

Abstract
Current methods for monitoring marine fish (including bony fishes and elasmobranchs) diversity mostly rely on trawling surveys, which are invasive, costly, and time‐consuming. Moreover, these methods are selective, targeting a subset of species at the time, and can be inaccessible to certain areas. Here, we used environmental DNA (eDNA), the DNA present in the water column as part of shed cells, tissues, or mucus, to provide comprehensive information about fish diversity in a large marine area. Further, eDNA results were compared to the fish diversity obtained in pelagic trawls. A total of 44 5 L‐water samples were collected onboard a wide‐scale oceanographic survey covering about 120,000 square kilometers in Northeast Atlantic Ocean. A short region of the 12S rRNA gene was amplified and sequenced through metabarcoding generating almost 3.5 million quality‐filtered reads. Trawl and eDNA samples resulted in the same most abundant species (European anchovy, European pilchard, Atlantic mackerel, and blue whiting), but eDNA metabarcoding resulted in more detected bony fish and elasmobranch species (116) than trawling (16). Although an overall correlation between fishes biomass and number of reads was observed, some species deviated from the common trend, which could be explained by inherent biases of each of the methods. Species distribution patterns inferred from eDNA metabarcoding data coincided with current ecological knowledge of the species, suggesting that eDNA has the potential to draw sound ecological conclusions that can contribute to fish surveillance programs. Our results support eDNA metabarcoding for broad‐scale marine fish diversity monitoring in the context of Directives such as the Common Fisheries Policy or the Marine Strategy Framework Directive.
Keywords: Actinopterygii, Elasmobranchii, environmental DNA, marine fish surveys, metabarcoding
eDNA samples provide information on fish diversity in a broad‐scale marine area, detecting almost ten times more fish species compared with pelagic trawling, including some considered elusive or difficult to capture with traditional fishing methods. The potential of eDNA is particularly relevant in a context of global change, where establishing efficient management actions based on numerous, continuous, and accurate biodiversity assessments is paramount.
![]()
1. INTRODUCTION
Monitoring of marine biodiversity provides a baseline for policy implementation toward a sustainable use of the marine environment and its resources. Among the traditional methods for surveying marine fauna, trawling has been widely used, as identification and quantification of large volumes of organisms are considered a reliable method for monitoring bony fishes and elasmobranchs (hereafter fishes) and other marine animal populations (ICES, 2015; Massé, Uriarte, Angélico, & Carrera, 2018). Fish surveys using trawls are conditioned by the gear's own characteristics (e.g., mesh size, area of opening) and deployment parameters (e.g., towing speed, depth, and diel variation) (Heino et al., 2011). Consequently, besides being invasive and time‐consuming, fish trawling in pelagic environments can be largely selective affecting diversity estimates and knowledge of species composition (Fraser, Greenstreet, & Piet, 2007; ICES, 2004). For instance, due to their large body size, fast swimming speed, and in some cases, scarcity, many elasmobranch species are not thoroughly surveyed (Rago, 2004). Therefore, alternative methods are needed, and advances in DNA sequencing and bioinformatics have opened new avenues to assess marine biodiversity in a noninvasive manner (Danovaro et al., 2016; Rees, Maddison, Middleditch, Patmore, & Gough, 2014).
In particular, the analysis of environmental DNA (eDNA), that is, the genetic material shed and excreted by organisms to the environment, to characterize the biological communities present in an environment (Taberlet, Coissac, Hajibabaei, & Rieseberg, 2012) is gaining increasing attention for monitoring aquatic environments (Thomsen & Willerslev, 2015). Community composition can be inferred from eDNA samples through metabarcoding, whereby the eDNA is collected from the water column through filtering, selectively amplified through PCR using primers targeting a given barcode from a particular taxonomic group and sequenced (Taberlet, Coissac, Pompanon, Brochmann, & Willerslev, 2012). The resulting sequences are then compared against a reference database to perform biodiversity inventories (Deiner, Bik, & Mächler, 2017). Besides the inherent biases of metabarcoding (Aylagas, Borja, Irigoien, & Rodríguez‐Ezpeleta, 2016), the use of eDNA adds additional biases due to the complex ecology of this molecule (Barnes & Turner, 2016) that might interfere with its potential use for biodiversity assessment. Thus, additional research is required to better understand the utility of eDNA for fish monitoring. Most studies using eDNA metabarcoding for monitoring fish communities are based on freshwater environments and have shown that eDNA metabarcoding provides overall estimates that are equivalent or superior to traditional methods such as visual surveys, trawling, or electrofishing (Hänfling et al., 2016; Minamoto, Yamanaka, Takahara, Honjo, & Zi, 2012; Pont et al., 2018).
As opposed to freshwater systems, the marine environment has in general a larger water volume to fish biomass ratio and is influenced by currents, implying that the eDNA is less concentrated and disperses quicker (Hansen, Bekkevold, Clausen, & Nielsen, 2018). This, coupled with a higher sympatric marine fish diversity, suggests that monitoring fish diversity through eDNA sampling could be particularly challenging in the marine environment. Indeed, only a handful of studies have applied eDNA metabarcoding for monitoring fish in natural marine environments (e.g., O’Donnell et al., 2017; Stat et al., 2017). Among them, only a few have compared eDNA and other traditional surveying methods and are based on a very small area of a few square kilometers either in ports (Jeunen et al., 2019; Sigsgaard et al., 2017; Thomsen et al., 2012) or in coastal areas (Andruszkiewicz et al., 2017; DiBattista et al., 2017; Yamamoto et al., 2017) or have performed comparisons at family level taxonomic assignments (Thomsen et al., 2016). Thus, although these studies envision eDNA metabarcoding as a promising method for noninvasive, faster, more efficient, and reliable marine surveys, this needs still to be tested in the context of a fishery survey covering a broad marine area.
The Bay of Biscay is a biogeographical area in the North Atlantic Region covering more than 220,000 km2, at which the main economic activities include commercial fishing. Large populations of species such as the European anchovy Engraulis encrasicolus, the European pilchard Sardina pilchardus, the European hake Merluccius merluccius, the Atlantic Mackerel Scomber scombrus, and the Atlantic horse mackerel Trachurus trachurus are dominant in the area (ICES, 2018). Fish diversity in the Bay of Biscay has been accounted using mainly observational methods, fish trawling, and acoustic surveys; thus, there is scope for incorporating and assessing the performance of eDNA‐based surveys. This paper aims to test the potential of eDNA metabarcoding to assess the fish community composition in a large marine area, such as the Bay of Biscay. For that aim, we have compared eDNA metabarcoding‐based biodiversity estimates with those derived from fishing trawls catches and have related eDNA metabarcoding‐based estimates with the known spatial distribution and ecological patterns of the species in the area.
2. METHODS
2.1. Sample collection
Fish and elasmobranchs catches and water samples were collected during the BIOMAN 2017 survey (Santos, Ibaibarriaga, Louzao, Korta, & Uriarte, 2018) between May 5 and May 29, 2017, covering the area of about 120,000 km2 between the French continental shelf and the Spanish shelf (Figure 1) on board the Emma Bardán and Ramón Margalef research vessels. Fish catches were obtained on board the R/V Emma Bardán pelagic trawler. The trawl had an 8 mm mesh size cod end, and towing time and speed were 40 min and 4 knots, respectively. A total of 44 stations were used for trawling. Although station depths varied between 26 and 3,000 m, the maximum fishing depth was 156 m. Onboard, fish were morphologically identified to species level or, when doubt, to the smallest taxonomic rank (e.g., family or genus). Biomass estimates were standardized as Kg caught per taxa and per station. In 44 additional stations (Figure 1), water samples were collected on board the R/V Ramón Margalef research vessel using the continuous circuit intake of the ship at 4.4 m depth, transferred to 5‐L plastic bottles and filtered through Sterivex 0.45 µm pore size enclosed filters (Millipore) with a peristaltic pump, using a 6 μm mesh size net in the incoming tube to avoid clogging. All material used for filtering, including tubes, net, and bottles were decontaminated by rinsing them once with 10% bleach solution, three times with Milli‐Q water and three times with the sampling water to be filtered. Filters were kept at −20°C until further processing.
FIGURE 1.
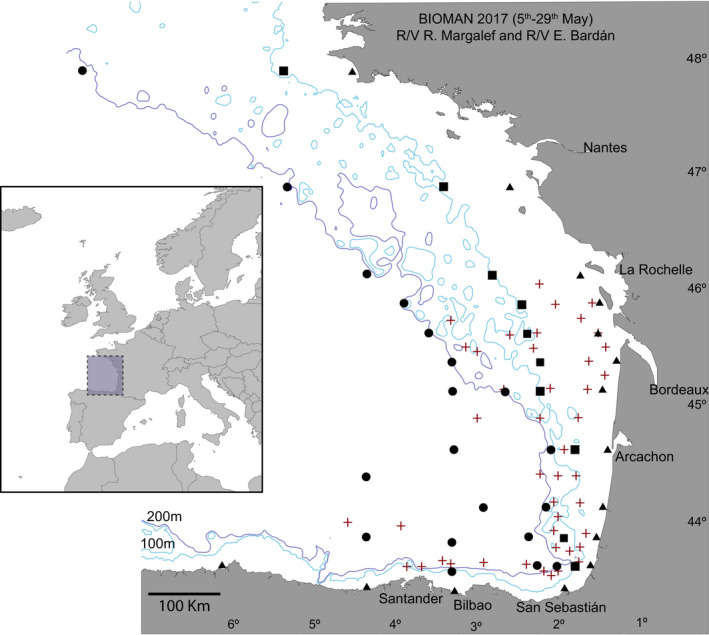
Study area and sampling sites for the BIOMAN 2017 survey in the Bay of Biscay. Triangles represent eDNA sampling sites where station depth was <90 m, squares, eDNA sampling sites with depths between 90 and 127 m, and circles, eDNA sampling sites with >127 m depths. Crosses are located where pelagic fishing trawls were deployed. 100 m and 200 m isobaths are shown
2.2. DNA extraction and amplicon library preparation
DNA extractions were performed in a dedicated pre‐PCR laboratory using the DNeasy® blood and tissue kit (Qiagen) following the modified protocol for DNA extraction from Sterivex filters without preservation buffer by Spens, Evans, and Halfmaerten (2017). DNA concentration was measured with the Quant‐iT dsDNA HS assay kit using a Qubit® 2.0 Fluorometer (Life Technologies, California, USA). DNA from all 44 samples was amplified with the teleo_F/telo_R primer pair (hereafter “teleo”), targeting a region (~60 bp) of the mitochondrial 12S rRNA gene, combined with the human blocking primer teleo_blk (Valentini et al., 2016). PCR mixtures were prepared under the hood in the pre‐PCR laboratory using dedicated micropipettes and disposable plastic ware that were previously decontaminated under the UV light, and all postamplification steps were carried out in the post‐PCR laboratory. Three replicate PCR amplifications were done per sample in a final volume of 20 µl including 10 µl of 2X Phusion Master Mix (Thermo Scientific, Massachusetts, USA), 0.4 µl of each amplification primer (final concentration of 0.2 µM), 4 µl of teleo_blk (final concentration of 2 µM), 3.2 µl of Milli‐Q water, and 2 µl of 10 ng/µl template DNA. Samples from 4 stations were also amplified (a) using the same procedure but without the blocking primer, and (b) using the mlCOIintF/dgHCO2198 primer pair (hereafter “mlCOI”), targeting a region (⁓310 bp) of the COI gene (Leray et al., 2013; Meyer, 2003). The thermocycling profile for PCR amplification included 3 min at 98°C; 40 or 35 cycles (for “teleo” and “mlCOI” as indicated in Valentini et al. (2016) and Leray et al. (2013), respectively) of 10 s at 98°C, 30 s at 55, or 46°C (for “teleo” and “mlCOI,” respectively) and 45 s at 72°C, and finally, 10 min at 72°C. Replicate PCR products were combined and purified using AMPure XP beads (Beckman Coulter, California, USA) following manufacturer's instructions and used as templates for the generation of 12 × 8 dual‐indexed amplicons in the second PCR following the “16S Metagenomic Sequence Library Preparation” protocol (Illumina, California, USA) using the Nextera XT Index Kit (Illumina, California, USA). PCR negative controls resulted in no visible amplification in agarose gels. Multiplexed PCR products were purified using the AMPure XP beads, quantified using Quant‐iT dsDNA HS assay kit using a Qubit® 2.0 Fluorometer (Life Technologies, California, USA), and adjusted to 4 nM. Five microlitre of each sample were pooled, checked for size and concentration using the Agilent 2100 bioanalyzer (Agilent Technologies, California, USA), sequenced using the 2 × 300 paired end protocol on the Illumina MiSeq platform (Illumina, California, USA), and demultiplexed based on their barcode sequences.
2.3. Reference database
Two reference databases were created for the “teleo” barcode. A first “global” database included all Chordata 12S rRNA and complete mitochondrial genome sequences available from GenBank (accessed in February 2018). By performing an all‐against‐all BLAST (Altschul, Gish, Miller, Myers, & Lipman, 1990), potential sources of contamination or erroneous taxonomic assignments were removed such as human contaminations (e.g., nonhuman labeled sequences that matched at 100% identity with the Homo sapiens 12S rRNA sequence) or cross‐contaminated sequences (e.g., sequences arising from the same study that, even when belonging to different genus, were 100% identical). All sequences were trimmed to the “teleo” region. Taxonomy for the GenBank sequences was retrieved using E‐utilities (Sayers, 2008) and modified to match that of the World Register of Marine Species: WoRMS (Horton, Kroh, & Ahyong, 2018), forcing for seven taxonomic levels, that is, Phylum, Subphylum, Class, Order, Family, Genus, and Species. This “global” reference database contains 10,284 “teleo” region sequences. For the second database, only sequences from target species were retrieved so that more exhaustive error checking was possible. The list of the 1,858 fish species expected in the Northeast Atlantic and Mediterranean areas was compiled from FishBase (http://www.fishbase.org), and their corresponding scientific names and sequences were obtained from NCBI (https://www.ncbi.nlm.nih.gov). For the retrieved records, only those covering the “teleo” region were selected and aligned. A phylogenetic tree was built with RAxML (Stamatakis, 2014) using the GTR‐CAT model and visualized with iTOl (Letunic & Bork, 2016). The tree was visually inspected, and the records corresponding to misplaced species were removed from the database. This “local” reference database contains “teleo” region sequences of 612 species. For the “mlCOI” barcode, the reference database consisted in the COI sequences and their corresponding taxonomy obtained from the BOLD (Ratnasingham & Hebert, 2007) database.
2.4. Read preprocessing, clustering, and taxonomic assignment
Overall quality of raw demultiplexed reads was verified with FASTQC (Andrews, 2010). Forward and reverse primers were removed with cutadapt (Martin, 2011) allowing a maximum error rate of 20%, discarding read pairs that do not contain the two primer sequences and retaining only those reads longer than 30 nucleotides. Paired reads were merged using pear (Zhang, Kobert, Flouri, & Stamatakis, 2014) with a minimum overlap of 20 nucleotides. Pairs with average quality lower than 25 Phred score were removed using Trimmomatic (Bolger, Lohse, & Usadel, 2014). mothur (Schloss et al., 2009) was used to remove reads (a) not covering the target region, (b) shorter than 40 or 313 nucleotides, for “teleo” and “mlCOI,” respectively, (c) containing ambiguous positions, and (d) being potential chimeras, which were detected based on the UCHIME algorithm (Edgar, Haas, Clemente, Quince, & Knight, 2011). Reads were clustered into OTUs using vsearch (Rognes, Flouri, Nichols, Quince, & Mahé, 2016) at 97% similarity threshold or using Swarm (Mahé, Rognes, Quince, de Vargas, & Dunthorn, 2014) with a d value of 1. In both cases, the LULU postclustering algorithm (Frøslev et al., 2017) was applied with a minimum threshold of sequence similarity for considering any OTU as an error of 97%. Taxonomic assignment of unique reads and of representative sequences for each OTU was performed using the naïve Bayesian classifier method (Wang, Garrity, Tiedje, & Cole, 2007) implemented in mothur using the 12S rRNA and COI databases described above. Reads with the same taxonomic assignment were grouped into phylotypes.
2.5. Biodiversity analyses
Analyses were performed in R v3.6.1 with the packages Phyloseq v1.22.3 (McMurdie & Holmes, 2013) and Vegan v2.5‐6 (Oksanen, Blanchet, & Friendly, 2019). Sampling stations were classified into three categories considering their depth (see Map in Figure 1) and grouped so that samples around the 100 isobath are grouped together: shallow stations where maximum station depth was <90 m, medium stations, when depth ranged between 90 and 127 m, and deep stations where depth was >127 m. To assess differences in fish diversity across categories (i.e., according to shallow, medium, and deep stations), we calculated the Bray–Curtis dissimilarity index for relative abundance of species with the function ordinate using only phylotypes with more than 10 reads. These distances were then ordinated using a nonmetric Multidimensional Scaling (NMDS) as implemented in Phyloseq and differences between stations were tested with PERMANOVA (1,000 permutations) using the function adonis within the R package Vegan previous testing for homogeneity of variance using the function betadisper. A linear model was used on species with more than 1,000 reads, to test for the effect of the abundance of reads (previously standardized according to the overall number of reads and stations per zone), and the distance from the coast. An overall correlation between the log‐transformed values (to deal with high variation on the relative scale) of the number of reads obtained and the biomass caught per species was explored with the Pearson correlation coefficient, using a t test to establish whether the correlation coefficient is significantly different from zero, as implemented in R package Stats v0.1.0. For an even geographic distribution between water and fish sampling sites, a total of nine water sampling sites north La Rochelle were removed for the comparison analyses. In addition, in order to compare eDNA and trawling‐based estimates at a smaller scale, we created groups of stations so that this comparison was possible. For that aim, we combined the data from all eDNA and trawling stations within <20 nautical miles of each eDNA station in what we call mega‐stations. A total of 30 mega‐stations resulted. A Mantel test as implemented in the R package ade4 v1.7‐13 (Dray & Dufour, 2007) was used to explore correlation between the mega‐station geographic and Bray–Curtis distance matrices of. The bias‐corrected Chao II species richness was estimated as in Olds et al. (2016). The list of species commonly reported from the Bay of Biscay was obtained mainly from (a) Basterretxea, Oyarzabal, and Artetxe (2012), (b) the AZTI’s database on fish bottom trawling discards in the area gathered according to EU regulation 2017/1004 of 17 May 2017, (c) the data obtained from fish pelagic trawling during BIOMAN surveys from 2003 until 2019, (d) the ICES database for International Bottom Trawling Surveys available from www.ices.dk, and (e) the 2017 Pélagiques Gascogne (PELGAS) integrated survey (Mathieu, Laurence, & Patrick, 2019).
3. RESULTS
3.1. Data quality and overall taxonomic composition
We obtained a total of 4,640,913 raw “teleo” reads from which 3,366,264 (72%) were retained after quality check for downstream analyses. The average number of “teleo” reads per sample was 70,131 (Table 1). Using the “global” database, 99.88% of the reads were classified as Actinopterygii or Elasmobranchii. The remaining were classified as mammals (40.16%) and birds (9.60%), with half of the reads (50.24%) not classified into Class level. Only 14 reads in eight samples were specifically assigned to H. sapiens. From these, two samples did not include the specific blocking primer used, suggesting that samples held very little contamination from external sources. Using the “local” database, 99.98% of the reads were classified either as Actinopterygii or Elasmobranchii and, depending on the clustering method used, the number of taxa recovered varied. swarm clustering yielded 90 OTUs identified at the species level (including 95.5% of the reads) and vsearch, 109 (including 95% of the reads), whereas not clustering reads into OTUs, but using phylotypes, resulted in 116 Actinopterygii and Elasmobranchii species (including 95% of the reads) identified. Further analyses were based on phylotypes assigned to the species level (Table 2) as no additional information is provided by using OTU clustered reads. From the 116 identified species, 50 included more than 10 reads.
TABLE 1.
Station depth, category, and number of reads obtained per sample after sequencing, removing primers, pair‐assembling, quality filtering, primer mapping, and chimera removal for the teleo region
| Sample | Station depth (m) | Category | Raw | Retained after primer checking | Retained after merging | Retained after quality filtering | Retained after mapping to teleo region | Retained after chimera removal | % of retained reads for analysis |
|---|---|---|---|---|---|---|---|---|---|
| Sample_01 | 27 | Shallow | 127,549 | 100,839 | 99,036 | 99,036 | 95,240 | 95,240 | 74.67 |
| Sample_02 | 1,315 | Deep | 99,724 | 96,995 | 90,080 | 90,080 | 89,206 | 89,206 | 89.45 |
| Sample_03 | 764 | Deep | 67,867 | 49,789 | 39,229 | 39,229 | 28,896 | 28,896 | 42.58 |
| Sample_04 | 46 | Shallow | 93,699 | 89,918 | 85,894 | 85,894 | 83,153 | 83,153 | 88.74 |
| Sample_05 | 43 | Shallow | 157,845 | 150,348 | 145,987 | 145,987 | 100,388 | 100,387 | 63.60 |
| Sample_06 | 180 | Deep | 120,961 | 116,014 | 103,982 | 103,982 | 101,419 | 101,418 | 83.84 |
| Sample_07 | 508 | Deep | 55,396 | 36,121 | 33,605 | 33,605 | 16,062 | 16,061 | 28.99 |
| Sample_08 | 1,373 | Deep | 104,158 | 77,717 | 71,716 | 71,716 | 65,091 | 65,091 | 62.49 |
| Sample_13 | 91 | Medium | 138,472 | 134,943 | 122,420 | 122,420 | 117,565 | 117,562 | 84.90 |
| Sample_14 | 735 | Deep | 66,247 | 50,282 | 21,624 | 21,624 | 18,813 | 18,813 | 28.40 |
| Sample_15 | 639 | Deep | 98,224 | 96,676 | 91,079 | 91,079 | 89,581 | 89,581 | 91.20 |
| Sample_16 | 25 | Shallow | 94,195 | 92,482 | 92,074 | 92,074 | 87,100 | 87,100 | 92.47 |
| Sample_17 | 741 | Deep | 60,308 | 38,492 | 24,125 | 24,125 | 20,355 | 20,355 | 33.75 |
| Sample_18 | 127 | Medium | 101,688 | 99,550 | 98,456 | 98,456 | 97,918 | 97,918 | 96.29 |
| Sample_19 | 38 | Shallow | 119,881 | 113,505 | 104,320 | 104,320 | 99,181 | 99,181 | 82.73 |
| Sample_20 | 1,285 | Deep | 111,757 | 107,998 | 103,660 | 103,660 | 96,993 | 96,993 | 86.79 |
| Sample_21 | 300 | Deep | 134,490 | 132,496 | 130,290 | 130,290 | 121,976 | 121,976 | 90.70 |
| Sample_22 | 33 | Shallow | 88,044 | 78,156 | 50,143 | 50,143 | 43,798 | 43,798 | 49.75 |
| Sample_23 | 968 | Deep | 52,240 | 39,687 | 16,584 | 16,584 | 12,090 | 12,090 | 23.14 |
| Sample_24 | 169 | Deep | 104,423 | 97,858 | 77,320 | 77,320 | 65,788 | 65,788 | 63.00 |
| Sample_25 | 23 | Shallow | 89,199 | 79,999 | 68,124 | 68,124 | 59,414 | 59,414 | 66.61 |
| Sample_26 | 132 | Deep | 110,206 | 106,817 | 106,547 | 106,547 | 105,436 | 105,436 | 95.67 |
| Sample_27 | 1,003 | Deep | 99,856 | 94,244 | 75,172 | 75,172 | 69,577 | 69,577 | 69.68 |
| Sample_28 | 112 | Medium | 100,002 | 98,722 | 97,656 | 97,656 | 97,462 | 97,462 | 97.46 |
| Sample_29 | 38 | Shallow | 87,452 | 73,824 | 61,161 | 61,161 | 51,590 | 51,590 | 58.99 |
| Sample_30 | 24 | Shallow | 155,459 | 153,556 | 153,517 | 153,517 | 152,877 | 152,877 | 98.34 |
| Sample_31 | 100 | Medium | 91,723 | 77,592 | 65,748 | 65,748 | 57,283 | 57,283 | 62.45 |
| Sample_32 | 185 | Deep | 76,396 | 64,566 | 52,863 | 52,863 | 35,175 | 35,175 | 46.04 |
| Sample_33 | 480 | Deep | 81,771 | 74,171 | 59,470 | 59,470 | 54,788 | 54,788 | 67.00 |
| Sample_34 | 104 | Medium | 70,037 | 56,967 | 54,193 | 54,193 | 45,712 | 45,712 | 65.27 |
| Sample_35 | 28 | Shallow | 124,438 | 122,670 | 122,076 | 122,076 | 113,812 | 113,812 | 91.46 |
| Sample_36 | 25 | Shallow | 108,071 | 107,183 | 107,166 | 107,166 | 106,898 | 106,898 | 98.91 |
| Sample_37 | 96 | Medium | 138,090 | 134,214 | 132,943 | 132,943 | 131,644 | 131,644 | 95.33 |
| Sample_38 | 590 | Deep | 89,791 | 77,675 | 72,468 | 72,468 | 59,562 | 59,562 | 66.33 |
| Sample_39 | 1,010 | Deep | 38,021 | 26,356 | 21,918 | 21,918 | 16,839 | 16,839 | 44.29 |
| Sample_40 | 108 | Medium | 79,924 | 74,147 | 67,941 | 67,941 | 64,152 | 64,152 | 80.27 |
| Sample_41 | 26 | Shallow | 119,488 | 117,958 | 117,881 | 117,881 | 116,414 | 116,414 | 97.43 |
| Sample_42 | 30 | Shallow | 135,958 | 133,840 | 46,726 | 46,726 | 46,640 | 46,640 | 34.30 |
| Sample_43 | 104 | Medium | 115,584 | 109,058 | 76,209 | 76,209 | 71,558 | 71,558 | 61.91 |
| Sample_44 | 185 | Deep | 25,519 | 24,976 | 8,064 | 8,064 | 7,755 | 7,755 | 30.39 |
| Sample_45 | 33 | Shallow | 115,109 | 113,582 | 99,454 | 99,454 | 93,219 | 93,219 | 80.98 |
| Sample_46 | 90 | Medium | 121,100 | 119,555 | 119,036 | 119,036 | 106,640 | 106,636 | 88.06 |
| Sample_47 | 675 | Deep | 68,697 | 55,896 | 19,019 | 19,019 | 6,716 | 6,716 | 9.78 |
| Sample_48 | 110 | Medium | 94,499 | 91,697 | 90,715 | 90,715 | 89,305 | 89,305 | 94.50 |
| Sample_01NOBP | 27 | Shallow | 115,003 | 97,454 | 93,987 | 93,987 | 82,749 | 82,748 | 71.95 |
| Sample_27NOBP | 1,003 | Deep | 88,793 | 78,114 | 51,755 | 51,755 | 43,902 | 43,902 | 49.44 |
| Sample_32NOBP | 185 | Deep | 57,276 | 38,142 | 29,015 | 29,015 | 26,686 | 26,686 | 46.59 |
| Sample_47NOBP | 675 | Deep | 46,283 | 35,228 | 7,304 | 7,304 | 1857 | 1857 | 4.01 |
| TOTAL | 4,333,558 | 3,989,131 | 3,497,691 | 3,497,691 | 3,211,081 | 3,211,071 | |||
| AVERAGE_all | 98,489.95 | 90,662.07 | 79,492.98 | 79,492.98 | 72,979.11 | 72,978.89 | 69.52 |
TABLE 2.
Number of reads, relative abundance, and taxonomic information recovered from eDNA by the 12S rRNA mitochondrial marker in the Bay of Biscay during the BIOMAN 2017 survey
| Number of reads | Relative abundance (%) | Class | Family | Species |
|---|---|---|---|---|
| 1,791,393 | 51.67 | Actinopterygii | Engraulidae | Engraulis encrasicolus |
| 959,248 | 27.67 | Actinopterygii | Clupeidae | Sardina pilchardus |
| 172,116 | 4.96 | Actinopterygii | Scombridae | Scomber scombrus |
| 119,672 | 3.45 | Actinopterygii | unclassified | unclassified |
| 81,658 | 2.36 | Actinopterygii | Gadidae | Micromesistius poutassou |
| 52,853 | 1.52 | Actinopterygii | Sparidae | Diplodus sargus |
| 41,467 | 1.20 | Actinopterygii | Sparidae | Pagellus acarne |
| 29,792 | 0.86 | Actinopterygii | Molidae | Mola mola |
| 25,536 | 0.74 | Actinopterygii | Moronidae | Dicentrarchus labrax |
| 22,982 | 0.66 | Actinopterygii | Lophiidae | Lophius piscatorius |
| 17,875 | 0.52 | Actinopterygii | Mugilidae | Chelon ramada |
| 17,307 | 0.50 | Actinopterygii | Scombridae | unclassified |
| 16,971 | 0.49 | Actinopterygii | unclassified | unclassified |
| 16,859 | 0.49 | Actinopterygii | Ammodytidae | Ammodytes dubius |
| 14,161 | 0.41 | Actinopterygii | Gobiidae | Gobius niger |
| 11,677 | 0.34 | Actinopterygii | Labridae | Ctenolabrus rupestris |
| 10,024 | 0.29 | Actinopterygii | Gobiidae | unclassified |
| 8,912 | 0.26 | Actinopterygii | Argentinidae | Argentina silus |
| 7,331 | 0.21 | Elasmobranchii | Somniosidae | Somniosus microcephalus |
| 7,158 | 0.21 | Actinopterygii | Gobiidae | Buenia affinis |
| 4,464 | 0.13 | Actinopterygii | Scombridae | Scomber colias |
| 4,456 | 0.13 | Actinopterygii | Merlucciidae | Merluccius merluccius |
| 3,577 | 0.10 | Actinopterygii | Clupeidae | Alosa fallax |
| 3,128 | 0.09 | Actinopterygii | Mugilidae | Chelon aurata |
| 2,527 | 0.07 | Actinopterygii | Sparidae | Pagellus bogaraveo |
| 2078 | 0.06 | Actinopterygii | Labridae | Labrus merula |
| 2075 | 0.06 | Actinopterygii | Cyprinidae | unclassified |
| 1921 | 0.06 | Actinopterygii | Alepocephalidae | Xenodermichthys copei |
| 1,284 | 0.04 | Elasmobranchii | Carcharhinidae | Prionace glauca |
| 1,284 | 0.04 | Actinopterygii | Cyprinidae | Rutilus rutilus |
| 1,249 | 0.04 | Actinopterygii | Labridae | Coris julis |
| 1,189 | 0.03 | Actinopterygii | Myctophidae | unclassified |
| 1,096 | 0.03 | Actinopterygii | Sparidae | unclassified |
| 996 | 0.03 | Actinopterygii | Soleidae | Microchirus azevia |
| 989 | 0.03 | Actinopterygii | Bathylagidae | Bathylagus euryops |
| 986 | 0.03 | Actinopterygii | Cyprinidae | Blicca bjoerkna |
| 971 | 0.03 | Actinopterygii | Scombridae | Katsuwonus pelamis |
| 806 | 0.02 | Actinopterygii | Clupeidae | unclassified |
| 695 | 0.02 | Actinopterygii | Labridae | Symphodus melops |
| 654 | 0.02 | Actinopterygii | unclassified | unclassified |
| 653 | 0.02 | Actinopterygii | Cyprinidae | unclassified |
| 591 | 0.02 | Actinopterygii | Soleidae | Solea solea |
| 570 | 0.02 | unclassified | unclassified | unclassified |
| 527 | 0.02 | Actinopterygii | Clupeidae | Alosa alosa |
| 384 | 0.01 | Actinopterygii | Sparidae | unclassified |
| 350 | 0.01 | Elasmobranchii | Rajidae | Raja undulata |
| 338 | 0.01 | Actinopterygii | Gadidae | unclassified |
| 299 | 0.01 | Actinopterygii | Mugilidae | Chelon labrosus |
| 188 | 0.01 | Actinopterygii | Sparidae | Pagrus major |
| 167 | 0.00 | Actinopterygii | Scombridae | unclassified |
| 163 | 0.00 | Actinopterygii | Trachinidae | Trachinus draco |
| 70 | 0.00 | Actinopterygii | Scombridae | unclassified |
| 64 | 0.00 | Elasmobranchii | unclassified | unclassified |
| 62 | 0.00 | Elasmobranchii | Lamnidae | Lamna nasus |
| 57 | 0.00 | Actinopterygii | unclassified | unclassified |
| 53 | 0.00 | Actinopterygii | Gadidae | Gadus morhua |
| 50 | 0.00 | Actinopterygii | Gobiidae | unclassified |
| 46 | 0.00 | Actinopterygii | Gadidae | Gadiculus thori |
| 43 | 0.00 | Elasmobranchii | unclassified | unclassified |
| 35 | 0.00 | Actinopterygii | Gobiidae | Neogobius melanostomus |
| 34 | 0.00 | Actinopterygii | Carangidae | Trachurus trachurus |
| 29 | 0.00 | Actinopterygii | Myctophidae | Notoscopelus kroyeri |
| 28 | 0.00 | Actinopterygii | Sparidae | Stenotomus chrysops |
| 25 | 0.00 | Actinopterygii | Labridae | unclassified |
| 25 | 0.00 | Actinopterygii | Myctophidae | unclassified |
| 21 | 0.00 | Actinopterygii | Cyprinidae | Squalius cephalus |
| 19 | 0.00 | Actinopterygii | Clupeidae | unclassified |
| 17 | 0.00 | Actinopterygii | Myctophidae | Benthosema glaciale |
| 16 | 0.00 | Actinopterygii | Scombridae | Scomber australasicus |
| 13 | 0.00 | Actinopterygii | Gempylidae | Gempylus serpens |
| 13 | 0.00 | Actinopterygii | Scombridae | Thunnus orientalis |
| 12 | 0.00 | Actinopterygii | unclassified | unclassified |
| 11 | 0.00 | Actinopterygii | Eurypharyngidae | Eurypharynx pelecanoides |
| 10 | 0.00 | Elasmobranchii | unclassified | unclassified |
| 9 | 0.00 | Actinopterygii | Labridae | Tautogolabrus adspersus |
| 9 | 0.00 | Actinopterygii | Lotidae | Ciliata mustela |
| 8 | 0.00 | Actinopterygii | Carangidae | unclassified |
| 8 | 0.00 | Actinopterygii | unclassified | unclassified |
| 8 | 0.00 | Actinopterygii | Gempylidae | unclassified |
| 8 | 0.00 | Actinopterygii | Soleidae | unclassified |
| 8 | 0.00 | Actinopterygii | Pomacentridae | Abudefduf saxatilis |
| 8 | 0.00 | Actinopterygii | Clupeidae | Alosa sapidissima |
| 8 | 0.00 | Actinopterygii | Myctophidae | Lampanyctus crocodilus |
| 7 | 0.00 | Actinopterygii | unclassified | unclassified |
| 7 | 0.00 | Elasmobranchii | Glaucostegidae | Glaucostegus cemiculus |
| 7 | 0.00 | Actinopterygii | unclassified | unclassified |
| 7 | 0.00 | Actinopterygii | Gobiidae | Odondebuenia balearica |
| 7 | 0.00 | Actinopterygii | Sparidae | unclassified |
| 7 | 0.00 | Actinopterygii | Sparidae | Sparus aurata |
| 6 | 0.00 | Actinopterygii | Labridae | Symphodus cinereus |
| 6 | 0.00 | Actinopterygii | Mugilidae | unclassified |
| 6 | 0.00 | Elasmobranchii | Somniosidae | unclassified |
| 6 | 0.00 | Actinopterygii | Nettastomatidae | unclassified |
| 6 | 0.00 | Actinopterygii | Alepocephalidae | unclassified |
| 6 | 0.00 | Actinopterygii | Scombridae | Acanthocybium solandri |
| 5 | 0.00 | Actinopterygii | Sparidae | Pagellus erythrinus |
| 5 | 0.00 | Actinopterygii | Pomacentridae | unclassified |
| 5 | 0.00 | Actinopterygii | Gobiidae | Thorogobius ephippiatus |
| 5 | 0.00 | Actinopterygii | Scombridae | Thunnus obesus |
| 5 | 0.00 | Actinopterygii | Gadidae | Trisopterus minutus |
| 4 | 0.00 | Actinopterygii | Molidae | unclassified |
| 4 | 0.00 | Actinopterygii | Labridae | Bodianus speciosus |
| 4 | 0.00 | Actinopterygii | Gadidae | Merlangius merlangus |
| 4 | 0.00 | Actinopterygii | Mugilidae | Mugil bananensis |
| 4 | 0.00 | Actinopterygii | Moronidae | Dicentrarchus punctatus |
| 4 | 0.00 | Actinopterygii | unclassified | unclassified |
| 4 | 0.00 | Actinopterygii | Gempylidae | Nealotus tripes |
| 4 | 0.00 | unclassified | unclassified | unclassified |
| 3 | 0.00 | Actinopterygii | Paralepididae | Magnisudis atlantica |
| 3 | 0.00 | Actinopterygii | Macrouridae | unclassified |
| 3 | 0.00 | Actinopterygii | Cyprinidae | Leuciscus idus |
| 3 | 0.00 | Actinopterygii | Derichthyidae | unclassified |
| 3 | 0.00 | Actinopterygii | Scombridae | Auxis thazard |
| 3 | 0.00 | Actinopterygii | Gonostomatidae | Sigmops bathyphilus |
| 3 | 0.00 | Actinopterygii | Macrouridae | unclassified |
| 2 | 0.00 | Actinopterygii | Molidae | Ranzania laevis |
| 2 | 0.00 | Actinopterygii | Lutjanidae | Lutjanus argentimaculatus |
| 2 | 0.00 | Actinopterygii | Scombridae | Euthynnus alletteratus |
| 2 | 0.00 | Actinopterygii | Gonostomatidae | unclassified |
| 2 | 0.00 | Actinopterygii | Carangidae | Alectis ciliaris |
| 2 | 0.00 | Actinopterygii | Syngnathidae | unclassified |
| 2 | 0.00 | Actinopterygii | Molidae | Masturus lanceolatus |
| 2 | 0.00 | Actinopterygii | Labridae | unclassified |
| 2 | 0.00 | Actinopterygii | Mugilidae | unclassified |
| 2 | 0.00 | Actinopterygii | Liparidae | Paraliparis copei copei |
| 2 | 0.00 | Actinopterygii | Myctophidae | Lampanyctus macdonaldi |
| 2 | 0.00 | Actinopterygii | unclassified | unclassified |
| 2 | 0.00 | Actinopterygii | Luvaridae | Luvarus imperialis |
| 2 | 0.00 | Actinopterygii | Clupeidae | Brevoortia tyrannus |
| 2 | 0.00 | Elasmobranchii | Dalatiidae | Dalatias licha |
| 2 | 0.00 | Elasmobranchii | Carcharhinidae | unclassified |
| 2 | 0.00 | Actinopterygii | Cyprinidae | Phoxinus ujmonensis |
| 2 | 0.00 | Actinopterygii | Gempylidae | Diplospinus multistriatus |
| 2 | 0.00 | Actinopterygii | Echeneidae | unclassified |
| 1 | 0.00 | Actinopterygii | Pomacentridae | unclassified |
| 1 | 0.00 | Actinopterygii | Gobiidae | Vanneaugobius canariensis |
| 1 | 0.00 | Actinopterygii | Lethrinidae | Monotaxis grandoculis |
| 1 | 0.00 | Actinopterygii | Psychrolutidae | Cottunculus thomsonii |
| 1 | 0.00 | Actinopterygii | Gobiidae | Deltentosteus collonianus |
| 1 | 0.00 | Actinopterygii | unclassified | unclassified |
| 1 | 0.00 | Elasmobranchii | Myliobatidae | Rhinoptera bonasus |
| 1 | 0.00 | Actinopterygii | Centracanthidae | Spicara maena |
| 1 | 0.00 | Actinopterygii | Centrolophidae | Centrolophus niger |
| 1 | 0.00 | Actinopterygii | Gobiidae | Millerigobius macrocephalus |
| 1 | 0.00 | Actinopterygii | Myctophidae | Myctophum asperum |
| 1 | 0.00 | Actinopterygii | Balistidae | unclassified |
| 1 | 0.00 | Elasmobranchii | Carcharhinidae | unclassified |
| 1 | 0.00 | Actinopterygii | Gobiidae | Pomatoschistus knerii |
| 1 | 0.00 | Actinopterygii | Soleidae | Pegusa lascaris |
| 1 | 0.00 | Actinopterygii | Anguillidae | Anguilla anguilla |
| 1 | 0.00 | Actinopterygii | Moridae | Halargyreus johnsonii |
| 1 | 0.00 | Actinopterygii | Myctophidae | Lampadena atlantica |
| 1 | 0.00 | Actinopterygii | Gobiidae | Gobius cobitis |
| 1 | 0.00 | Actinopterygii | Cyprinodontidae | unclassified |
| 1 | 0.00 | Actinopterygii | Belonidae | Tylosurus crocodilus |
| 1 | 0.00 | Actinopterygii | Gobiidae | Periophthalmus barbarus |
| 1 | 0.00 | Actinopterygii | Myrocongridae | Myroconger compressus |
| 1 | 0.00 | Actinopterygii | Gigantactinidae | Gigantactis vanhoeffeni |
| 1 | 0.00 | Actinopterygii | unclassified | unclassified |
| 1 | 0.00 | Actinopterygii | Cyprinidae | Alburnus alburnus |
| 1 | 0.00 | Actinopterygii | Nettastomatidae | Venefica proboscidea |
| 1 | 0.00 | Actinopterygii | Pleuronectidae | unclassified |
| 1 | 0.00 | Actinopterygii | Lotidae | Molva dypterygia |
| 1 | 0.00 | Actinopterygii | unclassified | unclassified |
| 1 | 0.00 | Actinopterygii | Myctophidae | Myctophum nitidulum |
| 1 | 0.00 | Actinopterygii | Notacanthidae | Polyacanthonotus rissoanus |
| 1 | 0.00 | Actinopterygii | Gasterosteidae | unclassified |
| 1 | 0.00 | Actinopterygii | Pleuronectidae | Platichthys flesus |
| 1 | 0.00 | Actinopterygii | Chiasmodontidae | Dysalotus alcocki |
| 1 | 0.00 | Actinopterygii | Macrouridae | Trachonurus sulcatus |
| 1 | 0.00 | Actinopterygii | Clupeidae | Alosa pseudoharengus |
| 1 | 0.00 | Actinopterygii | Carangidae | Naucrates ductor |
| 1 | 0.00 | Actinopterygii | Anotopteridae | Anotopterus pharao |
| 1 | 0.00 | Actinopterygii | Gobiidae | unclassified |
| 1 | 0.00 | Actinopterygii | Cyprinidae | Alburnus chalcoides |
More than half of the reads are assigned to European anchovy, E. encrasicolus (51.67%), followed by European pilchard, S. pilchardus (27.67%), Atlantic mackerel, Scomber scombrus (4.96%), blue whiting, Micromesistius poutassou (2.36%), white seabream, Diplodus sargus (1.52%), and axillary seabream Pagellus acarne (1.20%), which together represent 89.38% of the reads (Figure 2a). A small percentage of the reads (0.27%) were classified as Elasmobranchii, including seven species such as the Greenland shark, Somniosus microcephalus, the blue shark, Prionacea glauca, and the undulate ray, Raja undulata (Figure 2b). The remaining reads were assigned to species that represent each less than 1% of the total number or reads.
FIGURE 2.
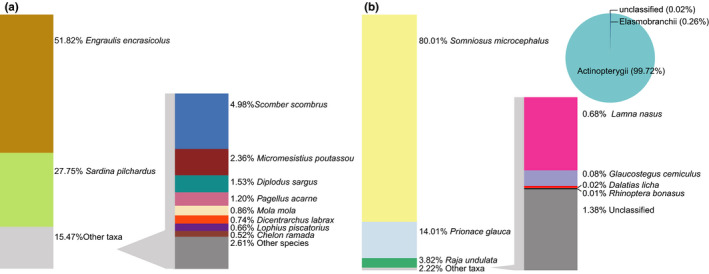
Relative number of “teleo” reads (%) assigned to (a) Actinopterygii and (b) Elasmobranchii species recovered from eDNA metabarcoding. Note that 4.96% Actinopterygii were not classified into species level
As for the four samples amplified with “mlCOI” primers, we obtained 389,665 raw reads from which, 324,731 (83%) were retained for downstream analyses. The average number of “mlCOI” reads per sample retained after quality filtering is 81,183 (Table 3). Using the BOLD database, 89.86% of the reads were classified into Phylum, 80.87% of which were metazoans, and among them 47.88% were classified as arthropods and 2.51% as chordates (Figure 3). Within chordates, 74.56% of the reads were classified as Actinopterygii (1.87% of the overall reads), resulting in only seven taxa classified into species (Figure 3).
TABLE 3.
Station depth, category, and number of reads obtained per sample after sequencing, removing primers, pair‐assembling, quality filtering, primer mapping, and chimera removal for the mlCOI region
| Sample | Station depth (m) | Category | Raw | Retained after primer checking | Retained after merging | Retained after quality filtering | Retained after mapping to coi region | Retained after chimera removal | % of retained reads for analysis |
|---|---|---|---|---|---|---|---|---|---|
| Sample_01 | 27 | Shallow | 103,773 | 103,171 | 102,988 | 102,988 | 86,259 | 82,595 | 79.59 |
| Sample_27 | 1,003 | Deep | 98,307 | 97,931 | 97,886 | 97,886 | 86,798 | 82,874 | 84.30 |
| Sample_32 | 185 | Deep | 98,873 | 98,095 | 98,013 | 98,013 | 87,716 | 83,993 | 84.95 |
| Sample_47 | 675 | Deep | 88,712 | 88,348 | 88,294 | 88,294 | 78,173 | 75,269 | 84.85 |
| TOTAL | 389,665 | 387,545 | 387,181 | 387,181 | 338,946 | 324,731 | |||
| AVERAGE_all | 97,416.25 | 96,886.25 | 96,795.25 | 96,795.25 | 84,736.50 | 81,182.75 | 83.42 |
FIGURE 3.
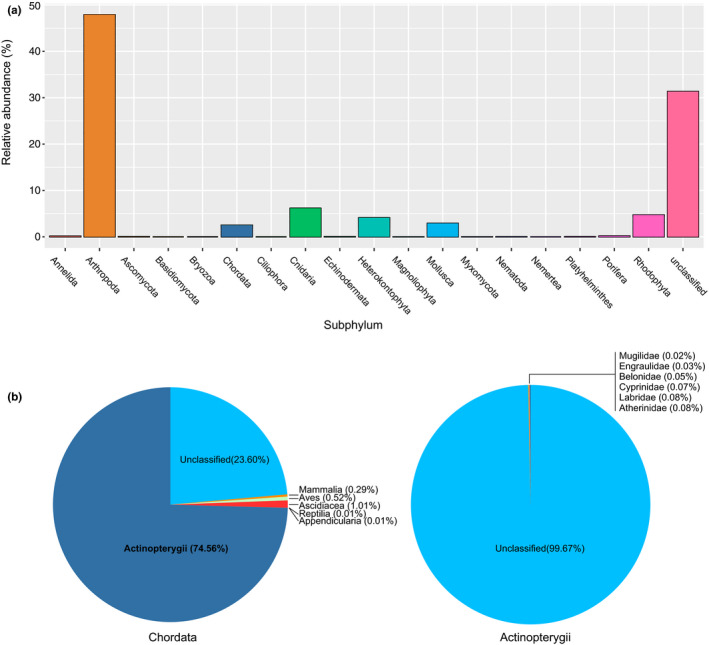
(a) Relative read abundance (%) of taxa classified to Subphylum, and (b) specifically classes within Chordata and families within Actinopterygii, respectively, from the four samples sequenced with the “mlCOI” primers
3.2. Comparison with fish trawling
Trawling operations during the BIOMAN survey resulted in a total of 18 taxa caught, from which lanternfishes (Fam. Myctophidae) and mullets (Mugil sp.) were the only ones not classified into species level. Qualitatively, a total of 10 species were identified both from the eDNA and trawling catches (Figure 4a) and even considering only the overlapping region between both sampling methods, eDNA resulted in 102 more species than catches. Six species were collected during catches and not detected through eDNA, namely Sprattus sprattus, Trachurus mediterraneus, Boops boops, Zeus faber, Trisopterus luscus, and Capros aper (Table 4); from these, there are no sequences for T. mediterraneus and B. boops in the reference database and the fact that we find T. minutus in eDNA suggest that this could be actually T. luscus. To assess the relationship between the biomass of fish caught and the number of reads obtained through eDNA, data from T. mediterraneus and T. trachurus were combined into Trachurus spp. and that from T. luscus and T. minutus into Trisopterus spp. There was an overall correlation between fish biomass and number of reads per species although not significantly different from 0 at p < .05 (Figure 4b). E. encrasicolus was the most abundant species for both methods, while the relative abundance for some species like Dicentrarchus labrax, M. poutassou, and S. pilchardus was higher when using eDNA. In contrast, the relative abundance of M. merluccius, S. scombrus, and Trachurus spp. was higher in catches than when using eDNA (Figure 4b; Table 4). At a local scale, no significant correlation between eDNA and trawling‐based abundances was found (Mantel test, r = −0.04 p = .646). In fact, eDNA data showed a more constant abundance of the three most abundant species (E. encrasicolus, S. pilchardus, and S. scombrus), compared to trawl data, which showed in general a higher number of species per station, except for those eight stations were E. encrasicolus was dominant (>94% of the catch) (Figure 5).
FIGURE 4.
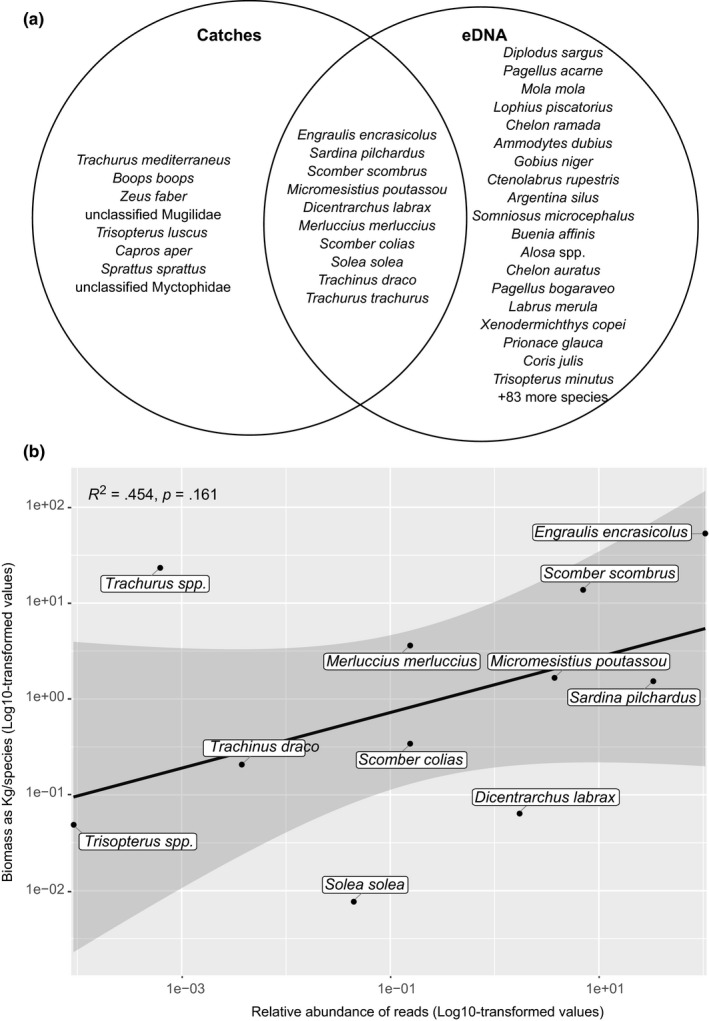
(a) Venn diagram showing fish species caught in trawls and detected through eDNA metabarcoding organized in decreasing order according to biomass or number of reads. (b) Relationship between the log10‐transformed values for the number of reads and biomass in kg from all fish species simultaneously found through eDNA and caught during fish trawling. Shaded area represents the 95% confidence interval of the linear regression
TABLE 4.
Biomass (Kg/species) caught in fishing trawls compared with the number of reads obtained through eDNA. The total number of reads does not include sites north La Rochelle
| Species | Number of reads | % | Biomass (kg) | % |
|---|---|---|---|---|
| Boops boops | 0 | 0.00 | 8.26 | 1.10 |
| Capros aper | 0 | 0.00 | 0.34 | 0.05 |
| Dicentrarchus labrax | 13,712 | 0.45 | 0.36 | 0.05 |
| Engraulis encrasicolus | 1,722,690 | 56.94 | 400.33 | 53.31 |
| Merluccius merluccius | 4,454 | 0.15 | 27.49 | 3.66 |
| Micromesistius poutassou | 81,649 | 2.70 | 12.44 | 1.66 |
| Mugil sp. | — | — | 0.90 | 0.12 |
| Myctophidae | — | — | 0.27 | 0.04 |
| Sardina pilchardus | 621,400 | 20.54 | 11.49 | 1.53 |
| Scomber colias | 4,464 | 0.15 | 2.57 | 0.34 |
| Scomber scombrus | 149,397 | 4.94 | 104.86 | 13.96 |
| Solea solea | 591 | 0.02 | 0.05 | 0.01 |
| Sprattus sprattus | 0 | 0.00 | 1.07 | 0.14 |
| Trachinus draco | 151 | 0.00 | 1.56 | 0.21 |
| Trachurus mediterraneus | 0 | 0.00 | 49.59 | 6.60 |
| Trachurus trachurus | 29 | 0.00 | 126.98 | 16.91 |
| Trisopterus luscus | 0 | 0.00 | 0.36 | 0.05 |
| Trisopterus minutus | 5 | 0.00 | 0.00 | 0.00 |
| Zeus faber | 0 | 0.00 | 2.07 | 0.28 |
FIGURE 5.
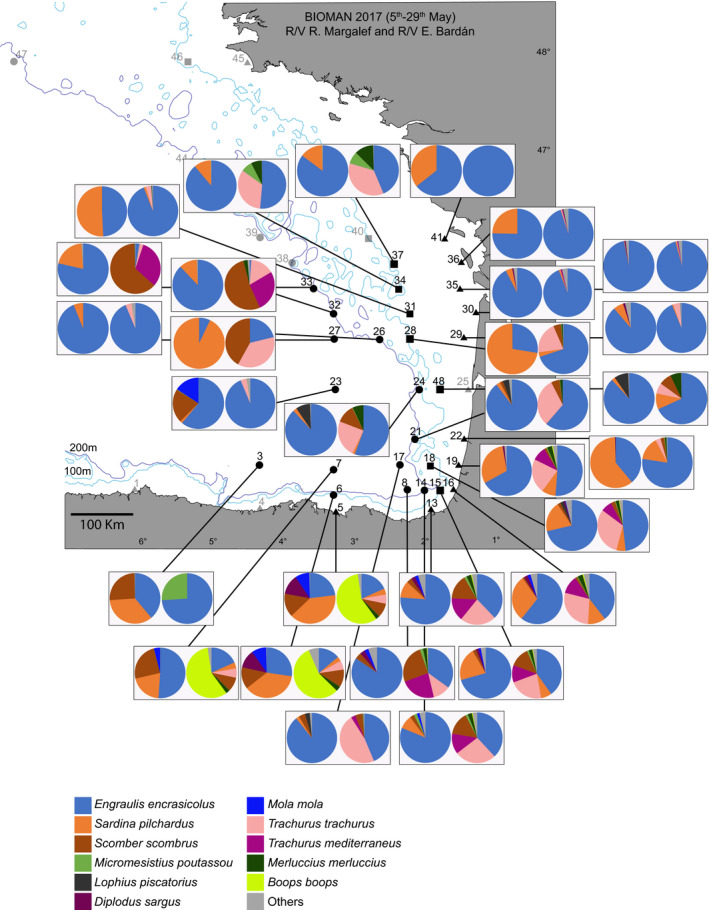
Pie charts showing the relative abundance of eDNA reads (first chart) and fish biomass caught (second chart) obtained from the 30 groups of stations within a 20 nm ratio. eDNA charts include species with >10 reads only. Species with >5% biomass caught/number of reads per station are coded by colors, the rest are grouped in “others”
3.3. Species distribution patterns
We found that correlation between compositional dissimilarities and geographic distances between stations was weak for both eDNA (R 2 = .38 p < .01) and trawling stations (R 2 = .20 p < .01). In both cases, pairs of stations that are less than about 100 nautical miles apart cover the full range of Bray–Curtis distances (Figure 6), whereas more distant stations differ more in taxonomic composition. This is particularly evident for eDNA samples, for which pairs of stations that are more than 200 nautical miles apart are available. Comparisons between samples within same or distinct depth category (shallow, medium, deep) or within same or distinct sampling methods (eDNA, trawling) had no effect over the observed patterns (Figure 7).
FIGURE 6.
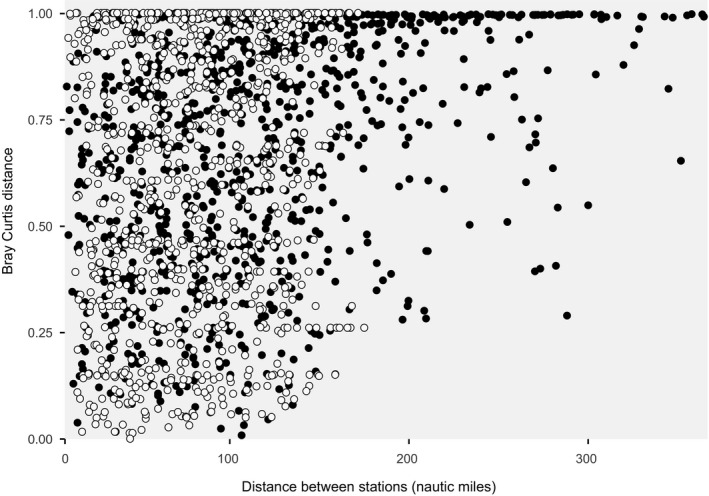
Scatterplot showing the overall relationship between Bray–Curtis distance and geographic distance between pairs of eDNA (black) and trawling (white) stations
FIGURE 7.
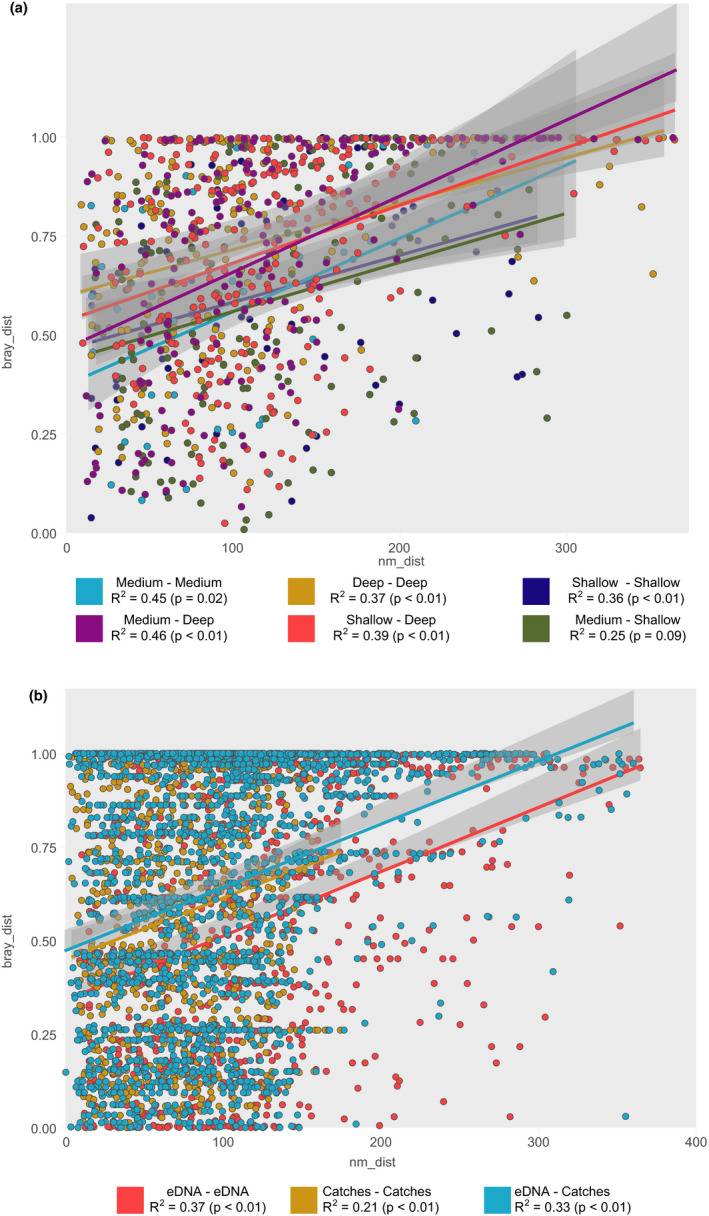
Scatterplot showing the relationship between Bray–Curtis distance and geographic distance between pairs of sampling points for (a) eDNA, (b) trawling, and (c) eDNA and trawling stations combined. Species included in c are only the common species detected by the two sampling methods. Pearson correlation is shown for each data group. Shaded area represents the 95% confidence interval of the linear regression
The overall compositional pattern of our data showed significant differences between species occurrence and sampling sites according to their zone (e.g., shallow, medium, and deep stations) (PERMANOVA F 2,43 = 2.24, p < .05) (Figure 8). Within the main species contributing to the spatial ordination of our data, two main groups can be broadly observed. On one side, species like E. encrasicolus, M. merluccius, Coris julis, S. scombrus, M. poutassou, Lophius piscatorius, S. microcephalus, Xenodermichthys copei, and P. glauca tended to be more abundant in deeper stations and their relative abundances increased in sites > 127‐m deep (Figure 9). In contrast, a second loop in the spatial ordination of the data include other species such as Gobius niger, Ammodytes dubius, D. sargus, Argentina silus, D. labrax, S. pilchardus, Mola mola, and Scomber colias (Figure 8). This information correlates with a pattern of higher abundance in <90 m‐deep sites for, for example, S. pilchardus, D. sargus, M. mola, A. dubius, D. labrax, and S. colias (Figure 9). Relatively to the abundance of reads and station depth, four species, namely A. silus, Glaucostegus cemiculus, G. niger, and Pagellus bogaraveo, remain unchanged between shallow and deep stations. Specifically, for elasmobranch species, a pattern correlated with higher relative abundances of typical demersal species like R. undulata in shallow sites and pelagic species like S. microcephalus and P. glauca in medium and deep sites (Figure 9). Species like Labrus merula and Buenia affinis were among the most abundant in number of reads (>1,000 per species) but have not been previously reported for the Bay of Biscay.
FIGURE 8.
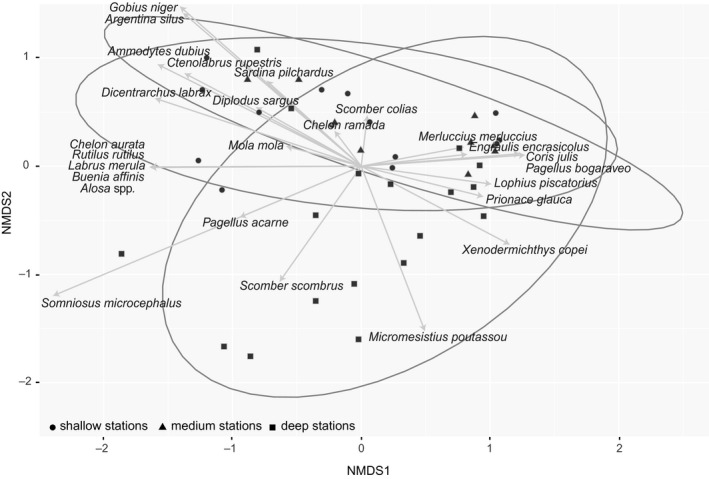
Nonmetric multidimensional scaling (NMDS) plot, with a stress of 0.15, showing the similarity of species from each sample based on their relative abundance. The ellipse shows the 95% distance based on the centroid of the three sampling zones groups (shallow, medium, and deep stations). Spatial patterns of the species with >1,000 reads are shown
FIGURE 9.
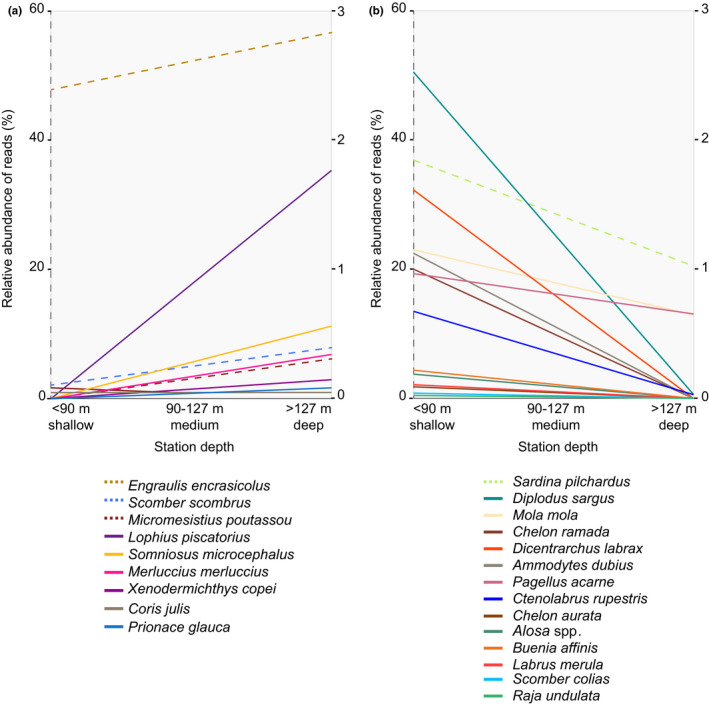
Linear relationship between depth and the relative abundance (in number of reads) obtained for those species with >1,000 reads, indicating those that increase (a) or decrease (b) with depth. For clarity, the more abundant species are represented with dashed lines on the left‐hand y‐axis, and the least abundant ones, with continuous lines to the right‐hand y‐axis
4. DISCUSSION
This study shows how eDNA metabarcoding provides a comprehensive overview of the fish diversity in a large‐scale marine area. Compared to fish trawling, eDNA metabarcoding was able to “capture” a larger number of fish species. Both, eDNA and trawling‐based estimates (in number of reads and biomass, respectively) indicate that E. encrasicolus represents half of the abundance, which is consistent to the known large and stable anchovy population in the Bay of Biscay (Erauskin‐Extramiana et al., 2019; Santos, Uriarte, Boyra, & Ibaibarriaga, 2018; Uriarte, Prouzet, & Villamor, 1996) and with the fact that the BIOMAN survey took place during the anchovy spawning season. The seven most abundant species in fish trawling representing > 1% of the total biomass were T. trachurus, S. scombrus, T. mediterraneus, M. merluccius, M. poutassou, S. pilchardus, and B. boops, which were all, except those not present in the reference database (B. boops and T. mediterraneus), also found in the eDNA metabarcoding data, and four of them (E. encrasicolus, S. pilchardus, S. scombrus, and M. poutassou) were also among the most abundant species from eDNA data. Thus, concerning the most abundant species in the Bay of Biscay, eDNA and trawling data provided comparable conclusions.
The following three species were caught during fish trawling but were absent from eDNA data despite being present in the reference database, Z. faber, S. sprattus, and C. aper. One possible explanation for this false‐negative detection could be the little abundance of this species’ DNA in the water, as suggested by the small and reduced number of catches (2.07 Kg in 3 sites, 1.07 kg in 2 sites, and 0.34 Kg in 2 sites, respectively). In fact, a small number of reads, that is, 591, was also detected for Solea solea, a species from which 0.05 kg were caught in a single station. If this is the case, filtering larger volumes of water and increasing sequencing depth could improve detection. Alternatively, reference sequences for Z. faber, S. sprattus, and C. aper could be undetected errors in the reference database (Li et al., 2018) or correspond to alternative intraspecific variants. On the other hand, in accordance with previous studies, eDNA data resulted in about 100 more species (35 with more than 10 reads) than trawling data collected simultaneously (Thomsen et al., 2012, 2016; Yamamoto et al., 2017). For example, species such as D. sargus, P. acarne, M. mola, D. labrax, L. piscatorius, Chelon ramada, A. dubius, G. niger, Ctenolabrus ruperstris, A. silus, S. microcephalus, and B. affinis were not found in catches, but were more abundant in eDNA reads than the 5th most abundant species (M. merluccius) in catches. The fact that eDNA results in a higher number of species could be partially attributed to the efficiency of the method to detect benthic or coastal species, difficult to catch by pelagic trawling nets, focused on small and medium‐size pelagic species. To check to what extent eDNA is able to detect in surface waters (4 m) demersal species, we compared the results with the ICES International Bottom Trawling Surveys (IBTS surveys) data for the Bay of Biscay from 2003 to 2019 (ICES, 2013) and with the 2017 Pélagiques Gascogne (PELGAS) integrated survey in the same area (Mathieu et al., 2019). eDNA metabarcoding data were able to detect at least 31 out of 164 species reported for the Bay of Biscay by IBTS surveys and 13 out of 45 species by PELGAS survey (Figure 10). Yet, according to the bias‐corrected Chao II estimator, the species richness obtained from eDNA would be around 161, which is closer to the IBTS based estimation. Although not being a thorough comparison, as time periods and sampling seasons at least from IBTS surveys are different, the comparison provides an overall sense of eDNA as a potential method for surveying a large marine area in a relatively simple way. Differences in eDNA and pelagic trawl catchability can also explain the differences in relative abundances of the species found by the two kind of sampling methods, such as S. pilchardus, M. poutassou, and D. labrax, with higher number of eDNA reads relative to the biomass caught, or T. trachurus, S. scombrus, and M. merluccius, showing the opposite. However, similarity between both eDNA and trawling stations suggests that stations further apart tend to be more different. The amount, quality, and stability of DNA molecules are largely affected by the production rate from each organism, diffusion of the molecules in the water, and its inherent degradation (Barnes & Turner, 2016; Collins et al., 2018; Murakami et al., 2019; Thomsen et al., 2012). But also, PCR amplification stochasticity and sequencing depth are known to affect the number of reads obtained from an eDNA sample (DiBattista et al., 2017; Zinger et al., 2019).
FIGURE 10.
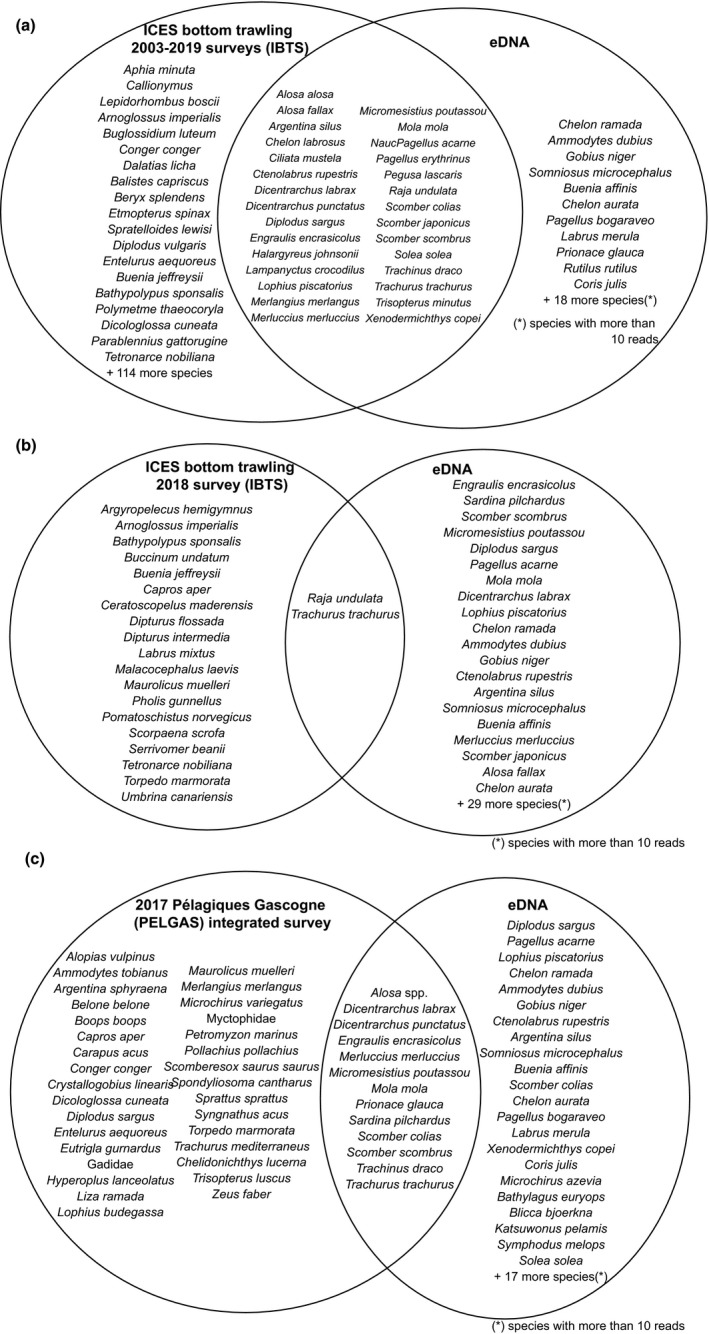
Venn diagrams showing fish caught in the ICES Bottom Trawling Survey carried out (a) between 2003 and 2019 and (b) in October 2018 available from ices.dk/marine‐data/data‐portals/ and (c) in the 2017 Pélagiques Gascogne (PELGAS) integrated survey compared to the fish species detected through eDNA metabarcoding
Trisopterus minutus, a morphologically similar species to T. luscus, was identified through eDNA, which make us raise the hypothesis that specimens collected from catches were misidentified as T. luscus, potentially being T. minutus as eDNA revealed. This would not be an isolated case where morphological characteristics difficult to observe hamper taxonomic identification, and other available data (e.g., DNA) are needed for species identification (Dayrat, 2005). A remarkable case are lanternfishes of the Myctophidae, where species identification is based on the morphology and the shape and size of photophores, which are extremely fragile and seldom recovered intact (Cabrera‐Gil et al., 2018). In this case, eDNA can play a major role for species identification as this study has shown, where at least five myctophid species were identified through eDNA. On the other hand, erroneous database records or missing sequences can bias eDNA‐based estimates. The quality and completeness of the reference database is crucial for taxonomic classification of eDNA data (Callahan, McMurdie, & Holmes, 2017). For example, two species were among the most abundant in our dataset, but not reported previously in the Bay of Biscay, namely L. merula and B. affinis. A careful examination suggests that, although L. merula could be misled by its close relative L. bimaculatus, occurring in the Bay of Biscay, the sequences attributed to B. affinis seem to be correctly assigned, suggesting that eDNA was able to detect species not previously reported in the area despite in low abundance.
Besides species diversity, eDNA also provides information on species distribution, which is comparable to that expected in the area. For instance, the number of reads assigned to the pelagic species M. poutassou and S. scombrus increased in stations deeper than 90m, where preferred habitats for these species occur (Ibaibarriaga et al., 2007) even if samples were collected from the surface. A contrasting pattern was observed for the greater argentine A. silus, a species commonly found at depths between 50 and 200 m (Basterretxea et al., 2012), but found in our data at shallower stations. This could also suggest an incongruence with species identification with a close relative, in this case A. sphyraena commonly found over the continental slope (Basterretxea et al., 2012), but with no 12Sr RNA sequence in our reference database, or DNA from A. silus (even in its form of egg or larvae) dispersed to shallower stations. Similarly, species like S. pilchardus, D. sargus, D. labrax, P. acarne, and Alosa spp. showed a distribution for this dataset in stations less than 90m depth, as our eDNA revealed. Available data on the diversity of elasmobranch species in the Bay of Biscay are limited, as most of these species are discarded from commercial fisheries and landing data are incomplete (ICES, 2017; Rodríguez‐Cabello, Pérez, & Sánchez, 2013; Rusyaev & Orlov, 2013). Hence, in agreement to previous studies, our data support eDNA as a potential mechanism for detecting and studying the distribution of elusive and deep‐water species, which normally go undetected in fish trawl surveys, for example, elasmobranchs (Thomsen et al., 2016). In any case, eDNA results also revealed an ecological pattern for elasmobranchs, for instance R. undulata, which has a high‐site fidelity occurred only in shallow waters (ICES, 2014), while large sharks as S. microcephalus, P. glauca and Lamna nasus predominantly occurred in deeper sites. Interestingly, these differences were observed even when collecting water from the surface.
Aside from biological factors (e.g., individual shedding rate, persistence of DNA in the water) that can alter the quantity of eDNA released to the environment, technical considerations can introduce biases on the quality and number of reads generated per species and hence inferences driven from them (Dejean et al., 2011; Lamb et al., 2019; Thomsen et al., 2016). Reference databases are crucial to secure taxonomic assignment for data derived from eDNA samples (Zinger et al., 2019). While recent analyses on the taxonomic annotation of metazoan GenBank sequences suggest their reliability for eDNA metabarcoding studies (Leray, Knowlton, Ho, Nguyen, & Machida, 2019; Li et al., 2018), we encountered the need of including a thorough curation step for our “global” database giving several mislabeled sequences. Species‐level annotations were not considered in Leray et al. (2019), and we found incorrectly annotated sequences at all taxonomic levels. As environmental samples contain highly complex DNA signal from various organisms, primer choice is critical for species‐level identification (Collins et al., 2019). We found that for our samples, the eukaryote universal COI primers result in a very small proportion of reads assigned to Actinopterygii. This is due to the fact that the primers target a large number of taxonomic groups, so larger coverage is needed for producing robust data (Alberdi, Aizpurua, Gilbert, Bohmann, & Mahon, 2018; Corse et al., 2019; Gunther, Knebelsberger, Neumann, Laakmann, & Martinez Arbizu, 2018; Stat et al., 2017). The use of more specific primers in our study allowed the specific detection of both Actinopterygii and Elasmobranchii. (Kelly, Port, Yamahara, & Crowder, 2014; Miya et al., 2015). Yet the amount of reads attributed to Elasmobranchii is small as “teleo” primers were not specifically designed for this taxa, for example, Kelly et al. (2014), and recent developments on elasmobranch‐specific primers (Miya et al., 2015) could potentially be a powerful tool to increase the elasmobranch diversity in future marine surveys. In addition, for closely related species such as Alosa alosa and Alosa fallax, the target barcode was exactly the same, so being cautious we consider them as Alosa spp. Another crucial methodological step is the clustering method. We showed that using a clustering method (i.e., vsearch and swarm) decreased the number of identified species, probably because the algorithm merged similar sequences from different species into singular OTUs. Recent studies have suggested that clustering techniques and the use of percentages of similarities specially in short (<100 bp) sequences might mislead diversity estimates (Calderón‐Sanou, Münkemüller, Boyer, Zinger, & Thuiller, 2019; Callahan et al., 2017; Xiong & Zhan, 2018). Thus, procuring a taxonomically comprehensive database with good quality sequences and accurate data curation steps is crucial for producing robust and reproducible ecological conclusions from eDNA metabarcoding methods (Collins et al., 2019; Weigand et al., 2019). Including a human‐specific blocking primer in our samples had little effect, as we indeed detect, although a small percentage (<0.01%), reads identified as H. sapiens. The use of blocking primers in metabarcoding analysis has been previously used to block dominant taxa in a specific samples, for instance host DNA from diet analysis (Jakubavičiūtė, Bergström, Eklöf, Haenel, & Bourlat, 2017), or human DNA from ancient samples (Boessenkool et al., 2012). Our results suggest that our samples held very little contamination from external sources such as human manipulation, air, or input from land.
Alternative ways to survey marine biodiversity and unbiased evaluations of the ecosystem components are needed as these provide the baseline for policy implementation in the context of global marine directives (e.g., Common Fisheries Policy or the Marine Strategy Framework Directive). eDNA metabarcoding is becoming a more accessible method that generates reliable information for ecosystem surveillance and invites its application on regular marine monitoring programs (Bohmann et al., 2014; Lacoursière‐Roussel, Rosabal, & Bernatchez, 2016; Takahara, Minamoto, Yamanaka, Doi, & Zi, 2012). However, there is still discussion on whether eDNA‐based approaches can be used to manage fisheries, and there is a demand of continuous research to build confidence in eDNA‐based results as evidence (Jerde, 2019). This study has shown that eDNA samples provide information on fish diversity in a broad‐scale marine area such as the Bay of Biscay, detecting almost ten times more fish species compared with pelagic trawling, including some considered elusive or difficult to capture with traditional fishing methods. These results show that, despite its inherent uncertainties, eDNA metabarcoding has the potential to become a routine technique for fisheries management as it can provide information on fish diversity and distribution in large oceanic areas, including less accessible locations and targeting rare and elusive species, in a cost‐effective and noninvasive manner. This is particularly relevant in a context of global change, where establishing efficient management actions based on numerous, continuous, and accurate biodiversity assessments is paramount.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Natalia Fraija‐Fernández: Conceptualization (lead); Formal analysis (lead); Methodology (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Marie‐Catherine Bouquieaux: Formal analysis (lead); Methodology (supporting); Writing‐review & editing (equal). Anaïs Rey: Formal analysis (supporting); Methodology (supporting); Supervision (supporting); Writing‐review & editing (equal). Iñaki Mendibil: Methodology (lead). Unai Cotano: Conceptualization (supporting); Funding acquisition (supporting); Resources (supporting); Writing‐review & editing (equal). Xabier Irigoien: Conceptualization (supporting); Funding acquisition (supporting); Resources (supporting); Writing‐review & editing (equal). María Santos: Conceptualization (supporting); Funding acquisition (supporting); Methodology (supporting); Resources (supporting); Writing‐original draft (supporting); Writing‐review & editing (equal). Naiara Rodríguez‐Ezpeleta: Conceptualization (lead); Formal analysis (equal); Methodology (lead); Project administration (lead); Resources (lead); Supervision (lead); Writing‐original draft (equal); Writing‐review & editing (equal).
ACKNOWLEDGMENTS
Authors are grateful to the crew of R/V Ramon Margalef and R/V Emma Bardán for their support during filtering and collection of samples, and specially to Luis Ferrer, Marina Chifflet, Bea Beldarrain, and Carlota Pérez for their support on filtering onboard. Thanks to Iker Pereda for bioinformatic support, Mikel Basterretxea and Estanis Mugerza for providing discard data, and Elisabete Bilbao for technical assistance. This project has been supported by the Department of Economic Development and Infrastructure of Basque Government (projects GENPES and ECOPES) and by the Spanish Ministry of Science, Innovation and Universities (project CTM2017‐89500‐R). This is contribution number 976 from the Marine Research Division (AZTI).
Fraija‐Fernández N, Bouquieaux M‐C, Rey A, et al. Marine water environmental DNA metabarcoding provides a comprehensive fish diversity assessment and reveals spatial patterns in a large oceanic area. Ecol Evol. 2020;10:7560–7584. 10.1002/ece3.6482
DATA AVAILABILITY STATEMENT
Raw sequencing reads are available at the NCBI SRA under Biosample accession numbers SAMN13489000‐SAMN13489051. Local database and scripts used for the preprocessing, clustering, and taxonomic assignment are available at https://github.com/rodriguez‐ezpeleta/fish_eDNAm.
REFERENCES
- Alberdi, A. , Aizpurua, O. , Gilbert, M. T. P. , Bohmann, K. , & Mahon, A. (2018). Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods in Ecology and Evolution, 9, 134–147. 10.1111/2041-210X.12849 [DOI] [Google Scholar]
- Altschul, S. F. , Gish, W. , Miller, W. , Myers, E. W. , & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. Retrieved from http://www.bioinformatics.babraham.ac.uk/projects/fastqc [Google Scholar]
- Andruszkiewicz, E. A. , Starks, H. A. , Chavez, F. P. , Sassoubre, L. M. , Block, B. A. , & Boehm, A. B. (2017). Biomonitoring of marine vertebrates in Monterey Bay using eDNA metabarcoding. PLoS One, 12, e0176343 10.1371/journal.pone.0176343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylagas, E. , Borja, Á. , Irigoien, X. , & Rodríguez‐Ezpeleta, N. (2016). Benchmarking DNA metabarcoding for biodiversity‐based monitoring and assessment. Frontiers in Marine Science, 3, 1–12. 10.3389/fmars.2016.00096 [DOI] [Google Scholar]
- Barnes, M. A. , & Turner, C. R. (2016). The ecology of environmental DNA and implications for conservation genetics. Conservation Genetics, 17, 1–17. 10.1007/s10592-015-0775-4 [DOI] [Google Scholar]
- Basterretxea, M. , Oyarzabal, I. , & Artetxe, I. (2012). Guía faunística de especies (comerciales y no comerciales) capturadas en el Golfo de Bizkaia por la flota vasca, Sukarrieta (Bizkaia). [Google Scholar]
- Boessenkool, S. , Epp, L. S. , Haile, J. , Bellemain, E. , Edwards, M. , Coissac, E. , … Brochmann, C. (2012). Blocking human contaminant DNA during PCR allows amplification of rare mammal species from sedimentary ancient DNA. Molecular Ecology, 21, 1806–1815. 10.1111/j.1365-294X.2011.05306.x [DOI] [PubMed] [Google Scholar]
- Bohmann, K. , Evans, A. , Gilbert, M. T. P. , Carvalho, G. R. , Creer, S. , Knapp, M. , … de Bruyn, M. (2014). Environmental DNA for wildlife biology and biodiversity monitoring. Trends in Ecology & Evolution, 29, 358–367. 10.1016/j.tree.2014.04.003 [DOI] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics (Oxford, England), 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera‐Gil, S. , Deshmukh, A. , Cervera‐Estevan, C. , Fraija‐Fernández, N. , Fernández, M. , & Aznar, F. J. (2018). Anisakis infections in lantern fish (Myctophidae) from the Arabian Sea: A dual role for lantern fish in the life cycle of Anisakis brevispiculata? Deep Sea Research Part I: Oceanographic Research Papers, 141, 43–50. 10.1016/j.dsr.2018.08.004 [DOI] [Google Scholar]
- Calderón‐Sanou, I. , Münkemüller, T. , Boyer, F. , Zinger, L. , & Thuiller, W. (2019). From environmental DNA sequences to ecological conclusions: How strong is the influence of methodological choices? Journal of Biogeography, 47, 193–206. [Google Scholar]
- Callahan, B. J. , McMurdie, P. J. , & Holmes, S. P. (2017). Exact sequence variants should replace operational taxonomic units in marker‐gene data analysis. The ISME Journal, 11, 2639–2643. 10.1038/ismej.2017.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins, R. A. , Bakker, J. , Wangensteen, O. S. , Soto, A. Z. , Corrigan, L. , Sims, D. W. , … Mariani, S. (2019). Non‐specific amplification compromises environmental DNA metabarcoding with COI. Methods in Ecology and Evolution, 10, 1985–2001. [Google Scholar]
- Collins, R. A. , Wangensteen, O. S. , O’Gorman, E. J. , Mariani, S. , Sims, D. W. , & Genner, M. J. (2018). Persistence of environmental DNA in marine systems. Communications Biology, 1, 185 10.1038/s42003-018-0192-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corse, E. , Tougard, C. , Archambaud‐Suard, G. , Agnèse, J.‐F. , Messu Mandeng, F. D. , Bilong Bilong, C. F. , … Dubut, V. (2019). One‐locus‐several‐primers: A strategy to improve the taxonomic and haplotypic coverage in diet metabarcoding studies. Ecology and Evolution, 9, 4603–4620. 10.1002/ece3.5063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danovaro, R. , Carugati, L. , Berzano, M. , Cahill, A. E. , Carvalho, S. , Chenuil, A. , … Borja, A. (2016). Implementing and innovating marine monitoring approaches for assessing marine environmental status. Frontiers in Marine Science, 3, 213. 10.3389/fmars.2016.00213 [DOI] [Google Scholar]
- Dayrat, B. (2005). Towards integrative taxonomy. Biological Journal of the Linnean Society, 85, 407–417. 10.1111/j.1095-8312.2005.00503.x [DOI] [Google Scholar]
- Deiner, K. , Bik, H. M. , Seymour, M. , Lacoursière‐Roussel, A. , Altermatt, F. , … Bernatchez, L (2017). Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Molecular Ecology, 26, 5872–5895. 10.1111/mec.14350 [DOI] [PubMed] [Google Scholar]
- Dejean, T. , Valentini, A. , Duparc, A. , Pellier‐Cuit, S. , Pompanon, F. , Taberlet, P. , & Miaud, C. (2011). Persistence of environmental DNA in freshwater ecosystems. PLoS One, 6, e23398 10.1371/journal.pone.0023398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiBattista, J. D. , Coker, D. J. , Sinclair‐Taylor, T. H. , Stat, M. , Berumen, M. L. , & Bunce, M. (2017). Assessing the utility of eDNA as a tool to survey reef‐fish communities in the Red Sea. Coral Reefs, 36, 1245–1252. 10.1007/s00338-017-1618-1 [DOI] [Google Scholar]
- Dray, S. , & Dufour, A. (2007). The ade4 Package: Implementing the duality diagram for ecologists. Journal of Statistical Software, 22, 1–20. [Google Scholar]
- Edgar, R. C. , Haas, B. J. , Clemente, J. C. , Quince, C. , & Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics (Oxford, England), 27, 2194–2200. 10.1093/bioinformatics/btr381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erauskin‐Extramiana, M. , Alvarez, P. , Arrizabalaga, H. , Ibaibarriaga, L. , Uriarte, A. , Cotano, U. , … Chust, G. (2019). Historical trends and future distribution of anchovy spawning in the Bay of Biscay. Deep Sea Research Part II: Topical Studies in Oceanography, 159, 169–182. 10.1016/j.dsr2.2018.07.007 [DOI] [Google Scholar]
- Fraser, H. M. , Greenstreet, S. P. R. , & Piet, G. J. (2007). Taking account of catchability in groundfish survey trawls: Implications for estimating demersal fish biomass. ICES Journal of Marine Science, 64, 1800–1819. 10.1093/icesjms/fsm145 [DOI] [Google Scholar]
- Frøslev, T. G. , Kjøller, R. , Bruun, H. H. , Ejrnæs, R. , Brunbjerg, A. K. , Pietroni, C. , & Hansen, A. J. (2017). Algorithm for post‐clustering curation of DNA amplicon data yields reliable biodiversity estimates. Nature Communications, 8, 1188 10.1038/s41467-017-01312-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther, B. , Knebelsberger, T. , Neumann, H. , Laakmann, S. , & Martinez Arbizu, P. (2018). Metabarcoding of marine environmental DNA based on mitochondrial and nuclear genes. Scientific Reports, 8, 14822 10.1038/s41598-018-32917-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hänfling, B. , Lawson Handley, L. , Read, D. S. , Hahn, C. , Li, J. , Nichols, P. , … Winfield, I. J. (2016). Environmental DNA metabarcoding of lake fish communities reflects long‐term data from established survey methods. Molecular Ecology, 25, 3101–3119. 10.1111/mec.13660 [DOI] [PubMed] [Google Scholar]
- Hansen, B. K. , Bekkevold, D. , Clausen, L. W. , & Nielsen, E. E. (2018). The sceptical optimist: Challenges and perspectives for the application of environmental DNA in marine fisheries. Fish and Fisheries, 19, 751–768. 10.1111/faf.12286 [DOI] [Google Scholar]
- Heino, M. , Porteiro, F. M. , Sutton, T. T. , Falkenhaug, T. , Godø, O. R. , & Piatkowski, U. (2011). Catchability of pelagic trawls for sampling deep‐living nekton in the mid North Atlantic. Laxenburg, Austria: IIASA. [Google Scholar]
- Horton, T. , Kroh, A. , Ahyong, S. et al. (2018). World Register of Marine Species (WoRMS). Retrieved from http://www.marinespecies.org [Google Scholar]
- Ibaibarriaga, L. , Irigoien, X. , Santos, M. , Motos, L. , Fives, J. M. , Franco, C. , … Reid, D. G. (2007). Egg and larval distributions of seven fish species in north‐east Atlantic waters. Fisheries Oceanography, 16, 284–293. 10.1111/j.1365-2419.2007.00430.x [DOI] [Google Scholar]
- ICES . (2004). Report of the Working Group on Fish Ecology (WGFE). p. 258. [Google Scholar]
- ICES . (2013). Report of the International Bottom Trawl Survey Working Group (IBTSWG). p. 278. [Google Scholar]
- ICES . (2014). Report of the Working Group on Elasmobranch Fishes (WGEF) (ed. edit) (p. 887). [Google Scholar]
- ICES . (2015). Manual for International Pelagic Surveys (IPS) In Series of ICES Survey Protocols SISP 9 – IPS (p. 92). [Google Scholar]
- ICES (2017). Report of the Working Group on Elasmobranchs (p. 1018). [Google Scholar]
- ICES . (2018). Bay of Biscay and the Iberian coast ecoregion In CIEM I (Ed.), Ecosystem overview (pp. 1–17). [Google Scholar]
- Jakubavičiūtė, E. , Bergström, U. , Eklöf, J. S. , Haenel, Q. , & Bourlat, S. J. (2017). DNA metabarcoding reveals diverse diet of the three‐spined stickleback in a coastal ecosystem. PLoS One, 12, e0186929 10.1371/journal.pone.0186929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerde, C. L. (2019). Can we manage fisheries with the inherent uncertainty from eDNA? Journal of Fish Biology.1–13. [DOI] [PubMed] [Google Scholar]
- Jeunen, G.‐J. , Knapp, M. , Spencer, H. G. , Lamare, M. D. , Taylor, H. R. , Stat, M. , … Gemmell, N. J. (2019). Environmental DNA (eDNA) metabarcoding reveals strong discrimination among diverse marine habitats connected by water movement. Molecular Ecology Resources, 19, 426–438. 10.1111/1755-0998.12982 [DOI] [PubMed] [Google Scholar]
- Kelly, R. P. , Port, J. A. , Yamahara, K. M. , & Crowder, L. B. (2014). Using environmental DNA to census marine fishes in a large mesocosm. PLoS One, 9, e86175 10.1371/journal.pone.0086175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoursière‐Roussel, A. , Rosabal, M. , & Bernatchez, L. (2016). Estimating fish abundance and biomass from eDNA concentrations: Variability among capture methods and environmental conditions. Molecular Ecology Resources, 16, 1401–1414. 10.1111/1755-0998.12522 [DOI] [PubMed] [Google Scholar]
- Lamb, P. D. , Hunter, E. , Pinnegar, J. K. , Creer, S. , Davies, R. G. , & Taylor, M. I. (2019). How quantitative is metabarcoding: A meta‐analytical approach. Molecular Ecology, 28, 420–430. 10.1111/mec.14920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leray, M. , Knowlton, N. , Ho, S.‐L. , Nguyen, B. N. , & Machida, R. J. (2019). GenBank is a reliable resource for 21st century biodiversity research. Proceedings of the National Academy of Sciences of the United States of America, 116(45), 22651–22656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leray, M. , Yang, J. Y. , Meyer, C. P. , Mills, S. C. , Agudelo, N. , Ranwez, V. , … Machida, R. J. (2013). A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: Application for characterizing coral reef fish gut contents. Frontiers in Zoology, 10, 34 10.1186/1742-9994-10-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic, I. , & Bork, P. (2016). Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Research, 44, W242–W245. 10.1093/nar/gkw290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Shen, X. , Chen, X. , Xiang, D. , Murphy, R. W. , & Shen, Y. (2018). Detection of potential problematic Cytb gene sequences of fishes in GenBank. Frontiers in Genetics, 9, 30 10.3389/fgene.2018.00030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahé, F. , Rognes, T. , Quince, C. , de Vargas, C. , & Dunthorn, M. (2014). Swarm: Robust and fast clustering method for amplicon‐based studies. PeerJ, 2, e593 10.7717/peerj.593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. (2011) Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet.journal, 17(1), 10 10.14806/ej.17.1.200 [DOI] [Google Scholar]
- Massé, J. , Uriarte, A. , Angélico, M. M. , & Carrera, P. (2018). Pelagic survey series for sardine and anchovy in ICES subareas 8 and 9 – Towards an ecosystem approach. ICES Cooperative Research Report. [Google Scholar]
- Mathieu, D. , Laurence, P. , Patrick, G. et al. (2019). Biotic data collected during identification trawls on the PELGAS integrated survey in the Bay of Biscay. SEANOE. [Google Scholar]
- McMurdie, P. J. , & Holmes, S. (2013). hyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLOS ONE, 8(4), e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, C. P. (2003). Molecular systematics of cowries (Gastropoda: Cypraeidae) and diversification patterns in the tropics. Biological Journal of the Linnean Society, 79, 401–459. 10.1046/j.1095-8312.2003.00197.x [DOI] [Google Scholar]
- Minamoto, T. , Yamanaka, H. , Takahara, T. , Honjo, M. N. , & Zi, K. (2012). Surveillance of fish species composition using environmental DNA. Limnology, 13, 193–197. 10.1007/s10201-011-0362-4 [DOI] [Google Scholar]
- Miya, M. , Sato, Y. , Fukunaga, T. , Sado, T. , Poulsen, J. Y. , Sato, K. , … Iwasaki, W. (2015). MiFish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: Detection of more than 230 subtropical marine species. Royal Society Open Science, 2, 150088 10.1098/rsos.150088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, H. , Yoon, S. , Kasai, A. , Minamoto, T. , Yamamoto, S. , Sakata, M. K. , … Masuda, R. (2019). Dispersion and degradation of environmental DNA from caged fish in a marine environment. Fisheries Science, 85, 327–337. 10.1007/s12562-018-1282-6 [DOI] [Google Scholar]
- O’Donnell, J. L. , Kelly, R. P. , Shelton, A. O. , Samhouri, J. F. , Lowell, N. C. , & Williams, G. D. (2017). Spatial distribution of environmental DNA in a nearshore marine habitat. PeerJ, 5, e3044 10.7717/peerj.3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. et al. (2019). vegan: Community Ecology Package R. package version 2.5‐5. Retrieved from https://CRAN.R‐project.org/package=vegan [Google Scholar]
- Olds, B. P. , Jerde, C. L. , Renshaw, M. A. , Li, Y. , Evans, N. T. , Turner, C. R. , … Lamberti, G. A. (2016). Estimating species richness using environmental DNA. Ecology and Evolution, 6, 4214–4226. 10.1002/ece3.2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pont, D. , Rocle, M. , Valentini, A. , Civade, R. , Jean, P. , Maire, A. , … Dejean, T. (2018). Environmental DNA reveals quantitative patterns of fish biodiversity in large rivers despite its downstream transportation. Scientific Reports, 8, 10361 10.1038/s41598-018-28424-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rago, P. J. (2004). Fishery‐independent sampling: survey techniques and data analyses. [Google Scholar]
- Ratnasingham, S. , & Hebert, P. D. N. (2007). BOLD: The Barcode of Life Data System (http://www.barcodinglife.org). Molecular Ecology Notes, 7, 355‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees, H. C. , Maddison, B. C. , Middleditch, D. J. , Patmore, J. R. M. , & Gough, K. C. (2014). REVIEW: The detection of aquatic animal species using environmental DNA – a review of eDNA as a survey tool in ecology. Journal of Applied Ecology, 51, 1450–1459. 10.1111/1365-2664.12306 [DOI] [Google Scholar]
- Rodríguez‐Cabello, C. , Pérez, M. , & Sánchez, F. (2013). New records of chondrichthyans species caught in the Cantabrian Sea (southern Bay of Biscay). Journal of the Marine Biological Association of the United Kingdom, 93, 1929–1939. 10.1017/S0025315413000271 [DOI] [Google Scholar]
- Rognes, T. , Flouri, T. , Nichols, B. , Quince, C. , & Mahé, F. (2016). VSEARCH: A versatile open source tool for metagenomics. PeerJ, 4, e2584 10.7717/peerj.2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusyaev, S. M. , & Orlov, A. M. (2013). Bycatches of the Greenland shark Somniosus microcephalus (Squaliformes, Chondrichthyes) in the Barents sea and the adjacent waters under bottom trawling data. Journal of Ichthyology, 53, 111–115. 10.1134/S0032945213010128 [DOI] [Google Scholar]
- Santos, M. , Ibaibarriaga, L. , Louzao, M. , Korta, M. , & Uriarte, A. (2018). Index of biomass of anchovy (Engraulis encrasicolus, L.) and sardine (Sardina pilchardus) applying the DEPM and sighting in the Bay of Biscay 2017 In ICES (Ed.), Working group on acoustic and egg surveys for sardine and anchovy in ICES Areas 7, 8 and 9 (WGACEGG) (pp. 158–198). ICES WGACEGG REPORT 2017, 3–17 November 2017. [Google Scholar]
- Santos, M. , Uriarte, A. , Boyra, G. , & Ibaibarriaga, L. (2018). Anchovy DEPM surveys 2003–2012 in the Bay of Biscay (Subarea 8): BIOMAN survey series In Massé J., Uriarte A., Angélico M. M., & Carrera P. (Eds.), Pelagic survey series for sardine and anchovy in ICES subareas 8 and 9 – Towards an ecosystem approach (pp. 85–102). ICES Cooperative Research Report. [Google Scholar]
- Sayers, E. (2008). E‐utilities Quick Start. Retrieved from www.ncbi.nlm.nih.gov/books/NBK25500/ [Google Scholar]
- Schloss, P. D. , Westcott, S. L. , Ryabin, T. , Hall, J. R. , Hartmann, M. , Hollister, E. B. , … Weber, C. F. (2009). Introducing mothur: Open‐source, platform‐independent, community‐supported software for describing and comparing microbial communities. Applied and Environmental Microbiology, 75, 7537 10.1128/AEM.01541-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigsgaard, E. E. , Nielsen, I. B. , Carl, H. , Krag, M. A. , Knudsen, S. W. , Xing, Y. , … Thomsen, P. F. (2017). Seawater environmental DNA reflects seasonality of a coastal fish community. Marine Biology, 164, 128 10.1007/s00227-017-3147-4 [DOI] [Google Scholar]
- Spens, J. , Evans, A. R. , Halfmaerten, D. et al. (2017). Comparison of capture and storage methods for aqueous macrobial eDNA using an optimized extraction protocol: Advantage of enclosed filter. Methods in Ecology and Evolution, 8, 635–645. [Google Scholar]
- Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post‐analysis of large phylogenies. Bioinformatics, 30, 1312–1313. 10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stat, M. , Huggett, M. J. , Bernasconi, R. , DiBattista, J. D. , Berry, T. E. , Newman, S. J. , … Bunce, M. (2017). Ecosystem biomonitoring with eDNA: Metabarcoding across the tree of life in a tropical marine environment. Scientific Reports, 7, 12240 10.1038/s41598-017-12501-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taberlet, P. , Coissac, E. , Hajibabaei, M. , & Rieseberg, L. H. (2012). Environmental DNA. Molecular Ecology, 21, 1789–1793. 10.1111/j.1365-294X.2012.05542.x [DOI] [PubMed] [Google Scholar]
- Taberlet, P. , Coissac, E. , Pompanon, F. , Brochmann, C. , & Willerslev, E. (2012). Towards next‐generation biodiversity assessment using DNA metabarcoding. Molecular Ecology, 21, 2045–2050. 10.1111/j.1365-294X.2012.05470.x [DOI] [PubMed] [Google Scholar]
- Takahara, T. , Minamoto, T. , Yamanaka, H. , Doi, H. , & Zi, K. (2012). Estimation of fish biomass using environmental DNA. PLoS One, 7, e35868 10.1371/journal.pone.0035868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen, P. F. , Kielgast, J. , Iversen, L. L. , Møller, P. R. , Rasmussen, M. , & Willerslev, E. (2012). Detection of a diverse marine fish fauna using environmental DNA from seawater samples. PLoS One, 7, e41732 10.1371/journal.pone.0041732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen, P. F. , Møller, P. R. , Sigsgaard, E. E. , Knudsen, S. W. , Jørgensen, O. A. , & Willerslev, E. (2016). Environmental DNA from seawater samples correlate with trawl catches of subarctic, deepwater fishes. PLoS One, 11, e0165252 10.1371/journal.pone.0165252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen, P. F. , & Willerslev, E. (2015). Environmental DNA – An emerging tool in conservation for monitoring past and present biodiversity. Biological Conservation, 183, 4–18. 10.1016/j.biocon.2014.11.019 [DOI] [Google Scholar]
- Uriarte, A. , Prouzet, P. , & Villamor, B. (1996). Bay of Biscay and Ibero Atlantic anchovy populations and their fisheries. Scientia Marina, 60, 237–255. [Google Scholar]
- Valentini, A. , Taberlet, P. , Miaud, C. , Civade, R. , Herder, J. , Thomsen, P. F. , … Dejean, T. (2016). Next‐generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Molecular Ecology, 25, 929–942. 10.1111/mec.13428 [DOI] [PubMed] [Google Scholar]
- Wang, Q. , Garrity, G. M. , Tiedje, J. M. , & Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Applied and Environmental Microbiology, 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigand, H. , Beermann, A. J. , Čiampor, F. , Costa, F. O. , Csabai, Z. , Duarte, S. , … Ekrem, T. (2019). DNA barcode reference libraries for the monitoring of aquatic biota in Europe: Gap‐analysis and recommendations for future work. Science of the Total Environment, 678, 499–524. 10.1016/j.scitotenv.2019.04.247 [DOI] [PubMed] [Google Scholar]
- Xiong, W. , & Zhan, A. (2018). Testing clustering strategies for metabarcoding‐based investigation of community–environment interactions. Molecular Ecology Resources, 18, 1326–1338. 10.1111/1755-0998.12922 [DOI] [PubMed] [Google Scholar]
- Yamamoto, S. , Masuda, R. , Sato, Y. , Sado, T. , Araki, H. , Kondoh, M. , … Miya, M. (2017). Environmental DNA metabarcoding reveals local fish communities in a species‐rich coastal sea. Scientific Reports, 7, 40368 10.1038/srep40368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Kobert, K. , Flouri, T. , & Stamatakis, A. (2014). PEAR: A fast and accurate Illumina Paired‐End reAd mergeR. Bioinformatics (Oxford, England), 30, 614–620. 10.1093/bioinformatics/btt593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinger, L. , Bonin, A. , Alsos, I. G. , Bálint, M. , Bik, H. , Boyer, F. , … Taberlet, P. (2019). DNA metabarcoding—Need for robust experimental designs to draw sound ecological conclusions. Molecular Ecology, 28, 1857–1862. 10.1111/mec.15060 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Raw sequencing reads are available at the NCBI SRA under Biosample accession numbers SAMN13489000‐SAMN13489051. Local database and scripts used for the preprocessing, clustering, and taxonomic assignment are available at https://github.com/rodriguez‐ezpeleta/fish_eDNAm.