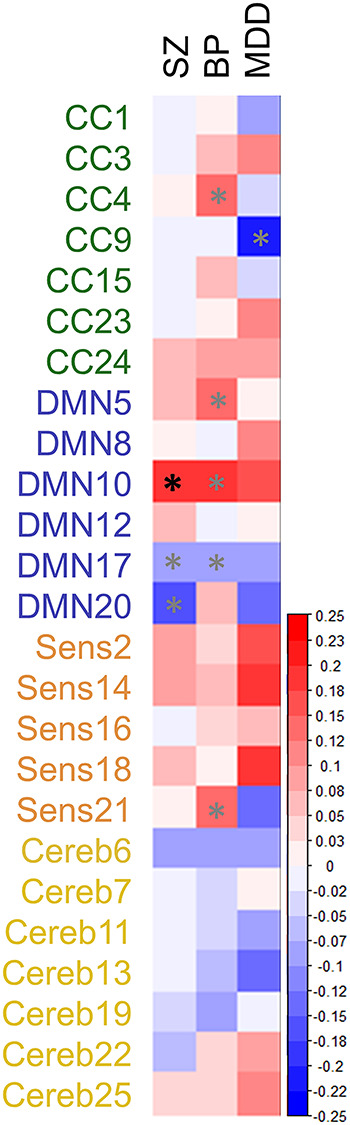
Results of the LDSC analysis. Plot shows genetic correlations between SBM networks and three psychiatric illnesses. Gray asterisks show significant association at the uncorrected threshold of P < 0.05. Black asterisk shows significant association after FDR correction. SZ, schizophrenia; BP, bipolar disorder; MDD, major depressive disorder; CC, cognitive control; DMN, default mode; Sens, sensory; Cereb, cerebellar.
Discussion
Using multivariate methods to derive novel neuroanatomic phenotypes in 20 306 individuals from the UK Biobank, we demonstrated that network-based phenotypes were heritable, shared genetic overlap with each other, and in a specific instance, were genetically associated with a psychiatric disorder (SZ) where neuroanatomical alterations are considered a hallmark of disease. Although there are many possible ways to parse the brain into neuroanatomical traits, we contend that network-based phenotypes can provide additional information about the genetic underpinnings of neuroanatomic variability and are supported by what we know about the brain from previous studies.
First, SNP-based heritability for all SBM networks was moderate and significant albeit variable from network to network. This is consistent with studies showing that heritability estimates vary across and within cortical, subcortical, and cerebellar regions (Thompson et al. 2001). Our estimates were also consistent with values obtained for regional volumes in GWA analyses from the UK Biobank (Elliott et al. 2018), which were likewise variable, with heritability estimates ranging between 0.009 and 0.57 and an average heritability of 0.28. However, regions in the UK Biobank analysis with relatively low heritability (h2snp estimates around 0.05), including parts of the inferior frontal cortex and supramarginal gyrus, were captured by some of our SBM networks, primarily in CC (4, 9, 23, and 24) and DMN networks (5, 17, and 25), where heritability was often above 0.25, indicating that genetics do indeed play a role in these regions.
Table 2.
Continued
| Phenotype | SNP ID | Chr | Position | Allele 1* | Allele 2 | P-value | β | se | Minor allele | MAF | INFO | Genes within 500 kb locus window | Locus No. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cereb13 | rs2699324 | 13 | 103 931 552 | G | A | 5.4 × 10−16 | -0.11 | 0.01 | G | 0.20 | 1.00 | SLC10A2, MIR548AS | 17 |
| Cereb7 | rs71446481 | 14 | 54 420 647 | ACC | A | 8.2 × 10−11 | 0.07 | 0.01 | ACC | 0.45 | 0.98 | MIR5580, BMP4 | 18 |
| Cereb6b | rs117332043 | 14 | 57 617 154 | T | C | 2.7 × 10−23 | 0.21 | 0.02 | T | 0.07 | 0.99 | OTX2-AS1, EXOC5, AP5M1, NAA30 | 19 |
| Cereb13b | rs76025319 | 14 | 57 606 103 | C | T | 7.2 × 10−26 | -0.21 | 0.02 | C | 0.07 | 1.00 | OTX2-AS1, EXOC5, AP5M1, NAA30 | 19 |
| DMN12c | rs74826997 | 14 | 59 628 609 | C | T | 1.3 × 10−11 | 0.11 | 0.02 | C | 0.12 | 1.00 | DAAM1 | 20 |
| Sens16c | rs4901904 | 14 | 59 624 317 | T | C | 9.4 × 10−12 | -0.11 | 0.02 | T | 0.12 | 0.99 | DAAM1 | 20 |
| Sens18c | 14:59631075 | 14 | 59 631 075 | G | GTTGT | 3.1 × 10−14 | -0.12 | 0.02 | G | 0.12 | 0.99 | DAAM1 | 20 |
| Cereb13 | rs535481763 | 14 | 69 278 573 | CT | C | 1.1 × 10−15 | 0.09 | 0.01 | CT | 0.46 | 0.94 | RAD51B, ZFP36L1, ACTN1, ACTN1-AS1, DCAF5 | 21 |
| Cereb25 | rs3742960 | 15 | 58 248 088 | T | C | 2.2 × 10−17 | -0.09 | 0.01 | C | 0.45 | 1.00 | MYZAP, GCOM1, POLR2M, ALDH1A2, AQP9 | 22 |
| Cereb13 | 15:67888314 | 15 | 67 888 314 | C | CT | 1.0 × 10−10 | -0.12 | 0.02 | CT | 0.09 | 0.93 | IQCH, IQCH-AS1, C15orf61, MAP2K5, SKOR1 | 23 |
| DMN8 | 16:51440331 | 16 | 51 440 331 | C | CA | 1.5 × 10−11 | 0.07 | 0.01 | CA | 0.44 | 0.99 | None | 24 |
| DMN8d | rs4843553 | 16 | 87 233 185 | C | A | 1.6 × 10−11 | 0.07 | 0.01 | A | 0.42 | 1.00 | LOC101928708, C16orf95, FBXO31, MAP1LC3B, ZCCHC14 | 25 |
| DMN10d | rs4843552 | 16 | 87 233 516 | A | G | 2.2 × 10−20 | 0.10 | 0.01 | G | 0.42 | 1.00 | LOC101928708, C16orf95, FBXO31, MAP1LC3B, ZCCHC14 | 25 |
| DMN17d | rs12920553 | 16 | 87 227 046 | T | G | 2.2 × 10−15 | -0.07 | 0.01 | G | 0.42 | 1.00 | LOC101928708, C16orf95, FBXO31, MAP1LC3B, ZCCHC14 | 25 |
| Cereb22 | rs78011262 | 17 | 43 837 917 | C | T | 5.6 × 10−11 | -0.07 | 0.01 | C | 0.30 | 0.97 | LRRC37A4P, ARL17A, LRRC37A3, LRRC37, LRRc37A, MAPK8IP1P2, CRHR1, LINC02210, MAPT-AS1, SPPL2C, MAPT, MAPT-IT1, STH | 26 |
| Cereb6 | rs4891262 | 18 | 73 017 380 | T | C | 7.4 × 10−16 | 0.09 | 0.01 | T | 0.35 | 1.00 | ZNF407, ZADH2, TSHZ1, SMIM21 | 27 |
Notes: Phenotypes with the same superscript letter had locus windows that overlapped significantly. SNPs without rs IDs are notated by the chromosome and base pair location. Chr, chromosome; CC, cognitive control; DMN, default mode; Sens, sensory; Cereb, cerebellum; GMV, gray matter volume.
*Reference allele.
As expected, there was some genetic overlap between networks, although this, like SNP-based heritability, was also variable with correlation coefficients ranging from −0.57 to 0.56. This wide range suggests that while some networks share over half their additive genetic influence, none show complete genetic overlap. Hierarchical clustering of genetic correlations also revealed an interesting pattern of genetic similarity across networks. Some clusters or genetic modules (GMs) contained networks from the same functional module. For example, GM4 included 5/7 of the cerebellar networks, consistent with known gene expression patterns which are more similar within the cerebellum than between the cerebellum and cortex (Hawrylycz et al. 2015). GM5 was composed of two visual-dominant networks, which likewise share high levels of gene co-expression (Anderson et al. 2018). In contrast, there were some GMs composed of networks from multiple functional modules (e.g., GM2 and GM6). Similar genetic contributions to networks with seemingly disparate functions could arise from similarity in functional connectivity profiles, which correlates with gene expression patterns (Krienen et al. 2016; Anderson et al. 2018). Correlation in gene expression is primarily observed across regions within the same network, although co-expression between networks is also apparent, especially for default mode and control networks, which composed the majority of our GM2. While our networks were constructed from anatomical images, they are not independent from spatially similar networks identified with functional connectivity analyses (Segall et al. 2012). Spatial proximity could also contribute to genetic overlap in that genetic contributions may be partially constrained by factors that are similar in neighboring locations, such as laminar organization and/or developmental origin (Zapala et al. 2005; Bernard et al. 2012; Chen et al. 2012). This may be true for CC1 and Cereb25 which include proximal regions that span the cerebellum and cortex.
Genetic overlap among networks was also reflected in the loci identified via GWA analyses. We detected seven loci significantly associated with more than one anatomical phenotype. There was also evidence that most loci impacted at least two phenotypes, as the statistical support for some associations was quite strong despite not exceeding our multiple comparison correction threshold (see dark green squares in Fig. 4). This one-to-many mapping, in addition to our genetic correlation results, suggests there are nontrivial pleiotropic effects on neuroanatomic variation. These shared genetic influences could arise from processes that have brain-wide effects, including gliogenesis, (Lim et al. 2000), astrocyte proliferation and branching (D'Alessandro et al. 1994; Gomes et al. 2003), and blood–brain barrier formation (Araya et al. 2008). In fact, some loci identified via our GWA analyses included genes that play a role in similar brain-wide events such as axon elongation and branching (NUAK1 and FXBO31) (Courchet et al. 2013; Vadhvani et al. 2013) and cellular senescence (NFKB1) (Bernal et al. 2014), although these findings will need to be replicated in an independent sample.
Although pleiotropic effects were evident, they did not seem to be completely accounted for by overlap with GMV. Only one locus was significant for both GMV and an SBM network, and many remaining loci were not strongly associated with GMV. Furthermore, most SBM networks did not show strong genetic correlations with GMV. Additionally, some loci were specific to a particular network (see lack of multiple dark green squares for loci 3, 9, 21, and 23 in Fig. 4), suggesting that network-specific genetic influences are also important. This balance between overlap and specificity is also reflective of genetic modeling of subcortical volumes, which show both common and unique genetic effects across structures (Rentería et al. 2014).
In addition to pleiotropic effects, SBM networks were also highly polygenic. Half of all phenotypes with significant genome-wide associations (9 of 18) were associated with more than one locus, and loci themselves were scattered throughout the genome both within and across traits. Additionally, we observed a linear relationship between chromosome length and proportion of heritability accounted for by each chromosome (see Supplementary Figure 2), similar to the polygenic architecture of other complex traits like height, BMI, and schizophrenia (Yang et al. 2013; Shi et al. 2016), where there appears to be small contributions from common SNPs across the genome. While present, the strength of the relationship between chromosome length and variance explained was variable across networks. Furthermore, there was a disproportionate amount of variance accounted for by chromosome 1 compared with others for DMN17 and Sens21, suggesting that there may be variants with larger effects on this chromosome for these networks. Indeed, we located genome-wide significant hits on chromosome 1 for both networks. It could be that using variance component and GWA analyses jointly could prioritize genomic locations for mapping at a finer scale.
We also found evidence of genetic overlap between one of our SBM networks and psychiatric disease. Alteration of brain structure is a hallmark of mood and psychotic disorders (Turetsky et al. 1995; Dotson et al. 2009), with genetic links between the two proposed (McIntosh et al. 2006; Lee et al. 2016). Indeed, reduced regional volumes are found in unaffected first-degree relatives of people with these disorders, although evidence in purely affective disorders is mixed (Campbell and MacQueen 2006; McDonald et al. 2006; Hajek et al. 2013). We found a significant genetic correlation between SZ and a DMN-related SBM network composed of the paracentral lobule and precuneus (DMN10). This association was positive meaning that the same genetic factors that contribute to disease status also contribute to larger volume in these regions. While volume reductions are more common in psychosis, studies primarily focus on frontal and temporal regions, whereas this component encompasses primarily posterior parts of the parietal lobe. This same network was also nominally significant for bipolar disorder but not MDD, supporting findings of greater genetic overlap between SZ and BPD than between SZ and MDD (Purcell et al. 2009; Lee et al. 2013; Bulik-Sullivan et al. 2015). However, this result should be interpreted with caution given that the bipolar association failed to reach the FDR significance threshold.
It should be noted that SNP-based heritability is a lower bound estimate, given that only the variance accounted for by common SNPs is estimated. This limitation extends to our genetic correlation analyses as well. It is possible that this shortcoming is reduced somewhat by the large number of imputed variants available (~9.7 million variants). We did not attempt to replicate our GWA analyses as the identification of individual SNPs was not the central focus of this work; it was, in principle, a way to look at more localized overlap among components. We did, however, replicate some SNP-level associations (rs13107325, rs72754248, and rs74826997) identified by the UK Biobank, which included approximately half of our current sample size. Yet, we refrain from making targeted claims about specific genes and limit our interpretation to genetic loci and patterns of pleiotropy and polygenicity within and across networks. We are also aware that the dimensionality of our ICA may impact our results, including our ability to capture subcortical networks. We were interested in large-scale brain networks that have been identified in other studies using a similar number of ICA components (Xu et al. 2009; Gupta et al. 2014; Abrol et al. 2017). Furthermore, large-scale networks would more closely approximate functional brain networks than individual regions identified with a higher-order ICA dimensionality and reduce the burden of multiple comparisons. Exploration of the genetic influences on such higher-order components, or how genetic influences change with changing ICA dimensionality, should be considered in future research. Finally, we acknowledge that volumetric SBM networks are only one of many phenotypes that could be used to probe the genetic effects on neuroanatomic variation, and other measures, such as surface area and cortical thickness (which are generated from surface-level data), should be examined in greater detail. However, a recent study from the UK Biobank showed that there is substantial overlap in variants contributing to surface area and cortical thickness (van der Meer et al. 2019). Additionally, although a surface-based strategy would undoubtedly contribute to understanding the genetic basis of neuroanatomy, our primary goal was to demonstrate that there are meaningful genetic contributions to volumetric SBM phenotypes, not only because of their potential to add to the information we know about the genetic underpinnings of neuroanatomy but also because these networks have been shown to be replicable and are linked to functional networks in addition to their growing use in neuroimaging (see Gupta et al. 2019 for a review).
We believe SBM phenotypes are well balanced between more commonly used aggregate phenotypes, which are likely an oversimplification of brain structure, and voxel-wise phenotypes (Hibar et al. 2011), which quickly become intractable given the required computational intensity and the need for multiple testing correction. Genetic influences on SBM networks also seem to reflect evidence of heterogeneity in heritability (Thompson et al. 2001) and gene expression (Hawrylycz et al. 2015) throughout the brain. Our study showed that this approach to phenotype generation can be successfully used to probe genetic influences on neuroanatomic variation and that some of these influences may or may not overlap with those discovered using traditional neuroanatomic phenotypes. Such an approach could enhance our ability to identify genetic factors that contribute to both brain structure and function, given the close relationship between SBM networks and networks identified by resting state and task-based MRI (Segall et al. 2012). This in turn may provide new avenues by which to find genetic overlap between neuroanatomic variation and other phenotypes that are commonly associated with it like cognitive ability and psychiatric diagnoses.
Funding
This work was supported by the National Institute of Mental Health (grant MH094524). The UK Biobank has received funding from the UK Medical Research Council, Wellcome Trust, Department of Health, British Heart Foundation, Diabetes UK, Northwest Regional Development Agency, the Scottish Government, and the Welsh Assembly Government.
Notes
The authors would like to acknowledge Amber Grant from Georgia State University for her assistance in generating Figure 1 and Supplementary Figure 3. The corresponding author can be contacted at the following address: Dr Amanda Rodrigue, 1 Autumn St., AU 108, Boston, MA 02215.
Conflict of Interest
None declared.
Supplementary Material
References
- Abrol A, Rashid B, Rachakonda S, Damaraju E, Calhoun VD. 2017. Schizophrenia shows disrupted links between brain volume and dynamic functional connectivity. Front Neurosci. 11:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander-Bloch A, Giedd JN, Bullmore E. 2013. Imaging structural co-variance between human brain regions. Nat Rev Neurosci. 14:322–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro-Almagro F, Jenkinson M, Bangerter NK, Andersson JL, Griffanti L, Douaud G, Sotiropoulos SN, Jbabdi S, Hernandez-Fernandez M, Vallee E. 2018. Image processing and quality control for the first 10 000 brain imaging datasets from UK biobank. NeuroImage. 166:400–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljondi R, Szoeke C, Steward C, Yates P, Desmond P. 2018. A decade of changes in brain volume and cognition. Brain Imaging Behav. 13:1–10. [DOI] [PubMed] [Google Scholar]
- Anderson KM, Krienen FM, Choi EY, Reinen JM, Yeo BTT, Holmes AJ. 2018. Gene expression links functional networks across cortex and striatum. Nat Commun. 9:1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya R, Kudo M, Kawano M, Ishii K, Hashikawa T, Iwasato T, Itohara S, Terasaki T, Oohira A, Mishina Y et al. 2008. BMP signaling through BMPRIA in astrocytes is essential for proper cerebral angiogenesis and formation of the blood-brain-barrier. Mol Cell Neurosci. 38:417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ. 2000. Voxel-based morphometry—the methods. NeuroImage. 11:805–821. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Royal Stat Soc Series B (Methodol). 57:289–300. [Google Scholar]
- Bergsland N, Horakova D, Dwyer MG, Uher T, Vaneckova M, Tyblova M, Seidl Z, Krasensky J, Havrdova E, Zivadinov R. 2018. Gray matter atrophy patterns in multiple sclerosis: a 10-year source-based morphometry study. NeuroImage Clin. 17:444–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal GM, Wahlstrom JS, Crawley CD, Cahill KE, Pytel P, Liang H, Kang S, Weichselbaum RR, Yamini B. 2014. Loss of Nfkb1 leads to early onset aging. Aging. 6:931–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard A, Lubbers Laura S, Tanis Keith Q, Luo R, Podtelezhnikov Alexei A, Finney Eva M, McWhorter Mollie ME, Serikawa K, Lemon T, Morgan R et al. 2012. Transcriptional architecture of the primate Neocortex. Neuron. 73:1083–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briley DA, Tucker-Drob EM. 2017. Comparing the developmental genetics of cognition and personality over the life span. J Pers. 85:51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R, ReproGen C, Psychiatric Genomics C, Genetic Consortium for Anorexia Nervosa of the Wellcome Trust Case Control C, Duncan L et al. 2015. An atlas of genetic correlations across human diseases and traits. Nat Genet. 47:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, Motyer A, Vukcevic D, Delaneau O, O’Connell J et al. 2017. Genome-wide genetic data on ~500 000 UK biobank participants. bioRxiv. 166298. [Google Scholar]
- Calhoun VD, Adali T, Pearlson GD, Pekar JJ. 2001. A method for making group inferences from functional MRI data using independent component analysis. Hum Brain Mapp. 14:140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, MacQueen G. 2006. An update on regional brain volume differences associated with mood disorders. Curr Opin Psychiatry. 19:25–33. [DOI] [PubMed] [Google Scholar]
- Chen C-H, Gutierrez ED, Thompson W, Panizzon MS, Jernigan TL, Eyler LT, Fennema-Notestine C, Jak AJ, Neale MC, Franz CE et al. 2012. Hierarchical genetic organization of human cortical surface area. Science (New York, NY). 335:1634–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chételat G, Fouquet M. 2013. Neuroimaging biomarkers for Alzheimer's disease in asymptomatic APOE4 carriers. Rev Neurol. 169:729–736. [DOI] [PubMed] [Google Scholar]
- Ciarochi JA, Calhoun VD, Lourens S, Long JD, Johnson HJ, Bockholt HJ, Liu J, Plis SM, Paulsen JS, Turner JA. 2016. Patterns of co-occurring gray matter concentration loss across the Huntington disease prodrome. Front Neurol. 7:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GP. 2015. A global reference for human genetic variation. Nature. 526:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium SWGotPG 2018. Genomic dissection of bipolar disorder and schizophrenia, including 28 subphenotypes. Cell. 173, 1705–1715. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium UK 2015. The UK10K project identifies rare variants in health and disease. Nature. 526:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courchet J, Lewis Tommy L, Lee S, Courchet V, Liou D-Y, Aizawa S, Polleux F. 2013. Terminal axon branching is regulated by the LKB1-NUAK1 kinase pathway via presynaptic mitochondrial capture. Cell. 153:1510–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alessandro JS, Yetz-Aldape J, Wang EA. 1994. Bone morphogenetic proteins induce differentiation in astrocyte lineage cells. Growth Factors. 11:53–69. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Ségonne F, Fischl B, Quinn BT, Dickerson BC, Blacker D, Buckner RL, Dale AM, Maguire RP, Hyman BT. 2006. An automated labeling system for subdividing the human cerebral cortex on MRI scans into gyral based regions of interest. NeuroImage. 31:968–980. [DOI] [PubMed] [Google Scholar]
- Dotson VM, Davatzikos C, Kraut MA, Resnick SM. 2009. Depressive symptoms and brain volumes in older adults: a longitudinal magnetic resonance imaging study. J Psychiat Neurosci. 34:367–375. [PMC free article] [PubMed] [Google Scholar]
- Elliott LT, Sharp K, Alfaro-Almagro F, Shi S, Miller KL, Douaud G, Marchini J, Smith SM. 2018. Genome-wide association studies of brain imaging phenotypes in UK biobank. Nature. 562:210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes WA, Mehler MF, Kessler JA. 2003. Transgenic overexpression of BMP4 increases astroglial and decreases oligodendroglial lineage commitment. Dev Biol. 255:164–177. [DOI] [PubMed] [Google Scholar]
- Gupta CN, Calhoun VD, Rachakonda S, Chen J, Patel V, Liu J, Segall J, Franke B, Zwiers MP, Arias-Vasquez A. 2014. Patterns of gray matter abnormalities in schizophrenia based on an international mega-analysis. Schizophr Bull. 41:1133–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta CN, Turner JA, Calhoun VD. 2018. Source-based morphometry: Data-driven multivariate analysis of structural brain imaging data. Brain Morphometry. New York, NY: Humana Press, p. 105–120. [Google Scholar]
- Gupta CN, Turner JA, Calhoun VD. 2019. Source-based morphometry: a decade of covarying structural brain patterns. Brain Struct Funct. 1–14. [DOI] [PubMed] [Google Scholar]
- Hajek T, Cullis J, Novak T, Kopecek M, Blagdon R, Propper L, Stopkova P, Duffy A, Hoschl C, Uher R et al. 2013. Brain structural signature of familial predisposition for bipolar disorder: replicable evidence for involvement of the right inferior frontal gyrus. Biol Psychiatry. 73:144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrisberger F, Spalek K, Smieskova R, Schmidt A, Coynel D, Milnik A, Fastenrath M, Freytag V, Gschwind L, Walter A et al. 2014. The association of the BDNF Val66Met polymorphism and the hippocampal volumes in healthy humans: a joint meta-analysis of published and new data. Neurosci Biobehav Rev. 42:267–278. [DOI] [PubMed] [Google Scholar]
- Hawrylycz M, Miller JA, Menon V, Feng D, Dolbeare T, Guillozet-Bongaarts AL, Jegga AG, Aronow BJ, Lee C-K, Bernard A. 2015. Canonical genetic signatures of the adult human brain. Nat Neurosci. 18:1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibar DP, Stein JL, Kohannim O, Jahanshad N, Saykin AJ, Shen L, Kim S, Pankratz N, Foroud T, Huentelman MJ. 2011. Voxelwise gene-wide association study (vGeneWAS): multivariate gene-based association testing in 731 elderly subjects. NeuroImage. 56:1875–1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes AJ, Lee PH, Hollinshead MO, Bakst L, Roffman JL, Smoller JW, Buckner RL. 2012. Individual differences in amygdala-medial prefrontal anatomy link negative affect, impaired social functioning, and polygenic depression risk. J Neurosci. 32:18087–18100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen AG, Mous SE, White T, Posthuma D, Polderman TJC. 2015. What twin studies tell us about the heritability of brain development, morphology, and function: a review. Neuropsychol Rev. 25:27–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkinson M, Beckmann CF, Behrens T, Woolrich MW, Smith SM. 2012. FSL. Neuroimage. 62:782–790. [DOI] [PubMed] [Google Scholar]
- Kašpárek T, Mareček R, Schwarz D, Přikryl R, Vaníček J, Mikl M, Češková E. 2010. Source-based morphometry of gray matter volume in men with first-episode schizophrenia. Hum Brain Mapp. 31:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knickmeyer RC, Wang J, Zhu H, Geng X, Woolson S, Hamer RM, Konneker T, Lin W, Styner M, Gilmore JH. 2013. Common variants in psychiatric risk genes predict brain structure at birth. Cereb Cortex. 24:1230–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krienen FM, Yeo BT, Ge T, Buckner RL, Sherwood CC. 2016. Transcriptional profiles of supragranular-enriched genes associate with corticocortical network architecture in the human brain. Proc Natl Acad Sci USA. 113:E469–E478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubera KM, Sambataro F, Vasic N, Wolf ND, Frasch K, Hirjak D, Thomann PA, Wolf RC. 2014. Source-based morphometry of gray matter volume in patients with schizophrenia who have persistent auditory verbal hallucinations. Prog Neuro-Psychopharmacol Biol Psychiatry. 50:102–109. [DOI] [PubMed] [Google Scholar]
- Lee PH, Baker JT, Holmes AJ, Jahanshad N, Ge T, Jung JY, Cruz Y, Manoach DS, Hibar DP, Faskowitz J et al. 2016. Partitioning heritability analysis reveals a shared genetic basis of brain anatomy and schizophrenia. Mol Psychiatry. 21:1680–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, Perlis RH, Mowry BJ, Thapar A, Goddard ME, Witte JS. 2013. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 45:984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Yang J, Goddard ME, Visscher PM, Wray NR. 2012. Estimation of pleiotropy between complex diseases using single-nucleotide polymorphism-derived genomic relationships and restricted maximum likelihood. Bioinformatics (Oxford, England). 28:2540–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim DA, Tramontin AD, Trevejo JM, Herrera DG, García-Verdugo JM, Alvarez-Buylla A. 2000. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron. 28:713–726. [DOI] [PubMed] [Google Scholar]
- Luo L, Xu L, Jung R, Pearlson G, Adali T, Calhoun VD. 2012. Constrained source-based morphometry identifies structural networks associated with default mode network. Brain Connect. 2:33–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machiela MJ, Chanock SJ. 2017. LDassoc: an online tool for interactively exploring genome-wide association study results and prioritizing variants for functional investigation. Bioinformatics. 34:887–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy S, Das S, Kretzschmar W, Delaneau O, Wood AR, Teumer A, Kang HM, Fuchsberger C, Danecek P, Sharp K. 2016. A reference panel of 64 976 haplotypes for genotype imputation. Nat Genet. 48:1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald C, Marshall N, Sham PC, Bullmore ET, Schulze K, Chapple B, Bramon E, Filbey F, Quraishi S, Walshe M. 2006. Regional brain morphometry in patients with schizophrenia or bipolar disorder and their unaffected relatives. Am J Psychiatr. 163:478–487. [DOI] [PubMed] [Google Scholar]
- McIntosh AM, Job DE, Moorhead WJ, Harrison LK, Whalley HC, Johnstone EC, Lawrie SM. 2006. Genetic liability to schizophrenia or bipolar disorder and its relationship to brain structure. Am J Med Genet B Neuropsychiatr Genet. 141B:76–83. [DOI] [PubMed] [Google Scholar]
- Muhlert N, Lawrence AD. 2015. Brain structure correlates of emotion-based rash impulsivity. NeuroImage. 115:138–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappaianni E, Siugzdaite R, Vettori S, Venuti P, Job R, Grecucci A. 2018. Three shades of grey: detecting brain abnormalities in children with autism using source-, voxel-and surface-based morphometry. Eur J Neurosci. 47:690–700. [DOI] [PubMed] [Google Scholar]
- Peakman TC, Elliott P. 2008. The UK biobank sample handling and storage validation studies. Int J Epidemiol. 37:i2–i6. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, De Bakker PI, Daly MJ. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, Wray NR, Stone JL, Visscher PM, O'donovan MC, Sullivan PF, Sklar P. 2009. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 460:748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rentería ME, Hansell NK, Strike LT, McMahon KL, Zubicaray GI, Hickie IB, Thompson PM, Martin NG, Medland SE, Wright MJ. 2014. Genetic architecture of subcortical brain regions: common and region-specific genetic contributions. Genes Brain Behav. 13:821–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, Byrne EM, Blackwood DH, Boomsma DI, Cichon S et al. 2013. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 18:497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics C 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 511:421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall J, Allen E, Jung R, Erhardt E, Arja S, Kiehl K, Calhoun V. 2012. Correspondence between structure and function in the human brain at rest. Front Neuroinform. 6:6–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Kichaev G, Pasaniuc B. 2016. Contrasting the genetic architecture of 30 complex traits from summary association data. Am J Hum Genet. 99:139–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strike LT, Couvy-Duchesne B, Hansell NK, Cuellar-Partida G, Medland SE, Wright MJ. 2015a. Genetics and brain morphology. Neuropsychol Rev. 25:63–96. [DOI] [PubMed] [Google Scholar]
- Strike LT, Couvy-Duchesne B, Hansell NK, Cuellar-Partida G, Medland SE, Wright MJ. 2015b. Genetics and brain morphology. Neuropsychol Rev. 25:63–96. [DOI] [PubMed] [Google Scholar]
- Sudlow C, Gallacher J, Allen N, Beral V, Burton P, Danesh J, Downey P, Elliott P, Green J, Landray M. 2015. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PM, Cannon TD, Narr KL, Van Erp T, Poutanen V-P, Huttunen M, Lönnqvist J, Standertskjöld-Nordenstam C-G, Kaprio J, Khaledy M. 2001. Genetic influences on brain structure. Nat Neurosci. 4:1253. [DOI] [PubMed] [Google Scholar]
- Toga AW, Thompson PM. 2005. Genetics of brain structure and intelligence. Annu Rev Neurosci. 28:1–23. [DOI] [PubMed] [Google Scholar]
- Turetsky B, Cowell PE, Gur RC, Grossman RI, Shtasel DL, Gur RE. 1995. Frontal and temporal lobe brain volumes in schizophrenia: relationship to symptoms and clinical subtype. Arch Gen Psychiatry. 52:1061–1070. [DOI] [PubMed] [Google Scholar]
- Vadhvani M, Schwedhelm-Domeyer N, Mukherjee C, Stegmüller J. 2013. The centrosomal E3 ubiquitin ligase FBXO31-SCF regulates neuronal morphogenesis and migration. PLoS One. 8:e57530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meer D, Frei O, Kaufmann T, Chen C-H, Thompson WK, O'Connell KS, Sanchez JM, Linden DE, Westlye LT, Dale AM. 2019. Quantifying the polygenic architecture of the human cerebral cortex: extensive genetic overlap between cortical thickness and surface area. bioRxiv. 868307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Groth KM, Pearlson G, Schretlen DJ, Calhoun VD. 2009. Source-based Morphometry: the use of independent component analysis to identify Gray matter differences with application to schizophrenia. Hum Brain Mapp. 30(3):711–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW et al. 2010. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 42:565–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. 2011. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 88:76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee T, Kim J, Cho M-C, Han B-G, Lee J-Y, Lee H-J, Cho S, Kim H. 2013. Ubiquitous polygenicity of human complex traits: genome-wide analysis of 49 traits in Koreans. PLoS Genet. 9:e1003355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zapala MA, Hovatta I, Ellison JA, Wodicka L, Del Rio JA, Tennant R, Tynan W, Broide RS, Helton R, Stoveken BS et al. 2005. Adult mouse brain gene expression patterns bear an embryologic imprint. Proc Natl Acad Sci USA. 102:10357–10362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.