

SUMMARY:
The intronic C9orf72 G4C2 expansion, the most common genetic cause of ALS and FTD, produces sense- and antisense-expansion RNAs and six dipeptide repeat-associated, non-ATG (RAN) proteins, but their roles in disease are unclear. We generated high-affinity human antibodies targeting GA or GP RAN proteins. These antibodies cross the blood-brain barrier and co-localize with intracellular RAN aggregates in C9-ALS/FTD BAC mice. In cells, α-GA1 interacts with TRIM21 and α-GA1 treatment reduced GA levels, increased GA turnover, decreased RAN toxicity and decreased co-aggregation of proteasome and autophagy proteins to GA aggregates. In C9-BAC mice, α-GA1 reduced GA as well as GP and GR proteins, improved behavioral deficits, decreased neuroinflammation and neurodegeneration, and increased survival. Glycosylation of the Fc region of α-GA1 is important for cell entry and efficacy. These data demonstrate RAN proteins drive C9-ALS/FTD in C9-BAC transgenic mice and establish a novel therapeutic approach for C9orf72 ALS/FTD and other RAN-protein diseases.
INTRODUCTION
The intronic C9orf72 GGGGCC expansion is the most common genetic cause of both amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD) (DeJesus-Hernandez et al. 2011, Renton et al. 2011). This mutation results in decreased expression of the C9orf72 protein (DeJesus-Hernandez et al. 2011), the accumulation of sense (G4C2) and antisense (G2C4) RNA foci and six dipeptide repeat (DPR) proteins, (sense: GA, GP, GR and antisense: PA, PR, GP) (Ash et al. 2013, Mori et al. 2013, Zu et al. 2013) expressed by repeat associated non-ATG (RAN) translation (Zu et al. 2011). C9orf72 protein loss of function, RNA gain of function and RAN protein toxicity have been proposed to contribute to disease (Cleary and Ranum 2017, Taylor et al. 2016, Gitler and Tsuiji 2016, Cleary et al. 2018). While RAN proteins are found in a growing number of repeat expansion diseases including C9orf72 ALS/FTD, and have been shown to be toxic when overexpressed in model systems (Cleary and Ranum 2017, Taylor et al. 2016, Kwon et al. 2014, Mizielinska et al. 2014), their role in disease and the therapeutic value of targeting them remain unclear. We and others (Jiang et al. 2016, O’Rourke et al. 2015, Peters et al. 2015, Y. Liu et al. 2016) developed BAC transgenic mouse models of C9orf72 ALS/FTD. Three of the four models, produced on C57BL/6 genetic backgrounds, develop molecular phenotypes (Jiang et al. 2016, O’Rourke et al. 2015, Peters et al. 2015) and one of them also show mild neurodegenerative and behavioral phenotypes (Jiang et al. 2016). In contrast, the C9–500 BAC mice developed on the FVB background by Liu et al. 2016 show both the molecular and neurodegenerative features of ALS/FTD (Y. Liu et al. 2016). These mice, which use the endogenous human promoters to drive expression of sense and antisense transcripts, show the accumulation of sense and antisense RNA foci and dipeptide RAN proteins and develop key phenotypic and pathological features of the disease (e.g. increased neuroinflammation, behavioral deficits, motor neuron loss, and decreased survival) (Y. Liu et al. 2016). Comparative investigations show GP levels in the Liu et al., C9–500 mice are significantly higher than other C9 BAC mouse models (unpublished data).
There is considerable interest in the development of immunotherapies for neurodegenerative diseases including Alzheimer’s disease, Parkinson’s disease and SOD1 ALS (Yu and Watts 2013, Bittar et al. 2018). To test this approach in C9orf72 ALS/FTD and to understand the role of RAN proteins in disease we targeted GA and GP proteins in C9 cells and C9–500 mice using human antibodies. Human antibodies targeting GA RAN proteins increase GA protein turnover and reduce GA RAN protein levels and aggregates. In cells, α-GA1 treatment decreased GA-induced toxicity and in C9-BAC mice, α-GA1 improved behavioral deficits, increased survival and decreased neuroinflammation and neurodegeneration.
RESULTS
Human antibodies bind RAN proteins with high affinity and selectivity
We generated human recombinant antibodies against C9 RAN proteins from libraries of memory B-cells from healthy elderly subjects (Figure 1A) (Sevigny et al., 2016). Three antibody candidates (α-GA1, α-GP1, and α-GA2) showed high affinities in low nanomolar range for GA (EC50 = 0.21 nM for α-GA1 and EC50 = 0.3 nM for α-GA2) or GP (EC50 = 0.29 nM for α-GP1) dipeptide proteins (Figure S1). α-GA1 and α-GA2 showed different binding properties. α-GA1 has 2.6 and 28-fold faster association and dissociation rates, respectively, than α-GA2 (Figure S2). Additionally, α-GA1 binding is strongly dependent on bivalent target engagement (avidity). The estimated monovalent affinity constant (determined using the α-GA1 Fab fragment) is higher (KD = 4.6 ± 0.2 μM) than the bivalent affinity constant (KD = 1.7 ± 0.1 nM) (determined using the full-length α-GA1 antibody) (Figure S2A,D). α-GA1, α-GP1, and α-GA2 are highly selective and showed no binding against protein targets found in other neurodegenerative diseases including Aβ, α-syn, tau, or TDP-43 (Figures 1B, S3A–B). Chimeric versions of α-GA1, α-GP1, and α-GA2 (chα-GA1, chα-GP1, chα-GA2) were generated containing mouse IgG2a/lambda constant regions for chronic mouse efficacy studies. High binding affinity of these antibodies to RAN proteins was maintained in conditions mimicking the cell environment (e.g. cell lysates and different temperatures and pH) (Figures 1C, S3C).
Figure 1. Human antibodies show high binding affinities and engage C9 RAN targets in vitro and in vivo.
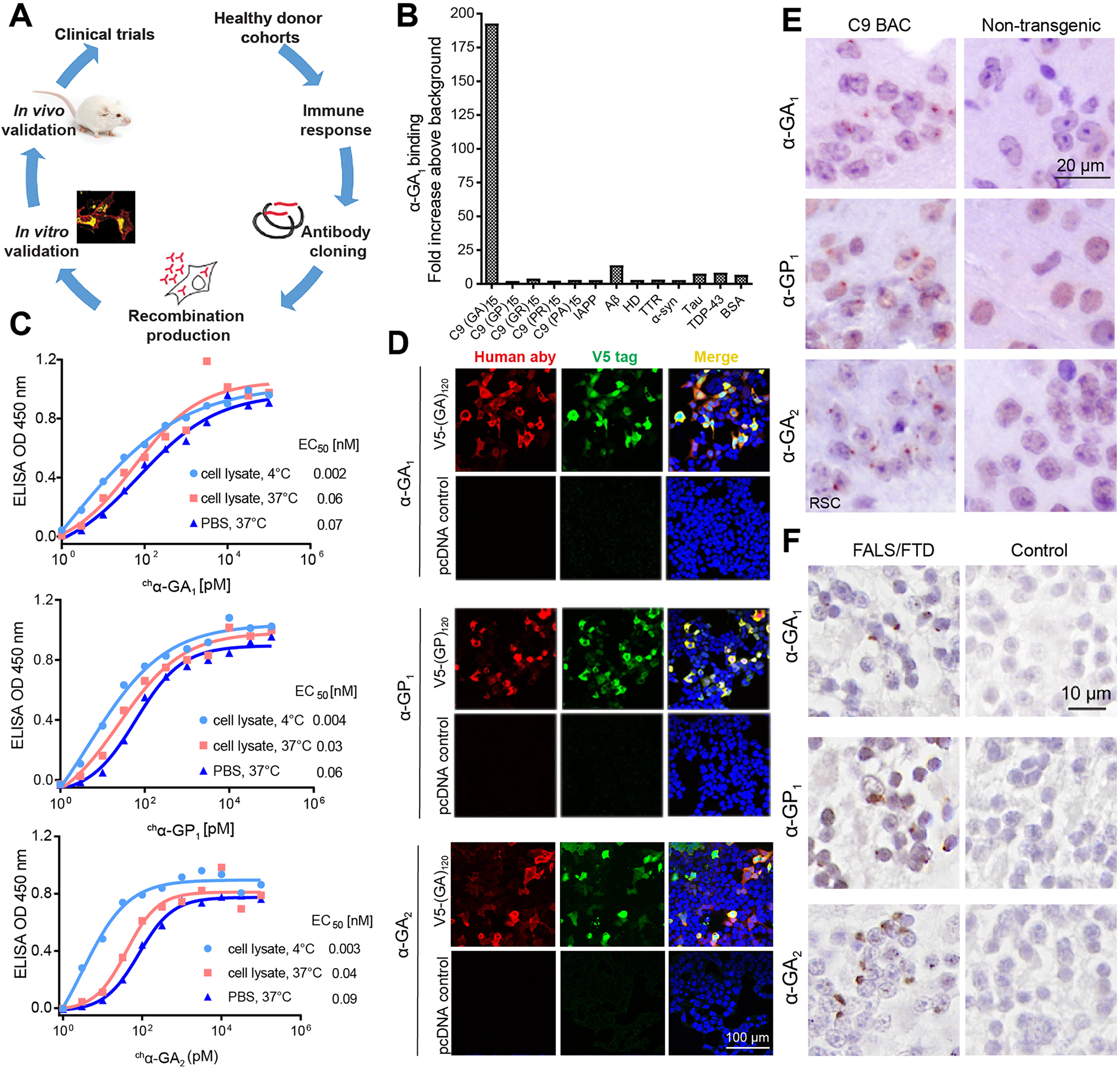
(A) Schematic diagram of human antibody generation, characterization and efficacy study. (B) Selectivity screening of α-GA1 against C9 DPRs and other proteins that contribute to other neurologic diseases. (C) ELISA binding for chα-GA1, chα-GP 1and chα-GA2 to (GA)15 or (GP)15 RAN peptides in SH-SY5Y cell lysate at 4 °C (light blue circle) or 37 °C (red square) or in PBS at 37 °C (dark blue triangle). Values presented as mean of duplicates. (D) Double labeling with IF showing that the human antibodies (a-human, red) and V5-epitope tagged protein (a-V5, green) GA or GP proteins co-localize. (E) IHC staining with α-GA1, α-GP1, or α-GA2 of the retrosplenial cortex (RSC) from 18-month-old C9 and non-transgenic (NT) mice. (F) IHC staining with α-GA1, α-GP1, or α-GA2 of human cerebellar tissue from a C9 expansion carrier and non-C9 control. See also Figures S1–S4.
Immunofluorescence (IF) and immunohistochemistry (IHC) experiments showed that α-GA1, α-GP1, and α-GA2 also recognize GA and GP repeat containing proteins expressed in HEK293T cells and C9 mouse and patient brains. In transfected cells, α-GA1, α-GA2, and α-GP1 staining co-localized with V5-tagged GA or GP proteins, respectively (Figures 1D, S4A–C). No or minimal staining by α-GA1, α-GP1, or α-GA2 was observed in cells overexpressing other C9 RAN proteins (Figure S4A–C). On protein blots of cell lysates expressing GA expansion proteins α-GA1 preferentially bound to GA proteins with longer repeat tracts (60 and 120 repeats), while α-GA2 recognized GA proteins with 30, 60 and 120 repeats (Figure S4D–F), further highlighting the different binding properties of α-GA1 and α-GA2. Characterization of the monovalent affinity and kinetics of α-GA1 and α-GA2 (Figure S2D–E) suggest that the selectivity of α-GA1 for longer GA repeats is likely a result strong bivalent engagement (avidity) (Figure S2A, D). In C9-BAC mice and human autopsy tissue, IHC using the α-GA1, α-GP1, and α-GA2 showed aggregate staining similar to previously reported RAN protein aggregates (Y. Liu et al. 2016, Zu et al. 2013) (Figure 1E, F).
In summary we generated high affinity human antibodies and chimeric derivatives that recognize GA or GP C9 RAN proteins with low nanomolar affinities and high selectivity. These antibodies recognize RAN targets in cells and in C9 mouse and human brains with α-GA1 showing preferential binding to longer GA repeat tracts.
chα-GA1 reduces GFP-GA levels in TRIM21-dependent manner and GFP-GA aggregates in transfected cells
Next, we tested if the human antibodies can reduce RAN proteins in cells. We focused on GA since GA has been reported to be more toxic than GP (Guo et al. 2018, Khosravi et al. 2017, May et al. 2014, Zhang et al. 2014a, Zhang et al. 2016, Freibaum et al. 2015, Lee et al. 2016). HEK293T cells transfected with non-hairpin-forming alternative codon constructs to overexpress ATG-initiated GFP-(GA)60 proteins (GFP-alt(GA)60) were treated with chα-GA1, chα-GP1, chα-GA2 or IgG isotype control antibodies. Protein blots show chα-GA1 treatment resulted in ~50% reduction of GFP-alt(GA)60 (p = 0.021) while no changes were observed in cells treated with chα-GP1 or IgG (Figure 2A). Surprisingly, chα-GA2 showed no effects on the levels of GFP-GA proteins, suggesting chα-GA1 and chα-GA2 differ in their effects on their GA target proteins at the 100 nM antibody concentration used in this overexpression system.
Figure 2. α-GA1 treatment reduces GFP-GA protein levels and increases GFP-GA turnover in cells.
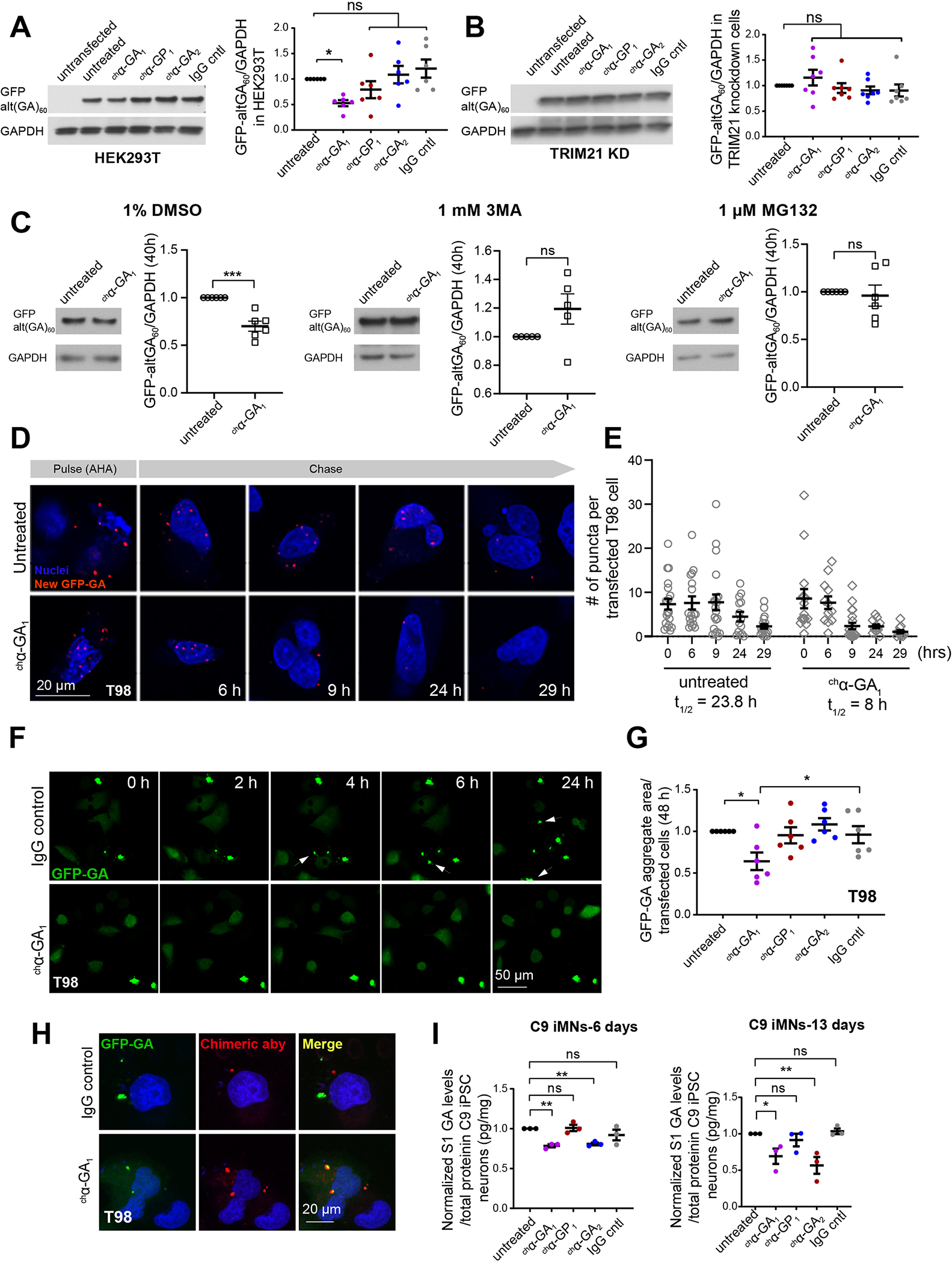
(A, B) Protein blot showing GFP-GA levels with and without antibody treatment (100 nM, 48 h) in HEK293T (A) and TRIM21 knockdown (KD) (TRIM21+/−) cells (B) (n ≥ 6, one-way ANOVA with Benjamin analyses for multiple comparisons). (C) GFP-GA levels in HEK293T cells after co-treatment with chα-GA1 (100 nM) plus DMSO, the autophagy inhibitor 3-methyl adenine (3MA), or the proteasome inhibitor MG132 (n ≥ 5, unpaired two-tailed t-test). (D) Pulse-chase experiment showing fluorescence images of azidohomoalanine (AHA) labeled GFP-GA (red puncta) and remaining label at various post-label chase times in T98 cells with and without chα-GA1 (100 nM) treatment. (E) Quantitative measurements of AHA-labeled GFP-GA signal in T98 cells with and without chα-GA1 treatment. (F) Live cell imaging of GFP-(GA)60 transfected T98 cells treated with IgG control or chα-GA1 (60 nM). Arrows highlight newly formed aggregates in individual frames. (G) GFP-GA aggregate quantification at 48h (n = 6, one-way ANOVA with Holm-Sidak analyses for multiple comparisons). (H) IF staining showing cellular uptake and target engagement of chα-GA1 (red) in T98 cells transfected with GFP-alt(GA)60 (green). (I) Quantitative measurements of polyGA in iPSC neurons using MSD (n = 3/group, one-way ANOVA with Sidak analyses for multiple comparisons). Graphs show mean ± SEM. ns p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. See also Figures S5 and S6.
To understand the mechanisms involved in chα-GA1 mediated protein clearance we tested the effects of the cytosolic Fc receptor tripartite motif 21 (TRIM21) pathway. TRIM21 links antigens recognized by Fc-mediated antibody recognition to the ubiquitin, proteasome and autophagy clearance mechanisms (Kimura et al. 2015, McEwan et al. 2013). We generated heterozygous knockout TRIM21 HEK293T cells (TRIM21+/−) using CRISPR/Cas9 (Figure S5A). In contrast to the results from TRIM21+/+ cells (Figure 2A), GFP-alt(GA)60 levels in TRIM21+/− cells were unchanged following chα-GA1 treatment (100 nM) compared to IgG treated cells, suggesting TRIM21 is required for GFP-alt(GA)60 clearance (Figure 2B). qRT-PCR showed no change in transcript levels (Figure S5B). To test if the GFP-(GA)60 clearance by the chα-GA1 antibody requires protein clearance pathways, we co-treated cells with chα-GA1 and DMSO, with or without the autophagy inhibitor 3-methyladenine (3MA), or the proteasome inhibitor MG132. In contrast to the results above and in cells treated with chα-GA1 and DMSO (Figures 2A, 2C), no change in GFP-(GA)60 levels were found when the autophagy or proteasome pathways were inhibited (Figure 2C). These data suggest clearance of GFP-(GA)60 by chα-GA1 is dependent on proteasome and autophagy mechanisms.
To test the effects of chα-GA1 on GA protein turnover, we performed a pulse-chase experiment using L-azidohomoalanine (AHA) (Dieterich et al., 2010; tom Dieck et al., 2015) to determine the turnover of newly synthesized GFP-(GA)60 using the proximity ligation assay (Figure 2D). The half-life (t1/2) of over-expressed GFP-(GA)60 in T98 cells is 23.8 h while chα-GA1 treated cells showed much faster clearance of GFP-(GA)60 (t1/2 = 8 h) (Figures 2D, 2E, S5C). Live cell imaging of T98 neuroblastoma cells expressing GFP-alt(GA)60 showed GFP-alt(GA)60 aggregates increased in number over time in IgG treated cells. In contrast in chα-GA1 treated cells GFP-GA remained diffuse and showed fewer aggregates (Figure 2F). At 48 hours, GA aggregates were reduced to 64% (p = 0.027) in with chα-GA1 treatment while chα-GP1, chα-GA2, and IgG showed no effects (Figure 2G). Co-staining of GFP-GA and chα-GA1 confirm target engagement in chα-GA1-treated cells. As expected, no co-localization of the IgG isotype control with GFP-GA was detected (Figure 2H).
chα-GA1 and chα-GA2 reduces C9 RAN proteins in C9 iPSC motor neurons
To examine antibody effects on RAN protein clearance in human neurons, we treated C9 patient derived iPSC motor neurons (iMNs) (Figure S6A–B) (Zhang et al. 2015) with α-GA1, α-GP1, α-GA2 or IgG control antibodies. Both the α-GA1 and α-GA2 but not the α-GP1 or IgG treatments reduced GA levels (Figure 2I). α-GA1 and α-GA2 treatment resulted in 21% (p = 0.002) and 16% (p =0.004) reductions in GA at day 6 and 31% (p = 0.025) and 43% (p = 0.004) at day 13, respectively (Figure 2I). In contrast to the results in GFP-alt(GA)60 overexpressing cells, both α-GA1 and α-GA2 treatment of patient derived iMNs resulted in a reduction of GA levels. These data suggest that the potency of α-GA1 is higher than that of α-GA2 and α-GA1 thus having beneficial effects in overexpression systems which express high poly(GA) levels.
Cellular fractionation further confirms that the α-GA1 and α-GA2 antibodies engage their GA protein target. GA levels were measured in protein samples obtained after a fractionation protocol in which protein lysates (S1) from iMNs were further fractionated by ultracentrifugation to generate soluble (S2) and pelleted (P2) fractions (Figure S6C). Nearly all of the GA protein from IgG and α-GP1 treated iMNs remained in the soluble S2 fraction (Figure S6D–E). In contrast, α-GA1 or α-GA2 treatment shifted the distribution of polyGA to the P2 fraction (Figure S6D–E). The observed decrease in S2 GA levels, which is accompanied by a corresponding increase in P2 GA levels in the anti-GA treatment groups suggests protein-antibody complexes shift the complexed GA proteins from the soluble S2 to the P2 fraction after high-speed centrifugation.
chα-GA1 interacts with TRIM21 in cells
To test if the chα-GA1 antibody interacts with TRIM21 in cells, we performed a proximity ligation assay (tom Dieck et al. 2015) on GFP-(GA)60 overexpressing cells treated with chα-GA1 (Figure 3A). In this assay, close proximity or interaction of TRIM21 and the treatment antibody allows the hybridization of probes conjugated with two secondary antibodies (~ 30–40 nm apart), resulting in the PCR amplification of ligated DNA templates (Figure 3A). A strong ligation signal was observed only in cells probed with α-TRIM21 antibody, indicating chα-GA1 and the TRIM21 protein are in close proximity or interact with each other (Figures 3B and S7). These data are consistent with previously studies showing TRIM21 binds to the Fc region of antibodies (Foss et al. 2015, Fletcher et al. 2015).
Figure 3. chα-GA1 reduces GA sequestration and improves cellular function.
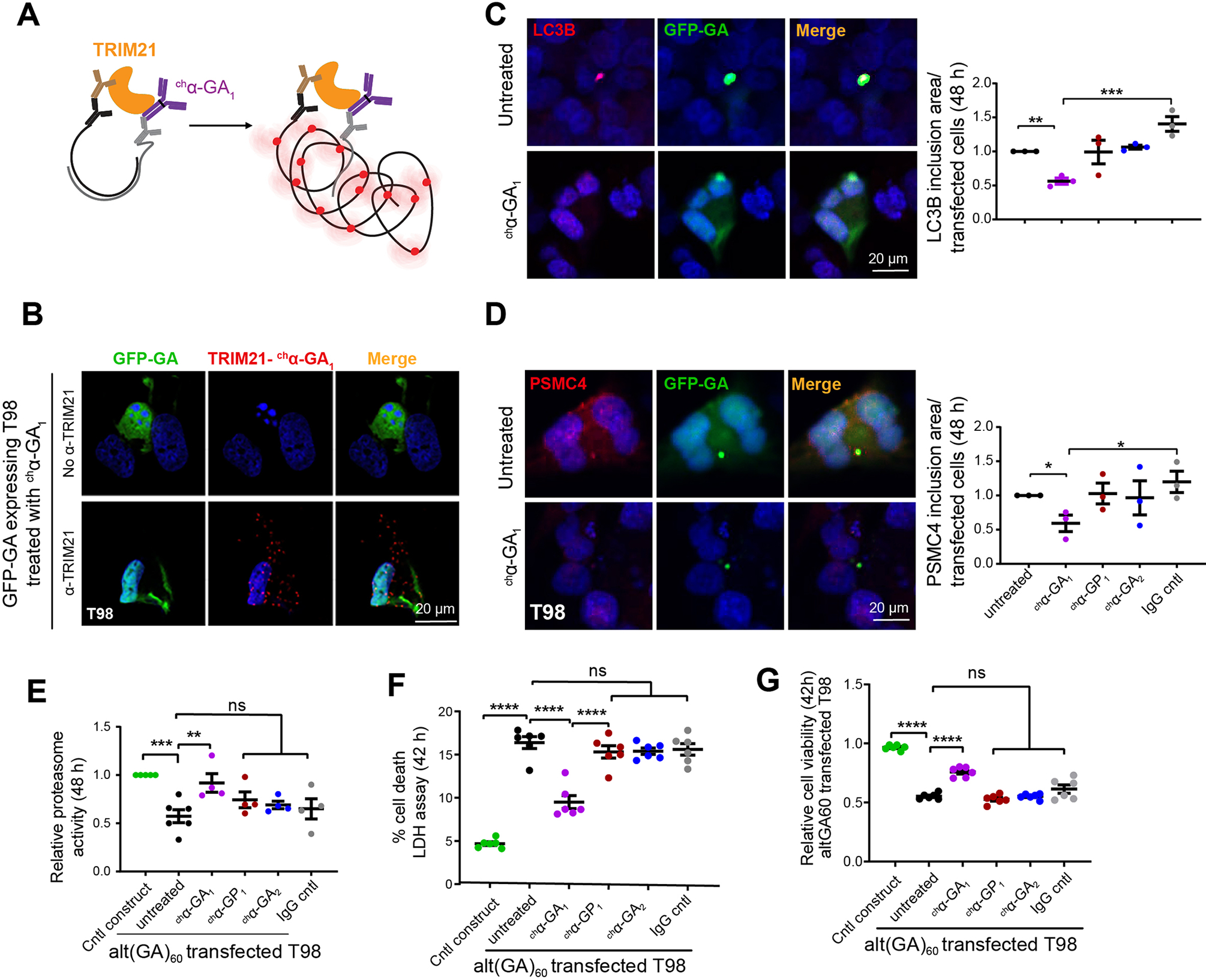
(A) Schematic diagram of the proximal ligation assay used to test TRIM21 and chα-GA1 interaction, α-TRIM21 (brown), secondary antibody for TRIM21 (black), secondary antibody for treated antibody (gray), amplified signals (red). (B) Double IF images of T98 cells expressing GFP-(GA)60 proteins show positive amplification signal (red puncta) showing TRIM21 and chα-GA1 interact or are in close proximity detected by proximity ligation assays. (C, D) Co-localization of GFP-GA with the autophagy marker LC3B (C) or the 26S proteasome subunit (D) in transfected T98 cells with or without antibodies (100 nM, 48 h) and quantification of LC3B (C) and 26S proteasome subunit (D) inclusions (n = 3, one-way ANOVA with Holm-Sidak analyses for multiple comparisons). (E) Proteasome activity in alt(GA)60 transfected T98 cells with or without chα-GA1 treatment (100 nM, 48h) (n ≥ 4, one-way ANOVA with Dunnett analyses for multiple comparisons). (F) Cell toxicity measured by LDH levels in cells transfected with alt(GA)60 with chα-GA1 treatment (1 μM, 42h), (n = 6). (G) Cell viability measured using MTT assays in cells overexpressing alt(GA)60 with chα-GA1 treatment (1 μM, 42h, n = 6). Graphs show mean ± SEM. One-way ANOVA with Sidak analyses for multiple comparisons. ns p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. See also Figures S7–S9.
chα-GA1 decreases sequestration of proteasome and autophagy proteins to GA aggregates and improves proteasome function in cells
Previous studies indicate autophagy and proteasome abnormalities contribute to C9orf72 ALS/FTD (Mann et al. 2013, May et al. 2014, Schludi et al. 2015) and additional studies show GA aggregates trap 26S proteasome complexes (Guo et al. 2018, Zhang et al. 2014b). To test if antibody treatment normalizes the cellular distribution of autophagy and proteasome markers that co-localize with GA aggregates, we performed co-staining of GFP-alt(GA)60 with the LC3B autophagy or PSMC4, a 26S proteasome marker. Consistent with previous studies, LC3B and PSMC4 co-localize with GFP-alt(GA)60 aggregates in transfected T98 cells (Figures 3C, D, S8). Treatment with chα-GA1 reduced LC3B inclusion to 47% (puntreated = 0.008, pIgG = 0.0004) (Figure 3C, S9A) and PSMC4 inclusions to 59% (puntreated = 0.044, pIgG = 0.018) (Figure 3D, S9B). Fluorescence-based proteasome function assays (Zhang et al. 2014a) showed overexpression of alt(GA)60 in T98 cells reduced proteasome activity by 44% (p = 0.0006) (Figure 3E). chα-GA1 treatment (100 nM, 48 h) increased proteasome activity by 54% in alt(GA)60 transfected cells (p = 0.0086) while chα-GP1, chα-GA2 and IgG antibodies showed no similar effects (Figure 3E).
In summary, chα-GA1 interrupts the co-aggregation of polyGA with proteins important for protein clearance (LC3B, proteasome 26S subunit) and rescues proteasome activity in polyGA overexpressing cells.
α-GA1 antibody reduces toxicity in cells
To test if α-GA1 treatment can impact cell death and viability we performed lactate dehydrogenase (LDH) and methylthiazol tetrazolium (MTT) assays in GA expressing cells. T98 cells expressing GFP-alt(GA)60 showed increased cell death (p < 0.0001) (Figure 3F) and decreased cell viability (p < 0.0001) (Figure 3G) compared with cells transfected with the control construct. Treatment with chα-GA1 decreased cell death by 41% (p < 0.0001) (Figure 3F) and increased cell viability by 50% (p = 0.0001) (Figure 3G). No rescue in cell death or viability was found in GA expressing cells treated with chα-GP1, chα-GA2 or IgG. In summary, chα-GA1 treatments reduced the toxicity associated with GA expansion proteins.
Human antibodies target in vivo RAN proteins in C9 BAC mice
To examine the effects of α-GA1, α-GA2 and α-GP1 in our C9-BAC ALS/FTD mouse model (C9–500 line) (Y. Liu et al. 2016), we first tested if peripherally [intraperitoneally (i.p.)] delivered human antibodies target the RAN protein aggregates in vivo (Figure 4A). Our data show that i.p. injected α-GA1, α-GP1, and α-GA2 human antibodies cross the blood-brain barrier (BBB) and localize as perinuclear aggregates in C9 mouse brains 10 days after a single injection with no similar staining found in IgG treated C9 animals (Figure 4B). Double labeling of the retrosplenial cortex (RSC) from α-GA2 injected C9 BAC mice show colocalization of the i.p. injected α-GA2 antibody (detected with α-human antibody) and GA aggregates stained with a previously reported mouse monoclonal GA 27B11A7 antibody (Y. Liu et al. 2016) (Figure 4C). No similar staining was found in IgG injected control animals.
Figure 4. Human antibodies engage in vivo targets in C9 BAC mice.
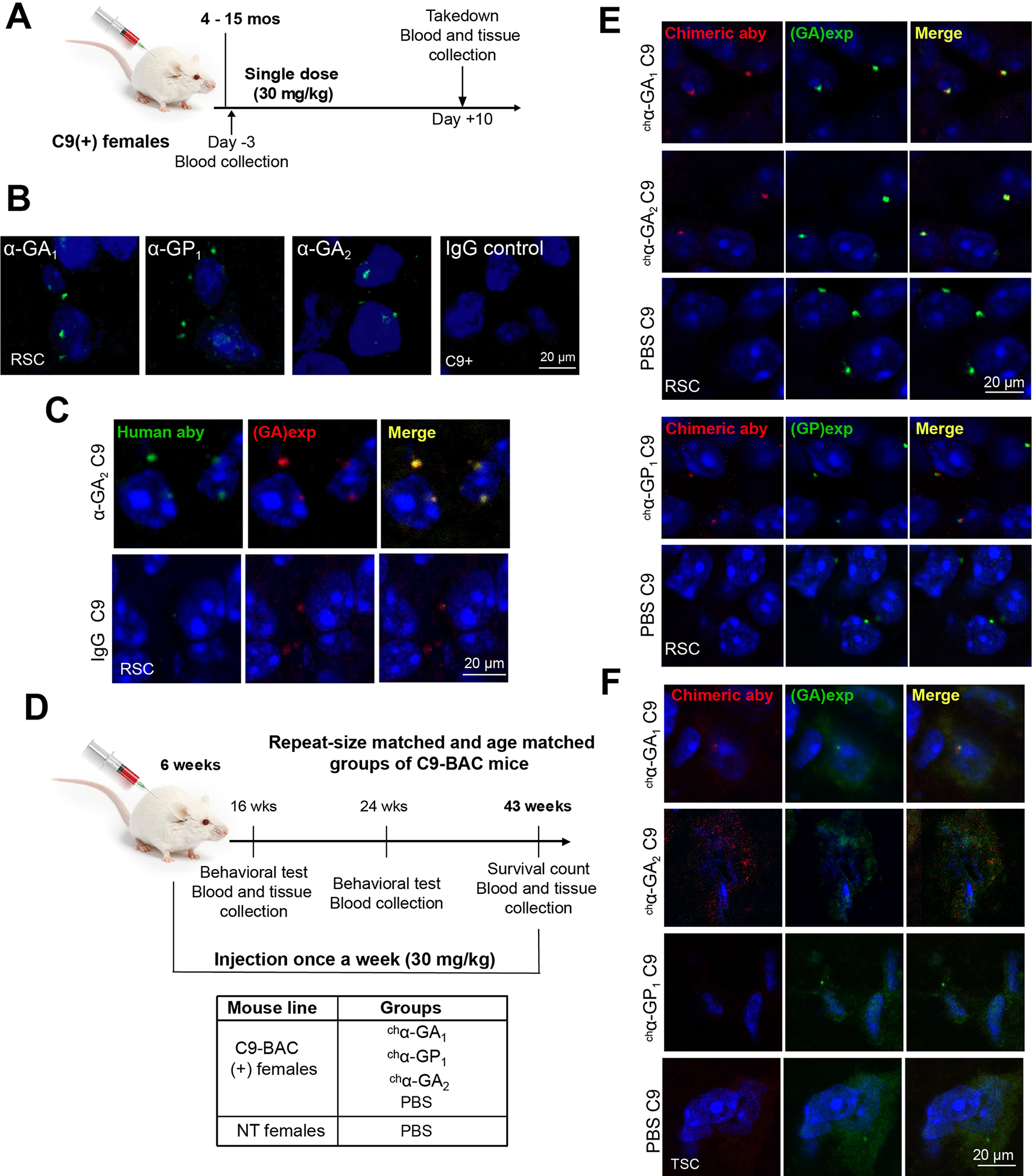
(A) Experimental design of target engagement study. (B) IF of human antibodies in the RSC 10 days after a single intraperitoneal injection. (C) Double IF staining of α-GA2 and GA aggregates 10 days after single injection. (D) Experimental design of a chronic treatment of antibodies in C9 BAC mice. (E, F) Double IF staining of the retrosplenial cortex (RSC) (E) or thoracic spinal cord (TSC) (F) from mice after chronic treatment with chimeric human antibodies chα-GA1, chα-GP1 or chα-GA2 (detected with α-human IgG, red) and GA or GP aggregates detected with human α-GA1 or α-GP1 antibodies (green). See also Figure S10.
Reduction of C9 RAN proteins in C9 BAC mice treated with GA targeting antibodies
To examine the impact of antibody treatment on the levels of C9 RAN proteins in C9 mice, we performed chronic treatment with chα-GA1, chα-GP1, chα-GA2 or PBS in cohorts of repeat-length and age-matched female C9-BAC mice (Y. Liu et al. 2016) (Figure S10A and STAR methods). Four cohorts of repeat-length matched female C9-BAC mice, containing 500 to 750 G4C2 repeats, (Figure 4D) were i.p. injected weekly with chα-GA1, chα-GP1, chα-GA2 (30 mg/kg) or PBS from 6 to 43 weeks of age. IF staining demonstrating GA aggregates in the retrosplenial cortex in 6-week old C9-BAC mice indicating that the molecular features of disease pathology are already evident in young C9-BAC mice at the time of treatment initiation (Figure S10B). An additional control group of non-transgenic (NT) mice were subjected to weekly injections of PBS (Figure 4D). To avoid mouse anti-human antibody responses, murine IgG2a chimeric derivatives of the human antibodies were used. The effects of antibodies on dipeptide protein levels were assessed using tissue from groups of repeat-size matched animals that were randomly selected for takedown at 16 and 43 weeks of age.
Similar to the results observed after a single i.p. injection, perinuclear aggregate signals were observed with murine IgG2a chimeric derivatives of the human antibodies (chα-GA1, chα-GP1, and chα-GA2) after weekly injections of C9 mice of over 37 weeks (Figure 4E, 4F). Double labeling of the RSC shows that chα-GA1, chα-GP1, and chα-GA2 co-localize to the perinuclear, intracellular GA or GP aggregates (Figure 4E). Similar to the in vivo target engagement data above, these results further demonstrate that the chimeric treatment antibodies against C9-RAN proteins engage their intracellular RAN protein targets in C9-BAC mice. To further characterize the molecular interactions of the GA targeting antibodies we performed Meso Scale Discovery (MSD) pulldown experiments. MSD plates coated with (GA)8 peptide or rabbit α-GA antibody showed antibody signal after exposure to brain lysates from mice treated with chα-GA1 or chα-GA2 (Figure S10B–C). These pulldown experiments show that antibodies isolated from treated mice bind to GA peptides (Figure S10C). Additionally, MSD plates coated with α-rabbit GA antibodies pull down complexes containing GA-chα-GA1 and GA-chα-GA2 (Figure S10D). Additionally, double IF staining performed on thoracic spinal sections showed that chα-GA1 and chα-GA2 co-localize with GA aggregates while no similar staining observed in the sections from chα-GP1 or PBS treated mice (Figure 4F). Taken together, these data show that IP injected chimeric human antibodies engaged their protein targets in both the brain and the spinal cord.
Compared to PBS treated C9 animals, GA aggregates detected with mAb GA 27B11A7 (Y. Liu et al. 2016) or the human antibody α-GA1 in the chα-GA1 treatment group were reduced to ~52% (16 weeks, p= 0.006,) and 45% (43 weeks, p < 0.0001,) in the retrosplenial cortex (RSC) and to 53% (43 weeks, p = 0.007) in the posterior lumbar spinal cord (Figure 5A, S11A–B). Treatment with chα-GA2 showed similar reductions at 16 weeks but less robust effects in older animals with ~71% of GA aggregates remaining in the RSC at 43 weeks (p = 0.012) (Figure 5A). An additional control experiment shows that pretreatment of RSC tissue from C9-BAC mice with the chimeric chα-GA1 antibody does not interfere with GA aggregate detection using the human α-GA1 antibody (Figure S11C), ruling out the possibility that the reductions seen in GA aggregate signal might be caused by epitope masking by the treatment antibodies.
Figure 5. chα-GA1 treatment reduces RAN levels in C9 BAC mice.
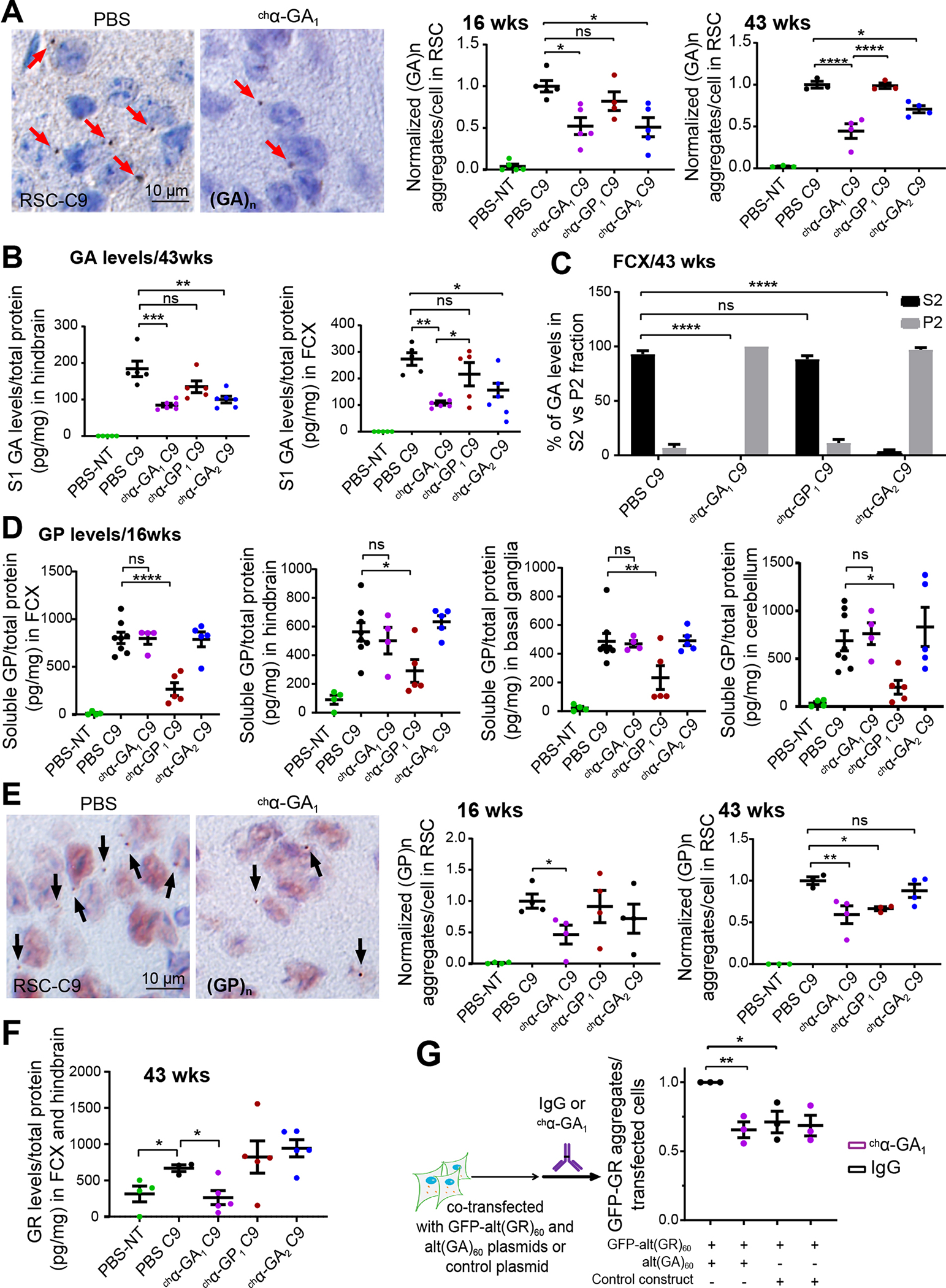
(A) GA aggregate quantification detected by mouse monoclonal GA or human α-GA1 antibodies at 16 (n ≥ 4/group) and 43 wks (n ≥ 3/group). (B) Quantitation of polyGA in S1 protein fractions from treated and untreated animals using MSD (n ≥ 5/group). (C) Percent GA protein in S2 and P2 fractions after ultra-centrifugation (100,000 x g). (D) GP protein levels detected by MSD using rabbit polyclonal α-GP antibodies in frontal cortex (FCX), hindbrain (HB), basal ganglia (BG), and cerebellum (CB) from treated animals at 16 wks (n ≥ 4/group). (E) Quantification of GP aggregates detected with human α-GP1 at 16 (n=4/group) and 43 wks (n ≥ 3/group), unpaired two-tailed t-test for 16 wk timepoint. (F) GR levels detected by MSD using rabbit polyclonal α-GR in PBS NT and C9 mice after chronic antibody treatment (43 wks, n ≥ 3, one-way ANOVA with Holm-Sidak analyses for multiple comparison). (G) GA-dependent reduction of GR aggregates after treatment with α-GA1 in cells transfected with alt(GA)60 and alt(GR)60 (n = 3). Graphs show mean ± SEM. One-way ANOVA with Sidak analyses for multiple comparisons. ns p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. See also Figures S11–S13.
Total GA levels were also measured biochemically by Meso Scale Discovery (MSD) in protein samples prepared from hindbrain (HB) and frontal cortex (FCX) that were pretreated with SDS/TCEP to denature and eliminate potential interference from the treatment antibodies (See STAR methods, Figure S11D–E). These experiments show GA levels in HB and FCX were reduced to ~ 50% in chα-GA1 (pHB = 0.0009, pFCX = 0.002) and chα-GA2 (pHB = 0.008 , pFCX = 0.012) treated mice at 43 weeks compared to PBS treated C9 mice (Figure 5B). Similar to the iMN data, treatment with chα-GA1 or chα-GA2 also resulted in the shift of GA protein from S2 soluble to P2 pellet fraction after high-speed centrifugation (Figure 5C, S11F).
Biochemical quantification by MSD assay revealed reductions of GP in S1 lysates from the chα-GP1 but not the GA targeting antibody groups in FCX (p = 0.0001), HB (p = 0.019), basal ganglia (BG) (p = 0.008), and cerebellum (CB) (p= 0.025) at 16 weeks (Figure 5D). A titration experiment shows that chα-GP1 incubated with brain lysates from C9 positive mice did not interfere with the GP signal (Figure S11G) in the GP MSD assay using rabbit polyclonal α-GP antibodies (Y. Liu et al. 2016). Similarly, treatment of the C9 mice with chα-GP1 lowered GP aggregate load to 62% compared to PBS controls at 43 weeks (p=0.046), but not at 16 weeks of age (Figure 5E, right panel). Although reductions in GP levels were not observed in S1 fractions from the chα-GA1 antibody group at 16 and 43 weeks (Figure 5D, S11H), unexpectedly, GP aggregates detected by IHC were reduced to 61% and 59% at 16 (p = 0.019) and 43 (p = 0.015) weeks, respectively, following chα-GA1 treatment (Figure 5E). Additionally, GR levels were also reduced in S1 lysates from chα-GA1 (p = 0.037) but not chα-GP1 or chα-GA2 treated animals compared to the PBS-C9 treatment group (Figure 5F). RT-PCR and FISH controls showed no changes in levels of sense or antisense transcripts or RNA foci in any of the treatment cohorts (Figure S12), indicating that the changes in RAN proteins (GA, GP and GR) observed in chα-GA1 treated mice were not caused by changes in RNA levels.
In summary, chronic treatment of the chimeric human antibodies chα-GA1, chα-GP1, or chα-GA2 in C9 BAC mice resulted in reduction of their RAN targets. Between the two GA-targeting antibodies tested, chα-GA1 is more potent in clearing GA aggregates compared to chα-GA2.
chα-GA1 antibody reduces GR aggregates in cells in a GA-dependent manner
Based on the unexpected finding that chα-GA1 treated mice reduced levels of GA as well as GP aggregates and GR protein, and previous studies that GA/GR can form complexes (Yang et al. 2015, Zhou et al. 2017), we tested the hypothesis that the α-GA1 antibody reduces aggregates of other C9-RAN proteins in a GA protein-dependent manner. Because GFP-alt(GR)60 but not GFP-alt(GP)60 form protein aggregates in T98 cells under our culture conditions, we tested if targeting the GA protein with the chα-GA1 antibody reduces GR aggregates. Cells co-transfected with GFP-alt(GR)60 and alt(GA)60 or control vector were treated with chα-GA1 or IgG control (60 nM) (Figure 5G). At 24 hrs, cells expressing both GR and GA showed a 30% increase (p = 0.012) in the number of GR positive aggregates compared to cells expressing GR alone. Consistent with previous studies (Zhou et al. 2017, Yang et al. 2015), these data indicate that the presence of GA proteins increase the formation of GR aggregates (Figure 5G, S13A). Similar to our in vivo results, targeting the GA protein with the chα-GA1 antibody reduced GR protein aggregates in T98 cells (p = 0.008). In contrast, in cells expressing GR alone, chα-GA1 showed no effect on GR aggregate number compared to IgG (Figure 5G, S13A). Furthermore, no changes in GFP-GR protein levels were observed between cell treatment groups (Figure S13B) indicating that changes in aggregate number upon chα-GA1 treatment cannot be explained by changes in GR protein levels, at least during this short (24 hour) treatment period. Taken together, these data demonstrate that the presence of the GA protein favors the formation of GR aggregates and that GR aggregates can be reduced by targeting GA protein in GA/GR expressing cells.
Reduction of inclusions of proteins involved in proteasome and autophagy in C9 BAC mice treated with chα-GA1
To test if chα-GA1 treatment also affects the GA aggresome in C9 BAC mice, we performed IF of p62, LC3B and the PSMC4 26S proteasomal subunit. Similar to the p62 positive GA inclusions previously reported (Mori et al. 2013, May et al. 2014), double labeling of tissue from PBS treated C9 animals (43 wks) (Figure 6A) with GA and p62 antibodies showed co-localization of p62 and GA aggregates in C9 BAC mice. In chα-GA1 treated C9 animals the p62 signal was reduced to 33% (p = 0.003) (Figure 6A). Additionally, double IF staining showed a subset of LC3B or PSMC4 signals co-localize with GA aggregates in PBS treated C9 BAC mice (Figure 6B, S14). Quantification showed an increased number of LC3B inclusions in PBS C9 mice compared with PBS treated NT controls (Figure 6C). These data, combined with the increased number of p62 inclusions in C9 animals, suggest proteasome and autophagy pathways are impaired in C9 BAC mice. Treatment of chα-GA1 significantly reduced co-localization of GA aggregates and LC3B (p = 0.005) and LC3B inclusions (p = 0.0009) compared with PBS treated C9 mice (Figure 6C).
Figure. 6. chα-GA1 treatment decreases GA aggresome and improves gait behaviors in C9 mice.
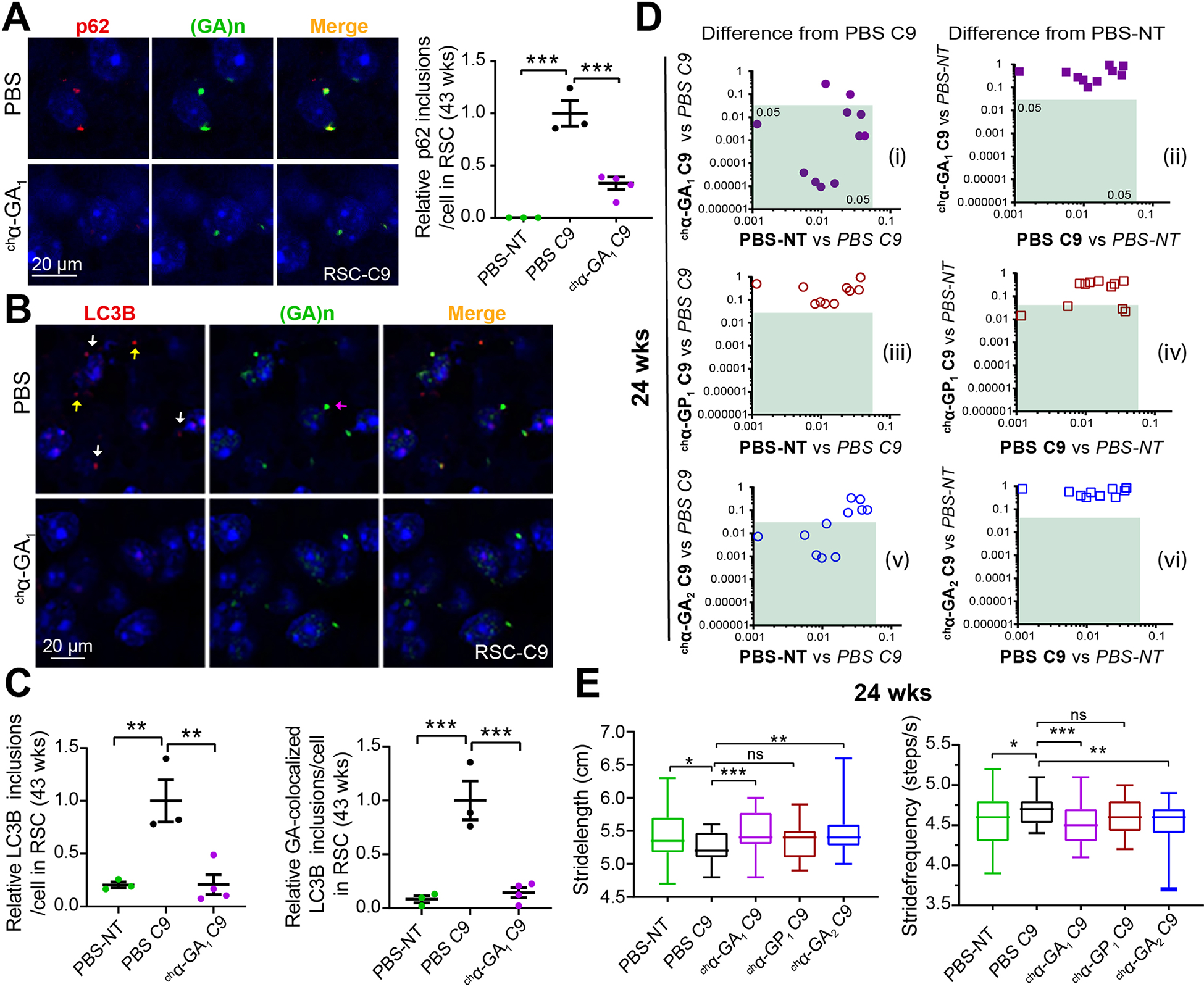
(A) Double IF images showing co-localization of p62 (a-p62, red) and GA aggregates (α-GA1, green) and quantification of p62 inclusion in PBS or chα-GA1 treated mice. (B) Double IF images showing a subset of LC3B inclusions (a-LC3B, red) and GA aggregates (α-GA1, green) co-localize (white arrows), non-colocalized LC3B (yellow arrows), and non-colocalized GA aggregates (purple arrow) and reductions in LC3B staining after chα-GA1 treatment (C) Quantification of double-positive LC3B/GA inclusions in PBS and chα-GA1 treated mice. (A, C) Graphs show mean ±SEM, n ≥ 3, one-way ANOVA with Sidak analyses for multiple comparisons. (D) Comparisons of 11 C9 relevant DigiGait parameters among antibody treatment groups compared to PBS-C9 or PBS-NT cohorts at 24 wks. Green boxes define regions of significant difference found in antibody treatment cohorts compared to PBS treated C9 or PBS treated NT controls (p ≤ 0.05), n ≥ 17/group. (E) Representative DigiGait parameters corrected in treated chα-GA1 C9 cohort at 24 wks. Data show min to max, interquartile, and median. One-way ANOVA with Holm-Sidak analyses for multiple comparison. ns p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. See also Figures S14 and S15.
Improved behavioral deficits and survival in chα-GA1 treated C9-BAC animals
To examine the effects of antibody treatment on behavioral phenotypes of C9 mice, we performed longitudinal DigiGait and open field assessments on mouse cohorts at 16, 24 and 32 weeks (Figure 4D, 6D–E, 7A–C). Cohorts of mice for these studies were randomly selected prior to the initiation of treatment and before the development of overt phenotypes. Consistent with previous observations (Y. Liu et al. 2016), PBS treated C9 animals showed pronounced gait abnormalities at both 16 and 24 weeks (Figure 6D, S15). Weekly i.p. treatment with chα-GA1 and chα-GA2 resulted in significant improvements in gait at both ages with fewer abnormal parameters at 24 versus 16 weeks of age (Figure 6D, E, S15A–C). Of the 11 DigiGait parameters that differed between the PBS-C9 and PBS-NT groups at 24 weeks, the chα-GA1 and chα-GA2 treatment groups showed improved performance in 9 and 6 of these parameters, respectively, (Figure 6D, left) and were indistinguishable from PBS-NT mice in all parameters (Figures 6D, right, S15D). In contrast, chα-GP1 treated animals showed no improvement in any of the 11 parameters (Figure 6D, left) and displayed 4 abnormal parameters compared to the PBS-NT control group (Figures 6D, right, S15D). Examples of two specific parameters, stride frequency and stride length are shown in Figure 6E. These data show that antibodies chα-GA1 and chα-GA2, but not chα-GP1 can rescue gait abnormalities in C9-BAC mice.
Open field assessment was performed at 32 weeks to determine if antibody treatment can impact anxiety-like behavior (Y. Liu et al. 2016). Similar to the improvements seen in gait, the chα-GA1 treatment group also showed significant improvements in anxiety-like behavior with increased center travel distance compared to PBS treated C9 animals, p = 0.033. In contrast, the chα-GP1 and chα-GA2 treated groups showed no similar improvement (Figure 7A–B).
Figure 7. Improvements in open-field, survival, and neurodegeneration in C9 mice treated with GA targeting chα-GA1 antibody.
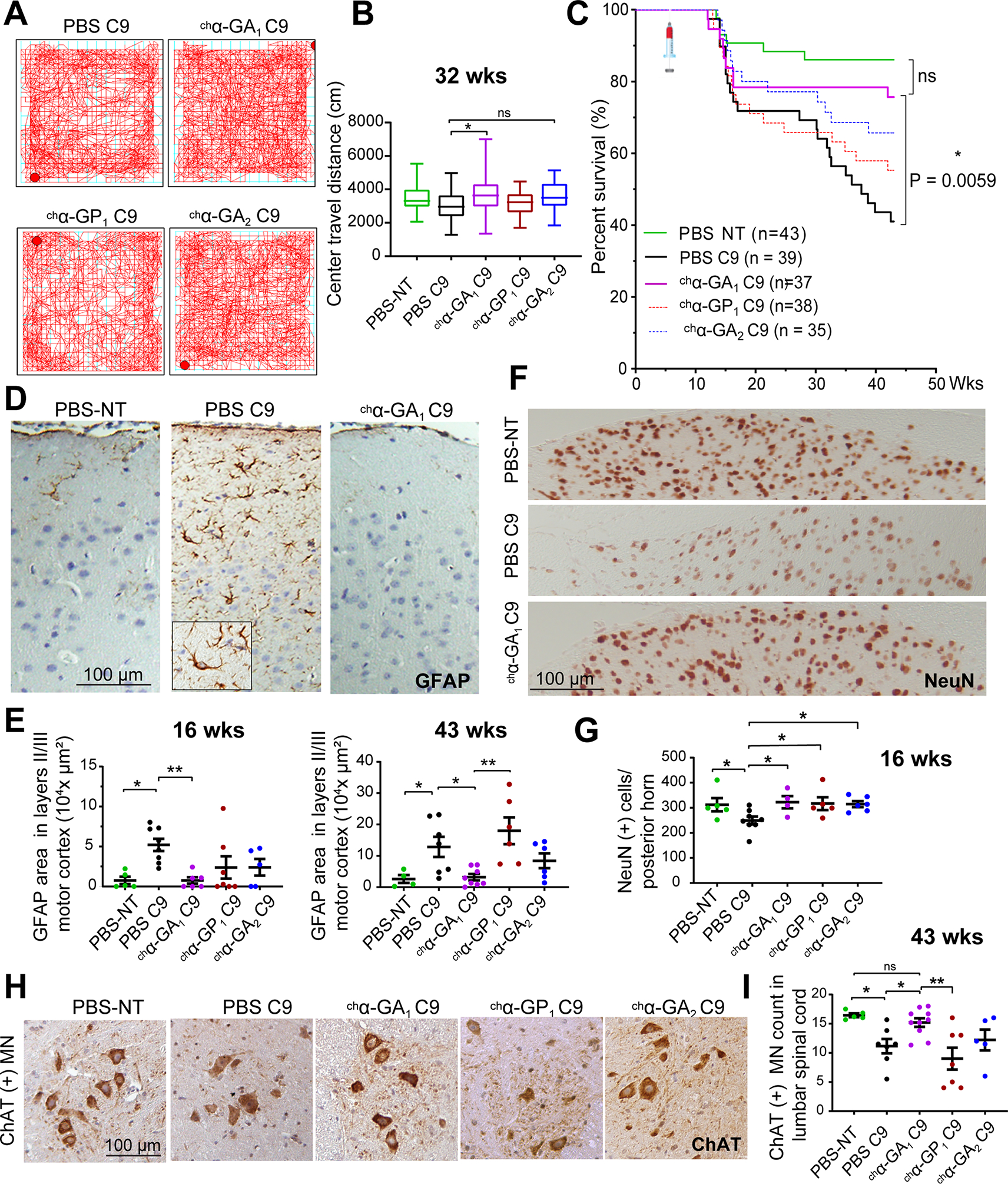
(A) Representative openfield traces of PBS, chα-GA1, ch α-GP1, and chα-GA2 treated animals taken after 15 min. (B) Center travel distance among treatment groups at 32 wks of age. n ≥ 17/group, one-way ANOVA with Sidak analyses for multiple comparisons. (C) Kaplan-Meier survival curvewith the Bonferroni analysis for multiple comparison, * p ≤ 0.025. (D, E) Representative GFAP staining (D) of PBS treated NT, PBS and chα-GA1 treated C9 animals with GFAP quantification (E) at 16 and 43 wks (n ≥ 5/group at 16 wks and for the 43 wk cohort n = 4 for PBS NT and ≥ 6/group for all other groups). (F, G) NeuN staining and NeuN(+) cell quantification at 16 wks (n ≥ 5 / group, one-way ANOVA, with Benjamini analyses for multiple comparisons). (H, I) ChAT+ motor neuron (MN) staining and quantification at 43 wks (n ≥ 5/group). One-way ANOVA with Holm-Sidak analyses for multiple comparison. ns p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤ 0.001. See also Figures S16 and S17.
Finally, Kaplan-Meier analyses showed that C9 mice treated with chα-GA1 had a significant increase in survival compared to the PBS C9 cohort (p = 0.0059) and no significant difference from the PBS treated NT group (p = 0.2321) (Figure 7C). The two other antibody treatment C9 groups (chα-GA2 and chα-GP1) were not significantly different from the PBS-C9 group at 43 wks (p = 0.0513 and p = 0.3163, respectively), although the chα-GA2 treatment group trended toward increased survival. Smaller cohorts of NT mice treated with chα-GA1 or chα-GP1 showed no difference in survival at 43 weeks (Figure S16A). All treatment antibodies showed similar pharmacokinetic properties in non-transgenic mice (Figure S16B) and antibody treated mice showed comparable levels of chimeric antibodies in blood and brain tissue over time, indicating comparable antibody exposure between the different treatment groups (Figure S16C–D).
In summary, chronic treatment of C9-BAC mice with both GA targeting antibodies (chα-GA1 and chα-GA2) resulted in reductions of GA protein aggregates and behavioral improvements with chα-GA1 displaying the highest potency. While the chα-GA2 treated mice showed corrections in gait at 24 weeks, the chα-GA1 treated mice showed more substantial improvements including improved gait, anxiety-like phenotypes, and survival. In contrast, the GP targeting antibody chα-GP1 did not improve behavior or survival phenotypes.
Reduced inflammation and increased neuronal survival in chα-GA1 treated C9-BAC mice
Next, we assessed the impact of antibody treatment on neuroinflammation and neuronal death at 16 and 43 weeks of age. GFAP, a marker of reactive astrocytosis, was increased in the motor cortex of ~60% of vehicle treated C9 animals at 16 and 43 weeks compared to NT controls respectively, (p = 0.012 and 0.020) (Figure 7D–E, S17A) (Sofroniew and Vinters 2010). GFAP staining was dramatically reduced in chα-GA1 mice at 16 and 43 weeks compared to PBS-C9 controls (p = 0.007 and 0.032, respectively) and chα-GP1 (p = 0.003, 43 weeks), respectively. While chα-GA1 and chα-GA2 treatments showed similar trends, there was no significant difference in GFAP staining with either chα-GA2 or chα-GP1 at 16 or 43 weeks compared to PBS treated C9 mice (Figure 7D–E).
In addition, we examined neurons in the anterior horn and the posterior horn of the lumbar spinal cord. We focused on these regions because they show consistent pathological changes in animals with both rapid decline and slowly progressive disease (Y. Liu et al. 2016). NeuN staining of the interneuron-rich posterior lumbar spinal cord in 16-week animals, showed a reduction of 21% (Figures 7F–G, S17B–C) in PBS-C9 compared to NT controls (p = 0.046). At 16 weeks of age, NeuN staining of animals from all three antibody treatment groups was similar to staining in the NT control cohort (Figures 7F–G, S17B–C). Similar to the previously reported data (Y. Liu et al. 2016), ChAT positive (ChAT+) motor neurons (MN) were reduced by ~30% in PBS-C9 animals compared to PBS-NT (p = 0.035) at 43 weeks. Here the chα-GA1 treatment group showed the best effects with increased numbers of ChAT+ MNs compared to PBS-C9 mice (p = 0.041) and chα-GP1 treated mice (p = 0.003) and no significant difference from the NT cohort (p = 0.508) (Figure 7H–I). In contrast, the chα-GA2 and chα-GP1 treated animals showed no similar increase in ChAT+ MNs (Figure 7H–I).
In summary, chα-GA1 treated animals showed decreased neuroinflammation and increased neuronal survival compared to PBS treated C9 mice. The improvements in disease pathology including the increased numbers of ChAT positive spinal motor neurons is consistent with the observed corrections in behavioral abnormalities and the increased survival in the chα-GA1 cohort. The second GA targeting antibody, chα-GA2, showed similar trends and increased interneuron survival at 16 weeks but no increase in MN survival. In contrast, although chα-GP1 antibody treated mice showed early trends toward decreased inflammation and significant increases in interneuron survival compared to PBS treated mice at 16 weeks, these early benefits were not observed in motor neurons at 43 weeks.
Glycosylation of the Fc region of chα-GA1 is required for cell entry and in vivo efficacy
Glycosylation of Fc antibody region has been shown to facilitate Fcγ receptor binding and associated immune effector functions as well as the internalization of antibodies into cells (Congdon et al. 2013, Dithmer et al. 2016). To determine if a fully glycosylated Fc domain on chα-GA1 is important for cellular uptake and the therapeutic effects observed in the C9 models, a murine chimeric aglycosylated version of α-GA1 (chα-GA1agly) was engineered. Aglycosylating chα-GA1 reduces its binding affinity to Fc receptors mCD16, mCD32, mCD64, and mFcRn (Figure S18A–B). Three cohorts of repeat-size matched C9-BAC mice were injected weekly with 30 mg/kg IgG control, chα-GA1 or chα-GA1agly beginning at 6 weeks of age (Figure 8A). DigiGait data were collected at 16 weeks, a timepoint at which a number of subtle but significant phenotypes are observed. Of the 8 gait abnormalities found between IgG C9 and IgG NT animals, 3 remained abnormal in the chα-GA1agly C9 compared to IgG-NT group while treatment with chα-GA1 fully rescued behavior compared to IgG-NT animals (Figure S18C, right panel). For example, stancewidth abnormalities in C9 mice were corrected in chα-GA1 treated mice (p = 0.026) but not in chα-GA1agly treated cohort (p = 0.699) (Figure 8B).
Figure 8. Glycosylation of Fc region of chα-GA1 required for cellular uptake and in vivo and in vitro efficacy.
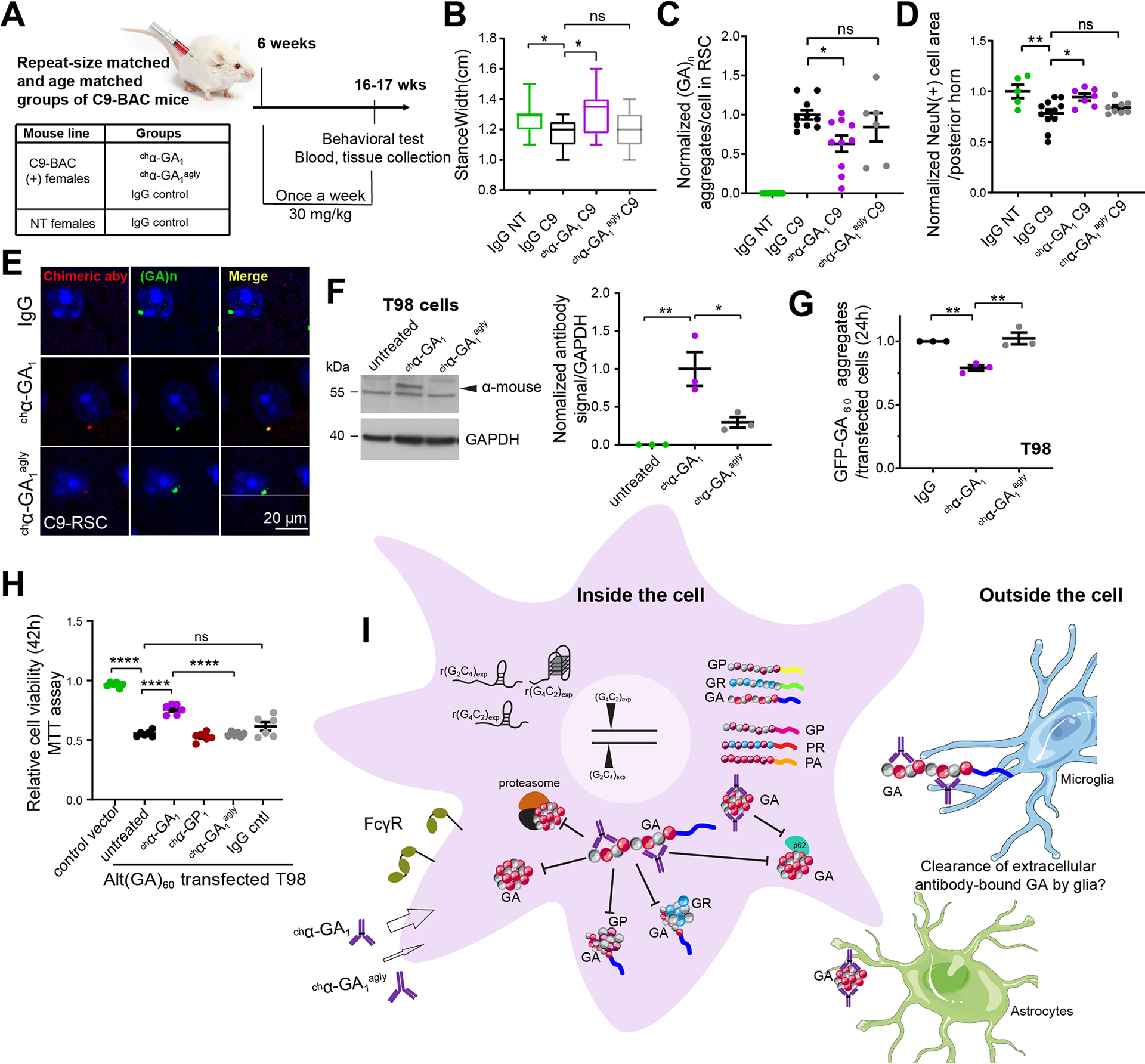
(A) Design of second antibody mouse study. (B) Representative DigiGait parameter, stancewidth, in 3 treatment groups, n ≥ 9/group. (C) GA aggregate quantification using α-GA1 (n ≥ 6/group). (D) NeuN(+) cell quantification at 17 wks (n ≥ 6/group). (E) Double labeling of GA aggregates with human α-GA1 and injected chα-GA1 aglycosylated chα-GA agly1 antibodies with α-human IgG. (F) Quantification of antibodies that bind to or are inside cells (n = 3, one-way ANOVA with Tukey analyses for multiple comparisons). (G) Quantification of GFP-GA aggregates in GFP-alt(GA)60 transfected T98 cells treated with chα-GA1 or chα-GA agly1 (60 nM, 24 h), n =3. (H) Cell viability measured by MTT for alt(GA)60 transfected T98 cells treated with antibodies (1 μM, 42h), n = 6. (I) Model of proposed mechanisms of action of α-GA1. Bars show mean ± SEM. (A-C, F, G) One-way ANOVA with Dunnett analyses for multiple comparisons. ns p > 0.05, * p ≤ 0.05, ** p ≤ 0.01, *** p ≤. 0.001, **** p ≤ 0.0001. See also Figure S18.
Next, we show that the glycosylated Fc portion of the chα-GA1 antibody is important for reducing GA aggregates in C9-BAC mice. We confirm in this independent study that GA aggregates are reduced in chα-GA1 treated mice compared to IgG controls (p = 0.029) but were unchanged following chα-GA1agly treatment (p = 0.539) (Figure 8C). In addition, while chα-GA1 treatment showed an increase in NeuN(+) neuron area in the posterior horn in lumbar spinal cord of C9 BAC mice (p = 0.018), chα-GA1agly did not show improvement (p = 0.551) compared with IgG treated C9 mice (Figure 8D). Double staining of GA aggregates and the murine chimeric antibody in C9 mouse brain tissue showed more pronounced staining of GA aggregates with the chα-GA1 compared to staining seen with chα-GA1agly (Figures 8E, S18D). We performed additional cell culture experiments to measure antibody uptake. Here, we incubated chα-GA1 and chα-GA1agly with Fc receptor-positive T98 cells and show that antibody levels in the chα-GA1 treated cells were 3.3-fold higher than the chα-GA1agly samples (24 h) (Figure 8F) (p = 0.023). Consistent with decreased uptake and efficacy of the chα-GA1agly treatment with chα-GA1agly showed no decrease in levels of GFP-GA aggregates (24 h) (Figure 8G) and no rescue of cell viability or cell death (Figure 8H, S18E) in T98 cells expressing GFP-alt(GA)60. In contrast, chα-GA1 treatment reduced GFP-GA aggregates (p = 0.006) by ~20% and significantly increased cell viability (p = 0.0001) (Figure 8H, S18E). These data demonstrate that glycosylation of the Fc region of chα-GA1 is important for cellular binding, antibody uptake, the reduction of GA aggregates in mice and cells, as well as improvements in survival in T98 alt(GA)60 expressing cells and behavior in the C9-BAC mice.
Taken together our data suggest a model in which α-GA1 antibody entry into the cell is facilitated by Fcγ receptors. Once inside the cell, α-GA1 interacts with TRIM21, increases GA protein turnover and reduces GA, GP and GR aggregates and the recruitment of p62, LC3B, and proteasome 26S subunit, resulting in increased cell survival (Figure 8I).
DISCUSSION
Our work shows that targeting a single RAN protein, GA, with the α-GA1 human-derived antibody improved behavior, decreased neuroinflammation and neurodegeneration, and increased survival of C9–500 ALS/FTD mice. These data demonstrate that peripherally delivered antibodies can cross the BBB, enter cells and target RAN protein aggregates. The chα-GA1 antibody, which targets long GA repeat tracts, was the most potent antibody, decreasing GA levels and GA aggregates, p62 recruitment, LC3B and 26S subunit proteasomal inclusions and, surprisingly, also GP aggregates and GR protein. In cell culture, chα-GA1 decreased total GA levels, GA aggregate formation, LC3B and proteasome 26S subunit inclusions, improved proteasome activity and rescued GA-induced toxicity. Additionally, we show chα-GA1 interacts with TRIM21 in cells, which may enhance the clearance of chα-GA1 and its complexes with polyGA via a TRIM21-depedent pathway (Fletcher et al. 2015, Pan et al. 2016). Using TRIM21 knockdown cells, we show the clearance of GFP-GA proteins is TRIM21 dependent. Additionally, we show GA-facilitated aggregation of GR is reduced by chα-GA1 treatment in transfected cells. Finally, we show that a fully glycosylated Fc region is required for α-GA1 therapeutic efficacy suggesting that FcγR-mediated cellular uptake is a central mechanism of action underlying the therapeutic effects of α-GA1.
Our C9-BAC model has several advantages for in vivo efficacy studies. The endogenous human sense and antisense promoters in the C9-BAC mice provide an important advantage compared to ectopic overexpression models because both sense and antisense transcripts are expressed at levels and in tissues more relevant to what is seen in patients. These are important considerations for drug stoichiometry and to ensure that improvements are seen in relevant cells and tissues affected by the disease. Reduced penetrance is frequently found in C9 families (Majounie et al. 2012, Galimberti et al. 2014) and in other neurodegenerative diseases (Conforti et al. 2011, Fernandez et al. 2017, Galimberti et al. 2014, Day et al. 2004, Ikeda et al. 2004). While the reduced penetrance found in our mice require larger cohorts than are typically used in animal studies, this also provides an opportunity to model the complex and variable features of ALS and FTD found in human C9orf72 patients. To address these challenges, we carefully controlled for repeat length among the different treatment groups and measured behavioral and histopathological phenotypes robustly detected in C9 vs. NT cohorts. While we selected female animals for the current study, because they are better characterized and develop more severe disease (Y. Liu et al. 2016), male C9-BAC mice also showed a number of behavioral phenotypes (Y. Liu et al. 2016). It will be interesting in the future to characterize the neurodegenerative changes in C9-BAC male mice to test if male mice also show improvements after α-GA treatment.
GA peptides can adopt amyloid-like structures that can sequester and inactivate proteasome complexes (Guo et al. 2018, Chang et al. 2016, Mackenzie et al. 2013, May et al. 2014). The reductions observed in GP, GR, p62, LC3B and proteasome 26S subunit aggregates following treatment with α-GA1 suggest that targeting the GA protein is effective because it reduces the sequestration of other aggregate-prone proteins to GA amyloid-like structures by reducing the GA aggregates themselves or by masking the binding sites. These data suggest that targeting other amyloid-like proteins with similar sequestration properties may be an effective approach for other disorders, including repeat expansion diseases that involve the formation of amyloid-like aggregates. Additionally, the reduction of GA and other aggregated RAN proteins may reduce ER stress which could in turn reduce additional RAN protein production (Cheng et al. 2018, Green et al. 2017), potentially interrupting a pernicious cycle. It is also possible that the intracellular α-GA1 antibody enhances protein clearance by the restoration of p62, LC3B and proteasome 26S subunit levels (Zhang et al. 2014a, W. J. Liu et al. 2016) and via Fcγ receptor TRIM21-mediated proteasome clearance (McEwan et al. 2017, Rhodes and Isenberg 2017). A number of studies have reported that C9 RAN proteins can be transmitted from cell to cell; in this context, extracellular GA protein may also be targeted by α-GA1 and cleared by glial cells (Westergard et al. 2016, Zhou et al. 2017). Future investigations into the mechanisms of both intracellular (autophagy and proteasome) as well as extracellular protein clearance mechanism will be important.
Of the two GA-targeting antibodies, α-GA1 treated cells and mice showed the most substantial reductions in GA and phenotypic improvements while α-GA2 treated C9 cells and animals showed significant but less pronounced reductions in GA protein levels as well as improvements in DigiGait performance and histopathological changes, but not a significant increase in survival. While these antibodies both bind to polyGA, differences that affect in vivo efficacy may include the preferential binding of α-GA1 to longer repeat proteins expressed in cells and its faster on and off rates compared to α-GA2. The more substantial decreases in GA aggregates as well as the decrease in GP and p62 aggregates found in α-GA1 compared to α-GA2 treated animals suggests that α-GA1 is more effective at reducing GA aggregates and also in interfering with GA protein-mediated recruitment of GP, p62, LC3B, proteasome 26S subunit and other proteins. Additionally, the selective binding of α-GA1 to longer repeat tracts may increase its relative binding to pathologically relevant target species.
The observation that GA aggregates are already detected in the retrosplenial cortex in C9 mice at 6 weeks of age, indicates that antibody treatment in animals with pre-existing GA aggregate pathology is efficacious. While α-GA1 improved the overall survival of C9-BAC mice, α-GA1 did not completely prevent or delay the first drop in the survival curve. These data suggest that a higher α-GA1 dose and/or an earlier treatment start may be required to rescue of the earliest-onset cases of disease in the C9-BAC mice. Alternatively, additional C9-dependent toxic pathways, which are not targeted by the α-GA1 antibody, may contribute to these early-onset cases. In the future, combinatorial treatment using antibody therapy and other approaches to lower RAN protein production could benefit patients.
In contrast to the GA targeting antibodies, α-GP1 showed no effects on GA levels and toxicity induced by polyGA, consistent with its high selectivity to polyGP. In C9 mice, reducing GP aggregates by 35% (43 wks) using α-GP1 was not sufficient to improve behavior, pathology or survival. This could be because further reduction would be needed to be efficacious or because GP does not play a critical role in disease pathology, which is in line with previously reported in vitro and in vivo toxicity studies (Mizielinska et al. 2014, Wen et al. 2014). Additionally, because antibodies used in both mouse studies have the identical IgG2a backbone, the demonstration that α-GA1 treatment but not α-GP1 or IgG treatment improved a number of disease phenotypes confirms that targeting of GA proteins, and not a general immune response triggered by an IgG2a backbone is mediating the observed therapeutic effects. Because α-GA1 treatment also reduces GP aggregates and GR protein levels the effects on these proteins may contribute to the improvements seen in the α-GA1 treated mice. It will be interesting to test in the future if α-GA1 treatment also reduces the levels of the PA and PR proteins.
In summary, our work shows that targeting GA RAN proteins improves cellular function, behavior, survival and neurodegeneration in C9-BAC mice without reducing RNA levels, thereby demonstrating RAN proteins drive disease in these mice. Additionally, this study provides the first proof of principle data that human antibody therapy may be a viable therapeutic approach for C9orf72 ALS/FTD and possibly other RAN protein diseases.
STAR Methods:
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Laura Ranum (ranum@ufl.edu). Data and material availability: All unique/stable reagents generated in this study are available from the Lead Contact with a completed Material Transfer Agreement.
Experimental Model and Subject Details
Animals
Animal studies were approved by the Institutional Animal Care and Use Committee at University of Florida. C9-BAC transgenic mice (Y. Liu et al. 2016) were bred with FVB mice (Jackson Laboratory; RRID:IMSR_JAX:001800) to generate age-matched cohorts for preclinical studies. Female C9 (+) pups and NT littermates were genotyped using DNA extracted from tail biopsies as previously described (Y. Liu et al. 2016). The number of G4C2 repeats in each animal was estimated using Southern blotting with molecular weight ladders to estimate repeat length. Because somatic repeat instability (e.g. tail vs brain) is limited in mice (Y. Liu et al. 2016), we used repeat size estimates from tail DNA to control for repeat length among the different treatment groups. Sized matched C9 (+) animals with repeats ranging from 500 to 750 repeats were distributed equally into the four treatment cohorts along with their non-transgenic littermates (Figure S10A). The treatment cohorts were also controlled by matching cohorts for age and by distributing pups from individual litters and sires among treatment and control groups. Additionally, animals from different treatment groups were co-housed to avoid cage specific effects that could affect individual treatment groups. Female mice were housed with five animals per cage. The animals had continuous access to food and water and were on a 12:12 hour light:dark cycle. Randomly selected repeat-length matched takedown cohorts were selected before treatment began and before phenotypes were detectable (< 6 weeks of age) for the 16-week behavioral studies and takedown. For the behavioral studies done at 24 and 32 weeks, all remaining animals were tested. Animals in the study were determined to reach humane endpoint at 20% weight loss or if requested by Animal Care Service at University of Florida.
Cell culture
HEK293T, SH-SY5Y or T98 (Zu et al. 2013) cells were cultured using DMEM supplemented with 10% FBS and incubated at 37 °C, 5% CO2. Transfection was performed using Lipofectamine 3000 reagent (Invitrogen) according to the manufacturer’s protocols.
iPS motor neuron maintenance differentiation.
Induced pluripotent (iPS) cell lines from C9orf72-ALS patients and non-neurological controls were obtained from the Cedars-Sinai Answer ALS repository and a generous gift from Kevin Talbot (Dafinca et al. 2016). iPSCs were maintained and differentiated into spinal motor neurons according to the publicly-available ‘diMNs’ (direct iMN) protocol, which generates motor neuron precursors by day 12 and 20–30% islet-1 positive motor neurons by day 18 (Figure S15A and http://neurolincs.org/pdf/diMN-protocol.pdf) (Sances et al. 2016).
Human tissue samples.
C9orf72 ALS/FTD patient and control tissue was collected with IRB approval at the University of California San Diego and kindly provided by Dr. J. Ravits. This was deidentified tissue and detailed information on age at death, disease duration and postmortem interval and sex were not provided.
Method Details
Antibody generation using Reverse Translational Medicine (RTM).
Human RAN protein antibodies were derived from a de-identified blood lymphocyte library collected from healthy elderly subjects as previously described (Sevigny et al. 2016). In brief, memory B cells, isolated from peripheral blood lymphocyte preparations by anti-CD22-mediated sorting were cultured on gamma-irradiated human peripheral blood mononuclear cell feeder layers. Supernatants from isolated human memory B cells were screened for their ability to bind (GA)15, (GP)15, (GR)15, (PA)15 or (PR)15 synthetic peptides. Positive hits were subjected to cDNA cloning of IgG heavy and kappa or lambda light chain variable region sequences, and sub-cloned in expression constructs using Ig-framework specific primers for human variable heavy and light chain families in combination with human J-H segment-specific primers. α-GA1, α-GA2 and α-GP1 were engineered to incorporate glycosylated human IgG1 heavy and human lambda light chain constant domain sequences. Murine chimeric IgG2a/lambda version of α-GA1, α-GA2 and α-GP1 (chα-GA1, chα-GA2 and chα-GP1) were generated for use in chronic efficacy studies in C9-BAC transgenic mice. An aglycosylated variant of chα-GA1 (chα-GA agly1), incorporating a single point mutation (N297Q, using standard Kabat EU numbering), which eliminates N-glycosylation of the Fc region and severely reduces FcγR binding was generated to test for Fc-related activities (Tao and Morrison 1989). Recombinant antibodies were transiently expressed in CHO cells, purified using standard Protein A affinity chromatography and desalted to PBS buffer. Endotoxin levels were confirmed to be <10 EU/ml.
Binding specificity and affinity to DPR peptides by indirect ELISA.
Synthesis and purification of DPR peptides was performed by Schafer-N (Copenhagen, Denmark) using the following sequences: (GA)15: H-CHHHHHH(GA)15-OH; (GP)15: H-C(GP)15-OH; (GR)15: H-C(GR)15-OH; (PA)15: H-C(PA)15-OH; (PR)15: H-C(PR)15-OH; GA20: H-(GA)20HHHHHH-NH2; (GA)10: H-(GA)10HHHHHH-NH2; (GA)6: H-(GA)6HHHHHH-NH2; (GA)5: H-(GA)5HHHHHH-NH2; (GA)4: H-(GA)4HHHHHH-NH2; (GA)3: H-(GA)3HHHHHH-NH2; (GA)2: H-(GA)2HHHHHH-NH2; (GP)20: H-(GP)20HHHHHH-NH2; (GP)10: H-(GP)10HHHHHH-NH2; (GP)6: H-(GP)6HHHHHH-NH2; (GP)5: H-(GP)5HHHHHH-NH2; (GP)4: H-(GP)4HHHHHH-NH2; (GP)3: H-(GP)3HHHHHH-NH2; (GP2): H-(GP)2HHHHHH-NH2. For determination of binding affinity, binding specificity and length-dependent affinity, 96-well microplates (Corning Incorporated) were coated with dipeptide repeat peptides at either 5 μg/ml (for binding specificity) or 50 μg/ml (for repeat length-dependent binding) in coating buffer (15 mM Na2CO3, 35 mM NaHCO3, pH 9.4). Non-specific binding sites were blocked for 1 h at RT with PBS containing 0.1 % Tween-20® and 2 % BSA (Sigma-Aldrich). Primary human anti-DPR antibodies were diluted to the indicated concentrations and incubated for 1 h at RT, followed by incubation with an HRP-conjugated donkey anti-human IgG Fcγ-specific antibody (Jackson ImmunoResearch Laboratories, Inc). Binding was determined by measurement of HRP activity in a standard colorimetric assay. EC50 values were calculated by non-linear regression using GraphPad Prism (Version 7.03). For determination of pH-dependent binding affinity, chimeric antibodies were incubated in 10 mM Tris-HCl at pH 4.5, 5.5, 6.5 or 7.5 for 1 h at RT. EC50 values were determined as described above, in 10 mM Tris-HCl at the respective pH, using a HRP-conjugated goat anti-mouse IgG (H + L)-specific secondary antibody (Jackson ImmunoResearch Laboratories, Inc). To determine effects of cytoplasmic proteins on antibody affinity, SH-SY5Y human neuroblast cells (#94030304, Sigma) were lysed by mechanical force in 10 mM Tris-HCl at pH 7.4 containing Deoxyribonuclease I (#SLBV1446, Sigma) and cOmplete Mini protease inhibitors (Roche Diagnostics, Indianapolis USA). Chimeric antibodies were incubated in fresh cell lysates or PBS for 16 h at 4 °C or 37 °C and EC50 values were determined as described above.
Characterization of antibody binding properties by bio-layer interferometry.
Poly-GA and Poly-GP DPR peptides were synthesized and purified by Schafer-N (Copenhagen, Denmark): (GA)15: H-CHHHHHH(GA)15-OH; (GP)15: H-C(GP)15-OH. Upon arrival from the manufacturer, lyophilized pure (GA)15 and (GP)15 were dissolved in DMSO (Sigma-Aldrich, Buchs, Switzerland) at 10 mg/ml and stored at −20°C. Bio-layer interferometry experiments (BLI) were conducted on an Octet RED96 instrument (Pall ForteBio LLC, Fremont, USA). For covalent immobilization of (GA)15 DPR peptides, Octet amine-reactive (AR2G) biosensors were used. AR2G biosensors were activated with EDC (1-Ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride at 20 mM in water; Pall ForteBio LLC, Fremont, USA) and s-NHS (N-hydroxysulfosuccinimide at 10 mM in water; Pall ForteBio LLC, Fremont, USA) for 300 s, followed by 600 s loading of the biosensor surface with 5 μg/ml of (GA)15 peptides in 10 mM acetate buffer pH 6 (Pall ForteBio LLC, Fremont, USA). After peptide loading, AR2G biosensors were quenched for 300 s using 1 M ethanolamine pH 8.5 (Pall ForteBio LLC, Fremont, USA) and rinsed in kinetics buffer (Pall ForteBio LLC, Fremont, USA) for 120 s (baseline). Human α-GA1 and α-GA2 antibody association was assessed for 600 s at different concentrations (for α-GA1: 10, 5, 2.5, 1.25 and 0.625 nM for α-GA2: 30, 15, 7.5, 3.75 and 1.875 nM) in diluted kinetics buffer (1:10 in PBS). Antibody dissociation was evaluated in kinetics buffer for 600 s. For replicate experiments AR2G biosensors were regenerated using 10 mM Glycine pH 2 (Sigma-Aldrich, Buchs, Switzerland) and 10x PBS (Fisher Scientific AG, Reinach, Switzerland). Biosensors with streptavidin surface (SA) were used for immobilization of biotinylated (GP)15 DPR peptides. Biotinylation and purification of (GP)15 peptides was performed using a commercial biotin labeling kit (Roche, Basel, Switzerland) according to the manufacturer’s protocol. For sensor loading, (GP)15 peptide-containing fractions from the purification column were pooled and diluted 1:3 in PBS/0.1% Tween® −20.SA sensors were activated using PBS/0.1% Tween® −20 and the biotinylated (GP)15 peptides were loaded on the sensors for 600 s. Upon peptide loading, SA biosensors were blocked for 300 s with 0.1% milk in PBS containing 0.1% Tween® −20 (assay buffer), rinsed in assay buffer for 180 s (baseline) and α-GP1 antibody association was assessed at different concentrations (20, 10, 5, 2.5 and 1.25 nM) in assay buffer for 600 s. Antibody dissociation was evaluated in assay buffer for 600 s. For replicate experiments SA biosensors were regenerated using 10 mM Glycine pH 2 (Sigma-Aldrich, Buchs, Switzerland) and 10x PBS (Fisher Scientific AG, Reinach, Switzerland). All binding data was referenced by collecting data with either a kinetics buffer or an assay buffer only reference. For competitive binding determination, AR2G biosensors were loaded with (GA)15 dipeptide repeat protein peptides, quenched and rinsed as described above. α-GA1 and α-GA2 antibodies were then assessed for target binding in a pairwise fashion: Binding (for 800 s) of reference antibody (15 nM, in kinetics buffer (Pall ForteBio LLC, Fremont, USA)) to (GA)15 peptides was directly followed by binding (for 600 s) of the competing antibody (15 nM, in kinetics buffer (Pall ForteBio LLC, Fremont, USA)). All experiments were performed at 25 °C. Kinetics data analysis was performed by using the Octet system software (Pall ForteBio LLC, Fremont, USA) with simultaneous ka/kd global fitting with 1:1 interaction model. BLI sensorgrams were plotted with the Prism software from GraphPad (San Diego, USA) upon fitting. Antibody affinity for murine Fc receptor subtypes was determined using His-tagged mouse CD64/FCΓR1 (#50086-M08H, Sino Biological), CD32/FCΓR2 (#50030-M08H, Sino Biological), CD16/FCΓR3 (#50326-M08H, Sino Biological) and FCΓRT & B2M heterodimer proteins (#CT029-M08H, Sino Biological) from solubilized stocks in water at 0.25 mg/ml. Before receptor immobilization Octet anti-His (HIS2) biosensors were washed with PBS pH 7.4 (Gibco) for 120 s. Fc receptor proteins were immobilized on the sensors for 500 s in PBS pH 7.4 or PBS pH 6 at a concentration of 10 μg/ml. After a 180 s baseline step, antibody association was assessed for 240 s at three different concentrations (500 nM, 250 nM and 125 nM). Wells containing PBS pH 7.4 without antibodies were used as a control. Dissociation of antibodies was measured for 300 s in the baseline wells. Experiments were performed at 25 °C and an instrument shaking speed of 500 rpm. BLI sensorgrams for qualitative binding analysis were plotted with the Prism software from GraphPad (San Diego, USA).
Binding specificity analysis to unrelated amyloidogenic proteins.
96-well microplates (Corning Incorporated, Corning, USA) were coated with (GA)15, (GP)15, (GR)15, (PA)15 or (PR)15 DPR peptides at 5 μg/ml per peptide or unrelated target proteins at a concentration of 5–10 μg/ml in coating buffer (15 mM Na2CO3, 35 mM NaHCO3, pH 9.42) or PBS. Non-specific binding sites were blocked for 1 h at RT with PBS/0.1% Tween® −20 containing 2% BSA (Sigma-Aldrich, Buchs, Switzerland). α-GA1, α-GA2 and α-GP1 antibodies were diluted at 4 nM concentration and incubated 1 h at RT. Binding was determined using donkey anti-human IgG Fcγ-specific antibody conjugated with HRP (Jackson ImmunoResearch Laboratories, Inc., West Grove, USA) followed by measurement of HRP activity in a standard colorimetric assay. Signals for target proteins were calculated in fold increase above background.
Affinity characterization by surface plasmon resonance (SPR)
To evaluate the intrinsic monovalent affinity of α-GA1 and α-GA2, the binding of antibody Fab fragments to (GA)8 peptides was measured by SPR. Biotinylated dipeptide repeat protein biotin-(GA)8 [H2NGGGS(K-lc-Biotin)WDGAGAGAGAGAGAGAGAS-OH] was synthesized and purified at New England Peptide (Gardner, MA USA). Bivalent binding of α-GA1 and α-GA2 to these peptides by ELISA showed similar sub-nM affinity (Figure S2) as seen with (GA)10 and (GA)20 peptides (Figure S3). Antibody Fab fragments with a c-terminal hexahistidine tag on the heavy chain were generated. Heavy chain fragment (VH-CH1–6xHIS) and light chain were transiently co-expressed in CHO cells and affinity purified using standard immobilized metal affinity chromatography (IMAC) followed by size-exclusion chromatography.
Monovalent binding affinity measurements were performed with a Biacore T200 instrument (GE Healthcare). Synthetic biotin-(GA)8 peptide (2.1 kDa) was captured on a Biotin CAPture chip (GE Healthcare, kit 28-9202-34) at 2–3 pg/mm2 from solutions at ~5 ng/mL in Biacore buffer (10 mM HEPES, pH 7.2, 150 mM NaCl, 3.4 mM EDTA, 0.05% BSA, 0.005% surfactant P20) using reagents and protocols provided by the manufacturer. Poly-GA antibody Fab fragments were injected for 3 min for α-GA1 (concentrations: 78, 156, 312, 625, 1250, 2500, 5000, 10000, or 20000 nM) and for 5 min for α-GA2 (concentrations: 3.1, 6.2, 12.5, 25, 50, 100, 200, or 400 nM) at 30 μL/min in Biacore buffer, and the binding response relative to a reference sensor with no captured peptide was recorded during and for ~10 min after the injections. The response to Fab at the highest concentrations, injected over sensor surfaces with no biotin-(GA)8 peptide, was <5% of the signal measured with biotin-(GA)8 peptide captured on the surface. Data were analyzed with Biacore T200 Evaluation Software v3.0 using a 1:1 binding model (Figure S2).
Plasma and brain drug levels determination by indirect ELISA
Plasma collection.
Blood was withdrawn from experimental animals 2 days post antibody dosing for the 16, 24, and 32 week time points, and 7 days post-injection for the 43 week time point. Whole blood samples were collected in BD Microtainer K2E tubes (Becton, Dickinson and Company, Franklin Lakes, USA) and mixed by inverting the tubes several times. If necessary, the withdrawn blood samples were stored on ice until further processing. Blood samples were centrifuged at 5000 rpm for 5 min in a refrigerated centrifuge. Following centrifugation, the liquid component (plasma) was immediately transferred into clean 0.5 ml Eppendorf tubes. The samples were maintained at 2–8 °C while handling and stored at −80°C until use.
Brain and spinal cord homogenates.
Various parts of mouse brain and spinal cord including hindbrain (HB), frontal cortex (FCX), basal ganglia (BG) and cerebellum (CB) for brain and thoracic (TR) spinal cord were collected during animal takedown and stored at −80 °C. Frozen tissues were homogenized using RIPA buffer (1% sodium deoxycholate, 150 mM NaCl, 1% Triton-X 100, 50 mM Tris pH 7.5, 1X proteinase inhibitors and 0.2 mg/mL DNase I). Soluble fractions were collected after spinning down the homogenates for 15 min at 14,000 rpm at 4 °C and the pellets were dissolved in 2% SDS. Samples were stored at −80 °C until use.
ELISA assay for determining drugs level in brain and plasma samples.
96-well microplates (Corning Incorporated, Corning, USA) were coated with either (GA)15 or (GP)15 dipeptide repeat protein peptides at a concentration of 0.5 μg/ml (for plasma drug levels) or 1 μg/ml (for brain drug levels) in coating buffer (15 mM Na2CO3, 35 mM NaHCO3, pH 9.42) overnight at 4 °C. Non-specific binding sites were blocked for 1 h at RT with PBS/0.1% Tween® −20 containing 2% BSA (Sigma-Aldrich, Buchs, Switzerland). Plasma samples diluted to 1:40000 in PBS or 30 μg total protein from brain homogenates were added and incubated for 2 h at RT, followed by incubation with a goat anti-mouse IgG2a-specific antibody conjugated with HRP (Southern Biotech, Birmingham, USA). Standard curves were prepared by two-fold serial dilutions of chα-GA1, chα-GA2 and chα-GP1 antibodies in PBS/mouse serum with initial antibody concentration at 1 nM. Binding was determined by measurement of HRP activity in a standard colorimetric assay. Drug plasma and brain levels were estimated by linear regression using Excel 2016 software (Microsoft, USA).
DNA constructs.
V5-(GA)120, V5-(GP)120, V5-(GR)120. FLAG-(GP)30 and FLAG-(GR)30 were synthesized as previously reported (Y. Liu et al. 2016). GAATTCAAGCTTGATATCCCCGGGGCA(GGAGCA)60GGAGCCGGCAATATTAAGACACTCGA GTCGACTAGTCTAGAGGATCC fragment (for alt(GA)60), GAATTCAAGCTTGATATCAGGCCT(GGACCA)60GGCGCCAATAACAGATATCTCGAGTCGACT AGTCTAGAGGATCC fragment (for alt(GP)60), GAATTCAAGCTTGATATCCCCGGG(GGACGA)60CTCGAGTCGACTAGTCTAGAGGATCC fragment (for alt(GR)60) were synthesized and cloned into pUC57 vector (GenScript). FLAG-alt(GA/GP/GR)60 constructs were generated by subcloning HindIII/BamHI fragments into p3xFLAG-Myc-CMV-24 vector (Sigma-Aldrich, ref# E9283). GFP-Alt(GA/GR)60 constructs were generated by subcloning NheI/HindIII fragment of peGFP-C1 into FLAG-alt(GA/GR)60 vector at EcoRV restriction enzyme site. GFP-Alt(GP)60 construct was generated by subcloning the NheI/HindIII fragment into FLAG-Alt(GP)60 vector at SfoI restriction enzyme site.
Western blotting.
Human antibodies were characterized using Western blotting following the previously reported protocol (Zu et al. 2013). Briefly, transfected cells were lysed using RIPA buffer (1% sodium deoxycholate, 1% Triton-X, 50 mM Tris pH 7.5, 150 mM NaCl, and proteinase inhibitor). Protein lysates (10 μl) were run on precast gel (4–12% Bis-Tris, Criterion) and transferred to a nitrocellulose membrane (Amersham). After the blocking step in 5% milk in PBS containing 0.05% Tween® −20, the membrane was probed with human antibody (1:1000) or with antibodies against V5 (Invitrogen, cat#R960–25, 1:1000) or FLAG (Sigma Aldrich, cat# F3165, 1:1000) tags expressed in frame with C9 RAN proteins of interest.
(GP)30 and (GR)30 expression and purification.
Recombinant FLAG-(GP)30 and FLAG-(GR)30 proteins were generated using the BAC-to-BAC Baculorius Expression System (Invitrogen) (Y. Liu et al. 2016). Proteins were purified using anti-FLAG M2 agarose bead (Sigma-Aldrich) following the manufacturer’s protocol.
Blinding procedures:
Researchers were blinded and did not know which groups of animals were receiving treatment antibodies, IgG or PBS controls. This blinding was done during the following experiments: 1) antibody screening including WB, IF, IHC; 2) the in vivo target engagement study; 3) both in vivo efficacy studies; 4) antibody testing in iPSC neurons; 5) cell toxicity studies; 6) quantification of IF and histological staining was performed using images taken by researchers who did not perform the experiments and images were analyze by researchers blinded to the treatment group.
Southern blotting.
Southern experiments were performed following the protocol reported previously (Y. Liu et al. 2016). Briefly, a 241bp DNA probe was generated by PCR using primers 5’AGAACAGGACAAGTTGCC (forward) and 5’-AACACACACCTCCTAAACC (reverse) using a construct with human C9orf72 DNA fragments as template (Y. Liu et al. 2016), with an initial denaturation at 94 °C for 3 min, then 32 cycles (94 °C for 45s, 58 °C for 45s, 72 °C for 1 min), followed by a final elongation at 72 °C for 6 min. α-32P-dCTPs were incorporated into 25 ng of the DNA probe using Random Primed DNA labeling kit (Invitrogen), purified with ProbeQuant™ G-50 Micro columns (GE Healthcare) and denatured at 100 °C in boiling water. A total of 8μg gDNA was digested with EcoRI and BamHI at 37 °C overnight and electrophoresed in 0.75% agarose gels in 1×TAE. The agarose gels were depurinated in 0.1 N HCl, denatured in 1.5 M NaCl, 0.5 M NaOH and neutralized in 1.5 M NaCl, 0.5 M Tris HCl pH 7.5 for 15 min at each step. DNA was transferred to a positively charged nylon membrane (GE Healthcare) by capillary blotting overnight and followed by crosslinking by UV irradiation. After two hours of prehybridization with Amersham™ Rapid-hyb buffer (GE Healthcare), the membrane was then hybridized with the probe for 3 hours at 65 °C in a rotating hybridization oven. The membrane was then washed once with 2×SSC, 0.1% SDS solution at room temperature for 20 min and two times with 0.2×SSC, 0.1% SDS solution at 65 °C for 15 min per wash. Radioactive signal was visualized on X-ray film after 1–2 days of exposure at −80 °C.
Immunofluorescence (IF) for human antibody characterization.
IF experiments were performed following the previously reported protocol (Zu et al. 2013). Briefly, transfected HEK293T cells in L-lysine coated 8 well chamber were fixed in 4% paraformaldehyde, and then permeabilized using 0.05% Triton X. Alternatively, cells were fixed and permeabilized using cold acetone/methanol (1:1). After a blocking step in 1% normal goat serum for 1 h at RT, cells were incubated with human antibody (60 nM) and rabbit anti-V5 (Sigma-Aldrich, 1:500) or mouse anti-FLAG (Sigma-Aldrich, 1:500) for 1 h at RT. After incubation with primary antibodies, cells were incubated with Dylight549 anti-human antibody (Jackson ImmunoResearch, 1:500) and A488 anti-mouse or anti-rabbit antibody (Invitrogen, 1:500) for 1 h RT. Cells were mounted with DAPI containing diamond Prolong mounting solution (Invitrogen). Images were taken using confocal microscopy LSM 880 (Zeiss).
IF on frozen mouse brain tissues.
Frozen tissues were warmed up to RT over ~ 2 h, fixed in 10% formalin for 15 min at RT, and then immediately permeabilized in cold acetone/methanol (1:1) for 5 min at −20 °C. After washing steps, tissues were blocked with background sniper solution (Biocare Medical) for 1 h at RT, and then incubated with primary antibody (see below for antibody information, 1:1000 in 1:10 background sniper) overnight at 4 °C. Next day, tissues were washed with PBS and probed with secondary antibody (see below for antibody information, 1:1000 in 1:10 background sniper) for 1–2 h at RT. Tissues were mounted with DAPI containing diamond Prolong mounting solution (Invitrogen). Images were taken using confocal microscopy LSM 880 (Zeiss).
For human antibody detection, Dylight549 conjugated anti-human antibody (Jackson ImmunoResearch). For target engagement study for animals injected with human antibodies, for GA aggregate staining: mouse monoclonal GA antibody (27B11A6) (Y. Liu et al. 2016), Cy5 conjugated anti-mouse antibody (Jackson ImmunoResearch), for human antibody staining: Dylight549 conjugated anti-human antibody (Jackson ImmunoResearch). For target engagement study for animals injected with chimeric human antibodies, for GA or GP aggregate staining: α-GA1, α-GA2 or α-GP1, Dylight549 conjugated anti-human antibody (Jackson ImmunoResearch), for chimeric human antibody staining: Cy5 conjugated anti-mouse antibody (Jackson ImmunoResearch). For p62 and GA aggregate co-staining, for GA aggregates: human antibody: α-GA1, Dylight549 conjugated anti-human antibody (Jackson ImmunoResearch), for p62 detection, rabbit anti-p62 (Biolegend, 1:100) and A488 conjugated anti-rabbit (Invitrogen, 1:1000). For LC3B or PSMC4 and GA aggregate co-staining, rabbit anti-LC3B (Cell Signaling, cat# 2775, 1:1000), rabbit anti-PSMC4 (Proteintech, cat# 11389–1-AP, 1:500), A488 conjugated anti-rabbit (Invitrogen, 1:1000).
Fluorescence in situ hybridization (FISH).
FISH experiments were performed following previously reported protocols (Y. Liu et al. 2016). Briefly, mice were perfused transcardially with PBS. Brain was excised and fresh frozen with OCT in 2-methylbutane on dry ice. Frozen sections (10 μm) were sectioned on the cryostat. Frozen sections were fixed in 4% PFA in PBS for 20 min and incubated in prechilled 70% ethanol for 30 min or longer at 4 °C. Following rehydration in 40% formamide in 2× SSC for 10 min, the slides were blocked with hybridization solution (40% formamide, 2× SSC, 20 μg/ml BSA, 100 mg/ml dextran sulfate, and 250 μg/ml yeast tRNA, 2 mM Vanadyl Sulfate Ribonucleosides) for 30 min at 55 °C and then incubated with 200 ng/ml of denatured DNA probe ((C2G4)×3-Cy-3 for sense foci and (G4C2)×3-Cy-3 for antisense foci) in hybridization solution at 55 °C for 3 h. After hybridization the slides were washed three times with 40% formamide in 2× SSC and briefly washed one time in PBS. Slides were washed and autofluorescence of lipofuscin was quenched by 0.25% of Sudan Black B in 70% ethanol.
Immunohistochemistry for human brain tissue.
Formalin fixed, paraffin-embedded cerebellar sections (5 μm) from a C9orf72 ALS/FTD patient and control subject were collected with IRB approval at the University of California San Diego and kindly provided by Dr. J. Ravits. This tissue was pretreated for antigen retrieval by heating in 1 mM EDTA buffer, pH 8.3, by microwave irradiation for 12 min (600 W). Quenching of endogenous peroxidase activity was achieved by treatment with 3% H2O2 in methanol for 10 min at RT. Non-specific binding sites were blocked for 1 h at RT with PBS/5% serum (horse/goat)/4% BSA. After the blocking step, sections were incubated with human-derived α-GA1, α-GA2 and α-GP1 antibodies at 20 nM concentration overnight at 4 °C. Detection was performed with biotinylated donkey anti-human IgG (H+L) (1:350, Jackson ImmunoResearch Laboratories, Inc., West Grove, USA) or anti-rabbit secondary antibody (1:250, Jackson ImmunoResearch Laboratories, Inc., West Grove, USA) and antibody signal was amplified with the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, USA) and detected with diaminobenzidine (DAB, Thermo Scientific, Rockford, USA). Slides were mounted using Eukitt® mounting medium (O. Kindler GmbH; Freiburg, Germany). Bright-field imaging was performed using a Dotslide VS120 slide scanner (Olympus Schweiz AG, Switzerland).
Immunohistochemistry for mouse tissue.
Animals were perfused transcardially with cold 1x PBS and fresh tissue was harvested and stored in 10% formalin (Fisher Diagnostics) for 48 hours. Tissue were then stored into 70% ethanol. Tissue processing and embedding was carried out by the histopathology core at the Molecular Pathology core at University of Florida. Five-micrometer sections were deparaffinized in xylene and rehydrated through graded ethanol. Antigen retrieval was performed by incubating the slides in a steamer with 10 mM citrate buffer (pH 6.0) for 30 min. The slides were washed for 10 min in running tap water. For C9 RAN protein detection, incubation in 95–100% formic acid for 5 min was performed followed by 10 min washing in running tap water. To block endogenous peroxidase activity, slides were incubated in 3% H2O2 (in 1X PBS) for 15 min, followed by washing in tap running water for 10 min. To prevent nonspecific binding and excessive background, slides were blocked with a serum free block (Biocare Medical) for 15 min. Primary antibody was applied on the slides and incubated overnight at 4 °C and 1 h at RT on the following day (see below for dilution information). Slides were washed with PBS and incubated with HRP conjugated secondary antibody (see below for dilution information) or with rabbit or mouse linking reagent (Biolegend, Ref# 93030 and 93029) for 30 min at RT. USA-HRP universal labeling reagent (Biolegend, Ref# 93028) was then applied to the slides for 30 min at RT. NovaRed or DAB (Vector Labs) was used to measure the peroxidase activity and the slides were counterstained with hematoxylin (modified Harris, Sigma Aldrich) or immediately rehydrated in graded alcohol and coverslipped for visualization. Images were taken on the Olympus BX51 microscope using the cellSens software. For hematoxylin and eosin staining, the slides were deparaffinized in xylene and dehydrated through graded ethanol. The slides were then soaked in hematoxylin (modified Harris, Sigma Aldrich) for 1 min and washed in running distilled water for 10 min. Next, the slides were immersed in Eosin Y (Sigma Aldrich) for 30 sec and washed in distilled water for 10 min. The slides were rehydrated and cover slipped before visualization.
For NeuN staining, anti-NeuN (1:500, Millipore). For ChAT staining, anti-ChAT (1:200, Millipore). For C9 RAN aggregate staining, mouse monoclonal 27B11A6 (1:1000) or α-GA1 (1:1000) (for GA), α-GP1 (1:1000) (for GP). For GFAP staining, anti-GFAP (1:5000, ProteinTech).
iPS motor neuron antibody dosing.
60 nM control and dipeptide antibodies were added to culture media at day 15, and replenished with media changes every three days. Cells were pelleted for DPR analysis at days 5–6 and days 12–13 of treatment.
Meso scale discovery (MSD) immunoassay.
RIPA soluble protein lysates from the brain were obtained to perform polyGP sandwich immunoassay using the Meso Scale Discovery electrochemiluminescence platform as described below or using the protocol previously reported (Su et al. 2014, Gendron et al. 2015). Rabbit polyclonal GP antibody (H3154) or GR antibody (H3148) (Zu et al. 2013) was used as the capture antibody. MSD plates were coated with the capture antibody, and was sealed and left overnight at 4 °C. Next day, the plates were washed with 1x Tris wash buffer and tapped dry. The plate was then blocked with blocking buffer as per manufacturer’s protocol. The plate was then sealed and incubated with shaking for 1 h at room temperature. Serial dilutions of recombinant (GP)8 or (GR)8 in TBS were used to prepare the standard curve. Approximately 100 μg of protein was loaded per well and the plate was sealed and allowed to shake at 300–1000 rpm for 1–2 h at room temperature. The wells were then washed with 1x Tris wash buffer and tapped dry. The detection antibody (rabbit polyclonal GP antibody H3155 or GR antibody H3148) (Zu et al. 2013) was coupled to a SULFO tag. The antibody was diluted to 1 μg/ml in 1% TBST and added to the wells, and incubated on a shaking plate at 300–1000 rpm for 1–2 hours. The plate was washed again to remove any unbound antibody and 2X read buffer was added to the well. Response values corresponding to the intensity of emitted light upon electrochemical stimulation of the assay plate using the Meso Scale Discovery QUICKPLEX SQ120 were acquired and background corrected using the response from the NT animals. Concentrations of poly(GP) or poly(GR) in lysates are presented as ng of polyGP or polyGR per mg protein in homogenate (pg/mg). All samples were tested in duplicate wells.
Control MSD experiments.
RIPA soluble brain lysates were obtained from C9-BAC mice at 16 weeks of age. Antibodies at concentrations of 1:10 serial dilutions starting from 450 μg/ml were added to the brain lysates and incubated for 1h or were added right before performing MSD assay. Interference from the injected antibodies to the MSD assay was calculated using this method.
Preparation of brain lysates and iPSC derived motor neurons for fractionation experiments.
Mouse brains or iPS motor neurons were thawed on ice in approximately 5 x (w/v) lysis buffer (150 mM NaCl, 20 mM tris, pH 7.5, 1 mM EDTA, 1 mM EGTA, 1% triton X-100, 0.2 % sodium deoxycholate) with protease inhibitor cocktail (cOmplete, Sigma). Homogenization was performed using MP BIO FastPrep-24 with Lysing matrix D 1.4 mm ceramic beads (MP Biomedicals, Solon, OH, USA) according to manufacturer’s protocol. Homogenates were sonicated for 10 seconds in 4 °C water bath and then separated from beads and debris removed by centrifugation at 850 x g for 10 minutes at 4 °C. The homogenate was clarified by centrifugation at 16,000 x g for 20 min at 4 °C. Total protein concentration was determined using BCA protein assay (Thermo Scientific) and used for normalization of polyGA and polyGP levels. Lysate was further fractionated by ultracentrifugation at 100,000 x g for 30 min at 4 °C. The pellet was resuspended in 2% SDS and heated to 95 °C for 5 minutes.
PolyGA immunoassays.
PolyGA in mouse brain lysates or in iPS motor neurons was measured using a Meso Scale Discovery sandwich immunoassay. In this assay, the human/murine chimeric form of anti-GA antibody chGA3 is used as capture antibody, and human anti-GA antibody GA4 along with a SULFO-tag anti-human secondary antibody is used for detection. Prior to measurement in assay, SDS and tris(2-carboxyethyl)phosphine (TCEP) were added to samples to final concentrations of 1% and 1 mM, respectively, and heated to 95 °C for 5 minutes to dissociate the treatment antibody from polyGA protein. This protocol eliminates possible masking of the GA protein with the GA treatment antibody. PolyGA concentrations were interpolated from the standard curve using 60X-GA expressed in HEK293 cells and expressed as pg/mg total protein.
SDS/TCEP methods for eliminating protein masking by the treatment antibody:
The murine chimeric chα-GA1 antibody was spiked into HEK293 cell lysates expressing 60 GA repeats. The samples were analyzed by an MSD sandwich assay using a third murinized human monoclonal antibody (GA3-mIgG2a) and a fourth human monoclonal GA antibody (GA4-hIgG1) to detect GA protein (Figure S11D). A decrease in signal was observed in samples pre-incubated with chGA1, indicating that the chGA1 antibody can mask epitopes (Figure S11E, red bars). In contrast, identical samples incubated in 2% SDS, 1 mM TCEP at 95 °C for 5 minutes showed comparable GA protein signals in the presence or absence of antibody, demonstrating that the SDS/TCEP treatment removed detection interference by the antibody (Figure S11E, blue and green bars). Interestingly, the signal is increased with this treatment (black bar: no treatment, blue bars: with treatment) suggesting that SDS/TCEP treatment exposes more GA epitopes for MSD detection, perhaps by unfolding/disrupting poly-GA aggregates. All processed tissue samples from mice taken down at 43 weeks or iPSC-derived motor neurons were subjected to SDS/TCEP treatment.
MSD assays for detecting GA targeting antibodies in mouse brain lysates.
Brain lysates were solubilized in RIPA lysis buffer and protein concentrations were assessed by BCA assay. Meso scale discovery (MSD) immunoassay was performed following manufacturer’s instructions. Briefly, MSD plates were coated with rabbit polyclonal GA antibody (H3164), rabbit polyclonal GP antibody (H3154) [Zu et al. 2013], or GA8 peptide and incubated at 4 °C overnight. The following day, plates were washed with 1X Tris buffered saline + 0.02% Tween (TBST), then blocked in 3% bovine serum albumin (BSA) in TBST for 1 hour at room temperature (RT) on a horizontal shaker at 600 rpm. The plates were washed in TBST, then ~150μg of protein was loaded to each well. The plates were sealed and incubated for 2 hours at RT on a horizontal shaker at 600rpm. Plates were then washed three times in TBST and thoroughly dried before the addition of the anti-mouse detection antibody (MSD Cat# R32AC-1) in 1% BSA. Plates were sealed and placed on a horizontal shaker for 1 hour at 600 rpm. Following three washes in TBST, 1X Read Buffer T (MSD Cat# R92TC-2) was added to each well and plates were immediately read using the Meso Scale Discovery QUICKPLEX SQ120. Background correction was performed through subtraction of value by lysate on 1X PBS-coated wells. Standardization was achieved by dividing resultant values by total protein concentration and this data was visualized using GraphPad Prism 8.
Transcript level determination.
RT-PCR was performed following the previously reported protocol (Y. Liu et al. 2016). Briefly, total RNA was isolated from mouse cerebellum and spinal cord tissue or GFP-alt(GA)60 expressing HEK293T cells and TRIM21 KD cells with different antibodies treatment with TRIzol (Invitrogen). DNA contamination was eliminated by DNase I (Ambion) treatment at 37 °C for 30 min and followed by heat inactivation at 75 °C for 10 min or with TURBO™ DNase (Invitrogen, cat# AM1907) and followed by inactivation with DNase inactivation reagent. Total RNA was reverse-transcribed using SuperScript III RT kit (Invitrogen) and random-hexamer primers (Applied Biosystems). Primers for Exon 1a and antisense transcripts were previously optimized and reported in (Y. Liu et al. 2016). Quantitative RT-PCR was performed using the PowerSYBR system (Applied Biosystems) using the AB Step One Plus Real time PCR system following manufacturer’s protocol.
Primers used in GFP-alt(GA)60 transcript quantification:
eGFP(forward): 5’-AGTCCGCCCTGAGCAAAGA-3’;
eGFP (reverse): 5’-TCCAGCAGGACCATGTGATC-3’;
GAPDH (forward): 5’-CCTGCACCACCAACTGCTTA-3’;
GAPDH (reverse): 5’-GGCCATCCACAGTCTTCTGAG-3’.
Proteasome activity determination.
T98 cells in a 6-well plate were transfected with 2 μg of alt(GA)60 plasmid or control vector using Lipofectamine™ 3000 following the manufacturer’s protocol. Transfected cells were treated with antibodies at final concentration of 100 nM. After 48 h, cells were harvested and homogenized in 100–150 μl of lysis buffer containing 10 mM phosphate (pH 7.4), 5 mM MgCl2 and 0.5% triton X-100 without proteinase inhibitors. To measure proteasome activity, 10 μl of cell lysate was subjected to proteasome assay using proteasome activity fluorometric assay kit (BioVision, cat# K245) following the manufacturer’s protocol. Proteasome activity in antibody treated cells were normalized to control cells: untreated cells transfected with control vector or alt(GA)60 construct.
Generation of TRIM21 KD cell line.
sgRNAs against TRIM21 gene were designed and cloned into gRNA cloning vector from Addgene plasmid #41824. These vectors were transfected into HEK293T cells and later single-cell sorting into 96-well plates was performed. Screening of knockdown cells lines was done using western blotting against TRIM21 protein using antibody Ro/SSA antibody (D-12, Santa Cruz Biotechnology, 1:500). TRIM21 sgRNA: 5’-CATGTTGGCTAGCTGTCGAT-3’; 5’-CAATCGACAGCTAGCCAACA-3’; 5-GAAACACCGTGACCACGCCA-3’.
Effects of α-GA1 treatment on GFP-GA levels in HEK293T cells and TRIM21 KD cells.
HEK293T or TRIM21 KD cells were transfected with 500 ng of plasmids (GFP-alt(GA)60 or control vector). After 4 h transfection, cells were washed with 1x PBS and treated with antibodies at concentration of 100 nM. After 2 days, cells were harvested, washed three times with 1x PBS, then homogenized in RIPA buffer (1% sodium deoxycholate, 150 mM NaCl, 1% Triton-X 100, 50 mM Tris pH 7.5, 1X proteinase inhibitors). Approximately 5 μg of total protein was loaded on precast gel (4–12% Bis-Tris, Criterion) and transferred to a nitrocellulose membrane (Amersham). GFP signal was detected using α-GFP antibody (Santa Cruz Biotechnology, sc8334, 1:2000) and normalized to GAPDH signal (α-GAPDH antibody, Millipore, MAB374, 1:5000).
To examine the clearance of GFP-GA levels in HEK293T cells under the conditions in which proteasome or autophagy is inhibited, GFP-alt(GA)60 transfected HEK293T cells were co-treated with chα-GA1 (100 nM) and 1% DMSO which was used to dissolve autophagy and proteasome inhibitors, or 1 mM 3-methyladenine (3MA, Sigma Aldrich, M9281), or 1 μM MG132 (Sigma Aldrich, M8699).
Fluorescence non-canonical amino acid tagging and proximity ligation assay (FUNCAT-PLA) for protein turnover determination.
Protein turnover of GFP-GA was determined following the steps that were optimized based on previously published protocols (Dieterich et al. 2010, tom Dieck et al. 2015). Approximately 50,000 T98 cells were seeded in a well of 8-well chamber (Thermo Scientific, cat#154534). Cells were treated with chα-GA1 (100 nM) 16 h before transfection. Cells were transfected with 300 ng GFP-alt(GA)60 plasmids using Lipofectamine 3000 following the manufacturer’s protocol. After 4 h, transfection cocktail was replaced by DMEM supplemented with 10% FBS and chα-GA1 (100 nM) was added to cells. After overnight, cells were washed with warmed pulse medium (methionine-free DMEM supplemented with 10% FBS) (Gibco, cat# 21013024), and then treated with pulse medium containing 2 mM azidohomoalanine (AHA) (Invitrogen, cat# C10102). After 30 min pulsing, cells were washed with warm chasing medium (2 mM methionine containing DMEM supplemented with 10% FBS). Next cells were treated with chα-GA1 (100 nM) in chasing medium. Cells were fixed at t = 0, 6, 9, 24, and 29 h for determining half-life (t1/2) of labeled proteins. For fixation, cells were incubated with 10% neutral buffered formalin for 30 min at RT, washed three times with 1X PBS, and then stored in 1X PBS at 4 °C until samples were processed further.
All washing steps were performed at RT, 5 min for each wash. For detecting newly synthesized GFP-GA, cells were permeabilized with 0.5% Triton X-100 in 1X PBS for 30 min at RT and then washed three times with 1X PBS. Cells were incubated with 4% normal goat serum prepared freshly in 1X PBS at RT for 20 min. After washed three times with 1X PBS, cells were incubated with a click reaction mixture containing 200 μM triazole ligand Tris((1-benzyl-1H-1,2,3-triazol-4-yl)methyl)amine (TBTA), 25 μM biotin alkyne tag, 500 μM TCEP and 200 μM CuSO4 in 1X PBS (pH 7.5) for 16 h at RT in dark. Cells were washed three times with 0.5% Triton X-100 in 1X PBS. Cells were blocked with the blocking solution (Sigma Aldrich, cat# DUO92008) for 20 min at RT. Next, cells were incubated with mouse anti-GFP antibody (1:500, Santa Cruz, cat# sc9996) and rabbit anti-biotin antibody (1:500, Cell Signaling, cat# 5597S) prepared in 1% normal goat serum in 1X PBS for 1 h at RT. After three time washing with 1X PBS, cells were incubated with mouse minus PLA (1:10, Sigma Aldrich, cat# DUO92004) and rabbit plus PLA (1:10, Sigma Aldrich, cat# DUO92002) in blocking solution for 1 h at 37 °C in a wet chamber. Cells were washed three times with 1X PBS and exposed to ligation mixture (Sigma Aldrich, cat# DUO92008) for 40 min at 37 °C. Ligation was terminated by washing three time with PBST (1X PBS containing 0.05% Tween-20). Cells then were incubated with amplification mixture (Sigma Aldrich, cat# DUO92008) for 100 min at 37 °C in dark. Cells were washed three times with 1X PBS and mounted with gold Prolong containing DAPI. Images were taken using Zeiss LSM 880 and analyzed using ImageJ. Signal of AHA labeled GFP-GA proteins was quantitatively measured in ≥ 12 fields that were randomly taken for each time point of chasing.
Proximity ligation assay (PLA) for TRIM21 and human antibody interaction study.
Approximately 50,000 T98 cells were seeded in a well of 8-well chamber (Thermo Scientific, cat#154534). Cells were transfected with 300 ng GFP-alt(GA)60 plasmids using Lipofectamine 3000 following the manufacturer’s protocol. After 2.5 h, transfection cocktail was replaced with DMEM containing 10% FBS. Cells were treated with chα-GA1 (100 nM) for overnight. Cells then were fixed in 10% neural buffered formalin for 30 min at RT and washed three times with 1X PBS. All washing steps were performed with 1X PBS (pH 7.5) at RT with 5 min for each wash. Cells were permeabilized with 0.5% Triton X-100 diluted in 1X PBS (pH 7.5) and washed three times. Cells were blocked with 1% normal goat serum (NGS) diluted in 1X PBS for 20 min at RT and then incubated with or without rabbit anti-TRIM21 antibody (1:500, Abcam, cat# ab91423) diluted in 1% NGS for 1 h at 37 °C. Cells were washed three times and then incubated with mouse minus PLA (1:10, Sigma Aldrich, cat# DUO92004) and rabbit plus PLA (1:10, Sigma Aldrich, cat# DUO92002) in blocking solution (Sigma Aldrich, cat# DUO92008) for 1 h at 37 °C in a wet chamber. Cells were washed three times and then exposed to ligation mixture (Sigma Aldrich, cat# DUO92008) for 40 min at 37 °C. Ligation was terminated by washing three times with PBST (1X PBS containing 0.05% Tween-20). Cells then were incubated with amplification mixture (Sigma Aldrich, cat# DUO92008) for 100 min at 37 °C in dark. Cells were washed three times and mounted with gold Prolong containing DAPI. Images were taken using Zeiss LSM 880 and analyzed using ImageJ.
Live cell imaging and GFP-GA aggregates, LC3B and proteasome 26S subunit inclusion.
Approximately 40,000 of T98 cells per well were seeded in a grid-500 μ-slide 8-well chamber (Ibidi). In the following day, cells in each well were transfected with 300 ng GFP-alt(GA)60 constructs using Lipofectamine 3000 following the manufacturer’s protocols. After 4 h, transfection cocktail was removed, cells were washed 4 times with PBS and incubated with DMEM containing 10% FBS. Cells were treated with antibody chα-GA1, chα-GP1, chα-GA2, or IgG control (60 nM). For live cell imaging, cells in specific location in the chamber were tracked under confocal microscope LSM 880 (Zeiss) over 1, 2, 4, 6, and 24 h. For 24 h aggregate quantification, GFP-GA aggregates were measured by GFP aggregate signal normalized to total GFP signal. After 48 h incubation, cells were fixed with 4% PFA and stained to assess target engagement of the human antibody, GFP-GA aggregates, LC3B, PSMC4 inclusion. LC3B or proteasome 26S subunit were probed using α-LC3B antibody (Novus Biologicals, NB100–2220, 1:500) or α-PSMC4 antibody (Bethyl lab, A303–850A, 1:500). Quantification of GFP-GA aggregates, LC3B, or proteasome 26S subunit inclusion was measured by area or count of aggregate/inclusion signal normalized to transfected cells.
Effects of α-GA1 treatment on GFP-GR aggregates.
T98 cells were transfected with 300 ng total DNA constructs (GFP-alt(GR)60 and alt(GA)60 or GFP-alt(GR)60 and control construct). After 4 h incubation with transfection cocktail, cells were washed and treated with α-GA1 or IgG control (60 nM) in DMEM containing 10% FBS. After 24 h, cell images were taken using confocal microscope LSM 880 for GFP and bright-field channels. Levels of GFP-GR aggregates were quantitatively measured by GFP aggregate signal normalized to total GFP signal.
Toxicity and cell viability
T98 cells were transfected at 60% confluency with 700 ng of 3xFLAG-Alt(GA)60 or control vector using Lipofectamine 2000 (Invitrogen) following manufacturer’s protocol. Cells were plated in 96 well plates and treated with chimeric antibodies (1 μM), and experiments were performed in quadruplicate for each experimental condition. Cell death was measured by quantifying the amount of lactate dehydrogenase (LDH) released into the media, using the CytoTox 96 nonradioactive cytotoxicity assay (Promega) according to the manufacturer’s protocol. Absorbance was measured at 490 nm. Cell survival was measured using the MTT assay (Affymetrix) using the manufacturer’s protocol. The absorbance was measured at 595 nm.
Quantification and Statistical Analysis
Quantification.
All the quantification steps were performed by two blinded investigators and on multiple replicates for each experiment. α-GA, α-GP, GFAP, GFP-GR, GFP-GA, NeuN, RNA foci positive staining was counted by setting thresholds using ImageJ (National Institute of Health). α-ChAT positive motor neurons and α-GA and α-GP-positive aggregates at 16 weeks were quantified by two investigators using the cell counter plugin in Image J. Quantification for GA, GP, ChAT and NeuN was performed on three serial sections 20–35 μm apart for each animal.
Co-localization quantification.
Co-localization of LC3B inclusions and GA aggregates was quantitatively measured using the COLOC program (Zeiss LSM880) with the crosshair setting following the instruction in https://www.zeiss.com/content/dam/Microscopy/Downloads/Pdf/FAQs/zen-aim_colocalization.pdf. The pixel area reported in the region 3, which indicates co-localization, per number of cells was reported for each sample and then normalized to the PBS treated C9 sample.
Statistical analysis.
Information on number (n) values and what n represents (e.g. animals, biological replicates) and definition of center and dispersion and precision measures [e.g. mean, median, standard deviation (SD), standard error of the mean (SEM)] can be found in the figures and/or figure legends. GraphPad Prism 7 was used to perform all the statistical analyses listed in the manuscript with p<0.05 considered statistically significant. The significance values in figures were indicated as follows: ns=not significant p>0.05, * (p≤0.05); ** (p≤0.01); ***(p≤0.001); **** (p≤0.0001). Survival in different treatment cohorts of the C9-BAC mice was plotted using the Kaplan-Meier method and statistical comparison was performed using the Log Mantel-Cox test. One-way ANOVA with Tukey, Dunnett, Sidak, Holm-Sidak or Benjamini statistical hypothesis testing for multiple comparisons to compare differences amongst various treatment groups was used in DigiGait for comparison among treatment groups and control cohorts, open-field, GFAP, NeuN, ChAT, C9-RAN protein aggregates and toxicity experiments. Unpaired two-tailed t-test was used for identification of significant DigiGait parameters between each treatment group and the control group (PBS NT or PBS-C9).
Data and Code Availability
The published article includes all datasets generated or analyzed during this study.
Supplementary Material
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Donkey anti-Human IgG, Fcγ Specific, HRP | Jackson ImmunoResearch | Cat#709–035-098; RRID:AB_2340494 |
| Goat anti-Mouse IgG (H+L), HRP | Jackson ImmunoResearch | Cat#115–035-003; RRID:AB_10015289 |
| Goat anti-Mouse IgG2a, HRP | Southern Biotech | Cat#1081–05; RRID:AB_2736843 |
| Mouse anti-V5 | Thermo Fisher Scientific | Cat#R960–25; RRID:AB_2556564 |
| Mouse anti-FLAG | Sigma Aldrich | Cat#F3165; RRID:AB_256529 |
| Rabbit anti-V5 | Sigma Aldrich | Cat#V8137, RRID:AB_261889 |
| DyLight 549 Goat Anti-Human IgG | Jackson ImmunoResearch | Cat#109–505-097; RRID:AB_2337813 |
| Goat anti-Mouse IgG, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A11001; RRID:AB_2534069 |
| Donkey anti-Rabbit IgG, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A21206; RRID:AB_2535792 |
| Cy5 Goat anti-Mouse IgG | Jackson ImmunoResearch | Cat#115–175-146; RRID:AB_2338713 |
| Rabbit anti-p62 | BioLegend | Cat#647701, RRID:AB_2028561 |
| Rabbit anti-LC3B | Cell Signaling Technology | Cat#2775; RRID:AB_915950 |
| Rabbit anti-PSMC4 | Proteintech | Cat#11389–1-AP;RRID:AB_2300373 |
| Biotinylated Donkey anti-Human IgG | Jackson ImmunoResearch | Cat#709–065-149; AB_2340507 |
| Biotinylated Donkey anti-Rabbit IgG | Jackson ImmunoResearch | Cat#711–065-152; AB_2340593 |
| Mouse anti-NeuN | Millipore | Cat#MAB377; RRID:AB_2298772 |
| Goat anti-ChAT | Millipore | Cat#AB144P; RRID:AB_2079751 |
| Rabbit anti-GFAP | BioGeneX | Cat#PU020-UP |
| Rabbit anti-GA | (Liu et al. 2016) | Cat#27B11A6; RRID:AB_2571626 |
| Rabbit anti-GP | (Zu et al. 2013) | Cat#H3154; RRID:AB_2571627 |
| Goat anti-Mouse IgG | Meso Scale Discovery | Cat#R32AC; RRID:AB_2783819 |
| Mouse anti-Human TRIM21 | Santa Cruz Biotechnology | Cat#sc-25351; RRID:AB_628286 |
| Rabbit anti-GFP | Santa Cruz Biotechnology | Cat#sc-8334; RRID:AB_641123 |
| Mouse anti-GAPDH | Millipore | Cat#MAB374; RRID:AB_2107445 |
| Mouse anti-GFP | Santa Cruz Biotechnology | Cat#sc-9996; RRID:AB_627695 |
| Rabbit anti-Biotin | Cell Signaling Technology | Cat#5597; RRID:AB_10828011 |
| Donkey anti-Mouse MINUS PLA | Sigma Aldrich | Cat#DUO92004; RRID:AB_2713942 |
| Donkey anti-Rabbit PLUS PLA | Sigma Aldrich | Cat#DUO92002; RRID:AB_2810940 |
| Rabbit anti-Human SSA1 | Abcam | Cat#Ab91423; RRID:AB_2050342 |
| Rabbit anti-LC3 | Novus | Cat#NB100–2220; RRID:AB_10003146 |
| Rabbit anti-PSMC4 | Bethyl | Cat#A303–850A; RRID:AB_2620201 |
| Human α-GA1 | This paper | N/A |
| Human α-GA2 | This paper | N/A |
| Human α-GP1 | This paper | N/A |
| Chimeric α-GA1 (chα-GA1) | This paper | N/A |
| Chimeric α-GA2 (chα-GA2) | This paper | N/A |
| Chimeric α-GP1 (chα-GP1) | This paper | N/A |
| Aglycosylated chimeric α-GA1 (chα-GA1algy) | This paper | N/A |
| Biological Samples | ||
| Human autopsy tissue | University of California at San Diego | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ProbeQuant G-50 Micro Columns | GE Healthcare | Cat#28903408 |
| Amersham Rapid-hyb buffer | GE Healthcare | Cat#RPN1636 |
| Methionine-free DMEM | Gibco | Cat#21013024 |
| L-Azidohomoalanine (AHA) | Thermo Fisher Scientific | SKU#C10102 |
| TBTA | Sigma Aldrich | SKU#678937 |
| TCEP | Sigma Aldrich | SKU#C4706 |
| Critical Commercial Assays | ||
| Proteasome Activity Assay | BioVision | Cat#K245 |
| Duolink In Situ Detection Assay | Sigma Aldrich | Cat#DUO92008 |
| Experimental Models: Cell Lines | ||
| Human: SH-SY5Y | Sigma Aldrich | Cat#94030304; RRID:CVCL_0019 |
| Human: HEK293T | ATCC | Cat#CRL-3216; RRID:CVCL_0063 |
| Human: T98 | ATCC | Cat#CRL-1690; RRID:CVCL_0556 |
| Human iPSC: CS0NKCiALS-n1 | Cedars-Sinai repository | N/A |
| Human iPSC: CS0BUUiALS-n3 | Cedars-Sinai repository | N/A |
| Human iPSC: CS8PAAiCTR-n2 | Cedars-Sinai repository | N/A |
| Human iPSC: CS0594iCTR-LBCn2 | Cedars-Sinai repository | N/A |
| Human iPSC: OXC9–02-02 | (Dafinca et al. 2016) | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse, C9-BAC | (Liu et al. 2016), The Jackson Laboratory | Stock No: 029099 RRID:IMSR_JAX:029099 |
| Mouse, FVB/NJ | The Jackson Laboratory | Stock#001800; RRID:IMSR_JAX:001800 |
| Mouse, C57BL/6J | The Jackson Laboratory | Stock#000664; RRID:IMSR_JAX:000664 |
| Software and Algorithms | ||
| Prism | GraphPad | https://www.graphpad.com/ |
| Octet Systems Software | Pall ForteBio LLC | https://www.fortebio.com/ |
| Biacore T200 Evaluation v3.0 | GE Healthcare | https://www.gelifesciences.com/ |
| Excel, 2016 | Microsoft | https://www.microsoft.com |
| CellSens | Olympus | https://www.olympus-lifescience.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Other | ||
| Octet RED96 | Pall ForteBio LLS | https://www.fortebio.com/ |
| Biacore T200 | GE Healthcare | https://www.gelifesciences.com/ |
| LSM 800 | Zeiss | https://www.zeiss.com |
| VS120 slide scanner | Olympus | https://www.olympus-lifescience.com |
| BX51 | Olympus | https://www.olympus-lifescience.com |
| QuickPlex SQ 120 | MESO | https://www.mesoscale.com |
Acknowledgements:
Non-Author Contributions: We thank N. Cavegn, P. Borter and N. Glassl for technical assistance, Dr. Christoph Hock for blood samples, Dr. J. Ravits for tissue samples, D. Sareen, C. Svendsen, and the iPS team at Cedars-Sinai for guidance on iPS motor neuron differentiation, X. Tang, W. Zhou, and Y. Huo for technical assistance for iPS maintenance and differentiation.
Funding: We thank the National Institutes of Health (RO1 NS098819), Target ALS, the ALS Association, the Packard Center, and the Muscular Dystrophy Association for support.
References:
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus-Hernandez M, van Blitterswijk MM, Jansen-West K, Paul JW 3rd, Rademakers R, Boylan KB, Dickson DW and Petrucelli L (2013) ‘Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS’, Neuron, 77(4), 639–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittar A, Sengupta U and Kayed R (2018) ‘Prospects for strain-specific immunotherapy in Alzheimer’s disease and tauopathies’, NPJ Vaccines, 3, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YJ, Jeng US, Chiang YL, Hwang IS and Chen YR (2016) ‘The Glycine-Alanine Dipeptide Repeat from C9orf72 Hexanucleotide Expansions Forms Toxic Amyloids Possessing Cell-to-Cell Transmission Properties’, J Biol Chem, 291(10), 4903–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Wang S, Mestre AA, Fu C, Makarem A, Xian F, Hayes LR, Lopez-Gonzalez R, Drenner K, Jiang J, Cleveland DW and Sun S (2018) ‘C9ORF72 GGGGCC repeat-associated non-AUG translation is upregulated by stress through eIF2alpha phosphorylation’, Nat Commun, 9(1), 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JD, Pattamatta A and Ranum LPW (2018) ‘Repeat-associated non-ATG (RAN) translation’, J Biol Chem, 293(42), 16127–16141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JD and Ranum LP (2017) ‘New developments in RAN translation: insights from multiple diseases’, Curr Opin Genet Dev, 44, 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti FL, Barone R, Fermo SL, Giliberto C, Patti F, Gambardella A, Quattrone A and Zappia M (2011) ‘sporadic motor neuron disease in a familial novel SOD1 mutation: incomplete penetrance or chance association?’, Amyotroph Lateral Scler, 12(3), 220–2. [DOI] [PubMed] [Google Scholar]
- Congdon EE, Gu J, Sait HB and Sigurdsson EM (2013) ‘Antibody uptake into neurons occurs primarily via clathrin-dependent Fcγamma receptor endocytosis and is a prerequisite for acute tau protein clearance’, J Biol Chem, 288(49), 35452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dafinca R, Scaber J, Ababneh N, Lalic T, Weir G, Christian H, Vowles J, Douglas AG, Fletcher-Jones A, Browne C, Nakanishi M, Turner MR, Wade-Martins R, Cowley SA and Talbot K (2016) ‘C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia’, Stem Cells, 34(8), 2063–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day JW, Dalton JC, Liu HY and Ranum LPW (2004) ‘Reduced penetrance of spinocerebellar ataxia type 8 is partly explained by volumetric MRI of cerebellum’, Neurology, 62(7), A26–A27. [Google Scholar]
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J, Kouri N, Wojtas A, Sengdy P, Hsiung GY, Karydas A, Seeley WW, Josephs KA, Coppola G, Geschwind DH, Wszolek ZK, Feldman H, Knopman DS, Petersen RC, Miller BL, Dickson DW, Boylan KB, Graff-Radford NR and Rademakers R (2011) ‘Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS’, Neuron, 72(2), 245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterich DC, Hodas JJ, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA and Schuman EM (2010) ‘In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons’, Nat Neurosci, 13(7), 897–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dithmer M, Hattermann K, Pomarius P, Aboul Naga SH, Meyer T, Mentlein R, Roider J and Klettner A (2016) ‘The role of Fc-receptors in the uptake and transport of therapeutic antibodies in the retinal pigment epithelium’, Exp Eye Res, 145, 187–205. [DOI] [PubMed] [Google Scholar]
- Fernandez MV, Kim JH, Budde JP, Black K, Medvedeva A, Saef B, Deming Y, Del-Aguila J, Ibanez L, Dube U, Harari O, Norton J, Chasse R, Morris JC, Goate A, group N-L Ncrad f. s. and Cruchaga C (2017) ‘Analysis of neurodegenerative Mendelian genes in clinically diagnosed Alzheimer Disease’, PLoS Genet, 13(11), e1007045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher AJ, Mallery DL, Watkinson RE, Dickson CF and James LC (2015) ‘sequential ubiquitination and deubiquitination enzymes synchronize the dual sensor and effector functions of TRIM21’, Proc Natl Acad Sci U S A, 112(32), 10014–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foss S, Watkinson R, Sandlie I, James LC and Andersen JT (2015) ‘TRIM21: a cytosolic Fc receptor with broad antibody isotype specificity’, Immunol Rev, 268(1), 328–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez-Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC, Petrucelli L, Kim HJ, Gao FB and Taylor JP (2015) ‘GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport’, Nature, 525(7567), 129–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galimberti D, Arosio B, Fenoglio C, Serpente M, Cioffi SM, Bonsi R, Rossi P, Abbate C, Mari D and Scarpini E (2014) ‘Incomplete penetrance of the C9ORF72 hexanucleotide repeat expansions: frequency in a cohort of geriatric non-demented subjects’, J Alzheimers Dis, 39(1), 19–22. [DOI] [PubMed] [Google Scholar]
- Gendron TF, van Blitterswijk M, Bieniek KF, Daughrity LM, Jiang J, Rush BK, Pedraza O, Lucas JA, Murray ME, Desaro P, Robertson A, Overstreet K, Thomas CS, Crook JE, Castanedes-Casey M, Rousseau L, Josephs KA, Parisi JE, Knopman DS, Petersen RC, Boeve BF, Graff-Radford NR, Rademakers R, Lagier-Tourenne C, Edbauer D, Cleveland DW, Dickson DW, Petrucelli L and Boylan KB (2015) ‘Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers’, Acta Neuropathologica, 130(4), 559–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitler AD and Tsuiji H (2016) ‘There has been an awakening: Emerging mechanisms of C9orf72 mutations in FTD/ALS’, Brain Res, 1647, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KM, Glineburg MR, Kearse MG, Flores BN, Linsalata AE, Fedak SJ, Goldstrohm AC, Barmada SJ and Todd PK (2017) ‘RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response’, Nat Commun, 8(1), 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Q, Lehmer C, Martinez-Sanchez A, Rudack T, Beck F, Hartmann H, Perez-Berlanga M, Frottin F, Hipp MS, Hartl FU, Edbauer D, Baumeister W and Fernandez-Busnadiego R (2018) ‘In Situ Structure of Neuronal C9orf72 Poly-GA Aggregates Reveals Proteasome Recruitment’, Cell, 172(4), 696–705 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Dalton JC, Moseley ML, Gardner KL, Bird TD, Ashizawa T, Seltzer WK, Pandolfo M, Milunsky A, Potter NT, Shoji M, Vincent JB, Day JW and Ranum LPW (2004) ‘spinocerebellar ataxia type 8: Molecular genetic comparisons and haplotype analysis of 37 families with ataxia’, American Journal of Human Genetics, 75(1), 3–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis-Downes M, Seelman A, Stauffer JE, Jafar-Nejad P, Drenner K, Schulte D, Chun S, Sun S, Ling SC, Myers B, Engelhardt J, Katz M, Baughn M, Platoshyn O, Marsala M, Watt A, Heyser CJ, Ard MC, De Muynck L, Daughrity LM, Swing DA, Tessarollo L, Jung CJ, Delpoux A, Utzschneider DT, Hedrick SM, de Jong PJ, Edbauer D, Van Damme P, Petrucelli L, Shaw CE, Bennett CF, Da Cruz S, Ravits J, Rigo F, Cleveland DW and Lagier-Tourenne C (2016) ‘Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs’, Neuron, 90(3), 535–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosravi B, Hartmann H, May S, Mohl C, Ederle H, Michaelsen M, Schludi MH, Dormann D and Edbauer D (2017) ‘Cytoplasmic poly-GA aggregates impair nuclear import of TDP-43 in C9orf72 ALS/FTLD’, Hum Mol Genet, 26(4), 790–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura T, Jain A, Choi SW, Mandell MA, Schroder K, Johansen T and Deretic V (2015) ‘TRIM-mediated precision autophagy targets cytoplasmic regulators of innate immunity’, J Cell Biol, 210(6), 973–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y and McKnight SL (2014) ‘Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells’, Science, 345(6201), 1139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KH, Zhang P, Kim HJ, Mitrea DM, Sarkar M, Freibaum BD, Cika J, Coughlin M, Messing J, Molliex A, Maxwell BA, Kim NC, Temirov J, Moore J, Kolaitis RM, Shaw TI, Bai B, Peng J, Kriwacki RW and Taylor JP (2016) ‘C9orf72 Dipeptide Repeats Impair the Assembly, Dynamics, and Function of Membrane-Less Organelles’, Cell, 167(3), 774–788 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu WJ, Ye L, Huang WF, Guo LJ, Xu ZG, Wu HL, Yang C and Liu HF (2016) ‘p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation’, Cellular & Molecular Biology Letters, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Pattamatta A, Zu T, Reid T, Bardhi O, Borchelt DR, Yachnis AT and Ranum LP (2016) ‘C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD’, Neuron, 90(3), 521–34. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzl S, Mori K, Weng SM, Haass C, Kretzschmar HA, Edbauer D and Neumann M (2013) ‘Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico-pathological correlations’, Acta Neuropathol, 126(6), 859–79. [DOI] [PubMed] [Google Scholar]
- Majounie E, Renton AE, Mok K, Dopper EG, Waite A, Rollinson S, Chio A, Restagno G, Nicolaou N, Simon-Sanchez J, van Swieten JC, Abramzon Y, Johnson JO, Sendtner M, Pamphlett R, Orrell RW, Mead S, Sidle KC, Houlden H, Rohrer JD, Morrison KE, Pall H, Talbot K, Ansorge O, Chromosome, A. L. S. F. T. D. C., French research network on, F. F. A., Consortium I, Hernandez DG, Arepalli S, Sabatelli M, Mora G, Corbo M, Giannini F, Calvo A, Englund E, Borghero G, Floris GL, Remes AM, Laaksovirta H, McCluskey L, Trojanowski JQ, Van Deerlin VM, Schellenberg GD, Nalls MA, Drory VE, Lu CS, Yeh TH, Ishiura H, Takahashi Y, Tsuji S, Le Ber I, Brice A, Drepper C, Williams N, Kirby J, Shaw P, Hardy J, Tienari PJ, Heutink P, Morris HR, Pickering-Brown S and Traynor BJ (2012) ‘Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study’, Lancet Neurol, 11(4), 323–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann DM, Rollinson S, Robinson A, Bennion Callister J, Thompson JC, Snowden JS, Gendron T, Petrucelli L, Masuda-Suzukake M, Hasegawa M, Davidson Y and Pickering-Brown S (2013) ‘Dipeptide repeat proteins are present in the p62 positive inclusions in patients with frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72’, Acta Neuropathol Commun, 1, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May S, Hornburg D, Schludi MH, Arzberger T, Rentzsch K, Schwenk BM, Grasser FA, Mori K, Kremmer E, Banzhaf-Strathmann J, Mann M, Meissner F and Edbauer D (2014) ‘C9orf72 FTLD/ALS-associated Gly-Ala dipeptide repeat proteins cause neuronal toxicity and Unc119 sequestration’, Acta Neuropathol, 128(4), 485–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan WA, Falcon B, Vaysburd M, Clift D, Oblak AL, Ghetti B, Goedert M and James LC (2017) ‘Cytosolic Fc receptor TRIM21 inhibits seeded tau aggregation’, Proceedings of the National Academy of Sciences of the United States of America, 114(3), 574–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwan WA, Tam JC, Watkinson RE, Bidgood SR, Mallery DL and James LC (2013) ‘Intracellular antibody-bound pathogens stimulate immune signaling via the Fc receptor TRIM21’, Nat Immunol, 14(4), 327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Gronke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IO, Pietrzyk J, Cleverley K, Nicoll AJ, Pickering-Brown S, Dols J, Cabecinha M, Hendrich O, Fratta P, Fisher EM, Partridge L and Isaacs AM (2014) ‘C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins’, Science, 345(6201), 1192–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E, Schmid B, Kretzschmar HA, Cruts M, Van Broeckhoven C, Haass C and Edbauer D (2013) ‘The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS’, Science, 339(6125), 1335–8. [DOI] [PubMed] [Google Scholar]
- O’Rourke JG, Bogdanik L, Muhammad AK, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S, Ho R, Bell S, Carmona S, Simpkinson M, Lall D, Wu K, Daughrity L, Dickson DW, Harms MB, Petrucelli L, Lee EB, Lutz CM and Baloh RH (2015) ‘C9orf72 BAC Transgenic Mice Display Typical Pathologic Features of ALS/FTD’, Neuron, 88(5), 892–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan JA, Sun Y, Jiang YP, Bott AJ, Jaber N, Dou Z, Yang B, Chen JS, Catanzaro JM, Du C, Ding WX, Diaz-Meco MT, Moscat J, Ozato K, Lin RZ and Zong WX (2016) ‘TRIM21 Ubiquitylates SQSTM1/p62 and Suppresses Protein Sequestration to Regulate Redox Homeostasis’, Mol Cell, 62(1), 149–51. [DOI] [PubMed] [Google Scholar]
- Peters OM, Cabrera GT, Tran H, Gendron TF, McKeon JE, Metterville J, Weiss A, Wightman N, Salameh J, Kim J, Sun H, Boylan KB, Dickson D, Kennedy Z, Lin Z, Zhang YJ, Daughrity L, Jung C, Gao FB, Sapp PC, Horvitz HR, Bosco DA, Brown SP, de Jong P, Petrucelli L, Mueller C and Brown RH Jr. (2015) ‘Human C9ORF72 Hexanucleotide Expansion Reproduces RNA Foci and Dipeptide Repeat Proteins but Not Neurodegeneration in BAC Transgenic Mice’, Neuron, 88(5), 902–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita VM, Kaivorinne AL, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Consortium I, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaev Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ and Traynor BJ (2011) ‘A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD’, Neuron, 72(2), 257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes DA and Isenberg DA (2017) ‘TRIM21 and the Function of Antibodies inside Cells’, Trends in Immunology, 38(12), 916–926. [DOI] [PubMed] [Google Scholar]
- Sances S, Bruijn LI, Chandran S, Eggan K, Ho R, Klim JR, Livesey MR, Lowry E, Macklis JD, Rushton D, Sadegh C, Sareen D, Wichterle H, Zhang SC and Svendsen CN (2016) ‘Modeling ALS with motor neurons derived from human induced pluripotent stem cells’, Nat Neurosci, 19(4), 542–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schludi MH, May S, Grasser FA, Rentzsch K, Kremmer E, Kupper C, Klopstock T, German Consortium for Frontotemporal Lobar, D., Bavarian Brain Banking A, Arzberger T and Edbauer D (2015) ‘Distribution of dipeptide repeat proteins in cellular models and C9orf72 mutation cases suggests link to transcriptional silencing’, Acta Neuropathol, 130(4), 537–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM and Sandrock A (2016) ‘The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease’, Nature, 537(7618), 50–6. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV and Vinters HV (2010) ‘Astrocytes: biology and pathology’, Acta Neuropathol, 119(1), 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Zhang Y, Gendron TF, Bauer PO, Chew J, Yang WY, Fostvedt E, Jansen-West K, Belzil VV, Desaro P, Johnston A, Overstreet K, Oh SY, Todd PK, Berry JD, Cudkowicz ME, Boeve BF, Dickson D, Floeter MK, Traynor BJ, Morelli C, Ratti A, Silani V, Rademakers R, Brown RH, Rothstein JD, Boylan KB, Petrucelli L and Disney MD (2014) ‘Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS’, Neuron, 83(5), 1043–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao MH and Morrison SL (1989) ‘studies of Aglycosylated Chimeric Mouse-Human Igg - Role of Carbohydrate in the Structure and Effector Functions Mediated by the Human-Igg Constant Region’, Journal of Immunology, 143(8), 2595–2601. [PubMed] [Google Scholar]
- Taylor JP, Brown RH Jr. and Cleveland DW (2016) ‘Decoding ALS: from genes to mechanism’, Nature, 539(7628), 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- tom Dieck S, Kochen L, Hanus C, Heumuller M, Bartnik I, Nassim-Assir B, Merk K, Mosler T, Garg S, Bunse S, Tirrell DA and Schuman EM (2015) ‘Direct visualization of newly synthesized target proteins in situ’, Nat Methods, 12(5), 411–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB, Pasinelli P, Ichida JK and Trotti D (2014) ‘Antisense proline-arginine RAN dipeptides linked to C9ORF72-ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death’, Neuron, 84(6), 1213–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergard T, Jensen BK, Wen X, Cai J, Kropf E, Iacovitti L, Pasinelli P and Trotti D (2016) ‘Cell-to-Cell Transmission of Dipeptide Repeat Proteins Linked to C9orf72-ALS/FTD’, Cell Rep, 17(3), 645–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Abdallah A, Li Z, Lu Y, Almeida S and Gao FB (2015) ‘FTD/ALS-associated poly(GR) protein impairs the Notch pathway and is recruited by poly(GA) into cytoplasmic inclusions’, Acta Neuropathol, 130(4), 525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YJ and Watts RJ (2013) ‘Developing therapeutic antibodies for neurodegenerative disease’, Neurotherapeutics, 10(3), 459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S, Gupta S, Thomas MA, Hong I, Chiu SL, Huganir RL, Ostrow LW, Matunis MJ, Wang J, Sattler R, Lloyd TE and Rothstein JD (2015) ‘The C9orf72 repeat expansion disrupts nucleocytoplasmic transport’, Nature, 525(7567), 56–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Gendron TF, Grima JC, Sasaguri H, Jansen-West K, Xu YF, Katzman RB, Gass J, Murray ME, Shinohara M, Lin WL, Garrett A, Stankowski JN, Daughrity L, Tong J, Perkerson EA, Yue M, Chew J, Castanedes-Casey M, Kurti A, Wang ZS, Liesinger AM, Baker JD, Jiang J, Lagier-Tourenne C, Edbauer D, Cleveland DW, Rademakers R, Boylan KB, Bu G, Link CD, Dickey CA, Rothstein JD, Dickson DW, Fryer JD and Petrucelli L (2016) ‘C9ORF72 poly(GA) aggregates sequester and impair HR23 and nucleocytoplasmic transport proteins’, Nat Neurosci, 19(5), 668–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Jansen-West K, Xu YF, Gendron T, Bieniek K, Lin WL, Sasaguri H, Caulfield T, Hubbard J, Daughrity L, Chew J, Belzil VV, Prudencio M, Stankowski JN, Castanedes-Casey M, Whitelaw E, Ash PEA, DeTure M, Rademakers R, Boylan KB, Dickson DW and Petrucelli L (2014a) ‘Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress’, Acta Neuropathologica, 128(4), 505–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YJ, Jansen-West K, Xu YF, Gendron TF, Bieniek KF, Lin WL, Sasaguri H, Caulfield T, Hubbard J, Daughrity L, Chew J, Belzil VV, Prudencio M, Stankowski JN, Castanedes-Casey M, Whitelaw E, Ash PE, DeTure M, Rademakers R, Boylan KB, Dickson DW and Petrucelli L (2014b) ‘Aggregation-prone c9FTD/ALS poly(GA) RAN-translated proteins cause neurotoxicity by inducing ER stress’, Acta Neuropathol, 128(4), 505–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Lehmer C, Michaelsen M, Mori K, Alterauge D, Baumjohann D, Schludi MH, Greiling J, Farny D, Flatley A, Feederle R, May S, Schreiber F, Arzberger T, Kuhm C, Klopstock T, Hermann A, Haass C and Edbauer D (2017) ‘Antibodies inhibit transmission and aggregation of C9orf72 poly-GA dipeptide repeat proteins’, EMBO Mol Med, 9(5), 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Gibbens B, Doty NS, Gomes-Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA, Nan Z, Forster C, Low WC, Schoser B, Somia NV, Clark HB, Schmechel S, Bitterman PB, Gourdon G, Swanson MS, Moseley M and Ranum LP (2011) ‘Non-ATG-initiated translation directed by microsatellite expansions’, Proc Natl Acad Sci U S A, 108(1), 260–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Liu Y, Banez-Coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramony SH, Ostrow LW, Rothstein JD, Troncoso JC and Ranum LP (2013) ‘RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia’, Proc Natl Acad Sci U S A, 110(51), E4968–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all datasets generated or analyzed during this study.