

Abstract

Increasing resistance to presently available antimalarial drugs urges the need to look for new promising compounds. The β-carboline moiety, present in several biologically active natural products and drugs, is an important scaffold for antimalarial drug discovery. The present study explores the antimalarial activity of a β-carboline derivative (1R,3S)-methyl 1-(benzo[d][1,3]dioxol-5-yl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (9a) alone in vitro against Plasmodium falciparum and in vivo in combination therapy with the standard drug artesunate against Plasmodium berghei. Compound 9a inhibited both 3D7 and RKL-9 strains of P. falciparum with half-maximal inhibitory concentration (IC50) < 1 μg/mL, respectively. The compound was nontoxic (50% cytotoxic concentration (CC50) > 640 μg/mL) to normal dermal fibroblasts. Selectivity index was >10 against both the strains. The compound exhibited considerable in vivo antimalarial activity (median effective dose (ED50) = 27.74 mg/kg) in monotherapy. The combination of 9a (100 mg/kg) and artesunate (50 mg/kg) resulted in 99.69% chemosuppression on day 5 along with a mean survival time of 25.8 ± 4.91 days with complete parasite clearance. Biochemical studies indicated the safety of the HIT compound to hepatic and renal functions of mice. Molecular docking also highlighted the suitability of 9a as a potential antimalarial candidate.
Introduction
Malaria is a serious issue, a vector-borne parasitic disease1−3 inflicting mortality,4 morbidity, and huge economic loss globally. The disease is caused by protozoan parasite of the genus Plasmodium. The World Health Organization (WHO) report estimated 228 million cases and 405 000 deaths from malaria in 2018. The African region accounts for 91% of all malaria deaths, followed by the WHO South East Asia region.4 In India, the states of Odisha, Jharkhand, Chhattisgarh, Madhya Pradesh, West Bengal, and North Eastern states contribute bulk of malaria cases.
The use of multifaceted malaria control interventions such as the use of insecticide-treated bed nets, indoor residual spraying, rapid diagnostic tests, and large-scale use of artemisinin-based combination therapy (ACTs) led to a reduction of malaria incidences over the past few decades. Artemisinin derivatives exert potent antimalarial activity against the blood stages of Plasmodium.5 Dihydroartemisinin, the common active metabolite, is a very potent and rapid-acting antimalarial drug; however, its short plasma elimination half-life results in the recrudescence of the parasite even after the initial rapid reduction in the parasitemia.6 Moreover, the WHO banned the monotherapy of artemisinin derivatives in 2006 and recommended the artemisinin-based combination therapy (ACTs) with other partner antimalarial drugs, having long plasma elimination half-life to prevent the development and spread of drug resistance.7
The emergence of multidrug-resistant strains of the parasite is posing a great threat to global malaria control and elimination efforts. There are reports on the replacement of local parasite populations with the artemisinin- and piperaquine-resistant KEL1/PLA1 co-lineage of Plasmodium falciparum, which has further diversified into six different subgroups in the Greater Mekong region and is spreading to the neighboring countries in South East Asia.8 In addition, decreased efficacy of artemisinin partner drugs is another emerging threat for the containment of malaria. For instance, piperaquine and mefloquine in ACT (artesunate-mefloquine and dihydro-artemisinin-piperaquine) regimens depicted decreased efficacy against P. falciparum in Cambodia.9,10 Since there is no clear evidence for emerging resistance for artemisinin and its derivatives, there is an urgent need to find newer alternatives from natural (traditional medicinal plants and their products) and synthetic sources (analogues of different moieties) to combat the dreaded disease. There is also a surged demand of novel artemisinin partner drugs, which can ensure complete parasite clearance by exhibiting different mechanisms of action, improved pharmacokinetic properties, difference in structure, and improved bioavailability that can boost the half-life of partner drug, thereby preventing the development and spread of drug resistance.
Alkaloids are widespread in nature and have been isolated from various sources. β-Carbolines are an important pharmacological class of compounds that belong to the indole alkaloids and have been implicated in a wide variety of biological activities and represent an important structure in several marketed drugs as well as in potential drug candidates. Likewise, this scaffold also holds significant features for the discovery of potent antimalarial drugs.11 β-Carboline alkaloids possess a tricyclic pyrido[3,4-b]indole ring structure and are derived from the amino acid tryptophan or tryptamine derivatives. The antiplasmodial activity of β-carbolines has been illustrated in various studies, for instance, ochlorifuanine A (1), a tetrahydro-β-carboline (THβC) alkaloid isolated from Strychnos species, exhibited antimalarial activity in the nanomolar range (Figure 1).12 Fascaplycin (2), a pentacyclic β-carboline alkaloid, showed half-maximal inhibitory concentration (IC50) of 168 nM against P. falciparum K1 strain.13 Similarly, harmine (3) was isolated from the seeds of Penganum harmala.14 Cipargamin (4), a spiroindole derivative of β-carboline, is currently undergoing phase II clinical trial and exhibited potent antiplasmodial activity in the in vitro, ex vivo, and in vivo assays.15 (1R,3S)-MMV008138 (5), a potent antimalarial agent, is known to specifically inhibit 2-C-methyl-d-erythritol 4-phosphate cytidylyltransferase enzyme (PfIspD) of P. falciparum and does not interfere with the function of human IspD.16 Similarly, compounds 6–8 are the synthetic β-carboline derivatives displaying promising antiplasmodial efficacy.17−19 Further, in vitro antimalarial activity of various natural β-carboline alkaloids has been reviewed in detail by Ashok et al.20 It has been hypothesized that β-carboline alkaloids inhibit DNA synthesis through intercalation of DNA base pairs, and therefore compounds containing this scaffold can inhibit parasite growth by interfering with the parasite DNA synthesis. Gellis et al. reported significant antimalarial activity (0.7 < IC50 < 1.7 mM) of semisynthetic β-carboline derivatives against the W2 multidrug-resistant strain of P. falciparum.21
Figure 1.

Naturally occurring and synthetic β-carboline antimalarials.
On the basis of antimalarial activities of various β-carboline derivatives, in our previous study,22 we reported the antimalarial efficacy of some C1-benzodioxole-, pyridyl-, thienyl-, n-octyl-substituted THβCs (9a, 9b, 10, 11a, 11b, 12a, 12b, 13a, and 13b), as shown in Figure 2. These derivatives were synthesized via the classical Pictet–Spengler reaction of l-tryptophan methyl ester with various aldehydes such as piperonal, 3-pyridine carboxaldehyde, 2-thiophene carboxaldehyde, and nonal.
Figure 2.

Structures of previously published C1- and C3-substituted tetrahydro-β-carboline (THβC) derivatives.
One compound, (1R,3S)-methyl 1-(benzo[d][1,3]dioxol-5-yl)-2,3,4,9-tetrahydro-1H-pyrido[3,4-b]indole-3-carboxylate (9a), was identified as an advanced HIT, which exhibited the best activity against the rodent malaria parasite with enhancement of the mean survival time (MST) of the treated mice. As the identification of some novel, effective partner drug with artemisinin derivatives is the need of the hour, we attempted to test and cross-investigate the antiplasmodial efficacy of compound 9a against both chloroquine-resistant and -sensitive strains of P. falciparum and in vivo in combination with artesunate (AS) against lethal murine malaria [Plasmodium berghei (NK-65)].
In addition, an in silico approach was also employed to observe the interaction of β-carboline derivatives and artesunate with different enzymes essential for parasite metabolism to identify its probable mechanism of action. Keeping in mind the problem of drug resistance, we focused on multitargeting, which is an innovative and promising strategy.23 Hence, six different enzymes of P. falciparum were selected for docking studies. For instance, phosphoenolpyruvate carboxylase (PEPC) and phosphoethanolamine methyltransferase (PMT) are absent in mammals; however, the former has a promising role in CO2 fixation in P. falciparum, as studied by Storm et al., making the parasite enzyme a feasible drug target of interest,24 and the latter is essential for the biosynthesis of phosphotidylcholine of bio-lemma at sexual and asexual stages of Plasmodium.25 Likewise, lactate dehydrogenase (LDH) enzyme aids in the interconversion of pyruvate to lactate in glycolysis and is thus considered as an essential molecular target for the development of antimalarials.26 Cytosolic malate dehydrogenase (MDH) in P. falciparum helps in the transport of metabolites to the mitochondria and is known to play a role in the generation of reducing equivalents to feed the respiratory chain.27 On the other hand, falcipain-2 (FP2) and falcipain-3 (FP3) are cysteine proteases that catalyze the degradation of hemoglobin into hemozoin and are also targets for drugs/inhibitors.28−30
Results and Discussion
In Vitro Antiplasmodial Activity against P. falciparum
The synthesized tetrahydro-β-carboline (THβC) derivatives were tested for their in vitro antiplasmodial efficacy against 3D7 (chloroquine-sensitive) and RKL-9 (chloroquine-resistant) strains of P. falciparum. The compounds showed moderate to potent antimalarial activity. It was observed that the antimalarial potency of the compounds decreases as the pyridine ring at the C1 position of THβC 11b (IC50 4.82 μg/mL for 3D7 and 5.22 μg/mL for RKL-9) was replaced with the thiophene ring 12b (IC50 6.84 μg/mL for 3D7 and 8.66 μg/mL for RKL-9), whereas insertion of 1,3-dioxolane ring in the aromatic ring at C1 position enhanced the potency of THβC 9b (IC50 3.25 μg/mL for 3D7 and 4.01 μg/mL for RKL-9). Aromatization of compound 9b to 10 also hampered the observed antimalarial activity (IC50 7.16 μg/mL for 3D7 and 9.04 μg/mL for RKL-9). The antimalarial activity of THβC also decreases when the C1-aromatic ring was replaced with the n-octyl aliphatic hydrocarbon chain 13b (IC50 11.24 μg/mL for 3D7 and 13.84 μg/mL for RKL-9). It was interesting to note that stereochemistry at the C1 position of THβC with respect to the C3 position played a crucial role in antimalarial activity; therefore, as we moved from cis-diastereomers (9b, 11b, 12b, 13b) to trans-diasteromers (9a, 11a, 12a, 13a), the parasite inhibitory potency of THβCs increased. Overall, compound 9a (trans-diastereomer) exhibited IC50 < 1 μg/mL against both 3D7 (chloroquine-sensitive) and RKL-9 (chloroquine-resistant) strains, whereas for 9b (cis-diastereomer), IC50 was >1 μg/mL for both the strains. Similarly, the antimalarial activity of trans isomers 11a (IC50 3.07 μg/mL for 3D7 and 2.12 μg/mL for RKL-9), 12a (IC50 4.50 μg/mL for 3D7 and 6.95 μg/mL for RKL-9), and 13a (IC50 8.80 μg/mL for 3D7 and 9.79 μg/mL for RKL-9) was greater than that of their corresponding cis isomers 11b, 12b, and 13b. Further, the activity of 9a against both the strains of P. falciparum was observed to be dose-dependent and more than 75% inhibition was observed against both 3D7 and RKL-9 strains at the lowest tested concentration (1 μg/mL) (Figure 3). The standard drugs artesunate (2 μM) and chloroquine (10 μM) exhibited 100 and 96.75% inhibition, respectively, for both the strains (Figure 4). According to Lekana-Douki et al., any compound or drug exhibiting IC50 < 5 μg/mL in vitro is categorized as an active antimalarial, promising activity (IC50 between 5 and 15 μg/mL), moderately antimalarial (IC50 15–50 μg/mL), and inactive (IC50 > 50 μg/mL).31 Therefore, 11a, 9a, and 9b can be classified as highly active molecules against both resistant and sensitive strains of P. falciparum, which is also supported by in silico studies (Figure 12d).
Figure 3.

Percent schizont maturation inhibition of RKL-9 and 3D7 strains of P. falciparum on treatment with various concentrations of 9a (concentration ranges from 1 to 16 μg/mL, i.e., 2.85–45.69 μM).
Figure 4.
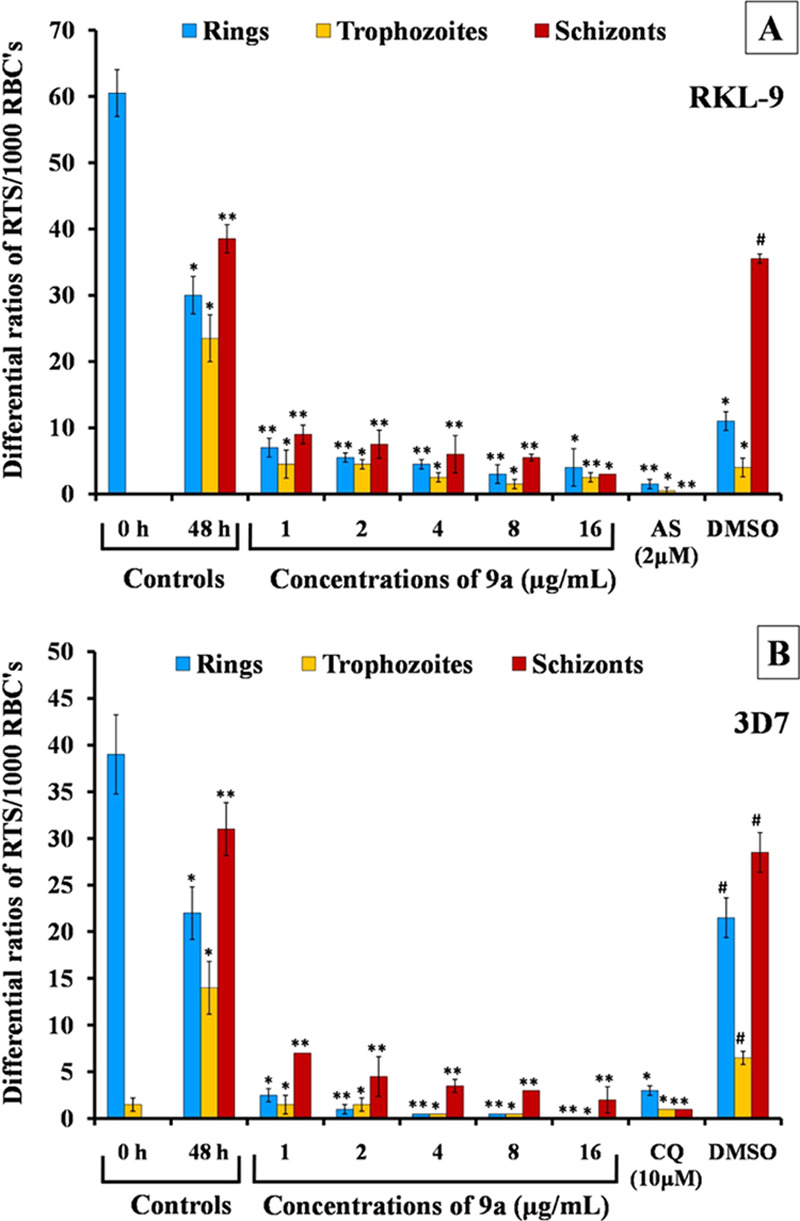
Differential R, T, S count after 48 h incubation on treatment with 9a in comparison to 0 h.
Figure 12.

Three-dimensional (3D) representations depicting molecular interactions of 9a with amino acid residues of 3BPM (a), Q8ILJ7 (b), and 1LDG (c); docking score of various ligand–receptor complexes (d); and details of docking parameters including ligand pose and forces involved in interaction with active amino acid residues (e).
In Vitro Cytotoxicity Assay
The evaluation of both in vitro and in vivo toxicity of any compound and calculation of its selectivity index (SI) are critical before it can be considered as a potential drug candidate.32 Cell toxicity primarily determines the range or dose of test substances (chemical/standard drugs) at which it starts killing the cells. HeLa cells are normally employed to find the toxicity profile. A big advantage of using cancer cell lines is that they provide progeny of cells with intact morphology (same genotype) and anatomical features, if passage is within limited ranges (20 passages). Besides, these cells are in constant rate of division and provide more targets in terms of expressing various receptors on the bio-lemma and presence of microtubules/filaments and metabolic factors, and are hence are vulnerable to chemicals/drugs.33,34 Compound 9a was slightly toxic to HeLa cells. The 50% cytotoxic concentration (CC50) was calculated to be 10 μg/mL.
On the contrary, 9a exhibited no toxicity toward normal (dermal fibroblasts) cells. Percent cell viability was observed to be 56.51 ± 5.07% even at a high concentration of 640 μg/mL (Figure 5). CC50 was calculated to be >640 μg/mL against the normal dermal fibroblasts. The selectivity of action of the different agents can be attributed to the fact that normal cells have distinct physiological or biochemical characteristics with the least above-mentioned attributes compared to the cancer cell line that influences drug actions.35 The results of cytotoxicity studies indicate the safety of the compound for human use.
Figure 5.

Percent viability of HeLa cells and normal dermal fibroblasts upon treatment with various concentrations of 9a (10–640 μg/mL).
The selectivity index (SI) of 9a was >10 for both the 3D7 and RKL-9 strains of P. falciparum. Therefore, according to the criteria given by Valdés et al., which classifies drugs/compounds possessing a high selectivity index (SI > 10) as highly efficacious antimalarial agents without general cytotoxic activity,36 compound 9a can be established as an active antimalarial.
Determination of Nitric Oxide
Nitric oxide has a diversified and multifunctional role in the host defense against infection. It is also involved in the process of platelet aggregation, immunoregulation, and signal transduction.37 However, its role in malaria is controversial. The nitrite production from macrophages treated with 9a (100 μg/mL) as well as artesunate (2 μM) was comparable to the normal cells. A slight increase in the NO concentration was evident at a higher concentration at 40 h (Figure 6). Even combination of 9a + AS did not stimulate the macrophages to seep NO into the media though after a prolong exposure (40 h) a slight rise was evidenced. These findings illustrate that drugs do not aid directly in starting off the immune response.
Figure 6.

NO production by macrophages upon treatment with various concentrations of 9a (100–200 μg/mL) and its combination with artesunate (2 μM).
In Vivo Suppressive Activity against P. berghei
After initial assessment of antimalarial activity and safety in the in vitro systems, it is essential to explore the antiplasmodial efficacy in vivo. Studies have shown the results of in vitro activity to be misleading in certain cases, wherein compounds exhibiting promising efficacy in vitro do not exhibit in vivo efficacy.32 Also, in vitro analysis aids in the initial screening of compounds, but confirmation of the observed efficacy is done by in vivo studies. The acute toxicity studies of 9a (LD50 > 4 g/kg) indicated its toxicological safety for oral administration at higher doses.22 The compound was further evaluated for its antimalarial potential in vivo in monotherapy to confirm our previous findings, and lower doses were also tested to determine the median effective dose (ED50) and its efficacy in combination therapy with artesunate. Rodent malaria parasite species are well-established experimental models for testing the efficacy of novel drug candidates.38P. berghei NK-65 was therefore used as an experimental model in the present study. In addition to being a lethal (not to human), the NK-65 strain of P. berghei also possesses a strong preference for invading the reticulocytes in contrast to P. bergheiANKA; thus, it also exhibits some resemblance with Plasmodium vivax.22 However, as the migration of the parasite in the reticulocytes causes variability in regular course of infection of the parasite, the authors attempted to restrict the parasite invasion into RBCs through regular passages, thereby providing uniformity in the results.
Compound 9a exhibited a dose-dependent inhibition of P. berghei infection on day 5. A chemosuppression of 91.19% was evident at a dose of 100 mg/kg in monotherapy (Table 1). The effective dose (ED50) of the compound was 27.74 mg/kg in monotherapy. A very low parasitemia level ranging between 2 and 3% was observed in the surviving mice (50 and 100 mg/kg) on day 28 post infection. Treatment with artesunate (4 mg/kg) resulted in parasite clearance on day 5 with 94.01% chemosuppression, but parasite recrudescence was evident, leading to mortality of the treated animals. However, at a higher dose (50 mg/kg), parasite recrudescence was observed in a few animals and only two mice survived till day 28 with complete parasite clearance.
Table 1. Percent Chemosuppression in Mice after Treatment with Various Concentrations (10–100 mg/kg) of 9a in Monotherapya.
| monotherapy groups | ||||
|---|---|---|---|---|
| groups, n = 5 | dose (D0–D3) (0.2 mL/(mouse OD oral)) D0—i.p. inoculation (1 × 106P. berghei-infected RBCs) | parasitemia (%), day 5 | chemosuppression (%), day 5 | parasitemia (%), day 28 |
| G1 | normal control | |||
| G2 | infected control | 29.09 ± 7.95 (n = 5) | – (n = 0) | |
| G3 | vehicle control | 30.7 ± 5.22 (n = 5) | ||
| G4 | 9a (10 mg/kg) | 17.31 ± 2.40** (n = 5) | 40.49 | – (n = 0) |
| G5 | 9a (20 mg/kg) | 15.47 ± 2.69** (n = 5) | 46.82 | – (n = 0) |
| G6 | 9a (50 mg/kg) | 10.25 ± 4.93*** (n = 5) | 64.76 | 3.43 ± 0.51 (n = 2) |
| G7 | 9a (100 mg/kg) | 2.56 ± 1.71*** (n = 5) | 91.19 | 2.41 ± 0.97 (n = 3) |
| G8 | artesunate (4 mg/kg) | 1.74 ± 0.83*** (n = 5) | 94.01 | – (n = 0) |
| G9 | artesunate (50 mg/kg) | 0.03 ± 0.01*** (n = 5) | 99.89 | 0 ± 0 (n = 2) |
| G10 | chloroquine (10 mg/kg) | 1.68 ± 1.06*** (n = 5) | 94.22 | 0 ± 0 (n = 5) |
Data are presented as mean ± standard deviation (SD) for five mice per group. n = total no. of mice in each group. p-Value in comparison to control is shown as ***p < 0.0005, extremely statistically significant; **p < 0.005, very statistically significant.
In the 9a-treated groups, mortality was evident at lower doses (10 and 20 mg/kg); however, at 50 and 100 mg/kg, a mean survival time (MST) of 21.8 ± 6.09 and 22.6 ± 7.79 days was observed, which was significant (p < 0.0005) compared to the infected control (6.8 ± 0.83 days) (Figure 7). In the artesunate 4 mg/kg-treated mice, MST of 18.6 ± 2.19 days was evident, whereas at a dose of 50 mg/kg, an MST of 22.4 ± 5.77 days was observed.
Figure 7.

Percent survival of mice treated with different concentrations (10–100 mg/kg) of 9a in monotherapy.
These observations highlight the potential of 9a to inhibit parasite development as well as to enhance the mean survival time of the treated animals. Also, these findings are similar to the observations made in previous studies showing parasite recrudescence after administration of artesunate in monotherapy due to the short half-life of the drug.6
The combination of the compound 9a (100 mg/kg) with artesunate was tested at two different doses (4 and 50 mg/kg) of the drug. At low doses of artesunate (4 mg/kg), though 90.89% chemosuppression was evident on day 5, parasite recrudescence resulted in the death of treated mice by day 20 post inoculation. When 9a was administered in combination with higher dose of artesunate (50 mg/kg), considerable reduction in the parasitemia levels was observed with 99.69% chemosuppression on day 5 (Table 2). On day 28, complete parasite clearance was observed in the surviving mice in this group (G5) with no recrudescence in the surviving mice.
Table 2. Percent Chemosuppression in Mice after Treatment with 9a in Combination Therapy with Artesunatea.
| combination
therapy groups | ||||
|---|---|---|---|---|
| groups, n = 5 | dose (D0–D3) (0.2 mL/(mouse OD oral)) D0—ip inoculation (1 × 106P. berghei-infected RBCs) | parasitemia (%), day 5 | chemosuppression (%), day 5 | parasitemia (%), day 28 |
| G1 | normal control | |||
| G2 | infected control | 29.09 ± 7.95 (n = 5) | – (n = 0) | |
| G3 | vehicle control | 30.7 ± 5.22 (n = 5) | – (n = 0) | |
| G4 | 9a (100 mg/kg) + artesunate (4 mg/kg) | 2.65 ± 2.44*** (n = 5) | 90.89 | – (n = 0) |
| G5 | 9a (100 mg/kg) + artesunate (50 mg/kg) | 0.09 ± 0.13*** (n = 5) | 99.69 | 0 ± 0 (n = 4) |
| G6 | artesunate (50 mg/kg) + sulfadoxine (25 mg/kg) – pyrimethamine (1.2 mg/kg) | 2.46 ± 0.1*** (n = 5) | 91.54 | 0 ± 0 (n = 5) |
Data are presented as mean ± SD for five mice per group. n = total no. of mice in each group. p-Value in comparison to control is shown as ***p < 0.0005, extremely statistically significant.
The group treated with combination of 9a (100 mg/kg) with a lower dose of artesunate (4 mg/kg) recorded a mean survival time of 19.2 ± 2.48 days. The combination of 9a (100 mg/kg) + AS (50 mg/kg) exhibited an MST of 25.8 ± 4.91 days (Figure 8), which was significant (p < 0.0005) compared to the infected control (G2) that died by day 6 post inoculation. According to the criteria given by Muñoz et al., any compound exhibiting ED50 < 100 mg/kg is classified as an active antimalarial.39 Hence, 9a can be categorized as a very good antimalarial on the basis of its in vivo antiplasmodial efficacy. Also, the combination of the compound with artesunate improved the efficacy of drug action with rapid parasite clearance, no recrudescence, and enhancement of the host’s survival. This further illustrates the suitability of 9a as a partner drug with artesunate
Figure 8.

Percent survival of mice treated with different concentrations (10–100 mg/kg) of 9a in combination therapy.
Measurements of Basic Parameters
Some physical signs of illness were seen in mice treated with lower doses (10 and 20 mg/kg) of the compound in monotherapy (Table 3), but these symptoms were not evident in mice treated with 50 and 100 mg/kg of the compound along with the positive control (PC). In the group treated with combination of 9a (100 mg/kg) + AS (4 mg/kg), some changes were observed along with the mortality of the treated animals. However, the combination of 9a (100 mg/kg) + AS (50 mg/kg) did not show any physical signs of illness. The morphometric features observed in the dose-administered surviving mice [no signs of pale yellow skin coat, dark urine, and lethargy, but a slight decrease in body weight was (data not mentioned) recorded] provide further evidence that treatment with the combination eliminated parasite in the early phase of infection in contrast to negative control (NC) that succumbed to the disease.
Table 3. Physical Signs of Illness in 9a-Treated Mice in Monotherapy and in Combination with Artesunatea.
| NC | monotherapy 9a |
combination therapy |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| clinical symptoms appeared in experimental animals | DPI | NC | 10 mg/kg | 20 mg/kg | 50 mg/kg | 100 mg/kg | PC | 9a + AS 4 mg/kg | 9a + AS 50 mg/kg | PC |
| behavioral lethargy | 5 | ++ | ++ | + | – | – | – | + | – | – |
| 7 | +++ | +++ | +++ | – | – | – | ++ | – | – | |
| 14 | ××× | ××× | ××× | + | – | – | +++ | – | – | |
| 21 | ××× | ××× | ××× | – | – | – | ××× | – | – | |
| 28 | ××× | ××× | ××× | – | – | – | ××× | – | – | |
| reduction in movement | 5 | ++ | ++ | + | – | – | – | + | – | – |
| 7 | +++ | +++ | +++ | – | – | – | ++ | – | – | |
| 14 | ××× | ××× | ××× | – | – | – | +++ | – | – | |
| 21 | ××× | ××× | ××× | – | – | – | ××× | – | – | |
| 28 | ××× | ××× | ××× | – | – | – | ××× | – | – | |
| passage of dark urine | 5 | ++ | ++ | + | – | – | – | + | – | – |
| 7 | +++ | +++ | ++ | – | – | – | ++ | – | – | |
| 14 | ××× | ××× | ××× | – | – | – | +++ | – | – | |
| 21 | ××× | ××× | ××× | – | – | – | ××× | – | – | |
| 28 | ××× | ××× | ××× | – | – | – | ××× | – | – | |
| impaired consciousness, convulsions, and seizures | 5 | – | – | – | – | – | – | – | – | – |
| 7 | – | – | – | – | – | – | – | – | – | |
| 14 | ××× | ××× | ××× | – | – | – | – | – | – | |
| 21 | ××× | ××× | ××× | – | – | – | ××× | – | – | |
| 28 | ××× | ××× | ××× | – | – | – | ××× | – | – | |
(−): absent; (+): mild; (++): moderate; (+++): severe; (×××): mortality; DPI: day post inoculation; NC: negative control; PC: positive control.
Differential Leukocyte Count (DLC)
The differential leukocyte count in P. berghei-infected mice reflected an increasing trend of immune cells along with rise in infection (Figure 9). The rise in the percentage of macrophages and neutrophils was evident first followed by the lymphocytes at a later stage. In mice administered with 9a (100 mg/kg) and AS (50 mg/kg), the number of all of the three immune cells was elevated from D5 onward, which might be contributing to the parasite clearance (Figure 9). The increased percentage of macrophages might trigger the immune response in terms of activation of lymphocytes and neutrophils. Instead of acquired immune response, innate immune cells play a pivotal role here.
Figure 9.

Differential leukocyte count in mice infected with 1 × 106P. berghei-infected erythrocytes along with the course of infection.
We also intended to focus on innate immune response while performing the experiments, and it was observed to be raised in mice treated with the current combination. Two things happen to reach such a threshold value of immune response: first, host exposed to repeated parasite infection over the years, but the demerit is that such activity promotes the blood stage infection and death is inevitable, and second, it is suitable to make such combinations that act in many ways, for instance, one therapeutic drug kills the parasite with its habitat and allows to recognize and trigger the antigen-presenting cells (APCs), activates M1/M2 macrophages, and if feasible, generates antibodies, and the other partner drug fills the voids of therapeutic drug such as low bioavailability, less plasma concentration, etc. The role of immunity has been illustrated by Stevenson and Riley, where mice depleting with interferon (IFN)-γ, NK-cells, B cell, and CD4+ cells confirmed the surge of parasitemia.40 Hence, it was noteworthy for us to look upon the increase in the number of immune cells (lymphocytes, neutrophils, and macrophages) in infected and treated groups in contrast to the normal ones.
In negative control, as the parasitemia moves up, three immune cells (macrophages, neutrophils, and lymphocytes) are raised, but the high parasite burden contributes to pathogenesis (anemia, metabolic acidosis), which ultimately led to the death of mice (Figure 10). It is well versed that in malaria, immunity is species- and stage-specific and depends upon antigenic variations, exposure of merozoites to antigen-presenting cells (APCs), recognition of schizonts via FC-receptor and CD36, etc.40 To cure the host, immunity ought to reach the threshold value, which is certainly possible when parasitemia reaches up to 20% in infected mice notwithstanding the above-mentioned attributes. Furthermore, in treated mice, it is probably hypothesized that after the drug treatment, dead merozoites are captured by APCs and presented to Th1 cells on one side that leads to the generation of IgG and IgM antibodies, and on the other hand, M1 macrophages initiate phagocytosis via complement activation as observed on day 21 and day 28, where lymphocytes were more in number successively followed by neutrophils and macrophages.
Figure 10.

Differential leukocyte count in mice treated with combination therapy of 9a (100 mg/kg) with artesunate.
Biochemical Assays
Biochemical assays were performed to assess the effect of drug treatment on the liver and kidney functions of the rodent host. Liver function tests (LFTs) are a mandatory screening tool to discern hepatic dysfunction. There are disparate sorts of tests, for instance, serum enzyme tests (for injury to hepatocytes) and for the biosynthetic capacity. But, in the current study, priorities are given only to those enzymes that are rather important in malaria and results can be compared to the percent parasitemia. Since Plasmodium spp. infects the parenchymal cells of host (either mice or man), merozoites accumulate in liver cells and starts perturbating the cells. The more the accumulation, the higher would be the elevation, thus engendering acute liver injuries (ALIs). Therefore, various enzymes seep into the blood, which can be easily detected using diagnostic kits.41 Therefore, SGOT, SGPT, and alkaline phosphatase (ALP) are good indicators of hepatocellular necrosis. SGOT and SGPT are main enzymes that transfer an amino group of aspartate and alanine to the keto group of ketoglutaric acid and exhibit elevated levels in malaria-infected mice in contrast to normal mice, depicting acute liver injury.42,43 Likewise, alkaline phosphatase (ALP) is also elevated in infected mice as it exuded from hepatic cells in bloodstream, which significantly illustrated parasite burden on liver. In malaria-induced anemia, heme is released after the hemolysis (IRBCs/NRBCs), which in turn converts into bilirubin (endogenous anion). Plasma levels of bilirubin have been reported to be increased in malaria infection.44 This elevation is multifactorial and can be due to the excessive destruction of the parasitized erythrocytes,45 hepatocellular damage, biliary tract obstruction, hemolysis, and jaundice.46 The observations made in the present investigation are in accordance with the aforementioned studies as the levels of ALP (152.55 ± 9.44 IU/L), SGOT (143.8 ± 5.31 IU/L), SGPT (106.79 ± 4.87 IU/L), and bilirubin (1.56 ± 0.28 mg/dL) were significantly (p < 0.0005) elevated in the untreated control due to high parasite burden. However, in the 9a (50 and 100 mg/kg)-treated groups, the levels were within normal range on day 10 and on day 28 (Figure 11A–D). In the group treated with the combination of 9a (100 mg/kg) + AS (50 mg/kg), also the ALP (96.65 ± 1.05 IU/L), SGOT (65.73 ± 1.83 IU/L), SGPT (82.66 ± 3.47 IU/L), and bilirubin (0.65 ± 0.2 mg/dL) levels were within the normal range (Table 4), indicating safety of the combination to the hepatic function of the host.
Figure 11.

Liver [ALP (A), bilirubin (B), SGOT (C), SGPT (D)] and kidney [urea (E), creatinine (F)] function tests in mice treated with 9a in monotherapy on D10 and D28. Data are presented as mean ± SD for five mice per group. p-Value in comparison to the infected control is shown as ***p < 0.0005, extremely statistically significant; **p < 0.005, very statistically significant; *p < 0.05, statistically significant; #p > 0.05, not statistically significant.
Table 4. Liver and Kidney Function Tests in Mice Treated with Combination of 9a and Artesunate on D10 and D28a.
| groups, n = 5 | dose (D0–D3) (0.2 mL/(mouse OD oral)) | ALP (IU/L) | bilirubin (mg/dL) | SGOT (IU/L) | SGPT (IU/L) | urea (mg/dL) | creatinine (mg/dL) |
|---|---|---|---|---|---|---|---|
| G1 | normal control | 90.41 ± 6.51 | 0.43 ± 0.14 | 38.40 ± 4.94 | 68.72 ± 8.05 | 28.68 ± 3.75 | 0.68 ± 0.3 |
| G2 | infected control | 152.55 ± 9.44*** | 1.56 ± 0.28*** | 143.8 ± 5.31*** | 106.79 ± 4.87*** | 45.21 ± 5.83*** | 1.92 ± 0.21*** |
| G3 | vehicle control (SSV) | 165.6 ± 7.87*** | 1.67 ± 0.20*** | 148.56 ± 4.28*** | 100.58 ± 4.75*** | 40.97 ± 5.17*** | 1.78 ± 0.31*** |
| G4 | 9a (100 mg/kg) + AS (4 mg/kg) | 104.62 ± 0.57*** | 0.97 ± 0.08** | 97.37 ± 2.03*** | 95.03 ± 1.61*** | 37.44 ± 1.65* | 1.02 ± 0.01*** |
| G5 | 9a (100 mg/kg) + AS (50 mg/kg) | ||||||
| day 10 | 96.65 ± 1.05*** | 0.65 ± 0.21*** | 65.73 ± 1.83*** | 82.66 ± 3.47*** | 25.51 ± 2.92*** | 0.81 ± 0.13*** | |
| day 28 | 94.51 ± 2.22 | 0.41 ± 0.02 | 83.19 ± 0.30 | 76.07 ± 2.92 | 31.05 ± 1.34 | 0.67 ± 0.04 | |
| G6 | AS + SP | ||||||
| day 10 | 93.21 ± 4.1*** | 0.33 ± 0.04** | 93.56 ± 4.90*** | 43.49 ± 1.6*** | 26.3 ± 0.75*** | 0.5 ± 0.13*** | |
| day 28 | 95.2 ± 9 | 0.33 ± 0.1 | 80.11 ± 2.84 | 25.93 ± 1.6 | 23.15 ± 1.1 | 0.4 ± 0.2 |
Data are presented as mean ± SD for five mice per group. n = total no. of mice in each group. p-Value in comparison to the infected control is shown as ***p < 0.0005, extremely statistically significant; **p < 0.005, very statistically significant; *p < 0.05, statistically significant.
Likewise, serum levels of creatinine and urea are of immense clinical importance for the assessment of kidney function. High levels indicate the renal inability to filter out creatinine and urea, which corresponds to the deposition of hemozoin in kidneys and renal impairment that further the apparent low glomerular filtration rate (GFR), the imbalance of electrolyte ratio, and acute renal failure (ARF).47 Taking together, creatinine (1.92 ± 0.21 mg/dL) and urea (45.21 ± 5.83 mg/dL) levels were found to be higher in negative control as high parasitemia results in impairment of renal function. However, in the groups treated with 9a in monotherapy (100 mg/kg) (Figure 11E,F) and in combination 9a (100 mg/kg + AS 50 mg/kg) (Table 4), the levels of these renal function biomarkers were within the normal range, indicating the safety of the compound to the kidneys of the rodent host. The results show that the treated mice exhibited less acute hepatic and renal injuries, which may be due to the reduction in parasite burden.43,48
Ligand Docking
Recent drug discovery approaches for combating malaria consist of combination therapy mainly to reduce the side effects and drug resistance of a single drug. Further research findings indicated the role of enlisted six enzymes [LDH (PDB ID: 1LDG), PMT (PDB ID: 3UJ9), MDH (PDB ID: 5NFR), FP3 (PDB ID: 3BPM), FP2 (PDB ID: 3BPF), and PEPC (PDB ID: Q8ILJ7)] involved directly and indirectly in the pathogenesis of malaria. In the current study, efforts have been made using known molecular docking tools to identify potential β-carboline derivative that showed binding interactions toward multiple proteins involved and can be considered for combination therapy. Artesunate drug is well reported for its mode of action in the previous studies with respect to artesunate combination therapy for malaria, but this is the first time that artesunate has been reported for its potential binding interaction to any of the six proteins under consideration.
Docking results indicated that all of the compounds (9a, 9b, 10, 11a, 11b, 12a, 12b, 13a, 13b) and artesunate exhibited a wide range of binding affinities to all of the receptors, as shown in Figure 12d. In drug–receptor interaction, the lowest D-scoring reflects the binding affinity, compactness, and stabilization of the different ligands toward a particular receptor.
High affinity has been indicated by all of the molecules for PMT; PDB ID: 3UJ9 (−51.57 to −63.10 kcal/mol) and FP3; PDB ID: 3BPM (0.09 to −46.76 kcal/mol). In contrast to weak affinity by almost all of the compounds against MDH; PDB ID: 5NFR, reasonably good affinity was observed for PEPC; amino acid sequences Q8ILJ7 (8.05 to −37.44 kcal/mol); LDH; PDB ID: 1LDG (−23.59 to −59.14 kcal/mol), and FP2; PDB ID: 3BPF (−30.74 to −60.32 kcal/mol), as indicated in Figure 12d.
The cumulative observations of in vitro and in vivo studies rendered us potential molecule 9a, with best activity against malaria parasite, as indicated by its increased mean survival time in the treated mice. A high negative D score, and thus high affinity, has been indicated by 9a toward three proteins: 3BPM −34.27 kcal/mol, Q8ILJ7 −37.44 kcal/mol, and 1LDG −59.14 kcal/mol, in comparison to artesunate (−8.64, −8.24, and −51.92 kcal/mol, respectively), and the observations are in agreement with in vitro studies. These high scores of 9a can be considered on account of the steady and strong interactions including hydrogen, hydrophobic, and van der Waals forces, as specified in Figure 12e. Consistent with our binding hypothesis, 9a was found to be engaged with different amino acid residues through hydrogen-bond interaction with Q8ILJ7 (GLU 173 Å 2.297 Å), 1LDG (PRO184A 2.581 Å), 3UJ9 (ARG 179B 2.503), and 3BPF (LYS 160A 2.332Å), except for the 9a–3BPM complex, where no hydrogen bond was observed. Figure 12e reflects the hydrogen, hydrophobic, and van der Waals forces in the aromatic and charge interactions involved in 9a with various receptor complexes through different amino acid residues, whereas Figures 12a–c and S1 show NewCartoon and 3D representations.
Docking simulations of artesunate indicated its good interactions into the active site of two but different proteins including 5NFR and 3BPF with binding energies of −54.62 and −62.45 kcal/mol, respectively. In contrast to this, low D scores of −17.24 and −60.32 kcal/mol of 9a against 5NFR and 3BPF in comparison to artesunate reflect the comparable less affinity for these two enzymes particularly.
The literature survey for the enlisted six enzymes and their role in malaria leads to the finding that multiple targeting approaches can play an important role in its management, and these predicted binding interactions findings have provided key insights into the possible mechanism of modulation of three proteins (Q8ILJ7, 1LDG, and 3BPM) by 9a, and two of the proteins (5NFR and 3BPF) by artesunate can be further utilized to design and optimize for a combination therapy.
In addition, compound 9a was identified as an advanced HIT with a molecular weight of 350 Da, two hydrogen-bond donors, two acceptors, and 2.3 SLog P. Prediction of druggability revealed that the druglike properties of 9a were found to obey Lipinski’s rule except for the hydrogen-bond acceptor count.
Inhibition of PfIspD Enzyme
Isopentenyl pyrophosphate (IPP) and dimethylallylpyrophosphate (DMAPP) are the essential components for the growth of P. falciparum produced via methylerythritol phosphate (MEP) pathway. Our HIT compound 9a was structurally similar to (1R,3S)-MMV008138 (5), which is known to stop the function of MEP pathway via inhibition of Pf-IspD enzyme, thereby leading to parasite growth inhibition. Furthermore, it was reported that the antimalarial activity of trans isomer of 5 was more than that of its cis isomer, and a similar pattern was also observed in our study, where trans isomers (9a, 11a, 12a, 13a) were more active than its cis isomers (9b, 11b, 12b, 13b). Both these observations point toward the fact that inhibition of the function of the PfIspD enzyme in the MEP pathway could be the probable mechanism for the observed antimalarial activity of our synthesized tetrahydro-β-carboline derivatives.16
Conclusions
Malaria is a serious infectious disease of the developing world with several challenges in drug discovery. Selection of appropriate HIT compounds for further lead development is an important criterion that demands thorough investigation at the early stage for the likelihood of clinical success. Acceptable in vitro potency, oral efficacy, and bioavailability; amendable synthetic chemistry for further optimization; good selectivity index without any toxicity; acceptable physicochemical properties; and manageable pharmacokinetic profile are important criteria for drug discovery research. Specifically, for malaria, a cellular potency (EC50) < 1 μM for sensitive and multiple resistant strains of Plasmodium spp. with 10-fold selectivity is recommended for the HIT compound.49
Our investigation reports β-carboline derivatives showing prominent antimalarial activity. The identified HIT (9a) was found to be active even against the drug-resistant P. falciparum. Compound 9a also exhibited efficacy in monotherapy as well as in combination therapy against P. berghei and may serve as a new partner drug with artesunate for the treatment of malaria. The docking studies of the test compounds are in agreement with the in vitro and in vivo studies supporting our hypothesis.
Experimental Section
Experimental Animal
White Swiss albino mice, Mus musculus of Laca strain (6–8 weeks, 25 ± 2 g), were used in this study. The experimental mice were obtained from and kept in the Central Animal House, Panjab University, Chandigarh, India. They were kept under controlled temperature (25 ± 2 °C) and humidity conditions and fed upon standard pellet diet and water ad libitum. All experimental procedures were in accordance with the guidelines of the committee for the purpose of control and supervision on experiments on animals (45/GO/ReBi/S/99/CPCSEA/PU/IAEC/S/16/50). In addition, the guidelines (ACC-2012-Tech09) of the Canadian Council on Animal Care (CCAC) for the acceptable oral dose volumes in rodents were also followed during the experimental procedures.
Parasite Strain
The NK-65 strain of P. berghei was maintained in vivo in Laca mice. The experimental infection was initiated by intraperitoneal (I.P) inoculation of 1 × 106P. berghei-infected erythrocytes/reticulocytes in citrate saline from infected to naive mice. While doing so, the mice were kept at 26 ± 1.5 °C.
In Vitro Antiplasmodial Activity against P. falciparum
The chloroquine-sensitive (3D7) and -resistant (RKL-9) strains of P. falciparum were obtained from the National Institute of Malaria Research (NIMR), New Delhi, India. P. falciparum culture was maintained by a modified method of Trager and Jensen in A+ human erythrocytes using Roswell Park Memorial Institute (RPMI)-1640 as culture medium supplemented with 10% human AB+ serum.50
The in vitro antiplasmodial activity of 9a was evaluated by schizont maturation inhibition assay.51 The compound (2 mg/mL) was dissolved in 1% dimethyl sulfoxide (DMSO) to prepare stock solution, which was further diluted in RPMI-1640 to make various concentrations [1–16 μg/mL (2.85–45.69 μM)] of the compound. Complete medium (90 μL) was added in duplicate in each well of a 96-well plate along with different concentrations of the compounds/drugs. Chloroquine (10 μM) and artesunate (2 μM) were used as positive control. Parasite culture synchronized as ring stages was added at 2–3% parasitemia and 1.8% final hematocrit. The plates were kept for 48 h at 37 °C under 5% CO2 atmosphere. After the incubation period, thin blood smears were prepared from each well, fixed in methanol, and stained with Giemsa stain. Inhibition of schizont development in comparison to the control wells was determined by the following formula
In Vitro Cytotoxicity Assay
In vitro cytotoxicity of the compound was assessed on HeLa cell lines and primary culture of normal human dermal fibroblasts employing the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.52HeLa cells and primary culture of dermal fibroblasts were obtained from Department of Dermatology, P.G.I.M.E.R, Chandigarh. The cells were maintained at 37 °C in 5% CO2 in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum, glutamine (2 mM), antibiotics streptomycin (100 μg/mL), and penicillin (100 μL/mL). The cells were subcultured every 5 days. As the major restrain of primary culture is that in culture conditions, there is a gradual loss of specialization with its phylogenic features and low homogeneity; therefore, whole results were determined within seven passages.
The cells in DMEM were seeded into a 96-well culture plate in 100 μL of complete medium containing 5000–10 000 cells/well. After 24 h, the cells were incubated with different concentrations (10–640 μg) of the compound in duplicate for 48 h. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was dissolved in DMEM to make 1 mg/mL stock solution. After 48 h incubation at 37 °C in 5% CO2, 10 μL of tetrazolium reagent was added into each well followed by further incubation at 37 °C for 1 h. The supernatant was decanted, and dimethyl sulfoxide (DMSO) (100 μL/well) was added to allow formazan solubilization. The optical density (OD) of each well was detected using a microplate reader at 450 nm. The selectivity index (SI), defined as the ratio of the CC50 determined on HeLa cells/dermal fibroblasts to the IC50 determined on P. falciparum, was also calculated.
Determination of Nitric Oxide
RAW 264.7 murine macrophage cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS). The cells were trypsinized and seeded in 24-well plates (10 000 cells/well) and were allowed to adhere for 1 h, after which they were incubated with 9a (100 and 200 μg/mL) for 16 and 40 h. After the specified intervals, the supernatant was collected. The production of NO was estimated from the accumulation of nitrite (NO2–) ions, the metabolic end product of NO metabolism, in the medium using the Griess reagent. Briefly, equal volumes (100 μL) of sample and Griess reagent (1:1 mixture of 1% sulfanilamide in 5% phosphoric acid and 0.1% α-naphthylamine in distilled water) were mixed in a 96-well culture plate and incubated at room temperature for 15 min, and absorbance was measured at 550 nm.53
In Vivo Suppressive Activity against P. berghei
The suppressive activity of the compound was evaluated by Peter’s 4 day test.54 The mice were divided into 10 experimental groups (G1–G10) of five mice each (Table 2). On D0, all of the mice were inoculated with 1 × 106P. berghei-infected erythrocytes (except G1), as done in our earlier study,55 followed by drug treatment 1 h post inoculation. The drug was administered once daily for 4 days (D0–D3). In the combination therapy group, the second drug was administered at an interval of 60 min. Twenty four hours after the completion of the 4 day dose regimen, thin blood smears were made from the tail of each mouse on D4. The slides were fixed in methanol followed by Giemsa staining. The percent chemosuppression in each experimental group was calculated using the following formula
where A is the average parasitemia in infected control and B is the average parasitemia in the treated group.
Measurements of Basic Parameters
The experimental mice in monotherapy and in combination with artesunate were observed throughout the experiment for changes in behavior and symptoms of illness visually.56 Various parameters were observed, including behavioral lethargy, reduction of movement, passage of dark urine, impaired consciousness, and mortality. These signs of illness were recorded as absent (−), mild (+), moderate (++), or severe (+++).
Differential Leukocyte Count (DLC)
Differential leukocyte count (DLC) was also studied in Giemsa-stained thin blood smears in the combination therapy treated group in contrast to the infected control.
Biochemical Assays
Biochemical assays were performed by taking blood from the mice of various experimental groups through tail vein drainage on day 10 and survivors of experimental groups on day 28. Serum levels of ALP,57 SGOT,58 SGPT,58 and bilirubin59 were checked for the assessment of hepatic function, while urea60 and creatinine61 concentrations were measured for the assessment of renal function. Biochemical assays were carried out using commercially available diagnostic kits (Reckon Diagnostic P. Ltd.).
Target Identification and Protein Preparation
In spite of the physiological and pathological importance of PEPC, 3D crystallographic structural information is still unknown. To address this problem and to understand ligand interaction with PEPC receptor, in silico homology modeling approach has been performed.
For PEPC, targeted amino acid sequence Q8ILJ7 was retrieved from the UniProtKB database (http://www.uniprot.org/). Alignment and selection of the sequence were performed considering the highest identity using blast tool and saved as PDB. Protein structure integrity was assessed and checked for its incomplete and missing residues, followed by insertion of loop regions and missing atoms near the active site. Furthermore, a model was analyzed for its correctness using an analytical tool, followed by local geometry check. Decisively, the prepared model was used for the docking studies.
The 3D crystallographic structures of LDH (PDB ID: 1LDG), PMT (PDB ID: 3UJ9), MDH (PDB ID: 5NFR), FP3 (PDB ID: 3BPM), and FP2 (PDB ID: 3BPF) were retrieved from a protein databank http://www.rcsb.org.
Protein structure was analyzed, reprocessed, and refined using the protein preparation protocol. The protocol was performed by the removal of water molecules and protonation of titratable residues to stabilize the receptor protein. Protein structure integrity was assessed and checked for its missing residues, followed by the insertion of loop regions and missing atoms near the active site, using Loop Builder tool. Final protein was saved in.mol format as a final receptor after the removal of co-factors and external ligand present in crystal structure.
Ligand Standardization
One of the important determinants for a successful docking is the structure of the ligand. ChemDraw Ultra 8.0 and VLifeMDS software were used for drawing the structures, energy calculation, and optimization of structural geometries of compounds. The two-dimensional (2D) structures of newly synthesized analogues were converted to their corresponding three-dimensional (3D) structures using the converter module of VLife. The 3D structures were then subjected to energy minimization and geometry optimization using the Merck Molecular Force Field (MMFF) method. The conformer with the lowest energy was selected for docking simulation studies. The comprehensive and integrated graphical user interface program of the VLifeMDS 4.6, i.e., “‘Bio Predicta module”’ was used to prepare, run, and analyze the docking simulations on the HP Pentium IV 2.80 GHz Processor/Microsoft Win XP Home Edition system.
Ligand Docking
Herein, we combined homology modeling and molecular docking studies using VLifeMDS tool to define and identify the critical binding mode of all of the optimized molecules against the active site of enlisted six proteins. Correct ligand pose assessment generally remains an important criterion for the optimal binding and affinity prediction.62,63 Further, docking results are being generated in a number of potential poses in association, and the best identified pose with the lowest energy (D score, Figure 12d) is generally considered for subsequent analysis and interaction involved with active amino acid residues.
Drug Likeness: Rule of Five
In recent years, extensive analysis of successful and high attrition rate in drug discovery programs has improved our awareness regarding the role of molecular properties for adequate oral bioavailability in the drug development process.64,65 Lipinski’s “rule of five” provides an adequate set of guidelines for selecting druglike compounds in terms of physicochemical parameters absorption, distribution, metabolism, and excretion (ADME),66 specifically molecular mass <500 Da, number of hydrogen-bond donors <5, number of hydrogen-bond acceptors <10, and calculated octanol–water partition coefficient <5.
Acknowledgments
D.B.S. is thankful to UGC New Delhi for start-up research grant and DBT New Delhi for the award of Ramalingaswami Fellowship. The support from UGC-CAS, DST-PURSE-II, DST-SAIF, and Panjab University Development fund is gratefully acknowledged. R.S. is thankful to CSIR, New Delhi, for the award of Research Fellowship. N.S.W. is thankful to CSIR, New Delhi, for the award of Research Associateship. The authors are grateful to Late Prof. Upma Bagai for her valuable guidance in the initial conception and design of this study.
Glossary
Abbreviations
- PEPC
phosphoenolpyruvate carboxylase
- PMT
phosphoethanolamine methyltransferase
- LDH
lactate dehydrogenase
- MDH
malate dehydrogenase
- FP2
falcipain-2
- FP3
falcipain-3
- AS
artesunate
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01256.
Accession Codes
PMT (PDB ID: 3UJ9), FP3 (PDB ID: 3BPM), PEPC (PDB ID: Q8ILJ7), LDH (PDB ID: 1LDG), FP2 (PDB ID: 3BPF), MDH (PDB ID: 5NFR).
Author Contributions
V.G., N.S.W., and S.K. contributed to the biological evaluation of the synthetic compounds. R.S. and D.B.S. synthesized and provided the compounds. M.C. and N.D. contributed to the in silico evaluation of the compounds.
The authors declare no competing financial interest.
Supplementary Material
References
- Sharma R.; Dutta A. K. Malaria and National Vector Borne Disease Control Programme. Indian J. Pediatr. 2011, 78, 1527–1535. 10.1007/s12098-011-0554-2. [DOI] [PubMed] [Google Scholar]
- Roberts D. R.; Andre R. G. Insecticide resistance issues in vector-borne disease control. Am. J. Trop. Med. Hyg. 1994, 50, 21–34. 10.4269/ajtmh.1994.50.21. [DOI] [PubMed] [Google Scholar]
- https://nvbdcp.gov.in/index4.php?lang=1&level=0&linkid=429&lid=3706 (assessed April 30, 2020).
- WHO . World Malaria Report 2019, Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, 2019. [Google Scholar]
- Tiwari M. K.; Chaudhary S. Artemisinin-derived antimalarial endoperoxides from bench-side to bed-side: Chronological advancements and future challenges. Med. Res. Rev. 2020, 1220. 10.1002/med.21657. [DOI] [PubMed] [Google Scholar]
- Gumede B.; Folb P.; Ryffel B. Oral artesunate prevents Plasmodium berghei ANKA infection in mice. Parasitol. Int. 2003, 52, 53–59. 10.1016/S1383-5769(02)00081-8. [DOI] [PubMed] [Google Scholar]
- Eastman R. T.; Fidock D. A. Artemisinin based combination therapies: a vital tool in efforts to eliminate malaria. Nat. Rev. Microbiol. 2009, 7, 864–874. 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton W. L.; Amato R.; van der Pluijm R. W.; Jacob C. G.; Quang H. H.; Thuy-Nhien N. T.; Hien T. T.; Hongvanthong B.; Chindavongsa K.; Mayxay M.; et al. Evolution and expansion of multidrug-resistant malaria in southeast Asia: a genomic epidemiology study. Lancet Infect. Dis. 2019, 943. 10.1016/S1473-3099(19)30392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaratunga C.; Lim P.; Suon S.; Sreng S.; Mao S.; Sopha C.; Sam B.; Dek D.; Try V.; Amato R.; et al. Dihydroartemisinin-piperaquine resistance in Plasmodium falciparum malaria in Cambodia: a multisite prospective cohort study. Lancet Infect. Dis. 2016, 16, 357–365. 10.1016/S1473-3099(15)00487-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaorattanakawee S.; Lon C.; Jongsakul K.; Gawee J.; Sok S.; Sundrakes S.; Kong N.; Thamnurak C.; Chann S.; Chattrakarn S.; et al. Ex vivo piperaquine resistance developed rapidly in Plasmodium falciparum isolates in northern Cambodia compared to Thailand. Malar. J. 2016, 15, 519 10.1186/s12936-016-1569-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo P. C.; Shi L. S.; Damu A. G.; Su C. R.; Huang C. H.; Ke C. H.; Wu J. B.; Lin A. J.; Bastow K. F.; Lee K. H.; et al. Cytotoxic and antimalarial β-Carboline alkaloids from the roots of Eurycoma longifolia. J. Nat. Prod. 2003, 66, 1324–1327. 10.1021/np030277n. [DOI] [PubMed] [Google Scholar]
- Frédérich M.; Jacquier M. J.; Thépenier P.; Mol P. D.; Tits M.; Philippe G.; Delaude C.; Angenot L.; Hanrot M. Z. Antiplasmodial activity of alkaloids from various Strychnos species. J. Nat. Prod. 2002, 65, 1381–1386. 10.1021/np020070e. [DOI] [PubMed] [Google Scholar]
- Lu Z.; Ding Y.; Li X.-C.; Djigbenou D. R.; Grimberg B. T.; Ferreira D.; Ireland C. M.; Wagoner R. M. V. 3-Bromohomofascaplysin A, a fascaplysin analogue from a Fijian Didemnum sp. ascidian. Bioorg. Med. Chem. 2011, 19, 6604–6607. 10.1016/j.bmc.2011.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astulla A.; Zaima K.; Matsuno Y.; Hirasawa Y.; Ekasari W.; Widyawaruyanti A.; Zaini N. C.; Morita H. Alkaloids from the seeds of Peganum harmala showing antiplasmodial and vasorelaxant activities. J. Nat. Med. 2008, 62, 470–472. 10.1007/s11418-008-0259-7. [DOI] [PubMed] [Google Scholar]
- Rottmann M.; McNamara C.; Yeung B. K. S.; Lee M. C. S.; Zou B.; Russell B.; Seitz P.; Plouffe D. M.; Dharia N. V.; Tan J.; et al. Spiroindolones, a potent compound class for the treatment of malaria. Science 2010, 329, 1175–1180. 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghavami M.; Merino E. F.; Yao Z. K.; Elahi R.; Simpson M. E.; Murga M. L. F.; Butler J. H.; Casasanta M. A.; Krai P. M.; Totrov M.; et al. Biological studies and target engagement of the 2-C-methyl-D-erythritol 4-phosphate cytidylyltransferase (IspD)-targeting antimalarial agent (1R, 3S)-MMV008138 and analogs. ACS Infect. Dis. 2018, 549. 10.1021/acsinfecdis.7b00159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yadav V.; Srivastava K.; Tripathi R.; Batra S. Synthesis of β-carboline-fused 1,4-oxazepines and their assessment as antiplasmodial agents. Tetrahedron 2017, 73, 5680. 10.1016/j.tet.2017.08.003. [DOI] [Google Scholar]
- Beghyn T. B.; Charton J.; Leroux F.; Laconde G.; Bourin A.; Cos P.; Maes L.; Deprez B. Drug to genome to drug: discovery of new antiplasmodial compounds. J. Med. Chem. 2011, 54, 3222–3240. 10.1021/jm1014617. [DOI] [PubMed] [Google Scholar]
- Gupta L.; Srivastava K.; Singh S.; Puri S. K.; Chauhan P. M. Synthesis of 2-[3-(7-Chloro-quinolin-4-ylamino)-alkyl]-1-(substituted phenyl)-2,3,4,9-tetrahydro-1H-β-carbolines as a new class of antimalarial agents. Bioorg. Med. Chem. Lett. 2008, 18, 3306–3309. 10.1016/j.bmcl.2008.04.030. [DOI] [PubMed] [Google Scholar]
- Ashok P.; Ganguly S.; Murugesan S. Manzamine alkaloids: isolation, cytotoxicity, antimalarial activity and SAR studies. Drug Discovery Today 2014, 19, 1781–1791. 10.1016/j.drudis.2014.06.010. [DOI] [PubMed] [Google Scholar]
- Gellis A.; Dumètre A.; Lanzada G.; Hutter S.; Ollivier E.; Vanelle P.; Azas N. D. Preparation and antiprotozoal evaluation of promising β-carboline alkaloids. Biomed. Pharmacother. 2012, 66, 339–347. 10.1016/j.biopha.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Gorki V.; Singh R.; Walter N. S.; Bagai U.; Salunke D. B. Synthesis and Evaluation of antiplasmodial efficacy of β-carboline derivatives against murine malaria. ACS Omega 2018, 3, 13200–13210. 10.1021/acsomega.8b01833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima M. N. N.; Cassiano G. C.; Tomaz K. C. P.; Silva A. C.; Sousa B. K. P.; Ferreira L. T.; Tavella T. A.; Calit J.; Bargieri D. Y.; Neves B. J.; et al. Integrative multi-kinase approach for the identification of potent antiplasmodial hits. Front. Chem. 2019, 7, 773 10.3389/fchem.2019.00773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm J.; Sethia S.; Blackburn G. J.; Chokkathukalam A.; Watson D. G.; Breitling R.; Coombs G. H.; Müller S. Phosphoenolpyruvate carboxylase identified as a key enzyme in erythrocytic Plasmodium falciparum carbon metabolism. PLoS Pathog. 2014, 10, e1003876 10.1371/journal.ppat.1003876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J.; Mansuri R.; Vijay S.; Sahoo G. C.; Sharma A.; Kumar M. Docking predictions-based Plasmodium falciparum phosphoethanolamine methyl transferase inhibitor identification and in-vitro antimalarial activity analysis. BMC Chem. 2019, 13, 43 10.1186/s13065-019-0551-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penna-Coutinho J.; Cortopassi W. A.; Oliveira A. A.; França T. C. C.; Krettli A. U. Antimalarial activity of potential inhibitors of Plasmodium falciparum lactate dehydrogenase enzyme selected by docking studies. PLoS One 2011, 6, e21237 10.1371/journal.pone.0021237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunev S.; Butzloff S.; Romero A. R.; Linzke M.; Batista F. A.; Meissner K. A.; Müller I. B.; Adawy A.; Wrenger C.; Groves M. R. Oligomeric interfaces as a tool in drug discovery: Specific interference with activity of malate dehydrogenase of Plasmodium falciparumin vitro. PLoS One 2018, 13, e195011 10.1371/journal.pone.0195011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musyoka T. M.; Kanzi A. M.; Lobb K. A.; Tastan-Bishop Ö. Structure based docking and molecular dynamic studies of plasmodial cysteine proteases against a South African natural compound and its analogs. Sci. Rep. 2016, 6, 23690 10.1038/srep23690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himangini; Pathak D. P.; Sharma V.; Kumar S. Designing novel inhibitors against falcipain-2 of Plasmodium falciparum. Bioorg. Med. Chem. Lett. 2018, 28, 1566–1569. 10.1016/j.bmcl.2018.03.058. [DOI] [PubMed] [Google Scholar]
- Chugh M.; Sundararaman V.; Kumar S.; Reddy V. S.; Siddiqui W. A.; Stuart K. D.; Malhotra P. Protein complex directs hemoglobin-to-hemozoin formation in Plasmodium falciparum. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 5392–5397. 10.1073/pnas.1218412110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekana-Douki J. B.; Bongui J. B.; Oyegue-Liabagui S. L.; Zang Edou S. E.; Zatra R.; Bisvigou U.; Druilhe P.; Lebibi J.; Toure Ndouo F. S.; Kombila M. In vitro antiplasmodial activity and cytotoxicity of nine plants traditionally used in Gabon. J. Ethnopharmacol. 2011, 133, 1103–1108. 10.1016/j.jep.2010.11.056. [DOI] [PubMed] [Google Scholar]
- Adebayo J. O.; Krettli A. U. Potential antimalarials from Nigerian plants: a review. J. Ethnopharmacol. 2011, 133, 289–302. 10.1016/j.jep.2010.11.024. [DOI] [PubMed] [Google Scholar]
- Ekwall B.; Silano V.; Paganuzzi-Stammati A.; Zucco F.. Short-Term Toxicity Tests for Non-genotoxic Effects. In Toxicity Tests with Mammalian Cell Cultures; Bourdeau P., Ed.; John Wiley & Sons Ltd., 1990. [Google Scholar]
- Chanput W.; Peters V.; Wichers H.. THP-1 and U937 Cells. The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Springer International Publishing, 2015; pp 147–159. [Google Scholar]
- Schwartz H. S.; Mihic E.. Species and Tissue Differences in Drug Selectivity. In Drug Resistance and Selectivity Biochemical and Cellular Basis; Mihic E., Ed.; Academic Press: New York, 1973; pp 413–449. [Google Scholar]
- Valdés A. F.-C.; Martínez J. M.; Lizama R. S.; Gaitén Y. G.; Rodríguez D. A.; Payrol J. A. In vitro antimalarial activity and cytotoxicity of some selected Cuban medicinal plants. Rev. Inst. Med. Trop. Sao Paulo 2010, 52, 197–201. 10.1590/S0036-46652010000400006. [DOI] [PubMed] [Google Scholar]
- Fang F. C. Perspectives series: host/pathogen interactions. Mechanisms of nitric oxide-related antimicrobial activity. J. Clin. Invest. 1997, 99, 2818–2825. 10.1172/JCI119473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janse C.General Introduction Rodent Malaria Parasites; Leids Universitair Medisch Centrum, 2020; https://www.lumc.nl/org/parasitologie/research/malaria/berghei-model/introduction-berghei/ (accessed Jan 09, 2020). [Google Scholar]
- Muñoz V.; Sauvain M.; Bourdy G.; Callapa J.; Rojas I.; Vargas L.; Tae A.; Deharo E. The search for natural bioactive compounds through a multidisciplinary approach in Bolivia. Part II. Antimalarial activity of some plants used by Mosetene Indians. J. Ethnopharmacol. 2000, 69, 139–155. 10.1016/S0378-8741(99)00096-3. [DOI] [PubMed] [Google Scholar]
- Stevenson M. M.; Riley E. M. Innate immunity to malaria. Nat. Rev. Immunol. 2004, 4, 169–180. 10.1038/nri1311. [DOI] [PubMed] [Google Scholar]
- Ignatius C. M.; Emeka E. N.; Blessing N. E. Effect of malaria parasitaemia on liver enzyme tests. Int. J. Trop. Med. 2008, 3, 49–52. [Google Scholar]
- Reichling J. J.; Kaplan M. M. Clinical use of serum enzymes in liver disease. Dig. Dis. Sci. 1988, 33, 1601–1614. 10.1007/BF01535953. [DOI] [PubMed] [Google Scholar]
- Al-Salahy M.; Shnawa B.; Abed G.; Mandour A.; Al-Ezzi A. Parasitaemia and its relation to hematological parameters and liver function among patients malaria in Abs, Hajjah, Northwest Yemen. Interdiscip. Perspect. Infect. Dis. 2016, 2016, 5954394 10.1155/2016/5954394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyewole I. O.; Senusie S.; Mansaray M. Plasmodium falciparum-induced kidney and liver dysfunction in malaria patients in Freetown, Sierra Leone. Sierra Leone J. Biomed. Res. 2010, 2, 70–74. 10.4314/sljbr.v2i1.56611. [DOI] [Google Scholar]
- Snow R. W.; Guerra C. A.; Noor A. M.; Myint H. Y.; Hay S. I. The global distribution of clinical episodes of Plasmodium falciparum malaria. Nature 2005, 434, 214–217. 10.1038/nature03342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoto U. S. C.; Calisei T. Malaria parasite and their relationships with their host. Malaria Res. 2006, 44, 265–273. [Google Scholar]
- Eiam-Ong S.; Sitprija V. Falciparum malaria and the kidney: A model of inflammation. Am. J. Kidney Dis. 1998, 32, 361–375. 10.1053/ajkd.1998.v32.pm9740151. [DOI] [PubMed] [Google Scholar]
- Severe malaria. Trop. Med. Int. Health 2014, 19, 7–131. 10.1111/tmi.12313_2. [DOI] [PubMed] [Google Scholar]
- Katsuno K.; Burrows J. N.; Duncan K.; Hooft van Huijsduijnen R.; Kaneko T.; Kita K.; Mowbray C. E.; Schmatz D.; Warner P.; Slingsby B. T. Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nat. Rev. Drug. Discovery 2015, 14, 751–758. 10.1038/nrd4683. [DOI] [PubMed] [Google Scholar]
- Trager W.; Jensen J. Human malaria parasites in continuous culture. Science 1976, 193, 673–675. 10.1126/science.781840. [DOI] [PubMed] [Google Scholar]
- World Health Organization . In Vitro Micro Test (Mark III) for the Assessment of Response of Plasmodium falciparum to Chloroquine, Mefloquine, Quinine, Amodiaquine, Sulfadoxine, Pyrimethamine and Artemisinin, CTD/MAL/97.20 Rev. 2; World Health Organization: Geneva, 2001. [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Migliorini P.; Corradin F.; Betz-Corradin S. Macrophage NO2– production as a sensitive and rapid assay for the quantitation of murine IFN-γ. J. Immunol. Methods 1991, 139, 107–114. 10.1016/0022-1759(91)90357-L. [DOI] [PubMed] [Google Scholar]
- Knight D. J.; Peters W. The antimalarial action of N-Benzyloxydihydrotriazines. The action of cycloguanil (BRL50216) against rodent malaria and studies on its mode of action. Ann. Trop. Med. Parasitol. 1980, 74, 393–404. 10.1080/00034983.1980.11687360. [DOI] [PubMed] [Google Scholar]
- Walter N. S.; Bagai U.; Kalia S. Antimalarial activity of Bergenia ciliata (Haw.) Sternb. against Plasmodium berghei. Parasitol. Res. 2013, 112, 3123–3128. 10.1007/s00436-013-3487-z. [DOI] [PubMed] [Google Scholar]
- Basir R.; Fazalul-Rahiman S. S.; Hasballah K.; Chong W. C.; Talib H.; Yam M. F.; Jabbarzare M.; Tie T. H.; Othman F.; Moklas M. A. M.; et al. Plasmodium berghei ANKA infection in ICR mice as a model of cerebral malaria. Iran. J. Parasitol. 2012, 7, 62–74. [PMC free article] [PubMed] [Google Scholar]
- Recommendations of the German Society for Clinical Chemistry. Standardization of methods for the estimation of enzyme activities in biological fluids. Experimental basis for the optimized standard conditions Z. Klin. Chem. Klin. Biochem. 1972, 10, 281–291.. [PubMed]
- Bergmeyer H. U. IFCC methods for the measurement of catalytic concentrations of enzymes. Clin. Chim. Acta 1980, 105, 147–154. 10.1016/0009-8981(80)90105-9. [DOI] [PubMed] [Google Scholar]
- Jendrassik L.; Grof P. Quantitative estimation of some biochemicals and biomolecules in serum. Biochem. Z. 1938, 297, 81–89. [Google Scholar]
- Chaney A. L.; Marbach E. P. Modified reagents for determination of urea and ammonia. Clin. Chem. 1962, 8, 130–132. 10.1093/clinchem/8.2.130. [DOI] [PubMed] [Google Scholar]
- Kaplan A.; Szabo L. L.. Clinical Chemistry: Interpretation and Techniques, 2nd ed.; Lea and Febiger: Philadelphia, 1983; p 157. [Google Scholar]
- Coleman R. G.; Carchia M.; Sterling T.; Shoichet B. K.; Irwin J. J. Ligand pose and orientational sampling in molecular docking. PloS One 2013, 8, e75992 10.1371/journal.pone.0075992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halperin I.; Ma B.; Wolfson H.; Nussinov R. Principles of docking: An overview of search algorithms and a guide to scoring functions. Proteins: Struct., Funct., Bioinf. 2002, 47, 409–443. 10.1002/prot.10115. [DOI] [PubMed] [Google Scholar]
- Ou-Yang S. S.; Lu J. Y.; Kong X. Q.; Liang Z. J.; Luo C.; Jiang H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. 10.1038/aps.2012.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Bickerton G. R.; Paolini G. V.; Besnard J.; Muresan S.; Hopkins A. L. Quantifying the chemical beauty of drugs. Nat. Chem. 2012, 4, 90. 10.1038/nchem.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.