

Abstract
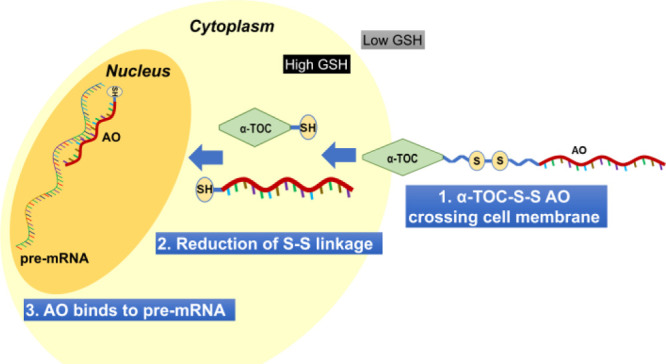
Splice-modulating antisense therapy has shown tremendous potential in therapeutic development in recent years with four FDA-approved antisense drugs since 2016. However, an efficient and nontoxic antisense oligonucleotide (AO) delivery system still remains as a major obstacle in nucleic acid therapeutics field. Vitamin-E (α-tocopherol) is an essential dietary requirement for human body. This fat-soluble compound is one of the most important antioxidants which involves in numerous biological pathways. In this study, for the first time, we explored the scope of using α-tocopherol-conjugated bioresponsive AOs to induce splice modulation in mouse muscle myotubes in vitro. Our results showed that the bioresponsive construct efficiently internalized into the cell nucleus and induced exon 23 skipping in mdx mouse myotubes. Based on our exciting new results, we firmly believe that our findings could potentially benefit toward establishing a delivery approach to advance the field of splice-modulating AO therapy.
1. Introduction
Synthetic antisense oligonucleotide (AO) has attracted extensive interest in therapeutic development for various genetic diseases after demonstrating successful inhibition of Rous sarcoma virus 35S RNA translation in 1978.1 Since then, eight AO drugs have been approved for clinical use including Vitravene, Kynamro, Tegsedi, and Waylivra utilizing RNase-H-dependent pathway and Exondys 51, Spinraza, Milasen, and Vyondys 53 using the splice modulation mechanism.2−6 In order to achieve high therapeutic efficacy, AO drug molecules need to be stable under nuclease conditions while possessing high binding specificity and affinity to the RNA target. In addition, tissue distribution and cellular uptake properties of AOs also play a pivotal role in the overall treatment outcome. However, despite the dramatic progress in recent years, establishing an effective AO delivery system still remains as one of the major challenges. Various delivery approaches have been studied for AOs over the last few years, and a comprehensive review of this topic can be found elsewhere.7
Conjugation of biomolecules to AOs has attracted significant interest in therapeutic development because of the simplicity in the synthesis and pharmacokinetic studies. Polyethylene glycol was one of the first molecules utilized in the delivery of an aptamer drug Macugen.8,9 In addition, the potential of N-acetylgalactosamine (GalNAc)10 has been studied for liver-specific uptake of oligonucleotides, and based on this development, an siRNA drug called Patisiran11 has been approved by the US FDA that offers a great promise for liver-specific delivery of AOs. Furthermore, the potential of various cell-penetrating peptides has been studied extensively particularly to enhance the delivery of phosphorodiamidate morpholino oligo-based AO drug molecules.12
Recently, vitamin-E, a group of eight naturally occurring fat-soluble compounds, has been emerged as a potential approach for oligonucleotide delivery.13,14 Vitamin-E, a widely used component in various cosmetic and dietary supplement products, has eight isoforms in which α-tocopherol (α-TOC, Figure 1) has been identified as the most abundant form in human body.14−16 α-TOC is believed to be beneficial for human health; however, there is no clear evidence on the positive effects as a treatment modality for disease prevention or therapy.16 Yokota and colleagues demonstrated the use of α-TOC for the delivery of siRNA and chimeric oligonucleotides in mouse liver.17−19 Another study from Kuwahara et al. also showed the efficacy of an α-TOC-conjugated heteroduplex oligonucleotide for the modulation of blood–brain barrier in mice.20 Inspired by these studies, we envisaged that the α-TOC-conjugated AO could also be applicable in the splice modulation scenario. However, unlike previous reports in which the AOs bind to the mature mRNA targets in the cell cytoplasm, splice-modulating AOs need to be delivered into the cell nucleus where it binds to the pre-mRNA and interfere with splicing events. Toward addressing this challenge, we established a novel construct by introducing a disulfide bridge that links between the AO and α-TOC (α-TOC-S-S AO) (Figure 1) and evaluated its efficiency to induce exon 23 skipping in a dystrophin gene (Dmd) transcript model system by utilizing mdx mouse myotubes in vitro. The designed AO construct is bio-reducible inside the cell, which can promote and enhance the ability of the AO to efficiently internalize into the cell nucleus and induce exon skipping.
Figure 1.
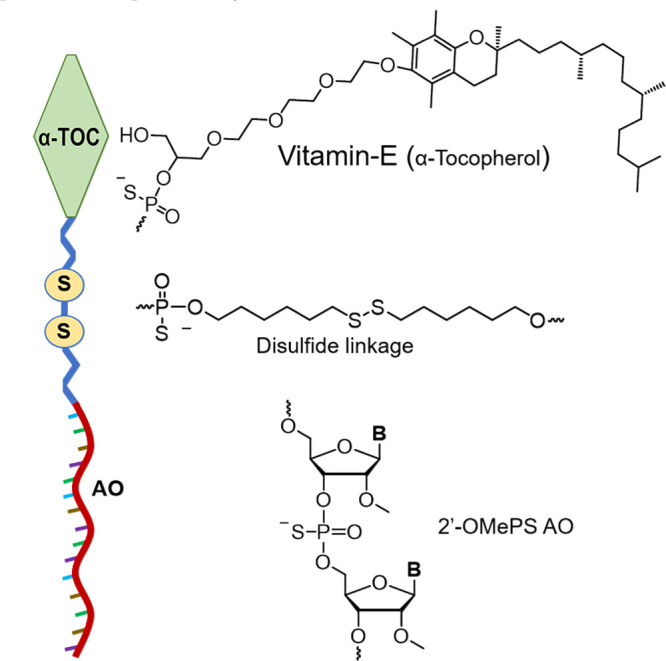
Schematic illustration of the bioresponsive α-TOC-S-S-AO established from three components, including α-TOC, S-S bridge, and 2′-O-methyl PS AO, and the structural representations of their monomeric units.
2. Results and Discussion
To demonstrate this rationale systematically, we first assessed the effects of a non-S-S-containing AO construct to induce exon 23 skipping in vitro. Accordingly, a 20-mer α-TOC-conjugated 2′-O-methyl (2′-OMe, Figure 1) AO construct was designed and synthesized on a phosphorothioate (PS) backbone (α-TOC-AO, Table 1). The AO sequence used in this study was based on a previously reported best performing AO in inducing exon 23 skipping in mdx mouse myotubes.21 A non-conjugated 2′-OMePS was also synthesized as a positive control. We then evaluated the potential to induce exon 23 skipping by directly incubating the AO candidates with H-2Kb-tsA58 mdx (H2K mdx)22 mouse myotubes at four different concentrations (100, 200, 400, and 800 nM). In parallel, the 20 mer 2′-OMePS control AO was transfected with Lipofectin reagent at 100 nM. After 24 h, the cells were collected, and the total RNA was extracted, followed by performing nested reverse transcription polymerase chain reaction (RT-PCR) to amplify the products as previously described.21
Table 1. AO Names and Sequences Used in This Studya.
| AO names | Sequences (5′–3′ direction) |
| α-TOC-AO | α-TOC-GGCCAAACCUCGGCUUACCU |
| α-TOC-S-S-AO | α-TOC-S-S-GGCCAAACCUCGGCUUACCU |
| α-TOC-AO-FAM | α-TOC-GGCCAAACCUCGGCUUACCU-FAM |
| α-TOC-S-S-AO-FAM | α-TOC-S-S-GGCCAAACCUCGGCUUACCU-FAM |
| control AO | GGCCAAACCUCGGCUUACCU |
All sequences were synthesized as 2′-OMe nucleotides on a PS backbone.
As expected, the positive control yielded an efficient (62.3%) exon 23 skipping product of 688 bp (Figure S1; Supporting Information), in line with our previous observations.23−27 On the other hand, the α-TOC AO failed to induce any exon skipping after 24 h (Figure S1). Not surprisingly, the 2′-OMePS control AO without any lipids (naked form) was also not efficient after 24 h of incubation. Therefore, we increased the incubation time to enhance the uptake and repeated the experiment with prolonged incubation times of 48 and 72 h using the α-TOC AO.
Notably, at 48 h time point, although the 100 nM concentration failed to induce any exon skipping, other tested concentrations yielded the expected exon-23-skipped product in a dose-dependent manner ranging from 13.3% at 200 nM to 22.6% at 800 nM (Figure 2A,G). Similar to the 48 h time point, the exon 23 skipping products were observed at 72 h time point with all concentrations except 100 nM, and the skipping efficiency was similar between 200 and 400 nM (10.3 and 10.1%, respectively) and increased to 22.4% at 800 nM (Figure 2B,H). The decrease in the yield of exon 23 skipping compared with the 48 h time point could possibly be due to the unfavorable dual exon 22–23 skipping (542 bp product; 1.8–12.2% from 800 to 200 nM; Figure 2B,H) upon prolonged incubation time. It is also encouraging that the potency of the AO increased proportionally to the time of incubation. However, the efficiency of exon 23 skipping (688 bp product) was very low in comparison with the positive control 2′-OMePS AO transfected using Lipofectin. Simultaneously, an independent control experiment was also performed by incubating the Lipofectin transfection reagent-free (naked transfection) 2′-OMePS AO control with the cells for 48 and 72 h. In this case, there was no exon skipping observed at 48 h (Figure 2E,K), while 72 h of incubation yielded exon 23 skipping in very low yields of 8.8 and 5.5% at 400 and 800 nM, respectively (Figure 2F,L), suggesting that a small amount of unconjugated AO could still be internalized and localized in the nucleus.
Figure 2.

RT-PCR analysis and transcript densitometry analysis of RNA prepared from H2K mdx mouse myotubes transfected with AOs; (A,G) α-TOC AO for 48 h of incubation; (B,H) α-TOC AO for 72 h of incubation; (C,I) α-TOC-S-S-AO for 48 h; (D,J) α-TOC-S-S-AO for 72 h; (E,K) control AO for 48 h of incubation; (F,L) control AO for 72 h of incubation. The triangles above the gel image indicate the increasing AO concentration (100, 200, 400, and 800 nM); FL (full length) = 901 bp, Δ23 (exon 23 skipped) = 688 bp, Δ22–23 (exons-22 + 23 skipped) = 542 bp, Ctrl = control AO transfected with Lipofectin at 100 nM, UT = untreated, and −ve = negative control. Green = exon 23 skipped, blue = exon 22–23 skipped, and gray = full length.
In line with our recent study (unpublished) using two different fatty-acid-conjugated AOs which failed to induce any exon 23 skipping, we speculated that α-TOC conjugation might also be limiting the nucleus localization of AOs after entering the cytoplasm. To investigate this hypothesis, we then explored the scope of a bioresponsive AO construct that can be cleaved in the cytoplasm by different mechanisms involving pH, glutathione level, or enzymatic reaction.28 For this purpose, incorporation of a reducible disulfide linkage (−S-S– linkage, Figure 1) could be a promising approach to cleave the AO from the α-TOC moiety. Higher concentration of glutathione (γ-glutamyl-cysteinyl-glycine; GSH) inside the cell compared to the extracellular matrix enables the cleavage of the disulfide bond by a redox reaction.29 Various studies, mainly in the field of cancer therapeutics, have utilized this biological mechanism for drug delivery. An update on this topic has been reviewed and published elsewhere.30 Notably, there were no previous reports on conjugating splice-modulating AOs with a disulfide linkage. Therefore, in this study, we envisioned the scope of a novel bioresponsive AO construct (α-TOC-S-S-AO; Figure 1; Table 1) by linking α-TOC (the delivery agent) and the AO via a disulfide linkage (to facilitate AO release in the cytosol).
To validate this rationale, we incubated α-TOC-S-S-AO (Table 1) with the H2K mdx myotubes at 100, 200, 400, and 800 nM for 48 and 72 h. Overall, the bioresponsive α-TOC-S-S-AO induced far more efficient exon 23 skipping compared with the α-TOC-AO and lipid-free 2′-OMePS controls at both time points in line with our hypothesis. At 48 h, although 100 and 200 nM of α-TOC-S-S-AO failed to induce any skipping, higher concentrations yielded the skipped products in a dose-dependent manner, with 18% at 400 nM and 36.4% at 800 nM (Figure 2C,I). A dual exon 22–23 skipping product was also observed at 800 nM, which accounted for 18.3% of the total band intensity. Remarkably, 72 h of incubation yielded very efficient exon 23 skipping at 400 nM (46.7%), with a minimal amount of dual skipping product at 3.6% (Figure 2D,J). The results also showed that the exon-23-skipped product was slightly dropped to 40.6% at 800 nM, mainly because of the increase of the dual skipped product (26.4%). Not surprisingly, 200 nM of the AO only induced a slight exon 23 skipping (13.4%), while 100 nM failed to yield any skipped product. Nonetheless, the data were truly encouraging as it supported our hypothesis in designing the bioresponsive α-TOC-S-S-AO construct in which the disulfide linkage possibly played an important role in the nucleus localization of AOs.
In addition, to further demonstrate the validity of our results, we synthesized two fluorescent constructs α-TOC-AO-FAM and α-TOC-S-S-AO-FAM in which we labeled the AOs with a fluorescent dye (6-carboxyfluorescein) at the 3′-end of the sequence (3′-FAM; Table 1). The purpose of this experiment was to visualize the localization of the AO part inside the cell nucleus after the disulfide cleavage. Toward this, we incubated the AOs with the cells at 400 and 800 nM concentrations, and the images were taken at 72 h time point. Remarkably, our results indicated that both AOs were able to internalize into various myotube cell nuclei at both concentrations (Figure 3). Quantification of the average fluorescence intensity in the nucleus (using ImageJ software) showed values of 72.1 and 56.1 for α-TOC-S-S-AO-FAM at 400 and 800 nM, respectively, in comparison to 20.5 and 40.3 of α-TOC-AO-FAM at 400 and 800 nM, respectively. Not surprisingly, this observation is in line with the exon skipping data.
Figure 3.
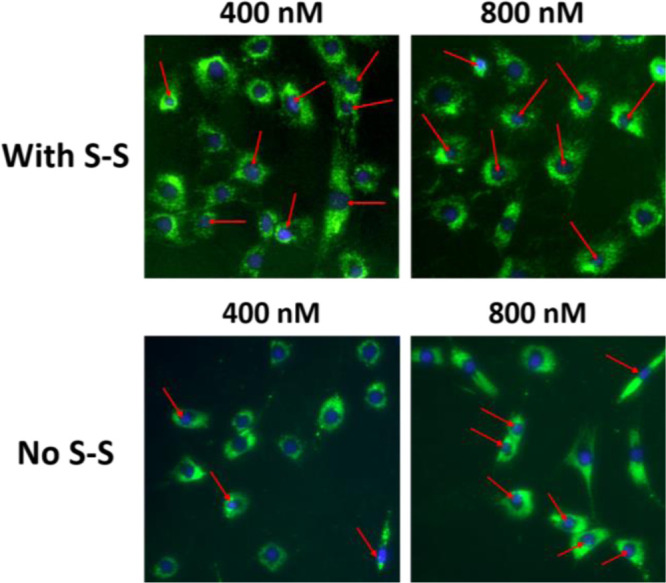
Fluorescence imaging analysis. Uptake of α-TOC-AO-FAM (No S-S) and α-TOC-S-S-AO-FAM (With S-S) inside the cell nucleus (green dots) at 72 h time point. Red arrows indicate cell nuclei with AO uptake.
3. Conclusions
In summary, our study has demonstrated the design and synthesis of a novel bioresponsive AO construct by conjugating α-TOC with a splice-modulating AO via a disulfide linkage (α-TOC-S-S-AO). The introduction of a bioreducible disulfide linkage certainly enhanced the uptake of the AO into the cell nucleus and therefore improved the exon 23 skipping potency in H2K mdx mouse myotubes. Based on our encouraging results, we firmly believe that these findings could certainly help toward establishing an efficient bioresponsive AO construct for inducing splice modulation.
4. Materials and Methods
4.1. Design and Synthesis of AOs
All 2′-OMe (A, C, G, and U) phosphoramidites, DMT-6-FAM phosphoramidite, and α-TOC (vitamin E) phosphoramidite were purchased from ChemGenes (USA). 5′-Thiol modifier C-6 disulfide modifier CED phosphoramidite was purchased from INNOVASYNTH Technologies (I) LTD (India). Glen UnySupportTM (Universal solid support) was purchased from Glen Research (USA).
Modified PS oligonucleotides were synthesized as DMT-off in 1 μmol scale on a Glen UnySupport using the AKTA oligopilot system. Standard procedures were used for the coupling of commercial 2′-OMe phosphoramidites, whereas α-TOC (vitamin E) phosphoramidite, DMT-6-FAM phosphoramidite, and 5′-thiol modifier C-6 disulfide modifier CED phosphoramidite were coupled with 5-(benzylthio)-1H-tetrazole (0.3 M) in CH3CN as an activator and an extended coupling time (20 min), the coupling efficiency was higher than 95% in all cases. Oligonucleotides were deprotected and cleaved from the solid support with 32% aq NH3 (1 mL) and left at 55 °C for 20 h. After deprotection, the oligonucleotides were desalted using an Illustra NAPTM-10 column (GE Healthcare). The purity of the final oligonucleotides was checked by ion-exchange chromatography using an HPLC system from Shimadzu on DNAPac PA200, a 250 × 4 mm analytical column. Buffers: [buffer A: MQ H2O; buffer B: 1 M NaClO4; and buffer C: 25 mM Tris-Cl, pH 8.0], flow rate: 1 mL min–1.
4.2. Cell Culture and Transfection
H-2Kb-tsA58 (H2K) mdx mouse myoblasts (provided by Prof. Sue Fletcher and Prof. Steve Wilton’s laboratory, Murdoch University, Australia) were cultured as described previously.21,31 Briefly, at 60–80% confluency, primary mdx myoblast cultures were treated with trypsin (Life Technologies) and seeded at a density of 2 × 104 cells/well into 24-well plates. The plate was pretreated with 50 μg/mL of poly-d-lysine (Sigma) and 100 μg/mL of Matrigel (Corning). Cultures were induced to differentiate into myotubes in Dulbecco’s modified Eagle’s medium containing 5% horse serum by incubation at 37 °C in 5% CO2 for 48 h. AOs were incubated directly with the cells, except the positive control which was complexed with Lipofectin (Life Technologies) at a ratio of 2:1 (Lipofectin/AO) and used in a final transfection volume of 500 μL/well in a 24-well plate as per the manufacturer’s instructions, except that the solution was not removed after 3 h.
4.3. RNA Extraction and RT-PCR
RNA was extracted from cells using the ISOLATE II RNA Mini Kit (Bioline) as per the manufacturer’s instructions. The dystrophin transcripts were then analyzed by nested RT-PCR across exons 20–26 as described previously.21 PCR products were separated on 2% agarose gels in Tris–acetate–EDTA buffer, and the images were captured on a Fusion Fx gel documentation system (Vilber Lourmat, Marne-la-Vallee, France). Densitometry was performed by ImageJ software.
4.4. Imaging of FAM AO Internalization
H2K mdx cells were cultured and differentiated as described previously. For internalization experiment, fluorescein (FAM)-labeled AOs were incubated at 400 and 800 nM concentrations in a final incubation volume of 500 μL/well in a 24-well plate. After 72 h of incubation, cell nuclei were stained with Hoechst for 10 min and washed five times with phosphate-buffered saline containing 10% fetal bovine serum before the images were captured using an Olympus TS-100 inverted fluorescence microscopy system.
Acknowledgments
We thank Prof. Steve Wilton and Prof. Sue Fletcher and their research group for providing H2K mdx cells.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01463.
RT-PCR and transcript densitometry analyses (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
R.N.V. acknowledges funding from the McCusker Charitable Foundation, Department of Health Western Australia Merit Award 2019 and the Perron Institute for Neurological and Translational Science. B.T.L. and T.R.K. acknowledge funding from Perron Institute for Neurological and Translational Science and Murdoch University.
The authors declare no competing financial interest.
Supplementary Material
References
- Stephenson M. L.; Zamecnik P. C. Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc. Natl. Acad. Sci. U.S.A. 1978, 75, 285–288. 10.1073/pnas.75.1.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C. A.; Castanotto D. FDA-Approved Oligonucleotide Therapies in 2017. Mol. Ther. 2017, 25, 1069–1075. 10.1016/j.ymthe.2017.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keam S. J. Inotersen: First Global Approval. Drugs 2018, 78, 1371–1376. 10.1007/s40265-018-0968-5. [DOI] [PubMed] [Google Scholar]
- Paik J.; Duggan S. Volanesorsen: First Global Approval. Drugs 2019, 79, 1349–1354. 10.1007/s40265-019-01168-z. [DOI] [PubMed] [Google Scholar]
- Kim J.; Hu C.; Moufawad El Achkar C.; Black L. E.; Douville J.; Larson A.; Pendergast M. K.; Goldkind S. F.; Lee E. A.; Kuniholm A.; Soucy A.; Vaze J.; Belur N. R.; Fredriksen K.; Stojkovska I.; Tsytsykova A.; Armant M.; DiDonato R. L.; Choi J.; Cornelissen L.; Pereira L. M.; Augustine E. F.; Genetti C. A.; Dies K.; Barton B.; Williams L.; Goodlett B. D.; Riley B. L.; Pasternak A.; Berry E. R.; Pflock K. A.; Chu S.; Reed C.; Tyndall K.; Agrawal P. B.; Beggs A. H.; Grant P. E.; Urion D. K.; Snyder R. O.; Waisbren S. E.; Poduri A.; Park P. J.; Patterson A.; Biffi A.; Mazzulli J. R.; Bodamer O.; Berde C. B.; Yu T. W. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. 10.1056/nejmoa1813279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo Y.-A. Golodirsen: First Approval. Drugs 2020, 80, 329–333. 10.1007/s40265-020-01267-2. [DOI] [PubMed] [Google Scholar]
- Juliano R. L. The delivery of therapeutic oligonucleotides. Nucleic Acids Res. 2016, 44, 6518–6548. 10.1093/nar/gkw236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonora G. M.; Ivanova E.; Zarytova V.; Burcovich B.; Veronese F. M. Synthesis and Characterization of High-Molecular Mass Polyethylene Glycol-Conjugated Oligonucleotides. Bioconjugate Chem. 1997, 8, 793–797. 10.1021/bc970082p. [DOI] [PubMed] [Google Scholar]
- Ng E. W. M.; Shima D. T.; Calias P.; Cunningham E. T.; Guyer D. R.; Adamis A. P. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat. Rev. Drug Discovery 2006, 5, 123–132. 10.1038/nrd1955. [DOI] [PubMed] [Google Scholar]
- Nair J. K.; Willoughby J. L. S.; Chan A.; Charisse K.; Alam M. R.; Wang Q.; Hoekstra M.; Kandasamy P.; Kel’in A. V.; Milstein S.; Taneja N.; O’Shea J.; Shaikh S.; Zhang L.; van der Sluis R. J.; Jung M. E.A.; Akinc A.; Hutabarat R.; Kuchimanchi S.; Fitzgerald K.; Zimmermann T.; van Berkel T. J.; Maier M. A.; Rajeev K. G.; Manoharan M. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J. Am. Chem. Soc. 2014, 136, 16958–16961. 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- Hoy S. M. Patisiran: First Global Approval. Drugs 2018, 78, 1625–1631. 10.1007/s40265-018-0983-6. [DOI] [PubMed] [Google Scholar]
- Amantana A.; Moulton H. M.; Cate M. L.; Reddy M. T.; Whitehead T.; Hassinger J. N.; Youngblood D. S.; Iversen P. L. Pharmacokinetics, Biodistribution, Stability and Toxicity of a Cell-Penetrating Peptide–Morpholino Oligomer Conjugate. Bioconjugate Chem. 2007, 18, 1325–1331. 10.1021/bc070060v. [DOI] [PubMed] [Google Scholar]
- Yang C.; Wu T.; Qi Y.; Zhang Z. Recent Advances in the Application of Vitamin E TPGS for Drug Delivery. Theranostics 2018, 8, 464–485. 10.7150/thno.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli F.; Azzi A.; Birringer M.; Cook-Mills J. M.; Eggersdorfer M.; Frank J.; Cruciani G.; Lorkowski S.; Özer N. K. Vitamin E: Emerging aspects and new directions. Free Radical Biol. Med. 2017, 102, 16–36. 10.1016/j.freeradbiomed.2016.09.017. [DOI] [PubMed] [Google Scholar]
- Evans H. M.; Bishop K. S. On The Existence Of A Hitherto Unrecognized Dietary Factor Essential For Reproduction. Science 1922, 56, 650. 10.1126/science.56.1458.650. [DOI] [PubMed] [Google Scholar]
- Jiang Q. Natural forms of vitamin E: metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radical Biol. Med. 2014, 72, 76–90. 10.1016/j.freeradbiomed.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina K.; Unno T.; Uno Y.; Kubodera T.; Kanouchi T.; Mizusawa H.; Yokota T. Efficient in vivo delivery of siRNA to the liver by conjugation of alpha-tocopherol. Mol. Ther. 2008, 16, 734–740. 10.1038/mt.2008.14. [DOI] [PubMed] [Google Scholar]
- Nishina T.; Numata J.; Nishina K.; Yoshida-Tanaka K.; Nitta K.; Piao W.; Iwata R.; Ito S.; Kuwahara H.; Wada T.; Mizusawa H.; Yokota T. Chimeric Antisense Oligonucleotide Conjugated to α-Tocopherol. Mol. Ther.--Nucleic Acids 2015, 4, e220 10.1038/mtna.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishina K.; Piao W.; Yoshida-Tanaka K.; Sujino Y.; Nishina T.; Yamamoto T.; Nitta K.; Yoshioka K.; Kuwahara H.; Yasuhara H.; Baba T.; Ono F.; Miyata K.; Miyake K.; Seth P. P.; Low A.; Yoshida M.; Bennett C. F.; Kataoka K.; Mizusawa H.; Obika S.; Yokota T. DNA/RNA heteroduplex oligonucleotide for highly efficient gene silencing. Nat. Commun. 2015, 6, 7969. 10.1038/ncomms8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara H.; Song J.; Shimoura T.; Yoshida-Tanaka K.; Mizuno T.; Mochizuki T.; Zeniya S.; Li F.; Nishina K.; Nagata T.; Ito S.; Kusuhara H.; Yokota T. Modulation of blood-brain barrier function by a heteroduplex oligonucleotide in vivo. Sci. Rep. 2018, 8, 4377. 10.1038/s41598-018-22577-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann C. J.; Honeyman K.; Cheng A. J.; Ly T.; Lloyd F.; Fletcher S.; Morgan J. E.; Partridge T. A.; Wilton S. D. Antisense-induced Exon Skipping and Synthesis of Dystrophin in the Mdx Mouse. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 42. 10.1073/pnas.98.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan J. E.; Beauchamp J. R.; Pagel C. N.; Peckham M.; Ataliotis P.; Jat P. S.; Noble M. D.; Farmer K.; Partridge T. A. Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev. Biol. 1994, 162, 486–498. 10.1006/dbio.1994.1103. [DOI] [PubMed] [Google Scholar]
- Le B. T.; Chen S.; Abramov M.; Herdewijn P.; Veedu R. N. Evaluation of anhydrohexitol nucleic acid, cyclohexenyl nucleic acid and d-altritol nucleic acid-modified 2’-O-methyl RNA mixmer antisense oligonucleotides for exon skipping in vitro. Chem. Commun. 2016, 52, 13467–13470. 10.1039/c6cc07447b. [DOI] [PubMed] [Google Scholar]
- Le B. T.; Filichev V. V.; Veedu R. N. Investigation of twisted intercalating nucleic acid (TINA)-modified antisense oligonucleotides for splice modulation by induced exon-skipping in vitro. RSC Adv. 2016, 6, 95169–95172. 10.1039/c6ra22346j. [DOI] [Google Scholar]
- Le B. T.; Murayama K.; Shabanpoor F.; Asanuma H.; Veedu R. N. Antisense oligonucleotide modified with serinol nucleic acid (SNA) induces exon skipping in mdx myotubes. RSC Adv. 2017, 7, 34049–34052. 10.1039/c7ra06091b. [DOI] [Google Scholar]
- Le B. T.; Adams A. M.; Fletcher S.; Wilton S. D.; Veedu R. N. Rational Design of Short Locked Nucleic Acid-Modified 2′-O-Methyl Antisense Oligonucleotides for Efficient Exon-Skipping In Vitro. Mol. Ther.--Nucleic Acids 2017, 9, 155–161. 10.1016/j.omtn.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le B. T.; Hornum M.; Sharma P. K.; Nielsen P.; Veedu R. N. Nucleobase-modified antisense oligonucleotides containing 5-(phenyltriazol)-2′-deoxyuridine nucleotides induce exon-skipping in vitro. RSC Adv. 2017, 7, 54542–54545. 10.1039/c7ra10964d. [DOI] [Google Scholar]
- Benizri S.; Gissot A.; Martin A.; Vialet B.; Grinstaff M. W.; Barthélémy P. Bioconjugated Oligonucleotides: Recent Developments and Therapeutic Applications. Bioconjugate Chem. 2019, 30, 366–383. 10.1021/acs.bioconjchem.8b00761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.; Fang Y.-Z.; Yang S.; Lupton J. R.; Turner N. D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- Guo X.; Cheng Y.; Zhao X.; Luo Y.; Chen J.; Yuan W.-E. Advances in redox-responsive drug delivery systems of tumor microenvironment. J. Nanobiotechnol. 2018, 16, 74. 10.1186/s12951-018-0398-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando T. A.; Blau H. M. Primary Mouse Myoblast Purification, Characterization, and Transplantation for Cell-Mediated Gene Therapy. J. Cell Biol. 1994, 125, 1275–1287. 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.