

SUMMARY
Recurrent copy-number alterations, mutations, and transcript fusions of the genes encoding CDKs/cyclins are characterized in >10,000 tumors. Genomic alterations of CDKs/cyclins are dominantly driven by copy number aberrations. In contrast to cell-cycle-related CDKs/cyclins, which are globally amplified, transcriptional CDKs/cyclins recurrently lose copy numbers across cancers. Although mutations and transcript fusions are relatively rare events, CDK12 exhibits recurrent mutations in multiple cancers. Among the transcriptional CDKs, CDK7 and CDK12 show the most significant copy number loss and mutation, respectively. Their genomic alterations are correlated with increased sensitivities to DNA-damaging drugs. Inhibition of CDK7 preferentially represses the expression of genes in the DNA-damage-repair pathways and impairs the activity of homologous recombination. Low-dose CDK7 inhibitor treatment sensitizes cancer cells to PARP inhibitor-induced DNA damage and cell death. Our analysis provides genomic information for identification and prioritization of drug targets for CDKs and reveals rationales for treatment strategies.
Graphical Abstract
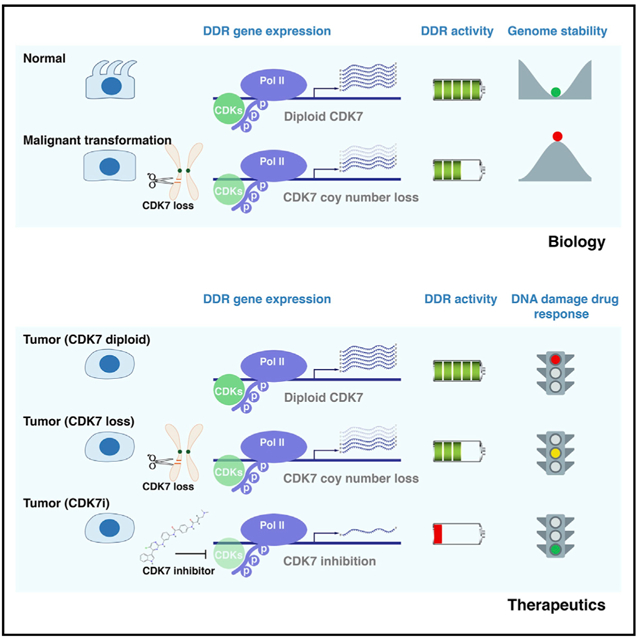
In Brief
Shan et al. characterize recurrent copy number alterations, mutations, and transcript fusions of the genes encoding CDKs/cyclins in >10,000 tumor specimens across common adult cancers. This analysis provides genomic information for identification and prioritization of drug targets for CDKs and reveals rationales for potential treatment strategies.
INTRODUCTION
Cyclin-dependent kinases (CDKs) make up an evolutionarily conserved serine-threonine kinase family and have central roles in controlling cell division and modulating transcription (Asghar et al., 2015; Ferguson and Gray, 2018; Malumbres et al., 2009; O’Leary et al., 2016; Otto and Sicinski, 2017; Sherr et al., 2016). A CDK, when bound with the regulatory protein cyclin, forms the cyclin-CDK complex, which activates multiple downstream proteins via phosphorylation. Consequently, these phosphorylated proteins are responsible for certain events during cell division and transcription. Given that tumor cells are always subject to uncontrolled proliferation and dysregulated transcription, CDKs have been historically considered attractive targets in cancer therapy (Asghar et al., 2015; Ferguson and Gray, 2018; Malumbres et al., 2009; O’Leary et al., 2016; Otto and Sicinski, 2017; Sherr et al., 2016). Although numerous CDK inhibitors (CDKis) have been developed during the past 20 years, most first-generation nonselective CDKis (pan-CDKis) failed in clinical trials because of toxicity and lack of efficacy. Recently, selective CDKis have been developed and are emerging as a class of anticancer agents. For example, several CDK4/6 inhibitors have been approved by the US Food and Drug Administration (FDA) for use in women with ER+ and HER2−, advanced, metastatic breast cancer (Asghar et al., 2015; Ferguson and Gray, 2018; O’Leary et al., 2016; Otto and Sicinski, 2017; Sherr et al., 2016). Thus, the development of selective CDKis to target certain CDKs is one of the keys to successfully translating CDK biology into clinical application. However, given that there are more than 20 distinct CDKs in humans (Malumbres et al., 2009), challenges in target identification and prioritization have resulted in a narrow focus in the development of drugs targeting CDKs for cancer treatment.
Transcriptional CDKs are master regulators of phosphorylation of the C-terminal domain (CTD) of RNA polymerase II (Pol II), dynamically coordinating transcription cycle and RNA processing (Bradner et al., 2017; Chou et al., 2020; Fisher, 2012; Zaborowska et al., 2016). Given that the transcriptional program is remarkably dysregulated in cancer (transcriptional addiction) (Bradner et al., 2017; Chou et al., 2020), inhibitors targeting transcriptional CDKs are emerging as a class of anti-cancer agents (Ali et al., 2009; Gao et al., 2018; Hu et al., 2019a; Kalan et al., 2017; Kwiatkowski et al., 2014; Minzel et al., 2018; Olson et al., 2019; Patel et al., 2018; Quereda et al., 2019; Zhang et al., 2016, 2018). CDK7 has critical roles in regulating both transcription and cell division (Bradner et al., 2017; Chou et al., 2020; Fisher, 2012; Zaborowska et al., 2016). As a component of the general transcription factor complex TFIIH, CDK7 modulates transcription initiation by phosphorylating the Pol II CTD (Ser 5 and Ser 7). Meanwhile, CDK7 also functions as a CDK-activating kinase (CAK), which controls cell division by phosphorylating other cell-cycle CDKs within the activation segment (T-loop). CDK12 modulates transcription elongation and mRNA processing by phosphorylating the Pol II CTD (Ser 5 and Ser 2) (Bradner et al., 2017; Chou et al., 2020; Fisher, 2012; Zaborowska et al., 2016) and preferentially affects transcription of the genes involved in DNA-damage repair (DDR) (Bajrami et al., 2014; Dubbury et al., 2018; Iniguez et al., 2018; Johnson et al., 2016; Krajewska et al., 2019). THZ1, a covalent inhibitor of CDK7 (CDK7i), was recently identified and forms a covalent link to cysteine-312 outside the kinase domain to inactivate CDK7 irreversibly (Kwiatkowski et al., 2014). Unlike other CDKis, the THZ1 binding site is located outside the kinase domain, providing an explanation for its specificity. Notably, because of the similarity of their protein structures, THZ1 also inhibits the activity of CDK12 at higher concentrations (Kwiatkowski et al., 2014). Therefore, THZ1 treatment leads to the loss of all three forms (Ser 2, 5, and 7) of CTD phosphorylation in Pol II (Kwiatkowski et al., 2014). Although CDK7 has dual roles in regulation of transcription and cell division (Fisher, 2012), increasing evidence suggests that the anti-tumor activity of THZ1 is primarily mediated through transcriptional inhibition rather than through cell-cycle regulation (Olson et al., 2019; Wang et al., 2015). By inhibiting activation of Pol II, THZ1 treatment broadly affects transcription on a genome-wide basis, subsequently disrupting the dysregulated transcriptional program that is required for tumor cell survival. Importantly, although THZ1 leads to global transcriptional suppression at higher doses, it also shows gene-selective repressive effects at lower doses of treatment (Kwiatkowski et al., 2014). The observation that THZ1 represses the transcription of a relatively selected subset of genes at lower doses indicates that CDK7 inhibition may preferentially impair the expression of a fraction of critical cancer-related genes with large, clustered enhancers (super enhancers), which are required to drive high-level transcription. THZ1 has shown to have promising therapeutic potential in preclinical models of multiple cancer types, including leukemia (Kwiatkowski et al., 2014; Wong et al., 2017), small-cell lung cancer (Christensen et al., 2014; Zhang et al., 2020), neuroblastoma (Chipumuro et al., 2014), breast cancer (Harrod et al., 2017; Li et al., 2017; Wang et al., 2015), glioma (Greenall et al., 2017; Nagaraja et al., 2017), melanoma (Eliades et al., 2018), lymphoma (Cayrol et al., 2017), chordoma (Sharifnia et al., 2019), and esophageal (Jiang et al., 2017), nasopharyngeal (Yuan et al., 2017), and ovarian (Francavilla et al., 2017; Zeng et al., 2018; Zhang et al., 2017) cancers. The combination therapy of THZ1 with other targeted drugs were also evaluated in cancer models (Cayrol et al., 2017; Kalan et al., 2017; Rusan et al., 2018; Terai et al., 2018). Notably, a medical-chemistry-optimized compound based on THZ1, SY-1365, has been advanced into early clinical trials for treatment of solid cancers, such as ovarian and breast cancers (Hu et al., 2019a).
RESULTS
Recurrent Genomic Alterations of Cell-Cycle-Related and Transcriptional CDKs/Cyclins Are Distinct across Cancers
A total of 47 genes (21 CDKs and 26 cyclins) (Malumbres et al., 2009) were analyzed, including 30 understudied CDKs/cyclins, as defined by their PubTator scores of <150 (Wei et al., 2013) (Table S1; Figure 1A). Among 21 CDKs, 2 and 16 were classified as Tclin and Tchem, respectively, based on their drug-target-development levels retrieved from the Target Central Resource Database (Nguyen et al., 2017) (Table S1; Figure 1A). To systematically characterize genomic alterations of CDKs/cyclins in cancers, we analyzed somatic copy-number alterations (SCNAs), mutations, and transcript fusions in The Cancer Genome Atlas (TCGA) (Table S2). We estimated the recurrently altered CDKs/cyclins driven by SCNAs in each cancer type using four criteria (Hu et al., 2019b) (Figure S1A) and identified 26 CDKs/cyclins that met all criteria in at least one cancer type (Figures 1A and S1B; Table S3). Notably, the genes encoding CDKs/cyclins showed significant enrichment for recurrent focal SCNAs compared with the other protein-coding genes (Figures S1C–S1E). As expected, we found that multiple cell-cycle-related CDKs/cyclins were focally amplified across cancers. For example, CCNE1, CCND1, CDK4, and CDK6 were recurrently amplified in 11, 8, 5, and 5 cancer types, respectively (Figure 1A). However, except for CCNK, which showed recurrent copy-number gain in lung squamous cell carcinoma (LUSC), copy-number losses of the transcriptional CDKs/cyclins were globally observed in multiple cancer types. For example, 7 of 10 transcriptional CDKs and 4 of 6 transcriptional cyclins showed recurrent focal copy number losses in at least one cancer type (Figure 1A). Notably, 6 of 16 transcriptional CDKs/cyclins were focally deleted in at least four cancer types, suggesting that copy-number loss in transcriptional CDKs/cyclins is a common genetic alteration during tumorigenesis. To estimate the SCNAs for CDKs/cyclins at a pan-cancer level, we also calculated an overall G score by an unweighted numeric sum of G scores estimated for each cancer type (Figure 1B; Table S3). Among CDKs, CDK4 and CDK6 showed the highest overall G scores for copy-number gain, whereas CDK7 had the highest overall G score for copy-number loss (Figures 1A and 1B). The recurrent CDK/cyclin SCNAs for each cancer type were summarized in Figure S2.
Figure 1. Recurrent Genomic Alterations of Cell-Cycle-Related and Transcriptional CDKs/Cyclins Are Distinct across Cancers.
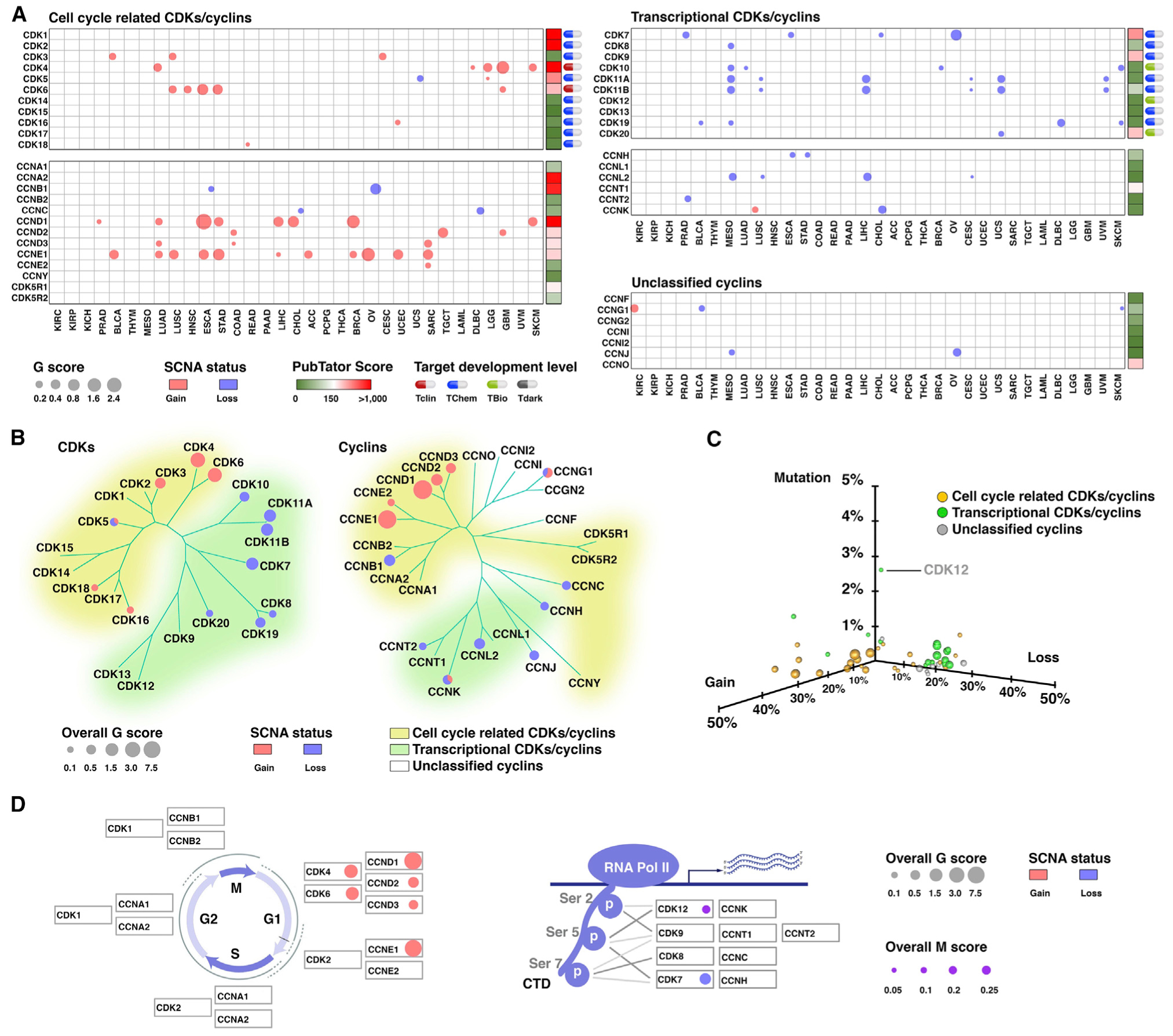
(A) Bubble plot shows the SCNA G scores, which consider both the amplitudes of the aberrations and the frequencies of their occurrence across samples, of the putative cancer-associated CDKs/cyclins driven by SCNAs in each cancer type. The size of the bubble G score: red, gain; blue, loss. PubTator scores, which represent the number of publications for a given gene, were retrieved from the PubTator database and are shown next to the G score plot: green, 1–150 (understudied genes); red, >150. Target-development levels for each gene, which were retrieved from the PHAROS database, are shown at the left. Red, Tclin; blue, TChem; green, Tbio; gray, Tdark.
(B) Summary of the overall G scores of the putative cancer-associated CDKs/cyclins driven by SCNAs. The size of the bubble represents the overall G score: red, gain; blue, loss. The phylogenetic trees were generated by multiple sequence alignments of the full-length sequences of the proteins.
(C) 3D plot shows the frequencies of the copy-number alterations and mutations for each CDK/cyclin at a pan-cancer level. Bubble size represents the total number of cancer types in which the genes were identified as recurrently amplified or deleted: yellow, cell-cycle-related CDKs/cyclins; green, transcriptional CDKs/cyclins; gray, unclassified cyclins.
(D) At a pan-cancer level, eight CDKs/cyclins recurrently altered in more than two cancer types. The G1-S CDKs/cyclins showed the most significant copy-number gains (left), whereas the CDKs/cyclins regulating phosphorylation of Pol II CTD showed the most significant copy-number losses and mutations (right). The size of the bubble represents the overall G score or overall M score: red, gain; blue, loss; violet, mutation.
Next, to characterize the somatic mutations of CDKs/cyclins in cancer, we analyzed whole exome sequencing (WES) profiles from TCGA (Table S2; Figure S3A). The hypermutated samples were excluded from the recurrent mutation analysis. In summary, the mutations in CDKs/cyclins exhibited much lower frequencies compared with SCNAs (Table S4; Figure 1C), and only seven CDKs/cyclins showed evidence for recurrent mutations (Figures S3B–S3E; Table S4). At a pan-cancer level, CDK12 was the most significantly mutated CDKs/cyclins with the highest mutation frequency (2.6%) among CDKs/cyclins. The recurrent CDK/cyclin mutations for each cancer type are summarized in Figure S4. Finally, to characterize transcript fusions of CDKs/cyclins in cancer, the gene fusion data of TCGA were retrieved from the TumorFusions database. We observed 145 fusion transcripts (133 fusion pairs) of 47 CDK/cyclin genes in 9,799 tumor specimens across 33 cancer types (Figures S5A, S5B, and S6; Table S5), suggesting that transcript fusion is a rare genetic alteration in CDKs/cyclins in common adult cancers. Only 21 of 145 (14.48%) of CDK/cyclin fusion transcripts, representing 9 of 133 fusion pairs, occurred at least twice across all cancer types. CCND3-TRERF1 (n = 4) and KIAA1109-CCNA2 (n = 3) were the most frequent fusions among the all cancer types (Table S5). Notably, among the 9,799 tumor specimens analyzed, fusion events for CDKs/cyclins were only detected in 133 of 9,799 (1.36%) of the tumor specimens (Figures S5C and S5D; Table S5).
Taken together, both cell-cycle-related and transcriptional CDKs/cyclins showed SCNAs with high frequencies. In contrast to cell-cycle-related CDKs/cyclins that were globally amplified in cancers, transcriptional CDKs/cyclins recurrently and focally lost copy numbers across multiple cancer types. Compared with SCNAs, mutations or transcript fusions are relatively rare genomic alterations, except CDK12, which showed the highest mutation frequencies and recurrently mutated in three cancer types. Notably, at a pan-cancer level, eight CDKs/cyclins exhibit recurrent genomic alterations in more than two cancer types. The G1-S CDKs/cyclins had the most significant gain-of-function genomic changes, whereas the CDKs regulating Pol II CTD phosphorylation showed the most significant loss-of-function alterations (Figure 1D).
Genomic Alterations of CDK7 and CDK12 Are Correlated with Increased Sensitivities to DNA-Damaging Drug Treatment
Among transcriptional CDKs, two of them showed significant recurrent loss-of-function alterations across cancers, i.e., CDK7 and CDK12 had the highest overall G score and M score among the deleted and mutant CDKs/cyclins, respectively (Figure 1D). Because of their tightly related functions, we sought to further characterize their genomic alterations and potential clinical significance in cancer. At a pan-cancer level, 27.32% (2,991 of 10,950) of tumor specimens showed CDK7 copy-number loss, as estimated with the GISTIC algorithm (Figure 2A). However, homozygous deletions, as estimated with the GISTIC algorithm or with the ABSOLUTE algorithm, were rare events for CDK7 (1.01% or 0.04%, respectively). Importantly, CDK7 showed significantly recurrent copy-number loss in four epithelial cancer types, including ovarian serous cystadenocarcinoma (OV), prostate adenocarcinoma (PRAD), esophageal carcinoma (ESCA), and cholangiocarcinoma (CHOL) (Figure 2B). Additionally, after further analyzing CDK7 deletion in subtypes of each cancer type, recurrent CDK7 deletion was also identified in triple-negative breast cancer (TNBC), which has been reported to share many genomic commonalities with high-grade serous ovarian cancer. In the five cancer types with CDK7 recurrent deletion, CDK7 mRNA expression was significantly and positively correlated with its predicted copy number (Figure 2C; p = 4.3e–93; R = 0.53). The CDK7-deleted tumors showed significantly lower mRNA expression compared with CDK7 diploid or non-deleted tumors (Figure 2D; p = 2.8e–66 and p = 2.3e–68, respectively). Similar results were also observed at a pan-cancer level for all TCGA tumor specimens (p = 2.82e–227; n = 10,029) as well as in a panel of cancer cell lines from the Cancer Cell Line Encyclopedia (CCLE) dataset (Barretina et al., 2012) (p = 8.5e–64; n = 977). At a pan-cancer level, CDK12 appeared to be widely mutated across cancers at a low frequency (average 2.6%; Figure 2E), and it was identified as recurrent mutation in uterine corpus endometrial carcinoma (UCEC), OV, and lung adenocarcinoma (LUAD) with frequencies of 7.2%, 4.6%, and 3.0%, respectively (Figure 2E). After analyzing the distributions of the mutations across the gene body, we found that CDK12 mutations were widely and spatially distributed along the entire coding regions (Figure 2F), slightly concentrated within the kinase domain (Figure 2G). The most common mutation category of CDK12 was the missense mutation (65.0%, pan-cancer; Figure 2E), and the dominant mutation type was heterozygous mutations (82.9%, pan-cancer; Figure 2H). Using the ABSOLUTE algorithm, we estimated the timing of the mutational processes and the clonal statuses of the mutations in CDK12. At a pan-cancer level, 47.6% of mutations in CDK12 were early genomic events (Figure 2I) and 59.4% of mutations in CDK12 were clonal mutations (Figures 2J and 2K). Taken together, the recurrence of CDK7 copy-number losses and CDK12 mutations strongly indicates the existence of selective pressures for loss-of-functions in the CDKs regulating Pol II CTD phosphorylation during tumor-cell evolution. Importantly, both CDK7 copy-number losses and CDK12 mutations appeared heterozygous, suggesting that they may have critical roles in cell survival, and complete loss might be lethal.
Figure 2. CDK7 Copy Number Loss Is Correlated with Increased Sensitivities to DNA-Damaging Drug Treatment.
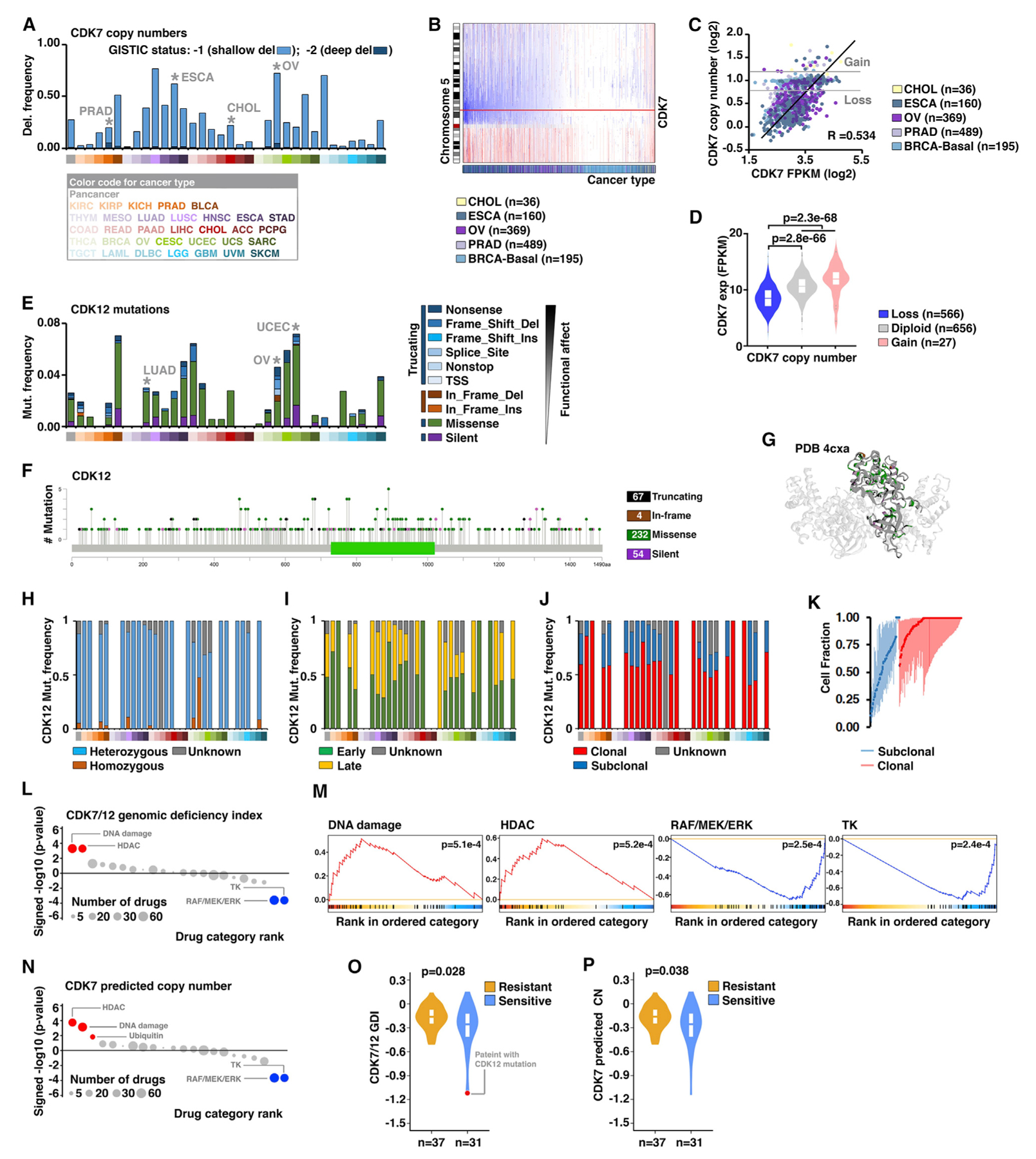
(A) The frequencies of CDK7 copy-number losses across 33 cancers: dark blue, deep deletion (GISTIC status, −2; possibly a homozygous deletion); light blue, shallow deletion (GISTIC status, −1; possible a heterozygous deletion). The cancer types with recurrent CDK7 deletions were indicated.
(B) Copy-number profiles of chromosome 5 across five cancer types, in which CDK7 copy number is recurrently lost. Each sample is represented with a vertical line, and the genomic locus of CDK7 is noted with a red horizontal line: blue, loss; red: gain.
(C) Scatterplot shows the correlation between CDK7 predicted copy number and expression across five cancer types with recurrent CDK7 loss.
(D) Violin plot shows the CDK7 expression levels among different CDK7 copy-number status groups across the five cancer types, using the same cancer types as in (C).
(E) The frequencies and types of CDK12 mutations across 33 cancer types. The cancer types with recurrent CDK7 mutations were indicated.
(F) The lollipop plots illustrate the distribution and categories of somatic mutations in the CDK12 gene-coding sequences across all cancer types. Note that, although mutations are randomly distributed along the entire coding sequence, the most frequent mutations are located within the catalytic domain.
(G) Ribbon drawings of the crystal structures of the kinase domain within CDK12 (4CXA). The different types of CDK12 mutations in the kinase domain were indicated by colors.
(H–J) The fractions of the mutation types (H), timing status (I), and clonal heterogeneity (J) of CDK12 in individual cancer types.
(K) Clonal heterogeneity of CDK12 across cancers. Based on the probability distributions of the cancer cell fractions, mutations were determined to be either clonal (red blocks) or subclonal (blue blocks).
(L) Dot plots show the significance of enrichment analysis (signed p value in the negative log10 scale) for all drug categories in association with CDK7/12 genomic-deficiency index (GDI). Drug categories are ordered according to their normalized enrichment score (NES). Drug categories with significant enrichment toward increased or decreased sensitivities in cell lines are shown in red or blue, respectively (false-discovery rate [FDR] < 10%). Drug categories with non-significant correlations are colored in gray. The size of each dot corresponds to the number of drugs in each category.
(M) Enrichment plots for selected drug categories in association with CDK7/12 GDI. Enrichment curves for drug categories with significant enrichment toward increased or decreased sensitivities in cell lines are colored in red or blue, respectively.
(N) Dot plots show the significance of enrichment analysis for all drug categories in association with CDK7 predicted copy number.
(O) The GDI of CDK7/12 between the chemo-sensitive and -resistant patients with ovarian cancer. The patient with a CDK12 mutation is indicated in red.
(P) The predicted copy numbers of CDK7 between the chemo-sensitive and -resistant patients with ovarian cancer.
To explore the potential clinical significance of CDK7/12 losses in cancer, we analyzed the correlation between CDK7/12 losses and drug-treatment response using large-scale drug-screening data generated by the Genomics of Drug Sensitivity in Cancer (GDSC) (Garnett et al., 2012) and the Cancer Therapeutics Response Portal (CTRP) (Barretina et al., 2012) projects. Using the SNP array and exome sequencing (exome-seq) profiles generated by GDSC/CTRP, we estimated a CDK7/12 genomic deficiency index (GDI) for each cancer cell line (n = 1,315), in which both CDK7 predicted copy numbers and CDK12 mutation impact on protein function were considered. After the effects of CDK7/12 GDI on the sensitivity profile of the compounds were assessed by linear-regression models adjusting for cancer types, the compounds were ranked by the strength of associations. We grouped the compounds into 22 general categories based on their mechanisms of action, and tested the enrichment of all drug categories against the ranked list of compounds. Among the compounds showing increased or decreased sensitivities in cancer-cell lines harboring CDK7/12 genomic losses, we found significant enrichment for four drug categories (Figures 2L and 2M). Increased sensitivity to DNA-damaging agents was the most significantly enriched drug category in the CDK7/12 loss group (p = 5.1e–4). Similar results were also observed when the compounds were ranked based on the strength of associations between CDK7 predicted copy numbers and sensitivity profiles assessed using the same datasets (Figure 2N), although the DNA-damaging agents showed slightly less significance compared with the histone deacetylase (HDAC) inhibitors. Because of the low mutation frequency of CDK12 (5.17%; 68 of 1,315), we were not able to perform statistical analysis using the CDK12 mutation alone. To further confirm the correlation between losses of CDK7/12 and increased sensitivity to DNA-damaging agents, we investigated chemotherapy response in patients with ovarian cancer, who were treated by standard platinum-based chemotherapy. In a cohort of ovarian cancer patients with detailed chemotherapy treatment and response information (Patch et al., 2015), we found that CDK7/12 GDI was significantly lower in the chemosensitive patients compared with the chemoresistant group (p = 0.029; Figure 2O). Identical result was observed, when we analyzed the predicted copy number of CDK7 in the same patients (p = 0.038; Figure 2P). Collectively, CDK7/12 genomic losses were correlated with increased sensitivity of cancer cells to DNA-damaging agent treatment.
Inhibition of CDK7 by THZ1 Represses Expression of Genes in the DDR Pathways
It has been reported that CDK7 inhibition preferentially represses distinct subsets of genes in cancers derived from different tissue lineages. For example, THZ1 treatment reduced the RUNX1 transcriptional regulatory circuitry and the transcription of a group of genes (called “Achilles cluster” genes) in T-cell-acute lymphoblastic leukemia (T-ALL) (Kwiatkowski et al., 2014) and TNBC (Wang et al., 2015), respectively. Meanwhile, a large-scale THZ1 treatment-response screen in >1,000 cancer cell lines showed that THZ1 had anti-cancer activity in ~53% of the cancer cell lines tested (Kwiatkowski et al., 2014), suggesting that THZ1 may also regulate common cancer-associated pathways. To test that hypothesis, the RNA sequencing (RNA-seq) data in three epithelial cancer types from independent groups (Jiang et al., 2017; Yuan et al., 2017; Zeng et al., 2018; Zhang et al., 2017) were analyzed (Figure 3A). We found that a large proportion of genes (4,277) were commonly down-regulated in at least two cancer types, including 1,924 genes down-regulated in all three cancer types (Figure 3B). Even when we expanded our analysis from epithelial cancers to hematologic malignancy (T-ALL) (Kwiatkowski et al., 2014), we can still identify a large group of genes that were shared among the distinct cancer lineages. For example, 2,355 of 4,277 and 1,298 of 1,924 of the above commonly down-regulated genes identified from epithelial cancers were also down-regulated in T-ALL (notably, ~6.3% of genes detected by the RNA-seq had no corresponding probes in the array that was used for T-ALL experiments). Interestingly, gene ontology (GO) analysis indicated that these common THZ1-response genes were enriched significantly in cancer-related pathways, including DDR, cell cycle, and apoptosis (Figure 3C; Table S6). Among them, genes in two closely related DDR pathways, homologous recombination (HR) and Fanconi anemia (FA) pathways, were most significantly enriched in these commonly down-regulated genes (Figures 3D and 3E). Furthermore, by analyzing the chromatin immunoprecipitation sequencing (ChIP-seq) data (Kwiatkowski et al., 2014), we found that Pol II binding to the promoter regions of these commonly regulated genes were remarkably reduced by two CDK7 inhibitors, THZ1 and THZ2, but not by flavopiridol (Figure 3F). For example, both THZ1 and THZ2 treatment reduced Pol II binding in the promoter regions of BRCA1, BRCA2, and RAD51, the essential genes in HR (Figure 3G). Given that CDK7 are master regulators of phosphorylation of Pol II CTD (Bradner et al., 2017; Chou et al., 2020; Fisher, 2012; Zaborowska et al., 2016), THZ1 treatment may lead to reduction of global transcription (Kwiatkowski et al., 2014). We then asked whether CDK7 inhibition preferentially reduced expression of genes in FA/HR pathways at low-dose or short-time treatment in cancer cells. To that end, we evaluated whether the FA/HR pathways were enriched in the genes down-regulated by THZ1 treatment in both a dose-dependent and a time-dependent manner in T-ALL experiment (Kwiatkowski et al., 2014). Interestingly, at all doses and time points, HR/FA genes were preferentially down-regulated compared with all other genes (Figure 3H). Importantly, at the low-dose treatment (50 nM, 4 h), the FA/HR genes were significantly enriched in the down-regulated genes compared with treatment at the higher dose (250 nM) at the same time point (50 nM: odds ratio [OR] = 4.27, p = 1.02e–6; 250 nM: OR = 2.94, p=3.83e–5). Our above analyses suggest that many essential genes in the HR/FA pathways are preferentially repressed by low-dose THZ1 treatment, providing an insight into the mechanism of CDK7 inhibition in cancer treatment.
Figure 3. Inhibition of CDK7 by THZ1 Represses the Expression of Genes in the DDR Pathways.
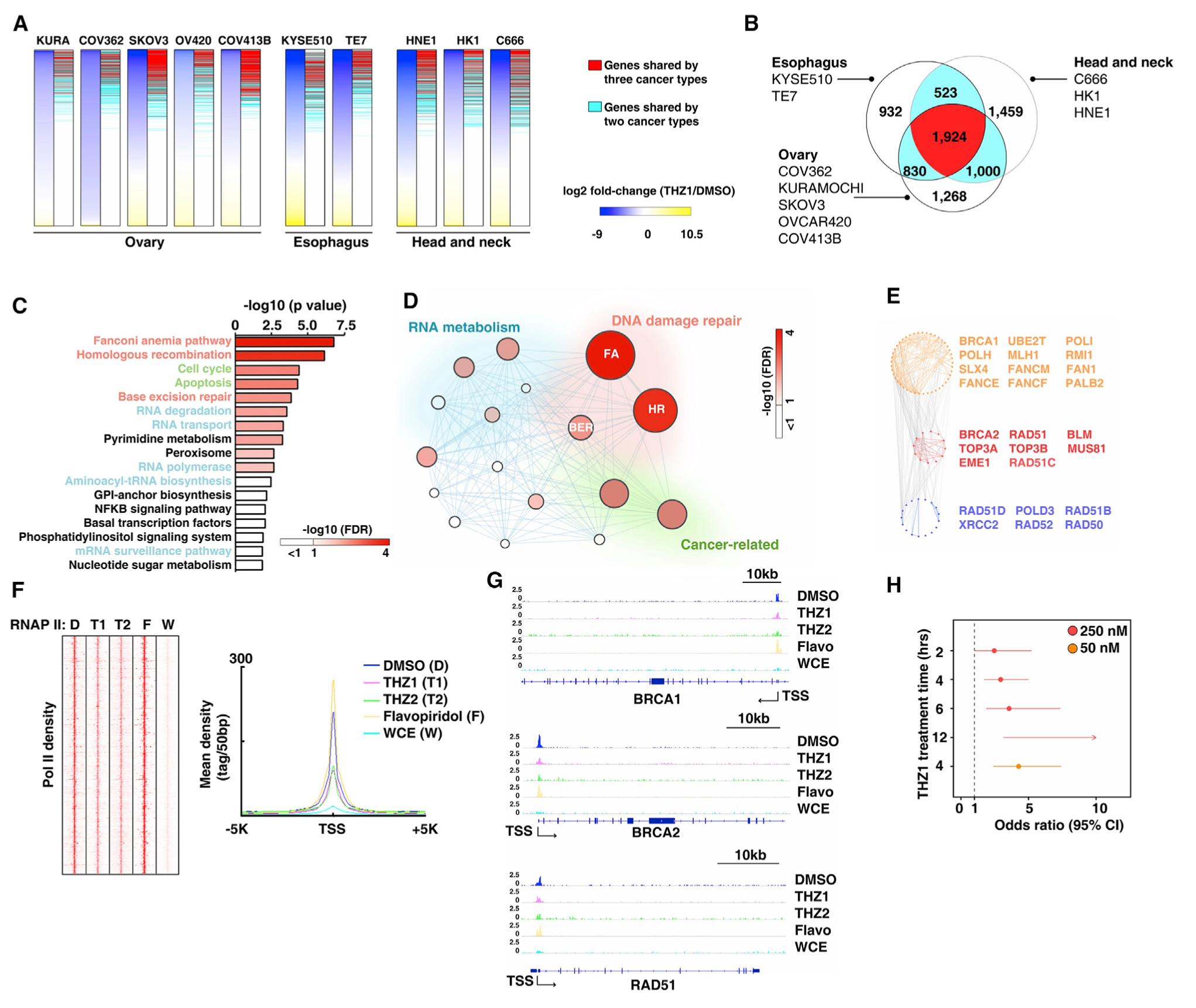
(A) Left columns of each panel show heatmaps of gene-expression changes measured by RNA-seq after THZ1 treatment in 10 epithelial cancer cell lines from ovarian (KURAMOCHI, COV362, SKOV3, OVCAR420, and COV413B), esophageal (KYSE510 and TE7), and head and neck (HNE1, HK1, and C666) cancers: blue, down regulated; yellow, upregulated. Right columns of each panel indicate the down-regulated genes shared among cancer types. Red bars indicate down-regulated genes shared by three cancer types; blue bars indicate down-regulated genes shared by two cancer types.
(B) Venn diagram shows overlap of down-regulated genes among three cancer types. Red indicates genes shared by all three cancer types, blue indicates genes shared by two cancer types, and white indicates genes in an individual cancer type.
(C) The pathways over-represented for shared genes down regulated by THZ1 treatment, according to a GO analysis.
(D) Diagram depicts the interaction of the pathways identified by a GO analysis. Node size: p value (−log10) in each pathway: node color, FDR value (−log10) in each pathway.
(E) Expanded view of genes from the FA and HR pathways, which were down regulated by THZ1 treatment.
(F) Left panel: heatmaps show the density of RNA polymerase II binding as measured by ChIP-seq at the promoter regions of the shared genes that were down regulated by THZ1 treatment in T-ALL cells. Abbreviations: D, DMSO; T1, THZ1; T2, THZ2; F, flavopiridol; W, whole cell extract. Right panel: mean density of RRNA polymerase II (RNAP II) ChIP-seq from left panel.
(G) RNAP II ChIP-seq signal at the promoter regions of BRCA1, BRCA2, and RAD51 in T-ALL cells treated with DMSO, THZ1, THZ2, and flavopiridol. Whole-cell extract DNA served as input.
(H) Forest plot shows the enrichment of the FA/HR pathway among genes down regulated by different concentrations of THZ1 at variant time points. Dots and horizontal lines represent OR values and their 95% confidence intervals (CI), respectively. Arrows indicate where the OR or CI values extend outside the range indicated.
To experimentally validate that finding, we analyzed the expression levels of three core genes in HR (BRCA1, BRCA2, and RAD51) during low-dose THZ1 treatment in a set of independent cancer cell lines. qRT-PCR analysis showed that low-dose THZ1 treatment (50 nM) indeed significantly reduced the mRNA levels of BRCA1, BRCA2, and RAD51 (Figure 4A). This result was further validated at the protein level in the same cell lines by western blots (Figure 4B). Notably, low-dose THZ1 treatment did not affect the G0/G1 phase of cell cycle, indicating THZ1 did not repress HR genes expression by inducing G0/G1-phase accumulation (Figure S7). Taken together, we demonstrate that low-dose THZ1 treatment (50 nM) significantly reduced the expression levels of core proteins in HR, such as BRCA1, BRCA2, and RAD51. Next, we took a genetic approach by knocking down CDK7 with two independent small interfering RNAs (siRNAs) to exclude potential off-target effects from the above pharmacological studies. Knockdown specificity was validated by individual siRNA transfection (Figures 4C and 4D). Consistently, a significant reduction in BRCA1, BRCA2, and RAD51 expression was observed in cells transfected with CDK7 siRNAs at both RNA (Figure 4C) and protein levels (Figure 4D). Taken together, we demonstrate that low-dose THZ1 treatment (50 nM) significantly reduced the expression levels of core proteins in HR, including BRCA1, BRCA2, and RAD51. To explore the mechanism by which THZ1 represses the expression of BRCA1, BRCA2, and RAD51 through RNA Pol II, we performed ChIP with antibodies against RNA Pol II followed by qPCR quantification. As expected, RNA Pol II was enriched in the promoter regions of BRCA1, BRCA2, and RAD51, and THZ1 treatment significantly reduced the recruitment of RNA Pol II to these regions (Figure 4E). Consistent with the ChIP-qPCR analyses, the luciferase activities of BRCA1, BRCA2, and RAD51 promoters were significantly repressed by THZ1 treatment (Figure 4F). Finally, knocking down CDK7 by siRNAs genetically mimicked the effects of THZ1 in repressing the binding ability of RNA Pol II (Figure 4G) and inhibiting the luciferase activities of BRCA1, BRCA2, and RAD51 promoters (Figure 4H).
Figure 4. CDK7 Inhibition and Depletion Repress the Expression of BRCA1, BRCA2, and RAD51.
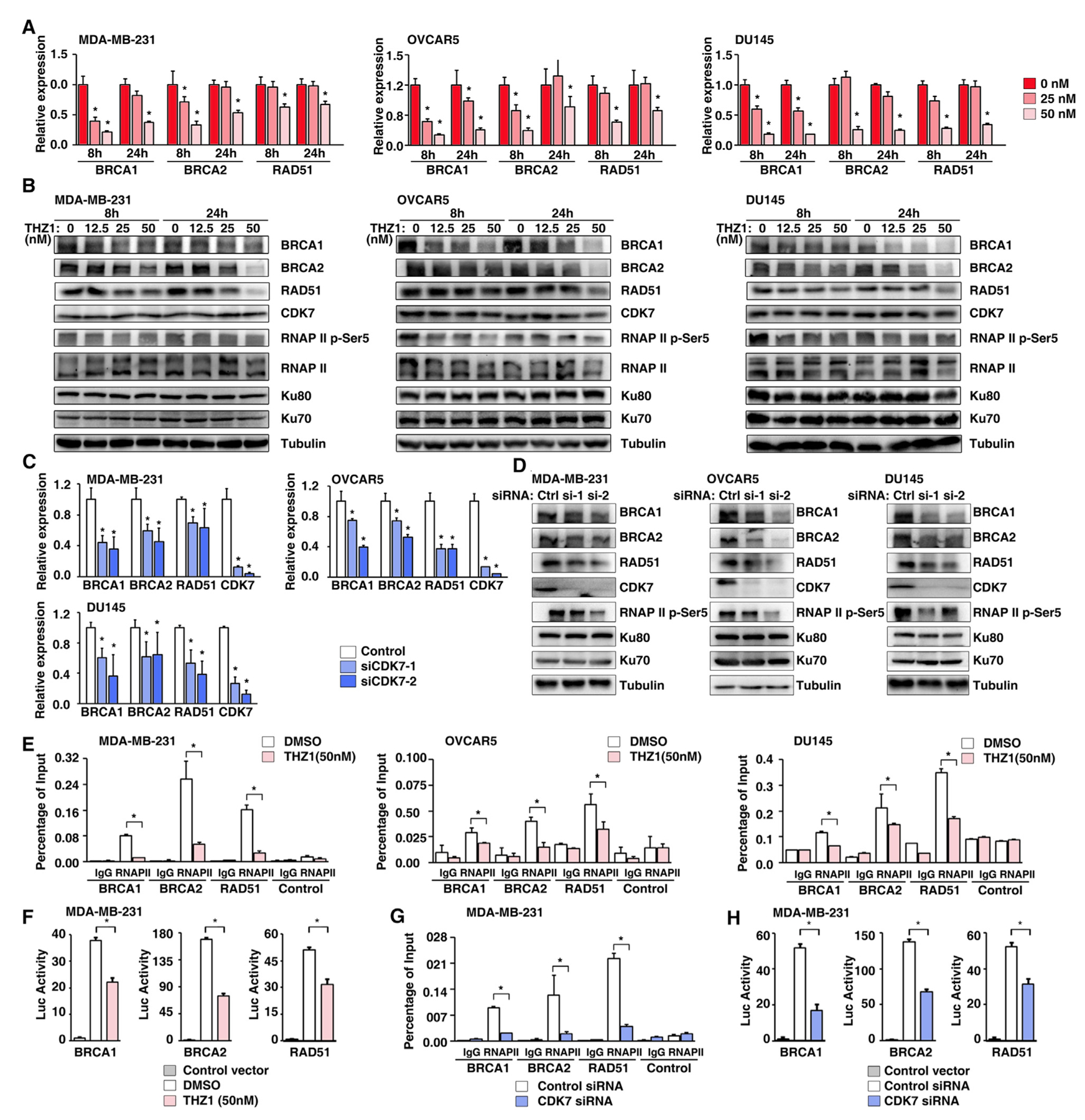
(A) The RNA expression levels of BRCA1, BRCA2, and RAD51 were measured by qRT-PCR in MDA-MB-231, OVCAR5, and DU145 cells treated with THZ1 at 25 nM and 50 nM for 8 h and 24 h. Data represented as means ± SD, n = 3 technical replicates, *p < 0.05 determined by two-tailed Student’s t test.
(B) The protein expression levels of BRCA1, BRCA2, RAD51, CDK7, RNAP II p-Ser5, RNAP II, Ku80, and Ku70 were detected by western blots in MDA-MB-231, OVCAR5, and DU145 cells treated with THZ1 at 12.5 nM, 25 nM, and 50 nM for 8 h and 24 h. Tubulin served as a loading control.
(C) The RNA expression levels of BRCA1, BRCA2, RAD51, and CDK7 were measured by qRT-PCR in MDA-MB-231, OVCAR5, and DU145 cells treated with CDK7 siRNAs for 72 h. Data represented as means ± SD, n = 3 technical replicates, *p < 0.05 determined by two-tailed Student’s t test.
(D) The protein expression levels of BRCA1, BRCA2, RAD51, CDK7, RNAP II p-Ser5, Ku80, and Ku70 were detected by western blots in MDA-MB-231, OVCAR5, and DU145 cells treated with CDK7 siRNAs for 72 h. Tubulin served as a loading control.
(E) Quantification of the amount of BRCA1, BRCA2, or RAD51 promoter bound to RNA Pol II in MDA-MB-231 (left), OVCAR5 (middle), and DU145 (right) cells treated with DMSO or THZ1. Data represented as means ± SD, n = 3 technical replicates, *p < 0.05 determined by two-tailed Student’s t test. IgG, immune-globulin G.
(F) Luciferase reporter assay of the activities of the BRCA1 (left), BRCA2 (middle), or RAD51 (right) core promoter reporters in MDA-MB-231 cells treated with DMSO and THZ1. Data represented as means ± SD, n = 3 biological replicates, *p < 0.05 determined by two-tailed Student’s t test.
(G) Quantification of the amount of BRCA1, BRCA2, or RAD51 promoter bound to RNA Pol II in MDA-MB-231 cells treated with control or CDK7 siRNAs. Data represented as means ± SD, n = 3 technical replicates, *p < 0.05 determined by two-tailed Student’s t test.
(H) Luciferase reporter assay of the activities of the BRCA1 (left), BRCA2 (middle), or RAD51 (right) core promoter reporters in MDA-MB-231 cells treated with control siRNA or CDK7 siRNA. Data represented as means ± SD, n = 3 biological replicates, *p < 0.05 determined by two-tailed Student’s t test.
Inhibition of CDK7 by THZ1 Impairs the Activity of HR in Cancer Cells
Given that THZ1 treatment preferentially represses expression of essential genes in the HR pathway, we hypothesized that CDK7 inhibition may impair the activity of HR in cancer cells. To test that hypothesis, we pre-treated three HR-proficient cancer cell lines with low-dose THZ1 (25 nM) for 12 h; then, we exposed the cancer cells to ionizing radiation (IR) to induce DSBs (Figure 5A). Immunofluorescent staining analyses indicated that THZ1 treatment significantly decreased the foci formation of BRCA1, BRCA2, and RAD51 compared with the control treatment during IR-induced DNA damage (Figures 5B and 5C). To further confirm that result, we used a GFP/Pem1-based HR reporter assay to measure HR activity in cancer cells (Figure 5D). After transfection with I-SceI-cut HR reporter plasmid, the cells were treated with low-dose THZ1 (25 nM and 50 nM) for 48 h, then HR activity was quantified by fluorescence-activated cell sorting (FACS) analysis. Consistent with the findings of our foci formation assay, we found that low-dose THZ1 treatment significantly decreased the activity of HR in all three cancer cell lines compared with the control treatment (Figure 5E). Collectively, our results indicate that repression of CDK7 impairs HR in cancer cells, providing a potential explanation for the observation that cancer cell lines with CDK7 losses were sensitive to DNA-damaging drug treatment.
Figure 5. CDK7 Inhibition Reduces HR Repair Activity.
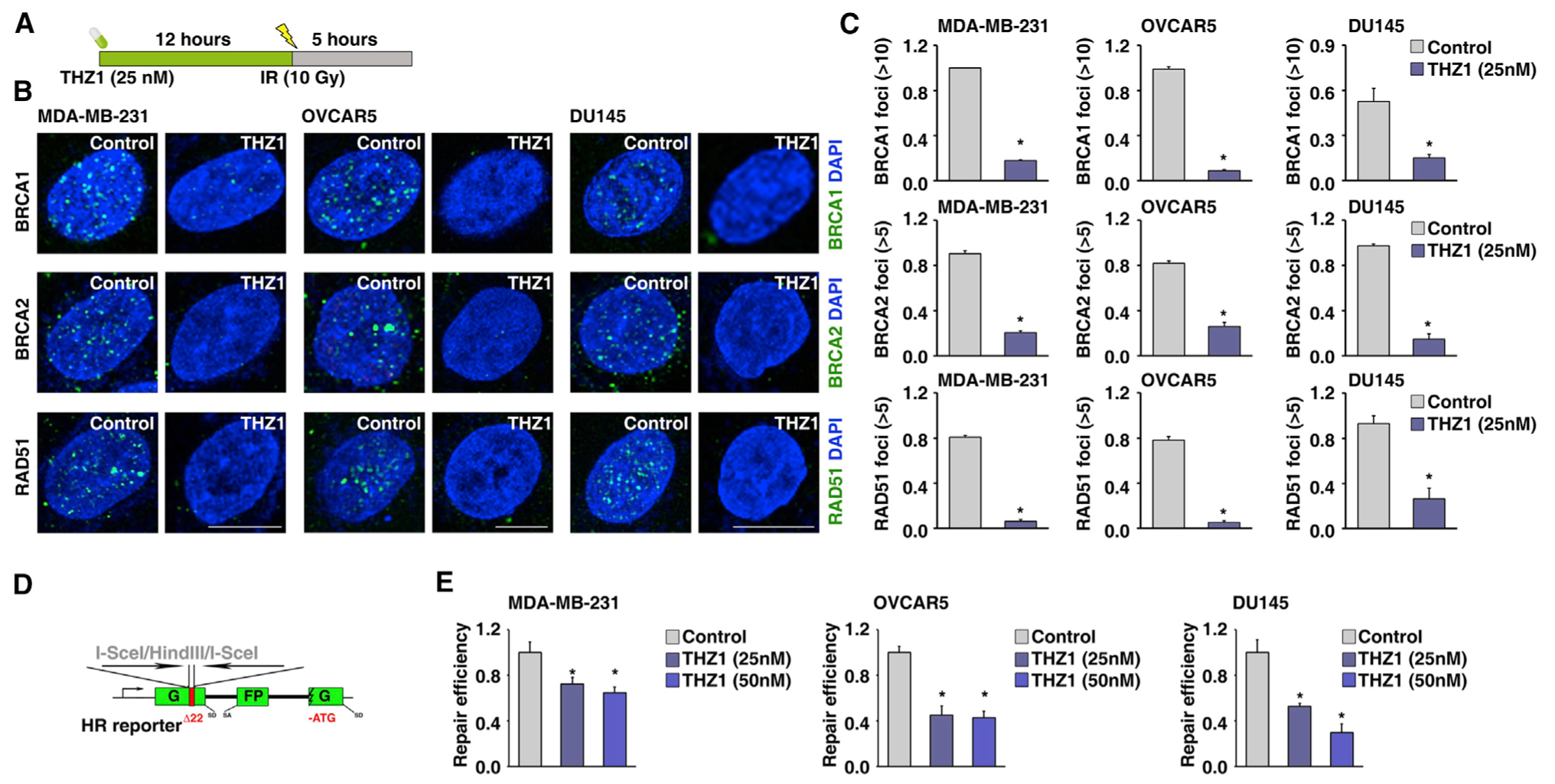
(A) Illustration of time schedule of the THZ1 and IR treatment.
(B) IR-induced BRCA1, BRCA2, and RAD51 foci formation in MDA-MB-231, OVCAR5, and DU145 cells treated with or without THZ1 (25 nM).
(C) Quantification of BRCA1, BRCA2, and RAD51-foci positive cells in MDA-MB-231, OVCAR5, and DU145 cells treated with or without THZ1 (25 nM). Data represented as means ± SD, n = 3 biological replicates, *p < 0.05 determined by two-tailed Student’s t test.
(D) Schematic illustration of HR reporter assay.
(E) HR-mediated DNA repair activity, measured by HR reporter assay, in MDA-MB-231, OVCAR5, and DU145 cells treated with THZ1 at 25 nM and 50 nM. Data represented as means ± SD, n = 3 biological replicates, *p < 0.05 determined by two-tailed Student’s t test.
CDK7 Inhibitor THZ1 Acts Synergistically with PARP Inhibition in HR-Proficient Cancer Cells
Recent studies demonstrated that suppression of HR by epigenetic or targeted therapy drugs (Ibrahim et al., 2012; Iniguez et al., 2018; Johnson et al., 2016; Kaplan et al., 2019; Sun et al., 2018; Yang et al., 2017) can sensitize HR-proficient cancer to PARP inhibitor (PARPi) treatment. Given that repression of CDK7 leads to decreased function of HR (chemically inducing BRCAness phenotype), we hypothesized that CDK7i may act synergistically with DNA-damaging agents, such as PARPis and platinum-based anticancer drugs. To test that hypothesis, we examined the synergistic effect of the combination of THZ1 and olaparib in HR-proficient cell lines (BRCA1/2 wild type) originating from three cancer types (ovarian, TNBC, and prostate cancers), for which PARPi treatment has been approved or proposed in clinical trials (Konstantinopoulos et al., 2015; Lord and Ashworth, 2017; Pilié et al., 2019; Pommier et al., 2016; Scott et al., 2015). Low-dose THZ1 treatment (12.5–50 nM) showed strong synergistic activities with olaparib in all six cancer cell lines (Figures 6A–6C). This synergistic effect was also confirmed by an anchorage-independent soft agar assay (Figure 6D). In addition, we expanded our drug combination to an additional PARPi, veliparib, as well as a DNA-damaging agent, cisplatin; and a strong synergistic activity was found in all combinations examined in three cancer-cell lines. Finally, we tested the effect of combination of CDK7i and PARPi in xenograft tumors in vivo. In the MDA-MB-231 model, when tumors reached a size of approximately 30 mm3, mice were randomly assigned to one of four groups to receive vehicle, THZ1, olaparib, or a combination of THZ1 and olaparib via intraperitoneal (i.p.) injection. Mice were sacrificed after 4 weeks of treatment (Figure 6E). Although single-agent treatment with THZ1 (10 mg/kg per day [qd]) or olaparib (50 mg/kg qd) led to a moderate reduction in tumor volume, the combination of THZ1 (10 mg/kg qd) and olaparib (50 mg/kg qd) resulted in a significant inhibition of tumor growth (Figures 6F–6H). Similar results were also observed in an OVCRA5 ovarian cancer model (Figures 6I–6L). Taken together, the combination of THZ1 and olaparib showed strong synergistic lethality in preclinical cancer models.
Figure 6. CDK7 Inhibitor THZ1 Acts Synergistically with PARP Inhibition in HR-Proficient Cancer Cells.

(A and B) Sensitivity of MDA-MB-231, MDA-MB-468, OVCAR5, OVCAR3, DU145, and PC3 cells to olaparib alone or olaparib combined with low-dose THZ1 (12.5 nM and 25 nM or 50 nM) by cell viability assay; 1,000 cells were seeded in 96-well plates and treated with different concentration of inhibitors for 3 days. Survival fraction (A) and the combination index (B) are shown for each of the cell lines, respectively. Fa, fraction affected. Data represented as means ± SD, n = 3 biological replicates, *p < 0.05 determined by two-tailed Student’s t test.
(C) Crystal-violet staining of MDA-MB-231, OVCAR5, and DU145 cells treated with different conditions.
(D) Anchorage-independent soft agar assay results for MDA-MB-231, OVCAR5, and DU145 cells treated with DMSO, THZ1, olaparib, or olaparib combined with THZ1. Crystal violet staining (left) and its quantification (right) were shown for the above lines. Data represented as means ± SD, n = 3 biological replicates, *p < 0.05 determined by two-tailed Student’s t test.
(E) Schematic illustrating the MDA-MB-231 mouse xenograft experimental design. MDA-MB-231 cells were implanted subcutaneously in mice and grown until tumors reached a size of approximately 30 mm3. Xenografted mice were randomized and then received vehicle, 10 mg kg−1 THZ1, 50 mg kg−1 olaparib, or the combination of both agents, as indicated, 5 days/week for 4 weeks. Caliper measurements were taken every other day from the initiation of drug treatment.
(F) Mean tumor volume is shown; n = 10 mice/group. Statistical analysis by two-tailed Student’s t test. Error bars represent means ± SD.
(G) Images of tumors collected from animals receiving vehicle, THZ1, olaparib, or the combination of both agents.
(H) Individual tumor weights from different treatment groups are shown. Statistical analysis by two-tailed Student’s t test. Error bars represent means ± SD.
(I) Schematic illustrating the OVCAR5 mouse xenograft experimental design. OVCAR5 cells were implanted subcutaneously in mice and grown until tumors reached a size of approximately 30 mm3. Xenografted mice were randomized and then received vehicle, 10 mg kg−1 THZ1, 50 mg kg−1 olaparib, or the combination of both agents, as indicated, 5 days/week for 3 weeks. Caliper measurements were taken every other day from the initiation of drug treatment.
(J) Mean tumor volume is shown; n = 8 mice/group. Statistical analysis by two-tailed Student’s t test. Error bars represent means ± SD.
(K) Images of tumors collected from animals receiving vehicle, THZ1, olaparib, or the combination of both agents.
(L) Individual tumor weights from different treatment groups are shown. Statistical analysis by two-tailed Student’s t test. Error bars represent means ± SD.
Inhibition of CDK7 by THZ1 Sensitizes Cancer Cells to Respond to PARPi-Induced DNA Damage and Cell Death
To further characterize the mechanism of action behind the combination of THZ1 and olaparib, we used a comet assay to measure the level of DNA damage induced by the treatment with THZ1 or olaparib singly and in combination. Although DNA damage was slightly increased by olaparib alone, the level of DNA damage was significantly increased when cells were treated with a combination of THZ1 and olaparib (Figures 7A and 7B). Next, immunofluorescent staining was used to monitor the number of γH2AX-positive foci formed in cells. Consistently, the numbers of γH2AX-positive foci were significantly increased in cells treated with the THZ1/olaparib combination compared with cells subjected to olaparib alone (Figures 7C and 7D). Consistently, western blots also observed that γH2AX levels were significantly increased in cells treated with the THZ1/olaparib combination compared with cells subjected to olaparib alone (Figure 7E). Given that unrepaired DNA damage can activate proapoptotic pathways, leading to apoptosis-related cell death, we measured apoptosis in different treatment conditions by three independent assays: annexin V staining, caspase 3/7 activity, and western blots. Compared with single-agent treatment with THZ1 or olaparib, combination treatment significantly induced apoptosis as demonstrated by all three assays (Figures 7F–7H). Consistently, CDK7 siRNA transfection significantly increased olaparib-treatment-induced apoptosis of cancer cell lines (Figure 7I). Together, our results demonstrate that low-dose THZ1 treatment significantly enhances DNA damage and cell death induced by PARPi in HR-proficient cancer cell lines.
Figure 7. Inhibition of CDK7 by THZ1 Sensitizes Cancer Cells to PARPi-Induced DNA Damage and Cell Death.
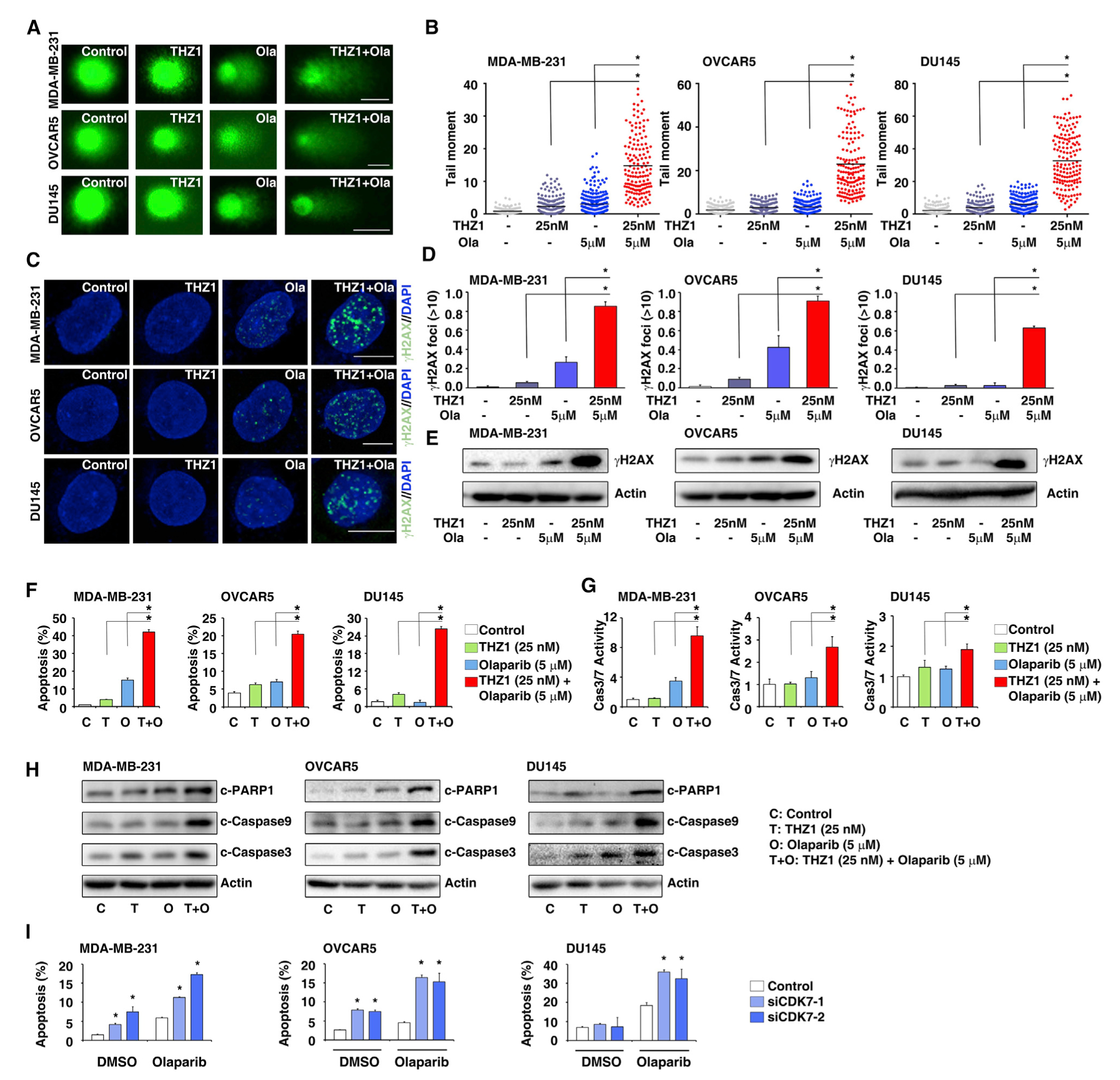
(A) DNA damage in MDA-MB-231, OVCAR5, and DU145 cells treated with DMSO, THZ1, olaparib, or THZ1 combined with olaparib, measured by the comet assay. Scale bars, 10 μm.
(B) Extent of DNA damage, quantified by the tail moment in the comet assay. Data represented as means ± SD, n = 100 cells measured, *p < 0.05 determined by two-tailed Student’s t test.
(C) Representative images of γH2AX foci in MDA-MB-231, OVCAR5, and DU145 cells treated with DMSO, THZ1, olaparib, or THZ1 combined with olaparib. Scale bars, 5 μm. DAPI, 4′,6-diamidino-2-phenylindole.
(D) Quantification of the number of γH2AX-foci-positive cells in MDA-MB-231, OVCAR5, and DU145 cells treated with DMSO, THZ1, olaparib, or THZ1 combined with olaparib. Data represented as means ± SD, n = 3 biological replicates, *p < 0.05 determined by two-tailed Student’s t test.
(E) Western blot analysis of γH2AX in MDA-MB-231, OVCAR5, and DU145 cells treated with DMSO, THZ1, olaparib, or THZ1 combined with olaparib.
(F) Percentage of annexin-V-positive cells in MDA-MB-231, OVCAR5, and DU145 cells treated with DMSO, THZ1, olaparib, or THZ1 combined with olaparib. Data represented as means ± SD, n = 3 biological replicates, *p < 0.05 determined by two-tailed Student’s t test.
(G) Caspase-3/7 activity, in MDA-MB-231, OVCAR5, and DU145 cells treated with DMSO, THZ1, olaparib, or THZ1 combined with olaparib, was measured by a caspase-3/7 assay. Data represented as means ± SD, n = 3 biological replicates, *p < 0.05 determined by two-tailed Student’s t test.
(H) Cleaved-PARP1, cleaved-caspase 9, and cleaved-caspase 3 expression assessed by western blots in MDA-MB-231, OVCAR5, and DU145 cells treated with DMSO, THZ1, olaparib, or THZ1 combined with olaparib are shown. Actin served as a loading control.
(I) Percentage of annexin-V-positive MDA-MB-231, OVCAR5, and DU145 cells treated with DMSO or olaparib combined with CDK7 siRNAs. Data represented as means ± SD, n = 3 biological replicates, *p < 0.05 determined by two-tailed Student’s t test.
DISCUSSION
We found a strikingly high prevalence of recurrent genomic alterations in the genes encoding CDKs/cyclins across cancers, and those genomic aberrations were dominantly driven by somatic copy-number alterations. In contrast, mutations and transcript fusions were rare events, except for CDK12, which showed recurrent mutations in multiple cancers and had relatively higher frequencies across cancers compared with the mutations of other CDKs/cyclins. These results indicate that aberrations of gene dos-ages of CDKs/cyclins may be a major strategy used by cancer cells to initiate and promote tumorigenesis. Supporting this hypothesis, significant and positive correlations were observed between the predicted copy numbers and mRNA expression levels of CDKs/cyclins in tumor specimens. Given that most TCGA specimens were treatment-naive primary tumors, we could not exclude the possibility that mutations or transcript fusions of CDKs/cyclins might also have roles during tumor progression. For example, recent studies on prostate cancer found that the mutation frequency of CDK12 was significantly increased in metastatic tumors compared with primary samples (Wu et al., 2018).
The CDKs/cyclins related to cell cycle and transcription showed distinct genomic alteration patterns across cancers, indicating that these two groups of CDKs/cyclins may have different roles in tumor development. As expected, we observed that many cell-cycle-related CDKs/cyclins showed recurrently and focally increased copy numbers. This is consistent with their critical functions in cell proliferation and is further supported by recent clinical successes from CDK4/6 inhibitor treatment in patients with breast cancer (O’Leary et al., 2016; Sherr et al., 2016). Unexpectedly, we found that transcriptional CDKs/cyclins were globally and recurrently deleted across cancers, and their copy-number losses were significantly correlated with lower levels of mRNA expression. It has been well demonstrated that the transcriptional program is remarkably dysregulated in cancer because of alterations of general epigenetic regulators (e.g., mutations in SWI/SNF genes) or specific transcriptional factors (e.g., amplification of MYC) (Bradner et al., 2017; Chou et al., 2020). However, the functional role of genomic alterations in transcriptional CDKs/cyclins during tumorigenesis is still largely unknown. Given that transcriptional CDKs (e.g., CDK7 and CDK12) preferentially regulate the expression of genes in DDR pathways, we hypothesize that dysregulated transcriptional CDKs/cyclins may reduce the expression of DDR genes during malignant transformation, consequently increasing genome instability and promoting tumorigenesis. Importantly, we found that homogeneous deletions of transcriptional CDKs/cyclins were very rare genomic events, suggesting that these CDKs/cyclins are essential genes for tumor cell growth and that complete losses may be lethal events for tumor cells.
Potent and selective CDKis have been developed to treat cancers by preventing proliferation or reprogramming transcription. An increasing number of inhibitors targeting the cell-cycle-related CDKs (e.g., CDK4/6) have been approved for the clinical care of patients with cancer, and inhibitors targeting transcriptional CDKs are also emerging as a class of anti-cancer agents that have shown promising therapeutic potential in preclinical models. Our genomic analysis provided an additional rationale for the clinical development of targeting transcriptional CDKs/cyclins, especially for CDK7-targeted therapy. Repression of HR gene transcription and suppression of their RNA processing, subsequently leading to HR-deficiency in HR-proficient tumors, has been proposed as a strategy to treat cancer in combination with PARPis or other DNA-damaging agents (Bajrami et al., 2014; Dubbury et al., 2018; Ibrahim et al., 2012; Iniguez et al., 2018; Johnson et al., 2016; Kaplan et al., 2019; Krajewska et al., 2019; Sun et al., 2018; Yang et al., 2017). Several combination approaches have been through early stages of clinical testing (Konstantinopoulos et al., 2015; Lord and Ashworth, 2017; Pilié et al., 2019; Pommier et al., 2016; Scott et al., 2015). Given that CDK7 preferentially regulates the transcription of genes in DDR pathways, including HR, CDK7i may suppress HR function, thereby sensitizing both intrinsic and acquired HR-proficient tumors to PARPi treatment.
The CDK family is a well-known druggable gene family for cancer treatment, and CDK-targeted therapy has shown promising results in the clinic. However, it has yet to be determined how a therapeutic window can be achieved and which patient population should be selected because CDK-mediated pathways have essential functions in cell cycles and transcription. We hypothesize that a systematic genomic analysis of the genes encoding CDKs/cyclins at pan-cancer and individual cancer levels can identify and prioritize cancer-associated events, providing a genomic view for further clinical development of CDK-targeted therapy. Among 21 CDKs in the human genome, the CDKs with recurrent copy-number alterations (e.g., CDK4, CDK6, and CDK7) may have important roles during tumorigenesis and serve as potential therapeutic targets for cancer treatment. In addition, the genomic status of both CDK and cyclin genes may be informative in identifying patients who may be more sensitive to CDK-targeted treatment. Genomic vulnerabilities of CDK/cyclin genes may also provide rationale for using CDKis as single agents or in combination with other therapeutic strategies. Finally, many CDKs/cyclins with recurrent genomic alterations are understudied genes (e.g., CDK19, CDK11, and CDK10); therefore, further characterization of the physiological and pathological functions of these understudied CDKs/cyclins is an urgent unmet medical need to develop next-generation cancer therapeutics.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Youyou Zhang M.D., Ph.D. (youyouzh@pennmedicine.upenn.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The data generated by this study are publicly available through the Functional Cancer Genome data portal (http://fcgportal.org/home) and the TCCCA website (http://fcgportal.org/TCCCA/).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
OVCAR3, MDA-MB-231, MDA-MB-468, DU145 and PC3 were purchased from ATCC and OVCAR5 was purchased from NCI Development Therapeutics Program without further authentication. MDA-MB-231, MDA-MB-468, DU145 and PC3 were cultured in RPMI1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS, Invitrogen). OVCAR5 and OVCAR3 were cultured in high glucose DMEM medium (Thermo Scientific) containing 10% FBS. Cells were routinely tested for mycoplasma contamination using Mycoplasma Plus PCR Primer Set (Agilent, Santa Clara, CA) and all cell lines were found to be negative.
Animals
Female nude mice, 6–8 weeks old, were purchased from Jackson Laboratory. All the mice were treated and maintained in accordance with the care and animal use committee of laboratory experimental animals under the NIH guidelines and in the form of IACUC approved animal protocol at the University of Pennsylvania. For MDA-MB-231 and OVCAR5 xenograft experiments, cells were implanted subcutaneously in female nude mice and grown until tumors reached a size of approximately 30 mm3. Xenografted mice were randomized into four groups and then received 1) vehicle; 2) 10 mg/kg THZ1, qd; 3) 50 mg/kg olaparib, qd; and 4) combination of both agents, qd. Treatments were administrated 5 days a week for 4 weeks for MDA-MB-231 (n = 10 per group) and 3 weeks for OVCAR5 (n = 8 per group). Tumors were measured by caliper every three days starting from the initiation of drug treatment. Tumor volumes were calculated according to the formula: tumor volume [mm3] = (1/6) × π × (tumor length) × (tumor width)2. Mice body weights were measured on each treatment day.
METHOD DETAILS
Genomic profiling collection and analysis
The genomic profiling data were generated by the TCGA project (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) supported by the NCI and NHGRI. The raw profiling data used for the current study were retrieved from the Genomic Data Commons (GDC) portal (https://portal.gdc.cancer.gov). The genomic data for RNA expression, somatic DNA copy number alteration, somatic mutation, and transcript fusion were retrieved, processed and analyzed through a master computational protocol developed by the Functional Cancer Genome project at University of Pennsylvania (FCG, http://fcgportal.org/home/) as described by our previous publication (Hu et al., 2019b).
Gene family annotation
The GENCODE comprehensive gene annotation version 22 (Djebali et al., 2012; Harrow et al., 2012) was downloaded from the GENCODE website (https://www.gencodegenes.org/human/release_22.html). It was used to define the gene features including chromosome position, transcript structure, as well as transcript and protein sequences. The human reference genome GRCh38/hg38 was downloaded from the UCSC Genome Browser website (http://hgdownload.cse.ucsc.edu/goldenPath/hg38/bigZips/) and used as the genome assembly. The CDK/cyclin gene family members (CDK: n = 21; cyclin: n = 26) were defined based on review articles (Malumbres, 2014; Malumbres et al., 2009), and further complemented by database searching by database searching via the Human Protein Reference Database (HPRD) (http://www.hprd.org/) (Keshava Prasad et al., 2009), the Pfam protein family database (http://pfam.xfam.org/) (Finn et al., 2014), and the SMART (Simple Modular Architecture Research Tool) database (http://smart.embl-heidelberg.de/) (Letunic et al., 2006).
Phylogenetic trees of the protein families
The full-length amino acid sequences of CDK/cyclin proteins were aligned using multiple alignment tool ClustalW (http://www.clustal.org/). The resulted sequence alignments were used to construct phylogenetic trees by MEGA7 (Kumar et al., 2016) with the maximum likelihood evolution algorithm. A Poisson correction was used for multiple substitution models. Initial trees for the heuristic search were obtained automatically by applying Neighbor-Join (NJ) and Bio-NJ algorithms to a matrix of pairwise distances estimated using a Jones-Taylor-Thornton (JTT) model, and then selecting the topology with superior log likelihood value (Kumar et al., 2016).
RNA-seq data processing and analysis
The poly(A)+ RNA-seq (Illumina) data were generated by the University of North Carolina and the British Columbia Cancer Agency Genome Sciences Centre as part of the TCGA project, and were processed by the TCGA Research Network using the NCI Genomic Data Commons (GDC) mRNA quantification analysis pipeline (https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/). Gene level RNA expression data (in Fragments Per Kilobase of transcript per Million mapped reads [FPKM]) from tumor specimens across 33 cancer types from 27 primary sites as well as corresponding normal adjacent specimens from 24 matched tissue types were downloaded from the GDC Data Portal (https://portal.gdc.cancer.gov/) (retrieved date: Oct, 27, 2017). If more than one sample existed for a participant, one single tumor sample (and matched adjacent sample, if applicable) was selected based on the following rules: (1) tumor sample type: primary (01) > recurrent (02) > metastatic (06); (2) order of sample portions: higher portion numbers were selected; and (3) order of plate: higher plate numbers were selected.
SNP array data processing and analysis
The single-nucleotide polymorphism (SNP) array data (Affymetrix Genome-Wide Human SNP Array 6.0) in CEL format across 33 cancer types were downloaded from the TCGA Data Portal (https://portal.gdc.cancer.gov/). Segmentation files of TCGA tumor samples processed by circular binary segmentation (CBS) algorithm (Olshen et al., 2004) were retrieved from the TCGA GDAC Firehose of the Broad Institute (http://gdac.broadinstitute.org/; retrieved date: Jan, 3, 2018). If multiple samples existed for one participant, one pair of tumor and matched control was selected for ABSOLUTE analysis (Carter et al., 2012) and one tumor sample was kept for focal SCNA analysis. Sample selection based on following rules: (1) sample type: for tumor tissues, primary (01) > recurrent (02) > metastatic (06); for normal control tissues, blood (10) > solid (11); (2) molecular type of analyte for analysis: prefer D analytes (native DNA) over G, W, or X (whole-genome amplified); (3) order of sample portions: higher portion numbers were selected; and (4) order of plate: higher plate numbers were selected.
Recurrent focal SCNA estimation
The Genomic Identification of Significant Targets in Cancer (GISTIC 2.0) algorithm (Mermel et al., 2011) (https://software.broadinstitute.org/cancer/cga/gistic) was used to identify significantly recurrent focal genomic regions that were gained or lost in a given tumor type. Segmentation files retrieved from the TCGA GDAC Firehose of the Broad Institute were used as input. GISTIC deconstructs copy number alterations into broad and focal events and applies a probabilistic framework to identify location and significance levels of SCNA. For the recurrent focal SCNA estimations, the significance levels (q values) were calculated by comparing the observed gains/losses at each locus to those obtained by randomly permuting the events along the genome. Tumors which have more than 2,000 segments were excluded from our analysis. Default parameters of GISTIC were used with the confidence level set to 0.99 (by -conf). Focal events with q-value below 0.25 were considered as significantly recurrent. Significant focal events in individual samples were then classified into four categories according to the amplitude threshold of GISTIC: GISTIC status = 0, below threshold; GISTIC status = 1, amplified (gain); GISTIC status = 2, highly amplified (amplification); GISTIC status = −1, deleted (loss); GISTIC status = −2, highly deleted (deletion). In each cancer type, a GISTIC score (G-score), which takes into account both frequency and amplitude of a given SCNA event (Mermel et al., 2011), was generated by GISTIC for each CDK/cyclin gene separately for gain or loss. Genes with G-score < 0.1 were excluded from downstream analysis due to low frequency and/or amplitude. For a given CDK/cyclin gene, an overall G-score across all cancer types was calculated by an unweighted sum of G-scores in every cancer type.
Enrichment analysis of recurrent focal SCNAs
We found 36/47 (76.60%) CDKs/cyclins located in a significantly high-level recurrent focal SCNA region in at least one cancer type. In order to discriminate this observation from random noise, we randomly chose a gene set with equal size (n = 47) and recorded a statistic assessing the degree to which the gene set was affected by SCNAs. A null distribution was then generated by repeating this procedure 1,000 times independently (Figure S1C). The sum of G-scores of all high-level SCNA events harboring genes from the chosen gene set was used as the statistic (s) to compare:
in which Gi,t represented G-score of the SCNA event harboring gene i in cancer type t and the SCNA event was altered at high level (Gi,t ≥ 0.1). Empirical p value assessing whether SCNA events were enriched on the genes encoding CDKs/cyclins was calculated comparing the observed statistic on CDK/cyclin pathway against the null distribution. To investigate whether the genes encoding CDKs/cyclins have significantly higher SCNA frequency compared to the background genomic alterations in the cancer genomes at pan-cancer level, we applied the Elbow method (Goutte et al., 1999; Thorndike, 1953) to overall G-scores at pan-cancer level and separated genes subject to selective pressure from the others (Figure S1D). Fisher’s exact test was applied to assess whether SCNA events on genes encoding CDKs/cyclins were more selective compare to the background, considering gain and loss events together.
Correlation between CN and expression
To identify CDK/cyclin genes that have positive correlations between their RNA expression levels and copy number alterations, the putative gene-level copy number of a given gene was estimated by the GISTIC algorithm. CDK/cyclin genes that were detectable in at least 10% of tumor specimens (90th percentile of FPKM value ≥ 1) in a given cancer type were subjected to correlation analysis. Pearson correlation analysis was performed by R software and the threshold of significant correlation between the estimated copy number and RNA expression level for each gene was set to p < 0.001.
Identification of the genes driven by SCNAs
At the individual cancer type level, we identified putative cancer-causing CDK/cyclin genes driven by SCNAs using four criteria as described by our previous publication (Hu et al., 2019b): 1) location in a peak region of a significantly recurrent focal SNCA locus estimated by GISTIC (q ≤ 0.25); 2) alteration with high frequency and large amplitude (G-score ≥ 0.1); 3) mRNA expression reliably detected in at least 10% of tumor specimens in a given cancer type (the 90th percentile of FPKM value ≥ 1); and 4) expression level of mRNA that was significantly and positively correlated with the estimated copy numbers (p value of Pearson’s correlation coefficient between log[FKPM+1] and logR < 0.001). To estimate SCNAs for these putative cancer-causing CDK/cyclin genes at a pan-cancer level, we calculated an overall G-score by an unweighted numeric sum of G-scores that met all four criteria in each individual cancer type.
WES data processing and analysis
Mutation Annotation Format (MAF) profiles for 33 cancer type were downloaded from the TCGA Multi-Center Mutation Calling in Multiple Cancers (MC3) project (https://repo-prod.prod.sagebase.org/repo/v1/doi/locate?id=syn7214402&type=ENTITY), a variant calling project of TCGA (Ellrott et al., 2018). The MC3 data was generated through seven independent mutation calling algorithms, including Pindel (INDEL) (Ye et al., 2009), MuSE (SNV) (Fan et al., 2016), Radia (SNV) (Radenbaugh et al., 2014), VarScan2 (SNV/INDEL) (Koboldt et al., 2012), MuTect (SNV) (Cibulskis et al., 2013), Indelocator (INDEL) (Chapman et al., 2011) and SomaticSniper (SNV) (Larson et al., 2012). Variants from each caller were merged, QC filtered and stored in MAF file (Ellrott et al., 2018). If multiple samples existed for a participant in the MAF, one single pair of tumor/matched control sample was kept following the rules: (1) sample type: for tumor tissues, primary (01) > recurrent (02) > metastatic (06); for normal tissues, blood (10) > solid (11); (2) molecular type of analyte for analysis: prefer D analytes (native DNA) over G, W, or X (whole-genome amplified); (3) order of sample portions: higher portion numbers were selected; and (4) order of plate: higher plate numbers were selected. We excluded all mutations that were not tagged with “PASS” or tagged with “WGA” alone in all cancer types.
Recurrent mutation gene estimation
As described by our previous publication (Hu et al., 2019b), to predict the putative cancer-causing CDK/cyclin genes driven by mutation, five independent methods were integrated and applied to identify recurrent mutations: (1) MutSigCV (Lawrence et al., 2013) (https://www.genepattern.org/modules/docs/MutSigCV), which identifies genes that are significantly mutated in cancer genomes, using a model with mutational covariates. It analyzes the mutations of each gene to identify genes that were mutated more often than expected by chance, given the background model; (2) Oncodrivefm (Gonzalez-Perez and Lopez-Bigas, 2012) (http://bg.upf.edu/group/projects/oncodrive-fm.php), which computes a metric of functional impact using three well-known methods (SIFT, Poly-Phen2 and MutationAssessor) and assesses how the functional impact of variants found in a gene across several tumor samples deviates from a null distribution to detect candidate driver genes; (3) OncodriveCLUST (Tamborero et al., 2013) (http://bg.upf.edu/group/projects/oncodrive-clust.php), which is designed to exploit the feature that mutations in cancer genes, especially oncogenes, often cluster in particular positions of the protein and change their functions, thus be used to nominate novel candidate driver genes; (4) ActiveDriver (Reimand and Bader, 2013) (http://reimandlab.org/software/activedriver/), which identifies post-translational modification (PTM) sites in proteins (i.e., “active” sites such as signaling sites, protein domains, regulatory motifs) that are significantly mutated in cancer genomes; and (5) HotSpot3D (Niu et al., 2016) (https://github.com/ding-lab/hotspot3d), which identifies mutation hotspots from linear protein sequence and correlate the hotspots with known or potentially interacting domains and mutations. MC3 MAF files were used as input for the above programs, and default parameters were used for all five programs. A mutation index “x” (range from 0 to 5) was assigned to a gene which has passed the threshold of “x” out of five programs in a given cancer type. In addition, a mutation score (M-score) was calculated for each mutated CDK/cyclin gene in a given cancer type, which takes into account both the mutation index and its frequency of mutation across samples (i.e., M score = mutation index × mutation frequency). Geneswith mutation index ≥ 2 (identified as positive by at least two programs) were considered as recurrently mutated. An overall M-score was generated to measure the recurrent mutation level of a given CDK/cyclin gene across all cancers, by unweighted sum of M-scores estimated for each individual cancer type. They hypermutated samples were excluded from the recurrent mutation analysis.
Fusion data processing and analysis
The gene fusion data of TCGA were retrieved from TumorFusions data portal (https://tumorfusions.org/), which analyzed transcript fusions across 33 cancer types from TCGA (Hu et al., 2018). The transcript fusion events were called by Pipeline for RNaseq Data Analysis (PRADA) (Torres-García et al., 2014), and fusions detected in normal samples were excluded. Six filters controlling for sequence similarity of the partner genes, transcriptional allelic fraction, dubious junctions, germline events and presence in non-neoplastic tissue were applied (Hu et al., 2018). If more than one sample existed for a participant, one single sample was kept following the rules: (1) sample type: for tumor tissues, primary (01) > recurrent (02) > metastatic (06); (2) order of sample portions: higher portion numbers were selected; and (3) order of plate: higher plate numbers were selected. The genome-wide view (Circos plot) of transcript fusion events in CDK/cyclin genes was generated by Circos (http://circos.ca/) (Krzywinski et al., 2009).
Definition of understudied genes
The PubTator/NCBI (https://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/PubTator/), a web-based text mining tool for assisting biocuration (Wei et al., 2013), was used to retrieve the publications related to the CDK/cyclin genes. The PubTator score retrieved from the Target Central Resource Database (TCRD) (http://juniper.health.unm.edu/tcrd/) was used to define the under-studied CDK/cyclin gene (i.e., PubTator score < 150).
Definition of drug target development levels
Target Central Resource Database (TCRD) (http://juniper.health.unm.edu/tcrd/), a central resource generated by the Illuminating the Druggable Genome Knowledge Management Center/NIH (Nguyen et al., 2017), was used to define the drug target development level (TDL) for each CDKs/cyclins. TDL was classified into four levels: Tclin, Tchem, Tbio and Tdark. Tclin: targets that have activities in DrugCentral (i.e., approved drugs) with known mechanism of action. Tchem: targets that have activities in ChEMBL or DrugCentral and satisfy the activity thresholds defined by TCRD. Tbio: targets that do not have known drug or small molecule activities that satisfy the activity thresholds defined by TCRD and satisfy one or more of the following criteria: (target is above the cutoff criteria for Tdark; target is annotated with a Gene Ontology Molecular Function or Biological Process leaf term(s) with an Experimental Evidence code; target has confirmed OMIM phenotype). Tdark: targets which virtually nothing is known. They do not have known drug or small molecule activities that satisfy the activity thresholds defined by TCRD and satisfy two or more of the following criteria: (a PubMed text-mining score from Jensen Lab < 5; ≤ 3 Gene RIFs; ≤ 50 antibodies available according to https://antibodypedia.com).
Analysis of drug screening data
The drug screening data were retrieved from the Genomics of Drug Sensitivity in Cancer (GDSC) (Garnett et al., 2012; Yang et al., 2013) and the Cancer Therapeutics Response Portal (CTRP) (Barretina et al., 2012; Basu et al., 2013; Seashore-Ludlow et al., 2015) projects. The sensitivity profile of the compounds screened across cancer cell lines were constructed based on the area under the curve (AUC) values. A CDK7/12 genomic deficiency index (GDI) for each cancer cell line was estimated from a combination of CDK7 predicted copy number and CDK12 mutation status. Briefly, a gene-level copy number of CDK7 (log2-scaled copy number relative to the normal copy number) established by GISTIC was used to represent CDK7 copy number alteration. A semiquantitative CDK12 mutation score was used to present CDK12 mutation, in which the putative function impact of each CDK12 mutation was scored as high (−1), moderate (−0.6) or low (−0.3) according to its functional annotation. A linear regression model was applied to determine whether the AUC values for a specific compound were significantly associated with CDK7/12 GDI and CDK7 predicted copy number. Cancer type was adjusted in the model. All compounds screened by either GDSC or CTRP were pooled and manually categorized according to the mechanisms of action. When a compound was screened in both projects, the AUC values measured by CTRP were preferentially used. To find drug categories associated with increased or decrease sensitivity in cell lines harboring CDK7/12 alteration or CDK7 loss, we used the R package fGSEA (https://bioconductor.org/packages/release/bioc/html/fgsea.html). The input for enrichment consisted of the significance statistics derived by regression models for all compounds tested. The fgsea algorithm was run against 22 drug categories manually defined according to the mechanisms of action. Normalized enrichment scores and p values were determined by a permutation test with 10,000 iterations and adjusted with the Benjamini and Hochberg method (Benjamini and Hochberg, 1995).
Analysis of chemotherapy response data
The clinical annotation and genomic profiles (copy number of recurrent focal peaks, and high-confidence mutation status of genes) of the ovarian cancer patients with chemotherapy response information were retrieved from a recent publication by Patch et al. (2015). 37 and 31 patients defined as chemotherapy sensitive and resistant by Patch et al. (2015) were used in our analysis. A CDK7/12 GDI for each patient was estimated based on predicated focal copy number of CDK7 locus and CDK12 mutation status. Welch two-sample t test were used to compare CDK7/12 GDI and focal copy number of CDK7 locus between the different patient groups.
Expression and ChIP-seq in cancer cell lines
RNA-seq data of cancer cells treated with or without THZ1 in esophagus (GSE76860), ovarian (GSE116282 and SRP106825) and head and neck (GSE95750) cancers were retrieved from GEO dataset, and further processed through a NCI Genomic Data Commons (GDC) mRNA quantification analysis pipeline (https://docs.gdc.cancer.gov/Data/Bioinformatics_Pipelines/Expression_mRNA_Pipeline/). Gene expression array data of T-ALL cells treated with or without THZ1 was obtained from GEO dataset (GSE50624). ChIP-seq data of T-ALL cells treated with or without THZ1 was retrieved from GEO dataset (GSE50622). BigWig files of ChIP-seq experiments were used for visualization in IGV.
GO and interaction network analyses
DAVID (https://david.ncifcrf.gov) (Huang et al., 2007) was used to identify Gene Ontology (GO) enriched in THZ1 downregulated genes. The KEGG pathway set was used in GO analysis. The network was exported and visualized in Cytoscape v.3.4.0 (https://www.cytoscape.org/) in which the node sizes and color presented p value and FDR, respectively.
Cell viability assay
Cells were seeded in 96-well plates at a concentration of 1 × 103 cells per well overnight. Cells were then treated with a single drug or a drug combination. For olaparib single drug viability assay: cells were treated with serial dilutions of olaparib (0 μM, 1.5 μM, 3.125 μM, 6.25 μM, 12.5 μM, 25 μM, 50 μM, 100 μM, 200 μM and 400 μM). For drug combination of olaparib and THZ1: serial dilutions of olaparib (0 μM, 1.5 μM, 3.125 μM, 6.25 μM, 12.5 μM, 25 μM, 50 μM, 100 μM, 200 μM and 400 μM) were used to combine with 12.5 nM, 25 nM or 50 nM THZ1 respectively. Cell were cultured for 3 days before harvest for MTT or crystal violet staining detection.
Soft agar assay
The bottom layer was prepared with a 0.8% agarose (Invitrogen) solution in culture medium in 6-well plates, and the gel was allowed to set for 20 min at room temperature. Cells (5 × 103) were resuspended in 0.4% top agarose solution (in culture medium) then carefully placed on top of the bottom agarose in the 6-well plates. Cells were cultured in normal medium for 3 days and then treated with DMSO, THZ1, olaparib or combination of THZ1 and olaparib. Culture medium was replaced every 3 days. After 2–4 weeks, cell colonies were stained using crystal violet (Sigma) and counted.
siRNA transfection
Cells were plated 24 h before transfection at 50% confluence. CDK7 siRNA and control siRNA were transfected with Lipofectamine RNAiMAX (Invitrogen). The following siRNA oligonucleotides from Sigma were used: CDK7 siRNA1: SASI_Hs01_00214780; CDK7 siRNA2: SASI_Hs01_00214781. Negative control siRNA was purchased from Sigma.
RNA isolation and qRT-PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen) and reverse transcribed using the High Capacity RNA-to-cDNA Kit (Applied Biosystems). cDNA was quantified by an ABI ViiA 7 System (Applied Biosystems).
Protein isolation and western blots
Cells were lysed in mammalian protein extraction reagent (Pierce). After quantification using a BCA protein assay kit (Pierce), total proteins were separated by SDS-PAGE under denaturing conditions and transferred to PVDF membranes (Millipore). Membranes were blocked in 5% non-fat milk (Bio-Rad) and then incubated with anti-CDK7 (Cat No: sc-7344, Santa Cruz Biotechnoloy); anti-BRCA1 (Cat No: sc-6954, Santa Cruz); anti-BRCA2 (Cat No: OP95, Millipore); anti-RAD51 (Cat No: ab213, Abcam); anti-Ku80 (Cat No: MA5–12933, Thermo); anti-Ku70 (Cat No: MA5–15110, Thermo); anti-RNAP II (Cat No: sc-17798, Santa Cruz Biotechnoloy); anti-RNAP II p-Ser5 (Cat No: A300–655A-2, Bethyl); or anti-phospho-H2AX(S139) (Cat No:05–636, Clone No: JBW301, Millipore), followed by incubation with secondary antibodies conjugated with horseradish peroxidase (HRP, GE Healthcare Life Sciences). Anti-b-Tubulin (Cat No: 2128, Clone No: 9F3, CST) or anti-Actin (Cat No: A3854, Sigma) was used for internal loading control. Immunoreactive proteins were visualized using the LumiGLO chemiluminescent substrate (Cell Signaling).
Comet assay
Cells were treated with DMSO, THZ1, olaparib or combination of THZ1 and olaparib for 72 h. Neutral comet assays with Sybr gold staining (Invitrogen) were performed. The quantification of tail DNA was done using CASP software.
Immunofluorescence
Cells were seeded on coverslips and treated with THZ1 and ionizing radiation, or with DMSO, THZ1, olaparib or combination of THZ1 and olaparib, then harvested for immunofluorescent staining. Cells were fixed in solution containing 3% paraformaldehyde and 2% sucrose for 10 min at room temperature, and subsequently permeabilized with 0.5% Triton solution for 5 min at 4°C. Permeabilized cells were incubated with anti-γH2AX antibody (Cat No: ab81299, Abcam), anti-BRCA1 (Cat No: sc-6954, Santa Cruz), anti-BRCA2 (Cat No: OP95, Millipore), or anti-RAD51 (Cat No: ab213, Abcam) in PBST buffer (PBS plus 0.1% Tween-20, 0.02% NaN3) overnight at 4°C. Cells were then washed three times with PBST, then incubated with secondary antibody for 1 hour at room temperature. After four washes with PBST, coverslips were mounted onto glass slides using Vectashield mounting medium containing DAPI (Vector Laboratories) and visualized using an Axiovert 200M inverted microscope (Zeiss).
HR activity assay
The HR activity reporter constructs were obtained from Dr. Gorbunova (Seluanov et al., 2010). THZ1 treated cells were transfected with I-SceI-digested plasmid with 0.1 μg of control pDsRed2-N1 plasmid. Expression of GFP and DsRed were monitored by fluorescence microscopy (Nikon, Eclipse TE2000-U). 48 h after transfection, cells were harvested, resuspended in 0.5 mL of PBS, pH 7.4 (GIBCO, Invitrogen), and analyzed by FACS (BD FACS Canto).
Chromatin immunoprecipitation (ChIP)
For ChIP-qPCR experiment, 2 × 107 cells treated with THZ1 for 24 hours, or 30nM control siRNA, and CDK7 siRNA1 for 72 hours were harvested for the following ChIP experiment. Cells were cross-linked with 1% formaldehyde at room temperature for 10 min, and then neutralized with 125 mM glycine for 5 min. Cells were rinsed with ice-cold PBS twice and scraped into 1 mL of ice-cold PBS. Cells were resuspended in 0.3 mL of lysis buffer and sonicated. After centrifugation, supernatants were collected and diluted in IP dilution buffer followed by immunoclearing with protein A-Sepharose for 2 hours at 4°C. 5 μg anti-RNA Pol II (Cat No: MMS-126R-200; Lot No: D13HF02305, Convance); or control IgG (Cat No: 2729S, CST) was used for immunoprecipitation. After immunoprecipitation, 45 μL protein A-Sepharose was added and incubated for another hour. Precipitates were washed, and DNA was purified after de-cross-linking for real-time PCR. Primers used are listed below:
RAD51 Promoter ChIP F TTGGCGGGAATTCTGAAAGC
RAD51 Promoter ChIP R ACGCTCCACTTCTCTACTCG
BRCA1 Promoter ChIP F GAATGAAGTTGCGGACCCTC
BRCA1Promoter ChIP R GCTGTAACACTCACCGCAAA
BRCA2 Enhancer ChIP F ATTGCTGTTTTGGGGAAGTG
BRCA2 Enhancer ChIP R CAGCAATTACCAGGGGCTTA
Negative control ChIP F TGTGGCAGTATGGAGTGAGG
Negative control ChIP R ACAGCCACTCACACCAGTAT
Luciferase assay
For drug treatment reporter assay, 500 ng of reporter vector plus 5 ng of the renilla luciferase plasmid were transfected to cells using FuGENE 6 (Promega); cells were then treated with DMSO and THZ1 for 24 hours. For CDK7 siRNA treatment reporter assay, cells transfected with CDK7 siRNA were then transfected with reporter vector and renilla, and cells were harvested 48 hours after transfection. Reporter assays were performed using a dual luciferase reporter assay system (Promega) by Fluoroskan Ascent FL fluorometer (Thermo Fisher Scientific).
Propidium iodide staining for cell cycle
MDA-MB-231, OVCAR5 and DU145 cells were treated with DMSO, 25 nM THZ1, and 50 nM THZ1 for 24 hours. Cells were trypsinized, washed with 1 × PBS and fixed with 70% (wt/vol) ethanol in PBS at 4°C overnight. Cells were washed with 1 × PBS twice, and then treated with 0.5 mg/ml RNase A for 1 hour at RT and incubated with 0.1 mg/ml propidium iodide (Sigma) for 45 min at 37°C. DNA content was determined by flow cytometry (BD LSR II Flow Cytometer). Data was analyzed with flowJo software package
Annexin V/7-AAD staining
Apoptotic cells were quantified by an Annexin-V-FLUOS Staining Kit followed the manufacturer’s protocol (Cat No: 11858777001, Roche).
Caspase3/7 activity assay
Caspase3/7 activity was measured by a Caspase-Glo® 3/7 Assay (Cat No: G8090, Promega) followed manufacture’s protocol.
QUANTIFICATION AND STATISTICAL ANALYSIS
The Pearson coefficient that was used to measure correlation between the estimated copy number and RNA expression level for each gene was calculated by R software. Coefficients with p values < 0.001 were considered statistically significant. Selectivity of SCNA events on genes encoding CDKs/cyclins compared to the background was assessed by Fisher’s exact test. A p value < 0.05 was considered statistically significant. The significance statistic that was used to assess the association of the response to a specific compound with CDK7/12 GDI and CDK7 predicted copy number was estimated by a linear regression model adjusting for cancer type. Enrichment for a given drug category among positive or negative associations was performed by the R package fGSEA (https://bioconductor.org/packages/release/bioc/html/fgsea.html). Normalized enrichment scores and p values were determined by a permutation test with 10,000 iterations and adjusted with the Benjamini and Hochberg method (FDR 10%) (Benjamini and Hochberg, 1995). CDK7/12 GDI and focal copy number of CDK7 locus between ovarian cancer patients sensitive and resistant to chemotherapy was compared by Welch two-sample t test. A p value < 0.05 was considered statistically significant. For all biological experiments, data were represented as mean ± standard deviation (SD) and were compared using two-tailed Student’s t test with p value < 0.05 suggesting significance unless otherwise indicated.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-gH2AX antibody | Abcam | Cat# ab81299; RRID:AB_1640564 |
| anti-BRCAl antibody | Santa Cruz Biotechnoloy | Cat# sc-6954; RRID:AB_626761 |
| anti-BRCA2 antibody | Millipore | Cat# OP95; RRID:AB_2067762 |
| anti-RAD51 antibody | Abcam | Cat# ab213; RRID:AB_302856 |
| anti-CDK7 antibody | Santa Cruz Biotechnoloy | Cat# sc-7344; RRID:AB_627243 |
| anti-Ku80 antibody | Thermo Fisher Scientific | Cat# MA5-12933; RRID:AB_10983840 |
| anti-Ku70 antibody | Thermo Fisher Scientific | Cat# MA5-15110; RRID:AB_10984110 |
| anti-RNAP II antibody | Santa Cruz Biotechnoloy | Cat# sc-17798; RRID:AB_677355 |
| anti-RNAP II p-Ser5 antibody | Bethyl Laboratories | Cat# A300-655A; RRID:AB_2268550 |
| anti-gH2AX antibody | Millipore | Cat# 05-636; RRID:AB_309864 |
| anti-b-Tubulin antibody | Cell Signaling Technology | Cat# 2128; RRID:AB_823664 |
| anti-Actin antibody | Sigma-Aldrich | Cat# A3854; RRID:AB_262011 |
| anti-RNA Pol II antibody | Convance | Cat# MMS-126R-200; RRID:AB_10013665 |
| normal rabbit IgG | Cell Signaling Technology | Cat# 2729S; RRID:AB_1031062 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| THZ1 | Selleckchem | Cat# S7549 |
| Olaparib | Selleckchem | Cat# S1060 |
| Veliparib | Selleckchem | Cat# S1004 |
| Cisplatin | Selleckchem | Cat# S1166 |
| Trizol | Thermo Fisher Scientific | Cat# 15596018 |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Fisher Scientific | Cat# 13778150 |
| FuGENE 6 | Promega | Cat# E2691 |
| Formaldehyde | Sigma-Aldrich | Cat# F8775 |
| Glycine | Sigma-Aldrich | Cat# G8898 |
| Protein A-Sepharose® 4B | Sigma-Aldrich | Cat# P9424 |
| Critical Commercial Assays | ||
| Dual-Glo Luciferase Assay System | Promega | Cat# E2920 |
| Annexin-V-FLUOS Staining Kit | Roche | Cat# 11858777001 |
| Caspase-Glo® 3/7 Assay | Promega | Cat# G8090 |
| Deposited Data | ||
| TCGA genomic profiling | TCGA project | https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga |
| TCGA Affymetrix SNP6.0 array data (CEL) | TCGA Data Portal | https://www.cancer.gov/tcga/ |
| TCGA Affymetrix SNP6.0 array data (segmentation) | TCGA GDAC Firehose | http://gdac.broadinstitute.org/ |
| TCGA whole exome sequencing data | TCGA MC3 project | https://repo-prod.prod.sagebase.org/repo/v1/doi/locate?id=syn7214402&type=ENTITY |
| TCGA RNA sequencing data | Genomic Data Commons | https://portal.gdc.cancer.gov/ |
| TCGA transcript fusion data | (Hu et al., 2018) | https://tumorfusions.org/ |
| Cancer Cell Line Encyclopedia (CCLE) | The Broad Institute | https://portals.broadinstitute.org/ccle |
| The Genomics of Drug Sensitivity in Cancer Project | The Wellcome Sanger Institute | https://www.cancerrxgene.org/ |
| The Cancer Therapeutics Response Portal (CTRP) Project | Broad Institute | https://portals.broadinstitute.org/ctrp/ |
| Human Protein Reference Database (HPRD) | (Keshava Prasad et al., 2009) | http://www.hprd.org/ |
| Pfam protein family database | (Finn et al., 2014) | http://pfam.xfam.org/ |
| SMART (Simple Modular Architecture Research Tool) database | (Letunic et al., 2006) | http://smart.embl-heidelberg.de/ |
| PubTator | (Wei et al., 2013) | https://www.ncbi.nlm.nih.gov/CBBresearch/Lu/Demo/PubTator/ |
| Target Central Resource Database (TCRD) | (Nguyen et al., 2017) | http://juniper.health.unm.edu/tcrd/) |
| THZ1 treatment RNA-seq profiles (esophageal squamous cell carcinoma) | (Jiang et al., 2017) | GEO: GSE76860 |
| THZ1 treatment RNA-seq profiles (ovarian cancer) | (Zeng et al., 2018) | GEO: GSE116282 |
| THZ1 treatment RNA-seq profiles (ovarian cancer) | (Zhang et al., 2017) | NCBI BioProject database: SRP106825 |
| THZ1 treatment RNA-seq profiles (T cell acute lymphoblastic leukemia) | (Kwiatkowski et al., 2014) | GEO: GSE50624 |
| THZ1 treatment RNA-seq profiles (nasopharyngeal carcinoma) | (Yuan et al., 2017) | GEO: GSE95750 |
| Genomic profiles of ovarian cancer patients | (Patch et al., 2015) | GEO: GSE61568 |
| THZ1 treatment ChIP-seq profiles | (Kwiatkowski et al., 2014) | GEO: GSE50622 |
| Functional Cancer Genome data portal | This paper | http://fcgportal.org/home |
| TCCCA | This paper | http://fcgportal.org/TCCCA/ |
| Experimental Models: Cell Lines | ||
| OVCAR3 | ATCC | HTB-161 |
| MDA-MB-231 | ATCC | HTB-26 |
| MDA-MB-468 | ATCC | HTB-132 |
| DU145 | ATCC | HTB-81 |
| PC3 | ATCC | CRL-1435 |
| OVCAR5 | NCI Development Therapeutics Program | N/A |
| Experimental Models: Organisms/Strains | ||
| Female mouse: Nude, Strain NU/J | Jackson Laboratory | Stock No: 002019 |
| Oligonucleotides | ||
| Primers used for PCR analyses | This paper | N/A |
| CDK7 siRNA-1 | Sigma-Aldrich | SASI_Hs01_00214780 |
| CDK7 siRNA-2 | Sigma-Aldrich | SASI_Hs01_00214781 |
| Recombinant DNA | ||
| HR activity reporter | (Seluanov et al., 2010) | N/A |
| Software and Algorithms | ||
| ClustalW | Science Foundation Ireland | http://www.clustal.org/ |
| MEGA7 | (Kumar et al., 2016) | https://www.megasoftware.net/ |
| ABSOLUTE | (Carter et al., 2012) | https://software.broadinstitute.org/cancer/cga/absolute |
| GISTIC 2.0 | (Mermel et al., 2011) | ftp://ftp.broadinstitute.org/pub/GISTIC2.0/ |
| MutSigCV | (Lawrence et al., 2013) | https://www.genepattern.org/modules/docs/MutSigCV |
| Oncodrivefm | (Gonzalez-Perez and Lopez-Bigas, 2012) | http://bg.upf.edu/group/projects/oncodrive-fm.php |
| OncodriveCLUST | (Tamborero et al., 2013) | http://bg.upf.edu/group/projects/oncodrive-clust.php |
| ActiveDriver | (Reimand and Bader, 2013) | http://reimandlab.org/software/activedriver/ |
| HotSpot3D | (Niu etal., 2016) | https://github.com/ding-lab/hotspot3d |
| Circos | (Krzywinski et al., 2009) | http://circos.ca/ |
| fGSEA | R package | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| DAVID | (Huang et al., 2007) | https://david.ncifcrf.gov |
Highlights.
Recurrent genomic alterations of the genes encoding CDKs/cyclins are characterized
CDKs/cyclins are dominantly driven by copy-number aberrations across cancers
CDK7 losses are correlated with increased sensitivities to DNA-damaging drugs
Low-dose CDK7i treatment sensitizes HR-proficient cancer cells to PARPi
ACKNOWLEDGMENTS
We thank Drs. Chi V Dang, Xin Lu, Congjian Xu, and Xiaojun Chen for their valuable comments. This work was supported, in whole or in part, by the US National Institutes of Health (R01CA225929, R01CA190415, R01CA142776, P50CA083638, and P50CA174523), the Harry Fields Professorship (L.Z.), the Ovarian Cancer Research Alliance, United States (X.H.), and the Foundation for Women’s Cancer, United States (X.H. and Y.Z.).
Footnotes
DECLARATION OF INTERESTS
Drs. Lin Zhang, Xiaowen Hu, and Youyou Zhang report having received research funding from Bristol-Myers Squibb/Celgene and Prelude Therapeutics.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107884.
REFERENCES
- Ali S, Heathcote DA, Kroll SHB, Jogalekar AS, Scheiper B, Patel H, Brackow J, Siwicka A, Fuchter MJ, Periyasamy M, et al. (2009). The development of a selective cyclin-dependent kinase inhibitor that shows anti-tumor activity. Cancer Res. 69, 6208–6215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asghar U, Witkiewicz AK, Turner NC, and Knudsen ES (2015). The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov 14, 130–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajrami I, Frankum JR, Konde A, Miller RE, Rehman FL, Brough R, Campbell J, Sims D, Rafiq R, Hooper S, et al. (2014). Genome-wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res. 74, 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, et al. (2013). An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell 154, 1151–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B, Stat. Methodol 57, 289–300. [Google Scholar]
- Bradner JE, Hnisz D, and Young RA (2017). Transcriptional addiction in cancer. Cell 168, 629–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayrol F, Praditsuktavorn P, Fernando TM, Kwiatkowski N, Marullo R, Calvo-Vidal MN, Phillip J, Pera B, Yang SN, Takpradit K, et al. (2017). THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat. Commun 8, 14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. (2011). Initial genome sequencing and analysis of multiple myeloma. Nature 471, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, Abraham BJ, Sharma B, Yeung C, Altabef A, et al. (2014). CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell 159, 1126–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou J, Quigley DA, Robinson TM, Feng FY, and Ashworth A (2020). Transcription-associated cyclin-dependent kinases as targets and biomarkers for cancer therapy. Cancer Discov. 10, 351–370. [DOI] [PubMed] [Google Scholar]
- Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, et al. (2014). Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell 26, 909–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubbury SJ, Boutz PL, and Sharp PA (2018). CDK12 regulates DNA repair genes by suppressing intronic polyadenylation. Nature 564, 141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliades P, Abraham BJ, Ji Z, Miller DM, Christensen CL, Kwiatkowski N, Kumar R, Njauw CN, Taylor M, Miao B, et al. (2018). High MITF Expression Is Associated with Super-Enhancers and Suppressed by CDK7 Inhibition in Melanoma. J. Invest. Dermatol 138, 1582–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Xi L, Hughes DS, Zhang J, Zhang J, Futreal PA, Wheeler DA, and Wang W (2016). MuSE: accounting for tumor heterogeneity using a sample-specific error model improves sensitivity and specificity in mutation calling from sequencing data. Genome Biol. 17, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson FM, and Gray NS (2018). Kinase inhibitors: the road ahead. Nat. Rev. Drug Discov 17, 353–377. [DOI] [PubMed] [Google Scholar]
- Finn RD, Bateman A, Clements J, Coggill P, Eberhardt RY, Eddy SR, Heger A, Hetherington K, Holm L, Mistry J, et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RP (2012). The CDK Network: linking cycles of cell division and gene expression. Genes Cancer 3, 731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francavilla C, Lupia M, Tsafou K, Villa A, Kowalczyk K, Rakownikow Jersie-Christensen R, Bertalot G, Confalonieri S, Brunak S, Jensen LJ, et al. (2017). Phosphoproteomics of primary cells reveals druggable kinase signatures in ovarian cancer. Cell Rep. 18, 3242–3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Zhang T, Terai H, Ficarro SB, Kwiatkowski N, Hao MF, Sharma B, Christensen CL, Chipumuro E, Wong KK, et al. (2018). Overcoming resistance to the THZ series of covalent transcriptional CDK inhibitors. Cell Chem. Biol 25, 135–142.e135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. (2012). Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Perez A, and Lopez-Bigas N (2012). Functional impact bias reveals cancer drivers. Nucleic Acids Res. 40, e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutte C, Toft P, Rostrup E, Nielsen F, and Hansen LK (1999). On clustering fMRI time series. Neuroimage 9, 298–310. [DOI] [PubMed] [Google Scholar]
- Greenall SA, Lim YC, Mitchell CB, Ensbey KS, Stringer BW, Wilding AL, O’Neill GM, McDonald KL, Gough DJ, Day BW, and Johns TG (2017). Cyclin-dependent kinase 7 is a therapeutic target in high-grade glioma. Oncogenesis 6, e336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, Lai CF, Metodieva G, de Giorgio A, Williams RL, Santos DB, et al. (2017). Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene 36, 2286–2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. (2012). GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Wang Q, Tang M, Barthel F, Amin S, Yoshihara K, Lang FM, Martinez-Ledesma E, Lee SH, Zheng S, and Verhaak RGW (2018). TumorFusions: an integrative resource for cancer-associated transcript fusions. Nucleic Acids Res. 46 (D1), D1144–D1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Marineau JJ, Rajagopal N, Hamman KB, Choi YJ, Schmidt DR, Ke N, Johannessen L, Bradley MJ, Orlando DA, et al. (2019a). Discovery and characterization of SY-1365, a selective, covalent inhibitor of CDK7. Cancer Res. 79, 3479–3491. [DOI] [PubMed] [Google Scholar]
- Hu Z, Zhou J, Jiang J, Yuan J, Zhang Y, Wei X, Loo N, Wang Y, Pan Y, Zhang T, et al. (2019b). Genomic characterization of genes encoding histone acetylation modulator proteins identifies therapeutic targets for cancer treatment. Nat. Commun 10, 733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Tan Q, Kir J, Liu D, Bryant D, Guo Y, Stephens R, Baseler MW, Lane HC, and Lempicki RA (2007). DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 35, W169–W175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim YH, García-García C, Serra V, He L, Torres-Lockhart K, Prat A, Anton P, Cozar P, Guzmán M, Grueso J, et al. (2012). PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov. 2, 1036–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iniguez AB, Stolte B, Wang EJ, Conway AS, Alexe G, Dharia NV, Kwiatkowski N, Zhang T, Abraham BJ, Mora J, et al. (2018). EWS/FLI confers tumor cell synthetic lethality to CDK12 inhibition in Ewing sarcoma. Cancer Cell 33, 202–216.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YY, Lin DC, Mayakonda A, Hazawa M, Ding LW, Chien WW, Xu L, Chen Y, Xiao JF, Senapedis W, et al. (2017). Targeting super-enhancer-associated oncogenes in oesophageal squamous cell carcinoma. Gut 66, 1358–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SF, Cruz C, Greifenberg AK, Dust S, Stover DG, Chi D, Primack B, Cao S, Bernhardy AJ, Coulson R, et al. (2016). CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 17, 2367–2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalan S, Amat R, Schachter MM, Kwiatkowski N, Abraham BJ, Liang Y, Zhang T, Olson CM, Larochelle S, Young RA, et al. (2017). Activation of the p53 Transcriptional Program Sensitizes Cancer Cells to Cdk7 Inhibitors. Cell Rep. 21, 467–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan AR, Gueble SE, Liu Y, Oeck S, Kim H, Yun Z, and Glazer PM (2019). Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci. Transl. Med 11, eaav4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, et al. (2009). Human Protein Reference Database—2009 update. Nucleic Acids Res. 37, D767–D772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, and Wilson RK (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Ceccaldi R, Shapiro GI, and D’Andrea AD (2015). Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov. 5, 1137–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewska M, Dries R, Grassetti AV, Dust S, Gao Y, Huang H, Sharma B, Day DS, Kwiatkowski N, Pomaville M, et al. (2019). CDK12 loss in cancer cells affects DNA damage response genes through premature cleavage and polyadenylation. Nat. Commun 10, 1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, and Marra MA (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, and Tamura K (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. (2014). Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 511, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson DE, Harris CC, Chen K, Koboldt DC, Abbott TE, Dooling DJ, Ley TJ, Mardis ER, Wilson RK, and Ding L (2012). SomaticSniper: identification of somatic point mutations in whole genome sequencing data. Bioinformatics 28, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. (2013). Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, Copley RR, Pils B, Pinkert S, Schultz J, and Bork P (2006). SMART 5: domains in the context of genomes and networks. Nucleic Acids Res 34, D257–D260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Ni Chonghaile T, Fan Y, Madden SF, Klinger R, O’Connor AE, Walsh L, O’Hurley G, Mallya Udupi G, Joseph J, et al. (2017). Therapeutic rationale to target highly expressed CDK7 conferring poor outcomes in triple-negative breast cancer. Cancer Res. 77, 3834–3845. [DOI] [PubMed] [Google Scholar]
- Lord CJ, and Ashworth A (2017). PARP inhibitors: synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M (2014). Cyclin-dependent kinases. Genome Biol. 15, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, Morgan DO, Tsai LH, and Wolgemuth DJ (2009). Cyclin-dependent kinases: a family portrait. Nat. Cell Biol 11, 1275–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, and Getz G (2011). GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 12, R41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minzel W, Venkatachalam A, Fink A, Hung E, Brachya G, Burstain I, Shaham M, Rivlin A, Omer I, Zinger A, et al. (2018). Small molecules co-targeting CKIα and the transcriptional kinases CDK7/9 control AML in preclinical models. Cell 175, 171–185.e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, Zhang L, Li M, Meng W, Ponnuswami A, Sun W, et al. (2017). Transcriptional dependencies in diffuse intrinsic pontine glioma. Cancer Cell 31, 635–652.e636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen DT, Mathias S, Bologa C, Brunak S, Fernandez N, Gaulton A, Hersey A, Holmes J, Jensen LJ, Karlsson A, et al. (2017). Pharos: collating protein information to shed light on the druggable genome. Nucleic Acids Res. 45 (D1), D995–D1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niu B, Scott AD, Sengupta S, Bailey MH, Batra P, Ning J, Wyczalkowski MA, Liang WW, Zhang Q, McLellan MD, et al. (2016). Protein-structure-guided discovery of functional mutations across 19 cancer types. Nat. Genet 48, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary B, Finn RS, and Turner NC (2016). Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol 13, 417–430. [DOI] [PubMed] [Google Scholar]
- Olshen AB, Venkatraman ES, Lucito R, and Wigler M (2004). Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5, 557–572. [DOI] [PubMed] [Google Scholar]
- Olson CM, Liang Y, Leggett A, Park WD, Li L, Mills CE, Elsarrag SZ, Ficarro SB, Zhang T, Duster R, et al. (2019). Development of a selective CDK7 covalent inhibitor reveals predominant cell-cycle phenotype. Cell Chem. Biol 26, 792–803.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto T, and Sicinski P (2017). Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 93–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patch AM, Christie EL, Etemadmoghadam D, Garsed DW, George J, Fereday S, Nones K, Cowin P, Alsop K, Bailey PJ, et al. ; Australian Ovarian Cancer Study Group (2015). Whole-genome characterization of chemoresistant ovarian cancer. Nature 521, 489–494. [DOI] [PubMed] [Google Scholar]
- Patel H, Periyasamy M, Sava GP, Bondke A, Slafer BW, Kroll SHB, Barbazanges M, Starkey R, Ottaviani S, Harrod A, et al. (2018). ICEC0942, an orally bioavailable selective inhibitor of CDK7 for cancer treatment. Mol. Cancer Ther 17, 1156–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilié PG, Tang C, Mills GB, and Yap TA (2019). State-of-the-art strategies for targeting the DNA damage response in cancer. Nat. Rev. Clin. Oncol 16, 81–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y, O’Connor MJ, and de Bono J (2016). Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med 8, 362ps17. [DOI] [PubMed] [Google Scholar]
- Quereda V, Bayle S, Vena F, Frydman SM, Monastyrskyi A, Roush WR, and Duckett DR (2019). Therapeutic targeting of CDK12/CDK13 in triple-negative breast cancer. Cancer Cell 36, 545–558.e7. [DOI] [PubMed] [Google Scholar]
- Radenbaugh AJ, Ma S, Ewing A, Stuart JM, Collisson EA, Zhu J, and Haussler D (2014). RADIA: RNA and DNA integrated analysis for somatic mutation detection. PLoS ONE 9, e111516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimand J, and Bader GD (2013). Systematic analysis of somatic mutations in phosphorylation signaling predicts novel cancer drivers. Mol. Syst. Biol 9, 637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusan M, Li K, Li Y, Christensen CL, Abraham BJ, Kwiatkowski N, Buczkowski KA, Bockorny B, Chen T, Li S, et al. (2018). Suppression of adaptive responses to targeted cancer therapy by transcriptional repression. Cancer Discov. 8, 59–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CL, Swisher EM, and Kaufmann SH (2015). Poly (ADP-ribose) polymerase inhibitors: recent advances and future development. J. Clin. Oncol 33, 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seashore-Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, Coletti ME, Jones V, Bodycombe NE, Soule CK, Gould J, et al. (2015). Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 5, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seluanov A, Mao Z, and Gorbunova V (2010). Analysis of DNA double-strand break (DSB) repair in mammalian cells. J. Vis. Exp (43), 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifnia T, Wawer MJ, Chen T, Huang QY, Weir BA, Sizemore A, Lawlor MA, Goodale A, Cowley GS, Vazquez F, et al. (2019). Small-molecule targeting of brachyury transcription factor addiction in chordoma. Nat. Med 25, 292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr CJ, Beach D, and Shapiro GI (2016). Targeting CDK4 and CDK6: from discovery to therapy. Cancer Discov. 6, 353–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, Vellano CP, Ju Z, Zhao W, Zhang D, et al. (2018). BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell 33, 401–416.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamborero D, Gonzalez-Perez A, and Lopez-Bigas N (2013). Oncodrive-CLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 29, 2238–2244. [DOI] [PubMed] [Google Scholar]
- Terai H, Kitajima S, Potter DS, Matsui Y, Quiceno LG, Chen T, Kim TJ, Rusan M, Thai TC, Piccioni F, et al. (2018). ER stress signaling promotes the survival of cancer “persister cells” tolerant to EGFR tyrosine kinase inhibitors. Cancer Res. 78, 1044–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorndike RL (1953). Who belongs in the family? Psychometrika 18, 267–276. [Google Scholar]
- Torres-García W, Zheng S, Sivachenko A, Vegesna R, Wang Q, Yao R, Berger MF, Weinstein JN, Getz G, and Verhaak RG (2014). PRADA: pipeline for RNA sequencing data analysis. Bioinformatics 30, 2224–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, et al. (2015). CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 163, 174–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CH, Kao HY, and Lu Z (2013). PubTator: a web-based text mining tool for assisting biocuration. Nucleic Acids Res. 41, W518–W522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RWJ, Ngoc PCT, Leong WZ, Yam AWY, Zhang T, Asamitsu K, Iida S, Okamoto T, Ueda R, Gray NS, et al. (2017). Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T-cell leukemia. Blood 130, 2326–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YM, Cieslik M, Lonigro RJ, Vats P, Reimers MA, Cao X, Ning Y, Wang L, Kunju LP, de Sarkar N, et al. (2018). Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 173, 1770–1782.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et al. (2013). Genomics of drug sensitivity in cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 41, D955–D961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Zhang Y, Shan W, Hu Z, Yuan J, Pi J, Wang Y, Fan L, Tang Z, Li C, et al. (2017). Repression of BET activity sensitizes homologous recombination-proficient cancers to PARP inhibition. Sci. Transl. Med 9, eaa1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, and Ning Z (2009). Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25, 2865–2871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Jiang YY, Mayakonda A, Huang M, Ding LW, Lin H, Yu F, Lu Y, Loh TKS, Chow M, et al. (2017). Super-enhancers promote transcriptional dysregulation in nasopharyngeal carcinoma. Cancer Res. 77, 6614–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborowska J, Egloff S, and Murphy S (2016). The pol II CTD: new twists in the tail. Nat. Struct. Mol. Biol 23, 771–777. [DOI] [PubMed] [Google Scholar]
- Zeng M, Kwiatkowski NP, Zhang T, Nabet B, Xu M, Liang Y, Quan C, Wang J, Hao M, Palakurthi S, et al. (2018). Targeting MYC dependency in ovarian cancer through inhibition of CDK7 and CDK12/13. eLife 7, e39030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Kwiatkowski N, Olson CM, Dixon-Clarke SE, Abraham BJ, Greifenberg AK, Ficarro SB, Elkins JM, Liang Y, Hannett NM, et al. (2016). Covalent targeting of remote cysteine residues to develop CDK12 and CDK13 inhibitors. Nat. Chem. Biol 12, 876–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Peng H, Wang X, Yin X, Ma P, Jing Y, Cai MC, Liu J, Zhang M, Zhang S, et al. (2017). Preclinical efficacy and molecular mechanism of targeting CDK7-dependent transcriptional addiction in ovarian cancer. Mol. Cancer Ther 16, 1739–1750. [DOI] [PubMed] [Google Scholar]
- Zhang H, Pandey S, Travers M, Sun H, Morton G, Madzo J, Chung W, Khowsathit J, Perez-Leal O, Barrero CA, et al. (2018). Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell 175, 1244–1258.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Christensen CL, Dries R, Oser MG, Deng J, Diskin B, Li F, Pan Y, Zhang X, Yin Y, et al. (2020). CDK7 inhibition potentiates genome instability triggering anti-tumor immunity in small cell lung cancer. Cancer Cell 37, 37–54.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated by this study are publicly available through the Functional Cancer Genome data portal (http://fcgportal.org/home) and the TCCCA website (http://fcgportal.org/TCCCA/).