

Abstract
The overexpression of immunomarker programmed cell death protein 1 (PD-1) and engagement of PD-1 to its ligand, PD-L1, are involved in the functional impairment of cluster of differentiation 8+ (CD8+) T cells, contributing to cancer progression. However, heterogeneities in PD-L1 expression and variabilities in biopsy-based assays render current approaches inaccurate in predicting PD-L1 status. Therefore, PD-L1 screening alone is not predictive of patient response to treatment, which motivates us to simultaneously detect multiple immunomarkers engaged in immune modulation. Here, we have developed multimodal probes, immunoactive gold nanostars (IGNs), that accurately detect PD-L1+ tumor cells and CD8+ T cells simultaneously in vivo, surpassing the limitations of current immunoimaging techniques. IGNs integrate the whole-body imaging of positron emission tomography with high sensitivity and multiplexing of Raman spectroscopy, enabling the dynamic tracking of both immunomarkers. IGNs also monitor response to immunotherapies in mice treated with combinatorial PD-L1 and CD137 agonists and distinguish responders from those nonresponsive to treatment. Our results showed a multifunctional nanoscale probe with capabilities that cannot be achieved with either modality alone, allowing multiplexed immunologic tumor profiling critical for predicting early response to immunotherapies.
Keywords: gold nanostar, immunoimaging, immunoPET, Raman spectroscopy, multiplexed detection, programmed cell death ligand 1, CD8
Graphical Abstract:
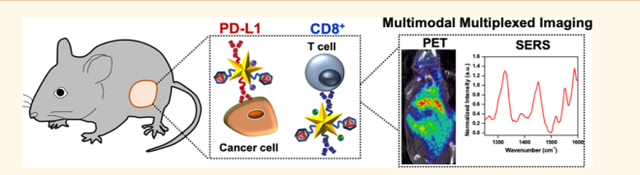
Upregulation of immune checkpoint PD-1 and subsequent binding of PD-1 to its ligand, PD-L1, impede effector T cell function, contributing to immunosup-pression.1,2 Inhibition of the PD-1/PD-L1 axis with immunotherapies has transformed the therapeutic landscape for a broad array of cancers. However, despite the clinical efficacy of these agents, only a fraction of patients respond in most tumor types, and identifying patients likely to benefit from these therapies remains challenging.3–5 Current predictive technologies rely on static measurement of PD-L1 levels in biopsies, which cannot adequately distinguish responders from nonresponders. The inaccuracy in PD-L1 detection could be in part due to limited tissue sampling or the spatial and temporal heterogeneity of PD-L1 among patients and even within the same patient’s primary and metastatic lesions.6,7 Therefore, noninvasive molecular imaging is critical in the immunotherapy drug development pipeline for multiple reasons. First, immunoimaging will allow accurate and dynamic measurement of PD-L1 status in vivo to identify patients who will respond to immunotherapies prior to treatment. Second, multiplexed detection will enable recognition of other immunomarkers that show engagement of the immune tumor microenvironment (TME) to determine alternative therapies for patients who are nonresponsive to PD-L1 blockade. Third, immunoimaging will establish end points for monitoring treatment efficacy early in the immunotherapy regimen and reflect the dynamic changes in immunomarker localization during therapeutic intervention.8,9
Dynamic tracking of both PD-L1 and cytotoxic CD8+ T cells in vivo is highly relevant to predict the complex interplay between the immune system and TME. The presence of CD8+ T cells both within the tumor and at the invasive margin is a positive prognostic marker that demonstrates active engagement of antitumor immunity.10,11 Studies have shown that tumors that are infiltrated with CD8+ T cells and simultaneously express PD-L1 are most likely to benefit from PD-L1 inhibitors.12–14 Further, dynamic changes occur in both PD-L1 level in tumor cells and CD8+ T cells during treatment cannot be captured by single biomarker imaging or by static measurement of receptor status.15,16 The strong correlation between PD-L1 inhibition and activation of CD8+ T cells motivates multiplexed detection of both markers to ultimately provide translatable methods for predicting clinical responses to immunotherapies.
In this work, we simultaneously detect PD-L1 and CD8 in vivo (Figure 1a) and monitor response to combinatorial immunotherapies with an innovative probe, immunoactive gold nanostars (IGNs), which combines positron emission tomography (PET) with surface-enhanced Raman spectroscopy (SERS). PET and SERS are complementary imaging techniques seamlessly integrated with IGNs, allowing depth-resolved whole-body imaging with PET to locate the macroscopic distribution of tumors to tissue depths of many centimeters. Multiplexed SERS is then followed to identify multiple immunomarkers that dynamically control local and systemic immunity in the TME. SERS, an optical technique, uses near-infrared (NIR) light to enhance the vibrational signal of Raman reporters and enables narrow spectral features amenable for multiplexing.17 Gold nanostars are ideal for SERS because they have shown to amplify the signal of Raman molecules by >109, enabling enhanced spatiotemporal resolution in vitro and in vivo.18–20 Here, we showed IGNs labeled with antibodies, Raman reporters, and 64Cu were targeted to BRAF mutant Yale University Mouse Melanoma cell line variant 2.1 (YUMM 2.1) tumors after systemic delivery in vivo in immunocompetent mice. IGNs detected both PD-L1 expressing cells and CD8+ T cells in the TME with high sensitivity and specificity via ImmunoPET-SERS imaging. Further, IGNs effectively monitored response to immunotherapies in mice treated with a combination of anti-PD-L1 and anti-CD137 monoclonal antibodies (mAbs). CD137 is a costimulatory receptor expressed on activated T cells and shown to have synergistic therapeutic benefits with PD-L1 blockade.21,22 IGNs also distinguished responders from those nonresponsive to immunotherapies by examining NRAS mutant YUMM 10.1 tumors that showed minimal change in PD-L1 and CD8 status post-treatment. We envision the findings of this work will catalyze a clinically translatable technology that will ultimately permit image-guided interventions ranging from noninvasive treatment planning to predicting therapeutic effectiveness and improve survival of cancer patients.
Figure 1.

Design and physicochemical properties of IGNs and bare gold nanostars (GNs). (a) Schematic representation of IGN-mediated multimodal multiplexed ImmunoPET-SERS imaging to detect both PD-L1 expression and CD8+ T cells in melanoma tumors. (b) The design of IGNs where gold nanostars were functionalized with Raman reporters (pMBA or DTNB) via a thiol-Au reaction followed by conjugation with PEG-stabilized antibodies (anti-PD-L1 or anti-CD8). IGNs were then bound to DOTA and chelated with 64Cu radiolabels. (c) Transmission electron micrograph of IGNs showing their star shape. (d) Hydrodynamic size of IGNs from dynamic light scattering. (e) Extinction spectra of bare gold nanostars and functionalized IGNs. (f) Raman spectra of a mixture of IGNs targeting both CD8 and PD-L1 via DTNB (1325 cm−1) and pMBA (1580 cm−1) reporters, respectively; the signature peaks of the reporters are highlighted. (g) Amount of Cu chelated per mg IGNs quantified via ICP-MS. (h) Zeta potential of the bare gold nanostars and IGNs showing surface charge.
RESULTS AND DISCUSSION
Synthesis of Immunoactive Gold Nanostars.
Gold nanostars were synthesized with a biological buffer, 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), as previously described by our group.23,24 HEPES binds weakly to gold surfaces, offering a straightforward surface chemistry that allows covalent conjugation of both PET and Raman labels for imaging and PEG ligands for biocompatibility to avoid recognition by the mononuclear phagocyte system (MPS). Multiplexed detection of both PD-L1 and CD8 was enabled by designing two sets of IGNs. To enable PD-L1 detection, nanostars were covalently conjugated with the Raman reporter para-mercaptobenzoic acid (pMBA), followed by covalent linking to anti-PD-L1 mAbs. Nanostars were then conjugated to chelators, (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) (DOTA), and finally radiolabeled with 64Cu (Figure 1b). We chose DOTA to chelate 64Cu because this complex has already been utilized in patients for PET imaging25–27 and is currently under numerous clinical trials (NCT03492762, NCT02827877, etc.). CD8 detection was facilitated by conjugating nanostars with anti-CD8 mAbs, the Raman reporter 5,5-dithiobis(2-nitrobenzoic acid) (DTNB), and 64Cu following a similar procedure. As PET cannot distinguish the signal between radiolabels, the two Raman reporters allowed multiplexed detection of the two immunomarkers. Covalent conjugation of IGNs with mAbs and chelated radiolabel was achieved via a bifunctional orthopyridyl-disulfide poly(ethylene glycol)-N-hydroxysuccinimide ester (OPSS-PEG-NHS) linker described in detail in Materials and Methods and in the Supporting Information (Scheme S1). Our previous work has shown this bifunctional linker not only provides stability to nanostars but also maintains bioactivity of the antibodies for successful in vivo detection.28 PEG with this bifunctional linker stabilized the mAbs and DOTA, and thiols in the orthopyridyl disulfide (OPSS) group enabled covalent conjugation to the nanostars’ surface. IGNs’ size regime, 50–80 nm (Figure 1c,d), is ideal for rapid accumulation in the TME, enabling longitudinal imaging and time-course study. Whereas this size can potentially limit efficient renal clearance of IGNs, recent studies have shown renal excretion of 50 nm mesoporous silica nanoparticles.29 In addition, the size of IGNs should not impede their clinical translation, as micrometer-sized particles have been shown to filtrate through the kidneys in large animals and humans.30,31
The increase in nanostar size postfunctionalization resulted in a ~30 nm red shift in their absorbance but remained resonant in the NIR (Figure 1e). NIR light has a 1–3 cm penetration depth, ideal for in vivo imaging.32–34 The SERS spectra of an equimolar mixture of IGNs consisting of IGNs/anti-PD-L1/pMBA/64Cu and IGNs/anti-CD8/DTNB/64Cu showed the dominant peak of DTNB at 1325 cm−1 and pMBA at 1580 cm−1 (Figure 1f). We also validated cold Cu chelation to IGNs with inductively coupled plasma mass spectrometry (ICP-MS) and quantified 0.99 ± 0.04 μg Cu/mg Au in the presence of DOTA relative to 0.035 ± 0.02 μg Cu/mg Au for bare nanostar control, indicating successful radiolabeling of IGNs (Figure 1g). Further, zeta potential measurements (Figure 1h) confirmed PEG conjugation on IGNs resulted in near-neutral surface charge in comparison to bare nanostars, which had a negative charge. The stability of IGNs in both water and cellular media supplemented with serum was studied by examining the intensity and the full width at half-maximum (FWHM) of the extinction spectra. The intensity remained unchanged (Figure S1a), and minimal broadening of extinction spectra was observed (Table S1), demonstrating IGNs did not flocculate over 4 days, which is the duration of longitudinal imaging in our study. In addition, the shelf life of IGNs was studied for 4 weeks. Aliquots of IGNs were dispersed in water, phosphate-buffered saline (PBS), media, and media supplemented with serum. Minimal change was observed in the normalized extinction of IGNs (Figure S1b) and only a slight (~1–6%) increase in the FWHM (Table S2) over the course of 4 weeks. These results indicate IGNs have a long shelf life and good stability in the time frame studied.
Preclinical Evaluation and Biodistribution of IGNs.
Prior to in vivo imaging, we performed an antibody–antigen binding assay similar to the enzyme-linked immunosorbent assay (ELISA) to validate IGNs’ ability to specifically bind to CD8 and PD-L1 receptors (Figure S2). This ex vivo study showed successful binding of IGNs to the respective antigen when conjugated with the corresponding mAbs. We evaluated the biocompatibility of IGNs in vivo at 5 and 15 days post-intraperitoneal (IP) delivery of IGNs (0.06 mg IGNs/g mouse weight) to examine both near-term and longer-term impact. This dosage of IGNs is comparable to or lower than other studies utilizing nanostars.35,36 We chose a murine model of melanoma for our study with YUMM 2.1 tumors that are highly immunogenic with intrinsically upregulated PD-L1 expression and high infiltration of CD8+ T cells.37 The toxicity of IGNs was studied by examining standard serum inflammatory markers to determine if IGNs elicit any immune response in mice. Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were used as indicators of liver function (Figure 2a), and total bilirubin (TBIL), creatinine (CREAT), and blood urea nitrogen (BUN) were used to measure renal function (Figure 2b).38 Further, complete blood count (CBC) analysis, including hemoglobin, red blood cells, white blood cells, platelet concentration, monocyte counts, and lymphocyte counts (Figure 2c–f), was also examined in mouse serum. These serum markers were comparable to mice that received IGNs relative to control mice, which received PBS. These observations were confirmed with hematoxylin and eosin (H&E) staining of major organs and tumors of mice that received IGNs, and no noticeable histopathological changes were observed (Figure S3). We note that the concentration of mAbs covalently bound to IGNs was <0.5 μg antibody/mouse (0.29 μg of anti-CD8 antibody/mouse and 0.22 μg of anti-PD-L1 antibody/mouse), which is a very low dose and should not contribute to any mAbs-related toxicities (Figure S4). Quantitative ICP-MS analysis of Au in tumor and major organs that were retrieved 5 and 15 day post-IP delivery of IGNs (Figure 2g) demonstrated IGNs were retained in tumors, indicative of active targeting in TME as well as accumulation through the enhanced permeability and retention (EPR) effect. Table S3 shows the concentration of IGNs in all organs. Only trace Au was found in brain tissue because the blood brain barrier prevents the entry of nanoparticles larger than 10 nm, and minimal Au was found in other organs. IGNs were predominantly cleared through the MPS organs, spleen and liver, via internalization by macrophages as expected for Au nanoparticles.29,39,40 Transmission electron micrographs (TEM) confirm that IGNs were localized in Kupffer cells in the liver (Figure 2h), and in the tumor IGNs were observed in intracellular vesicles or lysosome-like structures (Figure 2i), suggesting internalization through receptor-mediated endocytosis.
Figure 2.

Toxicity and biodistribution of IGNs. Serum inflammatory markers and complete blood count of tumor-bearing control mice without IGNs (n = 3) and mice that received intraperitoneal delivery of IGNs 5 days (n = 3) and 15 days (n = 3) postdelivery. Inflammatory markers including (a) liver enzyme, alanine aminotransferase (ALT), and aspartate aminotransferase (AST), and (b) kidney markers, total bilirubin (TBIL), CREAT (creatinine), and blood urea nitrogen (BUN), showed no significant differences between control and IGN-injected mice. Complete blood analysis also showed no abnormalities in hematological parameters, including (c) hemoglobin, (d) red blood cells, (e) white blood cell (WBC) and platelet concentration, and (f) the white blood cell profile (% monocytes and % lymphocytes). (g) Biodistribution and clearance of IGNs confirmed with ICP-MS showed Au in tumor, muscle, and major organs both 5 days (n = 3) and 15 days (n = 3) post-IGN delivery. TEM micrographs of IGNs in (h) Kupffer cells in liver and (i) intracellular vesicles in tumors.
Multimodal Multiplexed Immunoimaging.
Each diagnostic technology has both merits and drawbacks, and a single technique cannot simultaneously achieve all of the desired characteristics of an ideal imaging modality. Recent efforts in immunoPET with radiolabeled mAbs have been very effective in tracking single immunomarker in vivo.41,42 But multiplexing cannot be achieved with PET, as signals between radionuclides cannot be distinguished. Without the ability to multiplex, patients would undergo multiple dosing of radiolabeled mAbs, repeated radiation exposure, and discomfort. Further, dynamic changes in immunomarkers during treatment would be missed as sequential dosing of different mAbs would require >1 week wait time between doses to allow for decay of the radiotracers. Here, we show IGNs enable dynamic detection of both PD-L1+ tumor cells and CD8+ T cells in vivo by synergistically combining the advantages of immunoPET with SERS while overcoming limitations of each approach (Figure 3). Longitudinal immunoPET-SERS imaging was performed after IP delivery of 0.06 mg IGNs/g mouse weight at ~8 MBq of radioactivity in YUMM 2.1 tumor-bearing mice. The longer plasma half-time of IGNs relative to radiolabeled mAbs enabled extended longitudinal study of the two immunomarkers in the TME. First, PET and computed tomography (CT) images were acquired in mice post-IGN administration via whole body scans, which provided a depth-resolved view of the localization of IGNs in tumors with high sensitivity. The decrease in signal in PET images (Figure 3a) was reflective of the rapid decay of 64Cu (half-life ~12 h), and such a trend has been observed previously for chelated 64Cu conjugates.43,44 To account for the decay of 64Cu, the ratio of tumor to adjacent muscle (T/M) is obtained. T/M ratio has been shown in both mouse models and in patients as a more accurate measure of tracer uptake than absolute values in tumor.45,46 The specificity of IGNs in immunomarker detection was demonstrated with blocked control mice, where both PD-L1 and CD8 were preblocked by administering a saturating dose of anti-PD-L1 and anti-CD8 mAbs (200 μg each). Longitudinal PET images of blocked control mice showed lower signal in tumors (Figure S5), a trend supported by other PET imaging studies.47,48 Quantitative PET analysis of the T/M ratio showed high signal in tumors of experimental mice and statistically significant differences (SSD) relative to blocked control mice (Figure 3b). Note that the signal in liver and spleen is not entirely resulting from the use of nanoparticles, as the utility of 64Cu tracers has also shown high background activity in the liver of patients.49 We also observed minimal differences in PET analysis of liver/muscle, kidney/muscle, and spleen/muscle ratios (Figure S6) between experimental and blocked control mice, indicating that the biodistribution of IGNs was similar in both groups, and SSD were only observed in tumors.
Figure 3.

ImmunoPET-SERS imaging with IGNs to detect PD-L1 and CD8. (a) Whole body PET/CT images of mouse bearing YUMM 2.1 tumors at 6 h (1.02% ID/g), 18 h (0.48% ID/g), and 42 h (0.11% ID/g) post-IGN delivery. IGNs identify tumor location by targeting both immunomarkers. (b) Longitudinal PET by examining tumor to muscle (T/M) ratio showing statistically significant differences in the uptake of IGNs in experimental tumors (n = 7) relative to blocked control (n = 5). (c) Normalized SERS spectra of tumors before IGN delivery (0 h) and at maximum accumulation time (6 h) of experimental and blocked control mice. The dominant peaks for Raman reporters DTNB (1325 cm−1) and pMBA (1580 cm−1) are highlighted in yellow. (d) Longitudinal SERS analysis where pMBA and DTNB peaks were normalized to the intrinsic lipid peak at 1440 cm−1. (e) SERS quantification of PD-L1 and CD8 at maximum accumulation time indicating statistically significant differences (~49% difference in DTNB and ~38% in pMBA) between experiment (n = 4) and blocked control groups (n = 4). All ImmunoPET-SERS experiments were repeated 3 times. Here, * indicates p ≤ 0.05.
Immediately following PET, multiplexed detection with SERS was achieved with two different Raman labels, which delineated PD-L1 and CD8 in the tumor with high spatiotemporal resolution. SERS measurements were acquired using a custom portable Raman setup equipped with a 785 nm continuous-wave laser at 80 mW power and a fiber-optic probe. SERS spectra were acquired through the skin by placing the probe at different locations on the tumor; spectra were then averaged, smoothed using a Savitzky-Golay filter,50 and background subtracted. The SERS peaks indicating the detection of CD8+ T cells (DTNB, 1325 cm−1) and PD-L1+ cells (pMBA, 1580 cm−1) in the tumor were normalized to the 1440 cm−1 peak corresponding to lipids and proteins,51 which remained consistent within the same mouse during the time-course study. Individual spectra acquired at different locations on the tumor of mice in both experimental and blocked control groups did not show significant intramouse variability (Figure S7). The averaged SERS spectra of different mice (Figure 3c) before IGN delivery (0 h) and 6 h postdelivery (6 h) showed that IGNs accumulated in tumors, enabling highly specific multiplexed detection distinguishing both PD-L1 and CD8 in experimental tumors relative to the blocked control. It is noteworthy that labeling the mAbs directly with the Raman reporters (without the nanoparticles) would not have enabled any meaningful in vivo signal, as Raman scattering is intrinsically very weak and often overwhelmed by the fluorescence background of biological tissue. As shown in our previous work, the gold nanostars amplify the Raman signal by 109–1012, where the signal is higher at the protrusion tips necessary for highly sensitive signal-to-noise in vivo.19 Longitudinal SERS (Figure 3d) showed maximum accumulation of IGNs occurred at 6 h postdelivery and a decrease in SERS intensity at 42 h. The observed SERS intensity trend was attributed to the surface-weighted characteristics of SERS typical for most optical techniques, where signal was higher near the measurement surface (here mouse skin) closest to the Raman laser probe. Therefore, IGNs accumulating near the tumor surface were preferentially visualized in earlier time points. As IGNs transported from the peripheral vasculature and distributed within the tumor core at later time points, the SERS intensity decreased at 42 h. This trend follows literature evidence that nanoparticles enter solid tumors through leaky vasculatures via the EPR effect and concentrate near the peripheral vasculature rich in blood vessels. In the case of well-vascularized tumors (such as YUMM 2.1), in >24 h nanoparticles may transport into the tumor core via various pathways, such as intercellular or transcellular transport, or remain in the outer tumor layer in the case of necrotic or poorly vascularized solid tumors.52 TEM images taken from areas of the tumor core showed IGNs do transport beyond the periphery in the YUMM 2.1 model (Figure 2i). PET provided a depth-resolved field of view of IGN distribution in tumors even at 42 h. The specificity of our approach was demonstrated by revealing SSD between experimental and blocked control mice. Quantitative SERS at 6 h indicated SSD and lower signal in CD8 (49% lower DTNB signal) and PD-L1 (38% lower pMBA signal) in the blocked control mice relative to experimental mice (Figure 3e). In vivo end points with immunoPET-SERS were validated by flow cytometry analysis of excised tumors (Figure S8). Flow cytometry results showed robust blocking of both immunomarkers and a decrease in PD-L1+ tumor cells (42.6% to 0.34%) and CD8+ cells (7.47% to 0.086%) in the blocked control.
IGNs Monitor Response to Immunotherapies.
In addition to multiplexed detection of immunomarkers, we also demonstrated the utility of IGNs to monitor response to immunotherapies and distinguish responders from those nonresponsive to treatment. Unlike chemotherapy and radiation, accurate response to treatment is imperative in immunotherapies, as patients show distinct radiologic response patterns, including pseudoprogression,53 that are not adequately captured by the traditional Response Evaluation Criteria in Solid Tumors (RECIST).54 Therefore, dynamic imaging of both PD-L1+ tumor cells and CD8+ T cells is vital to accurately reflect the changes in immunomarker localization during the course of treatment since both of these cell types modulate the immune TME. Here, we delivered checkpoint blockade therapy to both BRAF mutant YUMM 2.1 and NRAS mutant YUMM 10.1 melanoma tumors treated with combinatorial PD-L1 and CD137 agonists. BRAF mutations are most prevalent in melanoma and responsive to immunotherapies,55 whereas NRAS mutations are most aggressive and nonresponsive to checkpoint blockade.56 It has been previously shown that costimulation with CD137 results in expansion of effector T cells, production of cytokines, and resistance to suppression by regulatory T cells21,22 and has demonstrated synergistic antitumor effects when combined with anti-PD-L1 mAbs in clinical trials (NCT02451982, NCT03414658).
First, YUMM 2.1 tumor-bearing mice were treated with 3 doses of combined anti-PD-L1 (200 μg/mouse) and anti-CD137 (110 μg/mouse) therapeutic antibodies. IGNs were delivered 24 h after the last treatment followed by ImmunoPET-SERS to monitor dynamic changes in CD8 and PD-L1 status (Figure 4a) post-treatment. Mouse weight did not decrease over the course of therapy, demonstrating minimal adverse effects (Figure S9). Mice with a tumor volume decrease of >20% from baseline to 22 d post-treatment were defined as responders, and all others were categorized as nonresponders. A decrease in tumor volume (Figure 4b) indicated that YUMM 2.1 tumors responded to combinatorial immunotherapy relative to control mice that received isotype-matched anti-IgG (310 μg/mouse). PET-CT images showed that IGNs accumulated in both tumors and major organs of mice, supporting our biodistribution studies (Figure 4c). Quantitative PET signal analysis of T/M ratios indicated an increase in PET signal for the experimental group relative to control group, and SSD between treatment and control mice (Figure 4d). To delineate if the observed increases in PET intensities corresponded to changes in PD-L1 or infiltration of CD8+ T cells, or both, multiplexed SERS analysis was performed. SERS showed (Figure 4e) proliferation of CD8+ T cells in tumors of the treated mice (indicated by a 1325 cm−1 DTNB peak), which corresponded well with PET results and the decrease in tumor volume in the treatment group. Success in immunotherapies in the tumor milieu is followed by recruitment and infiltration of activated CD8+ T cells in the TME. An increase in CD8+ cells gave rise to a higher accumulation of IGNs/anti-CD8/DTNB/64Cu in the tumor post-treatment. Thus, multimodal imaging of the tumors showed an increase in T/M in PET and higher DTNB signal in SERS in the treatment group relative to the IgG control group. SERS spectral analysis of PD-L1 (indicated by a 1580 cm−1 pMBA peak) showed minimal differences between treated and control mice (Figure 4f). This is not surprising, as YUMM 2.1 cells have a constitutionally high expression of PD-L1, and thus an upregulation of PD-L1 resulting from interferon gamma (IFN-γ) was not significant. Flow cytometry analysis of YUMM tumors showing the expression level of PD-L1 receptors (in CD45-negative subset cells) verified our SERS results (Figure 4g). In vivo imaging results were validated with immunohistochemistry (IHC) of excised tumors (Figure 4h) and spleen (Figure S10a,b) stained for CD8+ T cells. IHC images and quantification of the number of 3,3′-diaminobenzidine (DAB)-stained CD8+ cells (Figure 4i) confirmed the expansion of activated CD8+ tumor infiltrating lymphocytes (TILs) in mice treated with immunotherapies (23.7 ± 5.3% positive CD8+ cell) relative to the IgG control (7.6 ± 5.3%). We also quantified the intensity of DAB for the CD8+ stain (Figure S11a) and observed similar trends for treated (1.77 ± 0.73%) and IgG control mice (0.42 ± 0.18%). Localization of CD8+ T cells in the splenic T cell zones (Figure S10a) also suggested systemic T cell activation and expansion in the peripheral organs. H&E staining of tumor sections confirmed that immunotherapies did not alter tissue histomorphology in both treatment and IgG control groups (Figure S12a,b).
Figure 4.

ImmunoPET-SERS imaging to monitor immunotherapy response. (a) Mice bearing YUMM 2.1 tumors were treated with 3 doses of combinatorial immunotherapy of anti-CD137 + anti-PD-L1 followed by IGN delivery and imaging 24 h after the last treatment. Control mice received isotype-matched immunoglobulin G (IgG) treatment (n = 5 for both groups). (b) Tumor volumes decreased with immunotherapy. (c) PET-CT images of mouse revealed a higher localization of IGNs in tumors of treatment group (0.58% ID/g) relative to IgG control (0.31% ID/g). (d) Corresponding PET quantification showing statistically significant differences in tumor/muscle ratio between treatment and control groups. (e) Averaged SERS spectra of treatment and IgG control group. (f) Corresponding SERS quantification showing a statistically significant increase in CD8+ signal. (g) Flow cytometry showed minimal change in PD-L1 status after immunotherapy. (h) Immunohistochemistry images and (i) IHC DAB stain quantification of % CD8+ cells confirmed significantly higher CD8+ tumor-infiltrating lymphocytes in mice treated with immunotherapies (n = 5) relative to control group (n = 5). Here, * indicates p ≤ 0.05, ** indicates p ≤ 0.01, and n.s. indicates not significant. All in vivo and ex vivo experiments were repeated 3 times.
We further demonstrated the efficacy of IGNs in distinguishing responders from those nonresponsive to immunotherapies by examining NRAS mutant YUMM 10.1 murine melanoma tumors. YUMM 10.1 tumors were treated with a similar combinatorial immunotherapy regimen, and anti-IgG mAbs in control mice. YUMM 10.1 tumors were nonresponsive to combination anti-PD-L1 and anti-CD137 treatment, as observed in tumor volume measurement (Figure 5a). ImmunoPET-SERS imaging 24 h post-IGN delivery supported this trend, where whole-body PET-CT scans showed minimal differences in PET signal between treatment and IgG control groups (Figure 5b), verified with PET quantification of T/M ratio (Figure 5c). SERS measurement of tumors delineated both CD8+ (DTNB at 1325 cm−1) and PD-L1 (pMBA at 1580 cm−1) signals and showed minimal differences between experimental and IgG control mice (Figure 5d,e). In vivo end points were validated with flow cytometry analysis of PD-L1+ cells in CD45-subset cells (Figure 5f) and immunohistochemistry of CD8+ cells (Figure 5g,h), which supported our findings with ImmunoPET-SERS, showing minimal difference in the two immunomarkers between treatment and IgG control mice. We note that YUMM 10.1 is an immunogenic tumor model with constitutively high CD8+ T cell infiltration even without immunotherapy, as evidenced by IHC of control tumors (Figure 5h). Additional histopathology findings of YUMM 10.1 tumors treated with immunotherapies are provided in Figure S10c,d and Figure S12c,d.
Figure 5.

ImmunoPET-SERS imaging to distinguish nonresponders. Mice bearing NRAS mutant YUMM 10.1 melanoma tumors were treated with 3 doses of combinatorial immunotherapy of anti-CD137 + anti-PD-L1 followed by IGN delivery and imaging 24 h after the last treatment. Control mice received isotype-matched IgG treatment. (a) Tumor volumes did not decrease for mice receiving immunotherapy (n = 7) relative to IgG control (n = 7). (b) PET images of mice revealed similar localization of IGNs in tumors of treatment group (0.64% ID/g) relative to IgG control (0.7% ID/g). (c) Corresponding quantitative PET analysis showing tumor/muscle ratio. (d) Averaged SERS spectra shown for immunotherapy and control group. (e) Corresponding SERS quantification showed no difference in both CD8 and PD-L1 signals (n = 5 for both groups). (f) Flow cytometry showed minimal change in PD-L1 status in both groups (n = 5 for both groups). All ImmunoPET-SERS experiments were repeated 2 times. (g) Immunohistochemistry of tumors showed CD8+ TILs in both treatment and control groups. (h) IHC DAB quantification of % CD8+ cells showed no difference in cell counts between both groups. The differences were statistically not significant (n.s.) for (a), (c), (e), (f), and (h).
CONCLUSIONS
In summary, we demonstrated the design and in vivo validation of an innovative and clinically translatable nanoprobe, IGNs, for real-time immunological tumor profiling of multiple immunomarkers engaged in the TME. Our results demonstrated that ImmunoPET-SERS imaging with IGNs facilitated both biomarker screening before treatment to identify targetable pathways, and accurately monitored response to immunotherapies to improve the clinical outcome of PD-1/PD-L1 blockade. The results of this work will ultimately allow translation of IGNs from preclinical mouse models to clinically relevant systems because PET is already in the clinic and gold nanoparticles are in clinical trials.57 SERS is facilitated by NIR light that has a 2–4 cm penetration depth,32,33,58 enabling its utility in multiple organs including breast,59 brain,60 and liver,61 useful for both localized and metastatic disease. FDA-approved optical fibers can also now deliver light in deep tissues,62,63 allowing clinical translation of SERS in various tumor types beyond melanoma. Early detection of immunomarkers will improve therapeutic outcomes for responders and accelerate clinical decisions for those requiring alternative treatment as well as minimize toxicities and high costs of unsuccessful therapies for nonresponders. Further, our platform can be expanded beyond melanoma to a multitude of malignancies by targeting other inhibitory ligands (TIM3, LAG3, PSGL-1) and other immune cell populations (CD4+ T cells, NK cells). Moreover, whereas this proof of concept study enabled us to track two immunomarkers with multiplexed SERS, future work will expand the utility of IGNs to detect ~10 biomarkers64,65 for screening heterogeneous tumors and monitoring treatment response.
MATERIALS AND METHODS
Synthesis of Gold Nanostars.
IGNs were synthesized with a biological buffer, HEPES, through a one-step and seedless mediated method that was previously described by our group. Briefly, 18 mL of ultrapure water at 18 MΩ was added to 12 mL of 270 mM HEPES buffer at pH 7.40 ± 0.2. Next, 300 μL of 20 mM chloroauric acid was added. The solution was then mixed by gentle inversion and reacted for 75 min at room temperature. Both IGN synthesis materials, gold(III) chloride trihydrate (HAuCl4) and HEPES, were purchased from Sigma-Aldrich.
Functionalization of IGNs.
Raman reporters, pMBA and DTNB, were purchased from TCI America. Bifunctional linker OPSS-PEG-NHS ester (Mw 2000) was purchased from JenKem Technology. To conjugate Raman reporters to the IGN surface, 6 μL of 10 mM pMBA or DTNB (in 100% ethanol) was added to 60 mL of IGNs and reacted for 15 min at 4 °C. To remove excess Raman reporters, the IGNs were centrifuged at 6000 rpm for 10 min. To functionalize targeting antibodies to IGNs, OPSS-PEG-NHS ester linkers were first reacted with anti-PD-L1 (Bio X Cell BE0101, clone 10F.9G2) and anti-CD8 (Bio X Cell BE0004–1, clone 53-6.7) antibody. Briefly, 8 μL of 80 mg/mL OPSS-PEG-NHS was added to 72 μL of 1 mg/mL antibody and allowed to react in 100 mM (pH 8.4 ± 0.1) sodium bicarbonate (NaHCO3) buffer at 4 °C for 24 h. After, 80 μL of OPSS-PEG-anti-CD8 or OPSS-PEG-anti-PD-L1 was added to 6 mL of Raman-labeled-IGNs at 1.14 mg/mL. The IGN solution was then mixed on an inverter for another 24 h. Next, to conjugate the chelator, DOTA, to gold, OPSS-PEG-NHS ester linkers were reacted with 1.4 mg of DOTA-amine (Macrocyclics) at 1:1 ratio for 10 h. OPSS-PEG-DOTA was then reacted with IGNs for 12 h. Lastly, the fully functionalized IGNs (IGNs/anti-CD8/DTNB or IGNs/anti-PD-L1/pMBA) were centrifuged at 4000 rpm for 10 min twice and resuspended at a concentration of 5 mg/mL.
Characterization of Functionalized IGNs.
The plasmon resonance of a 1.5:1 mixture of anti-PD-L1-pMBA-IGNs and anti-CD8-DTNB-IGNs was measured with a Varian Cary 5000 UV–vis–NIR spectrophotometer. The size and shape of IGNs were visualized with an Osiris transmission electron microscope at 200 keV. The Raman spectra of an IGN mixture at 1.5:1 (IGNs/anti-PD-L1/pMBA and IGNs/anti-CD8/DTNB) were obtained with a custom Raman setup with a 785 nm laser at 80 nW. A Malvern Nano ZS dynamic light scattering apparatus was used to measure both the hydrodynamic size and the zeta potential of IGNs before and after functionalization.
ELISA Binding Assays and Antibody Quantification.
The ELISA binding assay of IGNs/anti-CD8/DTNB was performed with a mouse CD8 alpha ELISA kit (Abcam, ab238263), and that for IGNs/anti-PD-L1/pMBA was performed with a mouse PD-L1 DuoSet ELISA kit (R&D Systems, DY1019–05). All ELISA sandwich assays were performed according to the manufacturer-provided procedure, but detection antibodies were replaced with either IGNs/anti-CD8/DTNB or IGNs/anti-PD-L1/pMBA. ELISA quantification of anti-CD8 or anti-PD-L1 antibodies on IGNs was performed by using secondary antibody (ThermoFisher, 31470) conjugated with horseradish peroxidase and 3,3′,5,5′-tetramethylbenzidine (TMB) substrate. Briefly, IGNs/anti-PD-L1/pMBA or IGNs/anti-CD8/DTNB was blocked with bovine serum albumin in PBS for 1 h, then subsequently incubated with secondary antibody at 0.1 mg/mL for 1 h at room temperature. The sandwich complex was centrifuged and washed with washing buffer three times to remove excess free secondary antibody. TMB solution was then incubated with the sandwich complex for 15 min. The reaction was quenched with 2 N sulfuric acid. Colorimetric readings were performed at 450 nm.
YUMM 2.1 Tumor Model and in Vivo Multimodal Multiplexed Imaging.
Murine melanoma cell lines YUMM2.1 and YUMM10.1, generated by Dr. Marcus Bosenberg (Yale University), were provided by Ann Richmond Lab (Vanderbilt University School of Medicine) with permission from Dr. Richmond and were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco), supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich), 1% penicillin streptomycin (Gibco), and 1× MEM nonessential amino acid (Sigma-Aldrich). YUMM 2.1 cells were cultured at 37 °C and 5% CO2. To develop tumors in B6 (C57BL/6J, Jackson laboratory) mice, 1.5 million YUMM 2.1 cells per 100 μL were injected into the right flank of each mouse. The tumors were monitored with a caliper every 2 days. Once the tumor reached 5 mm in diameter, functionalized IGNs, IGNs/anti-PD-L1/pMBA and IGNs/anti-CD8/DTNB, at a 1.5:1 ratio were administered IP into mice for PET and SERS imaging experiments. Each mouse was injected with 1.2 mg of IGNs with 800 μCi of 64Cu activity. For blocked control, 200 μg of anti-PD-L1 (Bio X Cell, clone 10F.9G2) and 200 μg of anti-CD8 (Bio X Cell, clone 53–6.7) antibodies were injected (IP) concurrently with IGNs at the other side of the abdominal cavity. Note, the antibodies utilized for blocked control were the same clone as the antibodies used to functionalize IGNs.
Mice bearing YUMM 2.1 tumor (for both experiment and blocked control groups) were first placed in a small animal imaging PET/CT machine (Inveon microPET/CT from Siemens Preclinical, Knoxville, TN, USA). Mice were imaged at 6, 18, and 42 h post-IGN administration. The mice were imaged in an Inveon microPET/CT (Siemens Preclinical) while under 2% isoflurane anesthesia. All PET data sets were reconstructed using the MAP algorithm into 128 × 128 × 95 slices with a voxel size of 0.095 × 0.095 × 0.08 cm3 at a beta value of 0.01. The PET and CT images were uploaded in the medical imaging tool Amide (www.sourceforge.amide.com). The PET images were normalized to the injected dose. Regions-of-interest (ROIs) were drawn around the tumor, spleen, liver, kidneys, and muscle (hind limb) for reference. The mean radiotracer concentrations within these ROIs were measured in units of percent injected dose per unit volume (% ID/g).
Once the tumors were identified with PET, SERS imaging was then performed at the same time points as PET imaging (6, 18, and 42 h post-IGN administration) at eight different sites of the tumor with a custom portable Raman spectroscopy system. Measurements were taken for 10 s with a 785 nm diode laser (Innovative Photonics Solutions, Monmouth Junction, NJ, USA) that delivered 80 mW of power using a custom-made fiber-optic probe (EmVision, Loxahatchee, FL, USA), which was gently placed on the tumor. Wavelength calibration of the Raman system was performed using a neon–argon lamp, while acetaminophen and naphthalene standards were used to determine the exact excitation wavelength for calculating Raman shifts. Raman scattering from the samples was first collected from the fiber-optic probe and then by an imaging spectrograph (Holospec f/1.8i, Kaiser Optical Systems, Ann Arbor, MI, USA) coupled to a thermoelectrically cooled CCD camera (PIXIS: 256BR, Princeton Instruments, Princeton, NJ, USA).
The Raman system was corrected for spectral response using a National Institute of Standards and Technology (NIST) calibrated tungsten lamp. Spectra were smoothed with a Savitzky-Golay filter, background subtracted, and fluorescence subtracted using a modified polynomial fit method as previously described. At each time point, both DTNB (1325 cm−1) and pMBA (1580 cm−1) peaks were normalized to a 1440 cm−1 biological peak (corresponding to CH2 stretching), which did not change over time.
In Vivo Multimodal Multiplexed Imaging to Monitor Treatment Response.
A total of 1.5 million YUMM 2.1 or YUMM 10.1 cells per 100 μL were injected into the right flank of each mouse to develop tumors. Once the tumor reached 5 mm in diameter, immunotherapy treatment or IgG control injection commenced. Each treatment mouse received 3 doses of 115 μg of anti-CD137 antibodies (Bio X Cell BE0239, clone 3H3) and 200 μg of anti-PD-L1 (Bio X cell BE0101, clone 10F.9G2) antibodies every 3 days. Each IgG control mouse received 3 doses of 115 μg of IgG2a isotope control (Bio X Cell, BE0089) and 200 μg of IgG2b isotope control (Bio X cell, BE0090) every 3 days. Tumor sizes were measured with a caliper every 2 days. Mouse weight was also monitored to ensure the therapy did not cause any extraneous side effects. A day after the last treatment, mice were administrated with functionalized IGNs, IGNs/anti-PD-L1/pMBA and IGNs/anti-CD8/DTNB at 1.5:1 ratio, for PET/SERS imaging. PET imaging and Raman measurement were performed in the same manner as previously described.
Toxicity Study of IGNs in Vivo.
A total of 1.5 million YUMM 2.1 cells per 100 μL were injected into the right flank of each mouse to develop tumors. Once tumors reached 5 mm in diameter, functionalized IGNs, IGNs/anti-PD-L1/pMBA and IGNs/anti-CD8/DTNB at 1.5:1 ratio, were administered IP into mice. Mice were sacrificed either 5 or 15 day post-particle injection. Cardiac puncture was performed as soon as the mice were euthanized to obtain 500 μL of blood per mouse for both CBC and serum liver/kidney metabolite studies. In addition, tumor, heart, liver, kidney, and spleen of each mouse were retrieved and fixed in 6% formalin for H&E staining. The toxicity study was performed at Vanderbilt University Medical Center–Translational Pathology Shared Resources. Complete blood counts were performed in the Forcyte veterinary hematology analyzer manufactured by Oxford Science. Blood chemistries were performed on the Vet Axcel chemistry analyzer manufactured by Alfa Wassermann.
Inductively Coupled Plasma-Mass Spectrometry.
Mice bearing YUMM 2.1 tumor were injected with functionalized IGNs, anti-PD-L1-pMBA-IGNs and anti-CD8-DTNB-IGNs at 1.5:1 ratio. For each mouse, the tumor, stomach, liver, spleen, kidneys, heart, lungs, brain, and muscle were retrieved either 5 or 15 days post-particle injection. After dissection, the tissues were frozen immediately in liquid nitrogen. A lyophilizer was first used to remove any water in the tissues. Next, dried tissues were then placed in 75 vol % trace metal grade aqua regia (HCl from Fisher Scientific, A508-P500 and HNO3 from Fisher Scientific, A509-P500) for 72 h. Aqua regia was then boiled off, and the tissue samples were then redissolved in 10 mL of 2 vol % aqua regia. Filters (0.4 μm) were used to remove any impurities prior to ICP-MS readings.
ICP-MS measurement and analysis were performed at Vanderbilt University, Department of Civil and Environmental Engineering. The PerkinElmer model ELAN DRC II was operated in standard mode for all readings. The setting of the instrument was 1.5 kW radio frequency power, 15 L/min argon plasma flow, 1 L/min nebulizer flow, and 1 s integration time for 3 replicates. A six-point calibration curve was performed for gold isotope 197 between 0.05 and 500 μg/L. Analytical blanks and check standards (0.5 μg/L) were measured for every 3–5 samples to ensure the readings were within 15% of the specified value.
Transmission Electron Microscope Imaging of Tissues.
Mice bearing YUMM 2.1 tumor were injected with functionalized IGNs, IGNs/anti-PD-L1/pMBA and IGNs/anti-CD8/DTNB at 1.5:1 ratio. The mice were sacrificed at 6 h post-particle administration, and the tumor liver, and spleen were retrieved. All samples were sectioned into 1 mm by 1 mm pieces with razor blades and fixed in 2.5% gluteraldehyde in 0.1 M cacodylate buffer (pH 7.4 ± 0.1) first at room temperature for 1 h and then 24 h at 4 °C. The specimens were further processed for transition electron microscopy imaging by the Vanderbilt Cell Imaging Shared Resource facility. The tissue samples were further fixed with 1% osmium tetraoxide and washed with 0.1 M cacodylate buffer. Sample dehydration was done serially with graded ethanol. Three 100% ethanol exchanges and two exchanges of pure propylene oxide (PO) were performed. The samples were then filtrated with 25% Epon 812 resin and 75% PO for 30 min at room temperature, then with 50% Epon 812 resin for 1 h and 50% PO overnight. Subsequently, all samples went through an Epon 812 resin and PO (3:1) exchange and incubated with pure epoxy resin overnight. Before sample embedding, two exchanges of pure epoxy resin were performed. Lastly, polymerization was done at 60 °C for 48 h. Once the embedding process was complete, the samples were first sectioned at 500–1000 nm and then were cut into 70–80 nm ultrathin sections. The samples were placed on copper grids and stained with uranyl acetate (2%) and Reynold’s lead citrate. TEM imaging was performed with a Philips/FEI Tecnai T12 electron microscope.
Quantification of CD8 Immunohistochemistry.
Images were captured with a Leica SCN400 slide scanner automated digital image system from Leica Microsystems with 20× magnification to a resolution of 0.5 μm/pixel. Cell identification was performed either with standard Ariol analysis scripts (% cell) or by FIJI66 (ImageJ-based open-source software, % intensity). For Ariol analysis scripts, both brown (DAB) positive cells and blue (hematoxylin only) negative cells were distinguished by setting upper and lower thresholds for color, saturation, intensity, size, roundness, and axis length. For the FIJI algorithm, color deconvolution was used to extract and threshold positively stained areas. Resulting binary images were then used to calculate integrated density values.
Flow Cytometry Analysis.
YUMM 2.1 and YUMM 10.1 tumors were developed as previously described. Tumors were treated with blocking antibodies, anti-PD-L1 and anti-CD137 combination treatment, or IgG treatment. Harvested tumors were mechanically dissociated with an OctoMACS separator and digested in a solution of 125 μg/mL−1 deoxyribonuclease I (Worthington) and 500 μg/mL−1 collagenase III (Worthington) in RPMI media for 60 min at 37 °C. Tumors were then strained through a 40 μm cell strainer and further treated with ACK lysing buffer (Gibco). Cell suspensions (100 μL) for each sample were transferred into a 96-well plate and treated with FcX. Samples were stained with antibodies PE-PD-L1 (BioLegend 124307, clone 10F.9G2), B7-H1 (BioLegend 124307, clone 10F.9G2), FITC-CD8a (ThermoFisher Scientific 11-0081-82, clone 53-6.7), and APC/Cy7-CD45 (Biolegend 103115, clone 30-F11) (PE, phycoerythrin; APC, allophycocyanin; Cy, cyanine; FITC, fluorescein isothiocyanate). After staining, cells were washed with PBS twice and then suspended in PBS containing 2% FBS and 200 nM 4′,6-diamidino-2-phenylindole before analysis. Flow cytometry analysis was performed on a BD LSR Fortessa or BD LSR II flow cytometer.
Statistical Analysis.
All data are presented as mean ± standard deviation. The sample sizes were estimated based on our previously published work on SERS in vivo.20 Whereas that work was performed in immunocompromised nude mice, it directed us to the number of mice that should be used for good signal-to-noise ratio in vivo. Power analysis was performed, power level was set to 80%, and confidence level was set to 95%. Differences between groups were assessed using Excel with paired or unpaired two-sided Student’s t tests for the calculation of p values. Here, * indicates p ≤ 0.05 and ** indicates p ≤ 0.01.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Jennifer Bateman for her assistance with the portable Raman setup and maintenance of the equipment. We also acknowledge Eden Paul for animal welfare including tumor measurement, monitoring mice health, and sacrificing mice. This work is supported by American Cancer Society Institutional Research Grant (IRG-58-009-56), National Center for Advancing Translational Sciences CTSA award, and CDMRP Peer Reviewed Cancer Research Program Grant W81XWH1810139. X.W. and R.B. acknowledge support from National Science Foundation (NSF) grant CMMI-1634856. O.D.A. acknowledges support by a Department of Defense, Air Force of Scientific Research, National Defense Science and Engineering Graduate (NDSEG) Fellowship [32 CFR 168a]. R.C.D. acknowledges support from Vanderbilt’s Trans Institutional Programs grant: Materials Durability and Environmental Research Facilities Hub (Faculty PIs - Florence Sanchez and David Kosson). M.R. acknowledges support from the National Institutes of Health (NIH) R00CA201304. D.S. and J.T.W. acknowledge support from the NSF CBET-1554623 (J.T.W.), Vanderbilt Center for Immunobiology Pilot Grant, VICC Ambassador Discovery Grant, the Melanoma Research Alliance 503565, and Stand Up To Cancer Innovative Research Grant SU2C-AACR-IRG 20-17 (J.T.W.). A.E.V. acknowledges support from NIH (R37 CA233770-01), Breast Cancer Research Foundation (IIDRP-16-001), and Lloyd Foundation of Melanoma Research. A.R. acknowledges support from NIH grant CA116021. TEM micrographs of IGN were acquired with an instrument supported by NSF EPS 1004083. TEM images of tumor and organs were acquired by using VUMC Cell Imaging Shared Resource (supported by NIH grants CA68485, DK20593, DK58404, DK59637, and EY08126).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b07326.
Additional schematic for mAbs and DOTA conjugation to IGNs, IGN stability and shelf life studies, IGN antibody binding assays, quantitative ICP-MS analysis of biodistribution of IGNs, histopathology of tumor and major organs for IGN toxicity study, ELISA quantification of IGN antibodies, longitudinal PET images of blocked control mouse, quantification of PET signals in major orangs, examples of individual SERS spectra, flow cytometry of YUMM 2.1 cell line targeting CD8 and PD-L1, body weight of mice bearing YUMM 2.1 tumor treated with or without immunotherapy, quantification of tumor CD8 IHC by intensity, and CD8 IHC and histopathology of spleen and tumor of mice treated with or without therapy (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Bertucci F; Goncalves A Immunotherapy in Breast Cancer: the Emerging Role of PD-1 and PD-L1. Curr. Oncol. Rep 2017, 19, 64. [DOI] [PubMed] [Google Scholar]
- (2).Tumeh PC; Harview CL; Yearley JH; Shintaku IP; Taylor EJ; Robert L; Chmielowski B; Spasic M; Henry G; Ciobanu V; West AN; Carmona M; Kivork C; Seja E; Cherry G; Gutierrez AJ; Grogan TR; Mateus C; Tomasic G; Glaspy JA; et al. PD-1 Blockade Induces Responses by Inhibiting Adaptive Immune Resistance. Nature 2014, 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vonderheide RH; Domchek SM; Clark AS Immunotherapy for Breast Cancer: What Are We Missing? Clin. Cancer Res 2017, 23, 2640–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Jenkins RW; Barbie DA; Flaherty KT Mechanisms of Resistance to Immune Checkpoint Inhibitors. Br. J. Cancer 2018, 118, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Liang Y; Tang H; Guo J; Qiu X; Yang Z; Ren Z; Sun Z; Bian Y; Xu L; Xu H; Shen J; Han Y; Dong H; Peng H; Fu Y-X Targeting IFNα to Tumor by Anti-PD-L1 Creates Feedforward Antitumor Responses to Overcome Checkpoint Blockade Resistance. Nat. Commun 2018, 9, 4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rasmussen JH; Lelkaitis G; Håkansson K; Vogelius IR; Johannesen HH; Fischer BM; Bentzen SM; Specht L; Kristensen CA; von Buchwald C; Wessel I; Friborg J Intratumor Heterogeneity of PD-L1 Expression in Head and Neck Squamous Cell Carcinoma. Br. J. Cancer 2019, 120, 1003–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gniadek TJ; Li QK; Tully E; Chatterjee S; Nimmagadda S; Gabrielson E Heterogeneous Expression of PD-L1 in Pulmonary Squamous Cell Carcinoma and Adenocarcinoma: Implications for Assessment by Small Biopsy. Mod. Pathol 2017, 30, 530–538. [DOI] [PubMed] [Google Scholar]
- (8).Nishino M; Ramaiya NH; Hatabu H; Hodi FS Monitoring Immune-Checkpoint Blockade: Response Evaluation and Biomarker Development. Nat. Rev. Clin. Oncol 2017, 14, 655–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Broos K; Lecocq Q; Raes G; Devoogdt N; Keyaerts M; Breckpot K Noninvasive Imaging of the PD-1:PD-L1 Immune Checkpoint: Embracing Nuclear Medicine for the Benefit of Personalized Immunotherapy. Theranostics 2018, 8, 3559–3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Fumet J-D; Richard C; Ledys F; Klopfenstein Q; Joubert P; Routy B; Truntzer C; Gagné A; Hamel M-A; Guimaraes CF; Coudert B; Arnould L; Favier L; Lagrange A; Ladoire S; Saintigny P; Ortiz-Cuaran S; Perol M; Foucher P; Hofman P; et al. Prognostic and Predictive Role of CD8 and PD-L1 Determination in Lung Tumor Tissue of Patients Under Anti-PD-1 Therapy. Br. J. Cancer 2018, 119, 950–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hamanishi J; Mandai M; Iwasaki M; Okazaki T; Tanaka Y; Yamaguchi K; Higuchi T; Yagi H; Takakura K; Minato N; Honjo T; Fujii S Programmed Cell Death 1 Ligand 1 and Tumor-Infiltrating CD8+ T Lymphocytes Are Prognostic Factors of Human Ovarian Cancer. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 3360–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Huang AC; Postow MA; Orlowski RJ; Mick R; Bengsch B; Sasikanth M; Xu W; Harmon S; Giles JR; Wenz B; Adamow M; Kuk D; Panageas KS; Carrera C; Wong P; Quagliarello F; Wubbenhorst B; D’Andrea K; Pauken KE; Herati RS; et al. T-Cell Invigoration to Tumour Burden Ratio Associated with Anti-PD-1 Response. Nature 2017, 545, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Homet Moreno B; Zaretsky JM; Garcia-Diaz A; Tsoi J; Parisi G; Robert L; Meeth K; Ndoye A; Bosenberg M; Weeraratna AT; Graeber TG; Comin-Anduix B; Hu-Lieskovan S; Ribas A Response to Programmed Cell Death-1 Blockade in a Murine Melanoma Syngeneic Model Requires Costimulation, CD4, and CD8 T Cells. Cancer Immunol. Res 2016, 4, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Yao H; Lan J; Li C; Shi H; Brosseau J-P; Wang H; Lu H; Fang C; Zhang Y; Liang L; Zhou X; Wang C; Xue Y; Cui Y; Xu J Inhibiting PD-L1 Palmitoylation Enhances T-Cell Immune Responses Against Tumours. Nat. Biomed. Eng 2019, 3, 306–317. [DOI] [PubMed] [Google Scholar]
- (15).Kitano A; Ono M; Yoshida M; Noguchi E; Shimomura A; Shimoi T; Kodaira M; Yunokawa M; Yonemori K; Shimizu C; Kinoshita T; Fujiwara Y; Tsuda H; Tamura K Tumour-Infiltrating Lymphocytes Are Correlated With Higher Expression Levels of PD-1 and PD-L1 in Early Breast Cancer. ESMO Open 2017, 2, No. e000150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Altan M; Kidwell KM; Pelekanou V; Carvajal-Hausdorf DE; Schalper KA; Toki MI; Thomas DG; Sabel MS; Hayes DF; Rimm DL Association of B7-H4, PD-L1, and Tumor Infiltrating Lymphocytes with Outcomes in Breast Cancer. NPJ. Breast Cancer 2018, 4, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ding S-Y; Yi J; Li J-F; Ren B; Wu D-Y; Panneerselvam R; Tian Z-Q Nanostructure-Based Plasmon-Enhanced Raman Spectroscopy For Surface Analysis of Materials. Nat. Rev. Mater 2016, 1, 16021. [Google Scholar]
- (18).Ou Y-C; Wen X; Bardhan R, Cancer Immunoimaging with Smart Nanoparticles. Trends Biotechnol 2019, 10.1016/j.tibtech.2019.11.001. [DOI] [PubMed]
- (19).Webb JA; Aufrecht J; Hungerford C; Bardhan R Ultrasensitive Analyte Detection with Plasmonic Paper Dipsticks and Swabs Integrated With Branched Nanoantennas. J. Mater. Chem. C 2014, 2, 10446–10454. [Google Scholar]
- (20).Ou YC; Webb JA; O’Brien CM; Pence IJ; Lin EC; Paul EP; Cole D; Ou SH; Lapierre-Landry M; DeLapp RC; Lippmann ES; Mahadevan-Jansen A; Bardhan R Diagnosis of Immunomarkers In Vivo via Multiplexed Surface Enhanced Raman Spectroscopy with Gold Nanostars. Nanoscale 2018, 10, 13092–13105. [DOI] [PubMed] [Google Scholar]
- (21).Liu J; Blake SJ; Yong MCR; Harjunpää H; Ngiow SF; Takeda K; Young A; O’Donnell JS; Allen S; Smyth MJ; Teng MWL Improved Efficacy of Neoadjuvant Compared to Adjuvant Immunotherapy to Eradicate Metastatic Disease. Cancer Discovery 2016, 6, 1382–1399. [DOI] [PubMed] [Google Scholar]
- (22).Yonezawa A; Dutt S; Chester C; Kim J; Kohrt HE Boosting Cancer Immunotherapy with Anti-CD137 Antibody Therapy. Clin. Cancer Res 2015, 21, 3113–3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ou YC; Webb JA; Faley S; Shae D; Talbert EM; Lin S; Cutright CC; Wilson JT; Bellan LM; Bardhan R Gold Nanoantenna-Mediated Photothermal Drug Delivery From Thermo-sensitive Liposomes in Breast Cancer. ACS Omega 2016, 1, 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Webb JA; Erwin WR; Zarick HF; Aufrecht J; Manning HW; Lang MJ; Pint CL; Bardhan R Geometry-Dependent Plasmonic Tunability and Photothermal Characteristics of Multi-branched Gold Nanoantennas. J. Phys. Chem. C 2014, 118, 3696–3707. [Google Scholar]
- (25).Tamura K; Kurihara H; Yonemori K; Tsuda H; Suzuki J; Kono Y; Honda N; Kodaira M; Yamamoto H; Yunokawa M; Shimizu C; Hasegawa K; Kanayama Y; Nozaki S; Kinoshita T; Wada Y; Tazawa S; Takahashi K; Watanabe Y; Fujiwara Y 64Cu-DOTA-Trastuzumab PET Imaging in Patients With HER2-Positive Breast Cancer. J. Nucl. Med 2013, 54, 1869–1875. [DOI] [PubMed] [Google Scholar]
- (26).Mortimer JE; Bading JR; Park JM; Frankel PH; Carroll MI; Tran TT; Poku EK; Rockne RC; Raubitschek AA; Shively JE; Colcher DM Tumor Uptake of 64Cu-DOTA-Trastuzumab in Patients With Metastatic Breast Cancer. J. Nucl. Med 2018, 59, 38–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Persson M; Skovgaard D; Brandt-Larsen M; Christensen C; Madsen J; Nielsen CH; Thurison T; Klausen TL; Holm S; Loft A; Berthelsen AK; Ploug M; Pappot H; Brasso K; Kroman N; Højgaard L; Kjaer A First-in-Human uPAR PET: Imaging of Cancer Aggressiveness. Theranostics 2015, 5, 1303–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Webb JA; Ou YC; Faley S; Paul EP; Hittinger JP; Cutright CC; Lin EC; Bellan LM; Bardhan R Theranostic Gold Nanoantennas for Simultaneous Multiplexed Raman Imaging of Immunomarkers and Photothermal Therapy. ACS Omega 2017, 2, 3583–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Dogra P; Adolphi NL; Wang Z; Lin YS; Butler KS; Durfee PN; Croissant JG; Noureddine A; Coker EN; Bearer EL; Cristini V; Brinker CJ Establishing the Effects of Mesoporous Silica Nanoparticle Properties on In Vivo Disposition Using Imaging-Based Pharmacokinetics. Nat. Commun 2018, 9, 4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gatti AM; Rivasi F Biocompatibility of Micro-and Nanoparticles. Part I: In Liver and Kidney. Biomaterials 2002, 23, 2381–2387. [DOI] [PubMed] [Google Scholar]
- (31).Wiegand S; Heinen T; Ramaswamy A; Sesterhenn AM; Bergemann C; Werner JA; Lubbe AS Evaluation of the Tolerance And Distribution of Intravenously Applied Ferrofluid Particles of 250 and 500 nm Size in an Animal Model. J. Drug Target 2009, 17, 194–199. [DOI] [PubMed] [Google Scholar]
- (32).Farist F; Thornileyt M; Wickramasinghet Y; Houstont R; Rolfet P; Livers N; Spencer A Non-Invasive In Vivo Near-Infrared Optical Measurement of the Penetration Depth in the Neonatal Head. Clin. Phys. Physiol. Meas 1991, 12, 353–358. [DOI] [PubMed] [Google Scholar]
- (33).Henderson TA; Morries LD Near-Infrared Photonic Energy Penetration: Can Infrared Phototherapy Effectively Reach the Human Brain? Neuropsychiatr. Dis. Treat 2015, 11, 2191–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hong G; Diao S; Chang J; Antaris AL; Chen C; Zhang B; Zhao S; Atochin DN; Huang PL; Andreasson KI; Kuo CJ; Dai H Through-Skull Fluorescence Imaging of the Brain in a New Near-Infrared Window. Nat. Photonics 2014, 8, 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Liu Y; Maccarini P; Palmer GM; Etienne W; Zhao Y; Lee CT; Ma X; Inman BA; Vo-Dinh T Synergistic Immuno Photothermal Nanotherapy (SYMPHONY) for the Treatment of Unresectable and Metastatic Cancers. Sci. Rep 2017, 7, 8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Liu Y; Ashton JR; Moding EJ; Yuan H; Register JK; Fales AM; Choi J; Whitley MJ; Zhao X; Qi Y; Ma Y; Vaidyanathan G; Zalutsky MR; Kirsch DG; Badea CT; Vo-Dinh T A Plasmonic Gold Nanostar Theranostic Probe for In Vivo Tumor Imaging and Photothermal Therapy. Theranostics 2015, 5, 946–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Meeth K; Wang JX; Micevic G; Damsky W; Bosenberg MW The YUMM Lines: A Series of Congenic Mouse Melanoma Cell Lines with Defined Genetic Alterations. Pigm. Cell Melanoma Res 2016, 29, 590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Yang L; Kuang H; Zhang W; Aguilar ZP; Wei H; Xu H Comparisons of the Biodistribution and Toxicological Examinations After Repeated Intravenous Administration of Silver and Gold Nanoparticles in Mice. Sci. Rep 2017, 7, 3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Tsoi KM; MacParland SA; Ma X-Z; Spetzler VN; Echeverri J; Ouyang B; Fadel SM; Sykes EA; Goldaracena N; Kaths JM; Conneely JB; Alman BA; Selzner M; Ostrowski MA; Adeyi OA; Zilman A; McGilvray ID; Chan WCW Mechanism of Hard-Nanomaterial Clearance by the Liver. Nat. Mater 2016, 15, 1212–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Park S.-m.; Aalipour A; Vermesh O; Yu JH; Gambhir SS Towards Clinically Translatable In Vivo Nanodiagnostics. Nat. Rev. Mater 2017, 2, 17014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Bensch F; van der Veen EL; Lub-De Hooge MN; Jorritsma-Smit A; Boellaard R; Kok IC; Oosting SF; Schröder CP; Hiltermann TJN; van der Wekken AJ; Groen HJM; Kwee TC; Elias SG; Gietema JA; Bohorquez SS; de Crespigny A; Williams S-P; Mancao C; Brouwers AH; Fine BM; et al. 89Zr-Atezolizumab Imaging as a Non-Invasive Approach to Assess Clinical Response to PD-L1 Blockade in Cancer. Nat. Med 2018, 24, 1852–1858. [DOI] [PubMed] [Google Scholar]
- (42).Tavaré R; Escuin-Ordinas H; Mok S; McCracken MN; Zettlitz KA; Salazar FB; Witte ON; Ribas A; Wu AM An Effective Immuno-PET Imaging Method to Monitor CD8-Dependent Responses to Immunotherapy. Cancer Res 2016, 76, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Jiang L; Tu Y; Hu X; Bao A; Chen H; Ma X; Doyle T; Shi H; Cheng Z Pilot Study of 64Cu(I) for PET Imaging of Melanoma. Sci. Rep 2017, 7, 2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Sprague JE; Peng Y; Sun X; Weisman GR; Wong EH; Achilefu S; Anderson CJ Preparation and Biological Evaluation of Copper-64-Labeled Tyr3-Octreotate Using a Cross-Bridged Macro-cyclic Chelator. Clin. Cancer Res 2004, 10, 8674–8682. [DOI] [PubMed] [Google Scholar]
- (45).Sato Y; Oh M; Mori T; Kiyono Y; Fujieda S; Okazawa H Tumor-to-Muscle Ratio of Cu-ATSM Uptake May Reflect Outcome of Treatment in Head-and-Neck Cancers. J. Nucl. Med 2013, 54, 1526–1526. [Google Scholar]
- (46).Fleming IN; Manavaki R; Blower PJ; West C; Williams KJ; Harris AL; Domarkas J; Lord S; Baldry C; Gilbert FJ Imaging Tumour Hypoxia with Positron Emission Tomography. Br. J. Cancer 2015, 112, 238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ping Li W; Meyer LA; Capretto DA; Sherman CD; Anderson CJ Receptor-Binding, Biodistribution, and Metabolism Studies of 64Cu-DOTA-Cetuximab, a PET-Imaging Agent for Epidermal Growth-Factor Receptor-Positive Tumors. Cancer Bio-ther.Radiopharm 2008, 23, 158–171. [DOI] [PubMed] [Google Scholar]
- (48).Hettich M; Braun F; Bartholoma MD; Schirmbeck R; Niedermann G High-Resolution PET Imaging with Therapeutic Antibody-Based PD-1/PD-L1 Checkpoint Tracers. Theranostics 2016, 6, 1629–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Lee H; Shields AF; Siegel BA; Miller KD; Krop I; Ma CX; LoRusso PM; Munster PN; Campbell K; Gaddy DF; Leonard SC; Geretti E; Blocker SJ; Kirpotin DB; Moyo V; Wickham TJ; Hendriks BS (64)Cu-MM-302 Positron Emission Tomography Quantifies Variability of Enhanced Permeability and Retention of Nanoparticles in Relation to Treatment Response in Patients with Metastatic Breast Cancer. Clin. Cancer Res 2017, 23, 4190–4202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Lieber CA; Majumder SK; Ellis DL; Billheimer DD; Mahadevan-Jansen A In Vivo Nonmelanoma Skin Cancer Diagnosis Using Raman Microspectroscopy. Lasers Surg. Med 2008, 40, 461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Movasaghi Z; Rehman S; Rehman IU Raman Spectroscopy of Biological Tissues. Appl. Spectrosc. Rev 2007, 42, 493–541. [Google Scholar]
- (52).Wilhelm S; Tavares AJ; Dai Q; Ohta S; Audet J; Dvorak HF; Chan WCW Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater 2016, 1, 16014. [Google Scholar]
- (53).Cohen JV; Alomari AK; Vortmeyer AO; Jilaveanu LB; Goldberg SB; Mahajan A; Chiang VL; Kluger HM Melanoma Brain Metastasis Pseudoprogression After Pembrolizumab Treatment. Cancer Immunol. Res 2016, 4, 179–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Gerwing M; Herrmann K; Helfen A; Schliemann C; Berdel WE; Eisenblätter M; Wildgruber M The Beginning of the End for Conventional RECIST — Novel Therapies Require Novel Imaging Approaches. Nat. Rev. Clin. Oncol 2019, 16, 442–458. [DOI] [PubMed] [Google Scholar]
- (55).Cheng L; Lopez-Beltran A; Massari F; MacLennan GT; Montironi R Molecular Testing for BRAF Mutations to Inform Melanoma Treatment Decisions: A Move Toward Precision Medicine. Mod. Pathol 2018, 31, 24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Munoz-Couselo E; Adelantado EZ; Ortiz C; Garcia JS; Perez-Garcia J NRAS-Mutant Melanoma: Current Challenges and Future Prospect. OncoTargets Ther 2017, 10, 3941–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Rastinehad AR; Anastos H; Wajswol E; Winoker JS; Sfakianos JP; Doppalapudi SK; Carrick MR; Knauer CJ; Taouli B; Lewis SC; Tewari AK; Schwartz JA; Canfield SE; George AK; West JL; Halas NJ Gold Nanoshell-Localized Photothermal Ablation of Prostate Tumors in a Clinical Pilot Device Study. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 18590–18596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Ku G; Wang LV Deeply Penetrating Photoacoustic Tomography in Biological Tissues Enhanced with an Optical Contrast Agent. Opt. Lett 2005, 30, 507–509. [DOI] [PubMed] [Google Scholar]
- (59).Lyng FM; Traynor D; Nguyen TNQ; Meade AD; Rakib F; Al-Saady R; Goormaghtigh E; Al-Saad K; Ali MH Discrimination of Breast Cancer from Benign Tumours Using Raman Spectroscopy. PLoS One 2019, 14, No. e0216311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Jermyn M; Mok K; Mercier J; Desroches J; Pichette J; Saint-Arnaud K; Bernstein L; Guiot M-C; Petrecca K; Leblond F Intraoperative Brain Cancer Detection with Raman Spectroscopy in Humans. Sci. Transl. Med 2015, 7, 274ra19. [DOI] [PubMed] [Google Scholar]
- (61).Andreou C; Neuschmelting V; Tschaharganeh D-F; Huang C-H; Oseledchyk A; Iacono P; Karabeber H; Colen RR; Mannelli L; Lowe SW; Kircher MF Imaging of Liver Tumors Using Surface-Enhanced Raman Scattering Nanoparticles. ACS Nano 2016, 10, 5015–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Liang W; Hall G; Messerschmidt B; Li M-J; Li X Nonlinear Optical Endomicroscopy for Label-Free Functional Histology In Vivo. Light: Sci. Appl 2017, 6, No. e17082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Pisanello F; Leonardo Sileo A; Oldenburg I; Pisanello M; Luigi M; Assad J; Sabatini B; Vittorio M Multipoint-Emitting Optical Fibers for Spatially Addressable In Vivo Optogenetics. Neuron 2014, 82, 1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Harmsen S; Wall MA; Huang R; Kircher MF Cancer Imaging Using Surface-Enhanced Resonance Raman Scattering Nanoparticles. Nat. Protoc 2017, 12, 1400–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Laing S; Jamieson LE; Faulds K; Graham D Surface-Enhanced Raman Spectroscopy for In Vivo Biosensing. Nat. Rev. Chem 2017, 1, 0060. [Google Scholar]
- (66).Schindelin J; Arganda-Carreras I; Frise E; Kaynig V; Longair M; Pietzsch T; Preibisch S; Rueden C; Saalfeld S; Schmid B Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.