

Abstract
De novo protein design has been successful in expanding the natural protein repertoire. However, most de novo proteins lack biological function, presenting a major methodological challenge. In vaccinology, the induction of precise antibody responses remains a cornerstone for next-generation vaccines. Here, we present a protein design algorithm, termed TopoBuilder, with which we engineered epitope-focused immunogens displaying complex structural motifs. Both in mice and non-human primates, cocktails of three de novo-designed immunogens induced robust neutralizing responses against the respiratory syncytial virus (RSV). Furthermore, the immunogens refocused pre-existing antibody responses towards defined neutralization epitopes. Overall, our design approach opens the possibility of targeting specific epitopes for vaccine and therapeutic antibody development, and more generally will be applicable to design de novo proteins displaying complex functional motifs.
Introduction
The computational design of novel proteins from first principles has revealed a variety of rules for the accurate design of structural features in de novo proteins (1–4). However, the de novo design of functional proteins remains far more challenging (5, 6). A commonly used strategy to design functional proteins is to transplant structural motifs from other proteins to pre-existing or de novo protein scaffolds (7–9). In nearly all cases previously reported, the transplanted motifs mediated protein–protein interactions. These structural motifs are common in the natural protein repertoire, such as linear helical segments, which allows their grafting without extensive backbone adjustments (7, 8). Most protein functional sites, however, are not contained within regular single segments in protein structures, but arise from the three-dimensional arrangement of multiple and often irregular, structural elements supported by the overall architecture of the protein structure (10–12). Thus, the development of computational approaches to endow de novo proteins with irregular and multi-segment motifs is crucial to expand their function and the scope of their application.
Protein design has sparked hopes in the field of rational vaccinology, in particular, to elicit targeted neutralizing antibody (nAb) responses (9, 13). Although many potent nAbs have been identified and structurally characterized in complex with their target antigens, the design of immunogens that elicit precise and focused antibody responses remains a major challenge (14, 15). To date, structure-based immunogen design efforts have mostly focused on modifying viral fusion proteins, through conformational stabilization, silencing of non-neutralizing epitopes and germline-targeting approaches (16). Unlike respiratory syncytial virus (RSV), several major human pathogens only display a limited number of broadly neutralizing epitopes, surrounded by strain-specific, non-neutralizing, or in some cases, disease-enhancing epitopes (17–19). Thus, one of the central goals for vaccine development is to elicit antibody responses with precisely defined epitope specificities, and, in some cases, constrained molecular features (e.g. antibody lineage, CDR length or binding angle) (20–24).
The difficulty in developing immunogens that can elicit antibodies specific for a restricted subset of epitopes on a single protein continues to be a critical barrier to rational vaccine design. Previous studies have sought to elicit epitope-specific responses using peptide-based approaches (25) or epitope-scaffolds (9, 13, 26–28). Leveraging computational design, the antigenic site II of the RSV fusion protein (RSVF), a linear helix–turn–helix motif, was transplanted onto a heterologous protein scaffold, which was shown to elicit nAbs in non-human-primates (NHPs) after repeated boosting immunizations (9). Despite this proof-of-principle showing induction of functional antibodies using a computationally designed immunogen, several major caveats have emerged: the neutralization titers observed in immunogenicity studies are inconsistent; and the computational approach is not suitable for structurally complex epitopes.
To address these limitations, here we used de novo design approaches to engineer epitope-focused immunogens mimicking irregular and discontinuous RSV neutralization epitopes (site 0 (29) and IV (30), Fig 1). We designed a trivalent cocktail formulation (“Trivax”) consisting of a previously published immunogen for site II (13), and newly designed immunogens mimicking sites 0 and IV. In vivo, Trivax induced a balanced antibody response against all three epitopes, resulting in consistent levels of serum neutralization in six out of seven NHPs. Upon priming with RSVF, the computationally designed immunogens boosted site-specific antibodies, resulting in an improved antibody quality. Our approach enables the targeting of specific epitopes for vaccine and therapeutic antibody development and more broadly will be applicable to design de novo proteins displaying complex functional motifs.
Fig 1. Computational design of RSV epitope-focused immunogens.
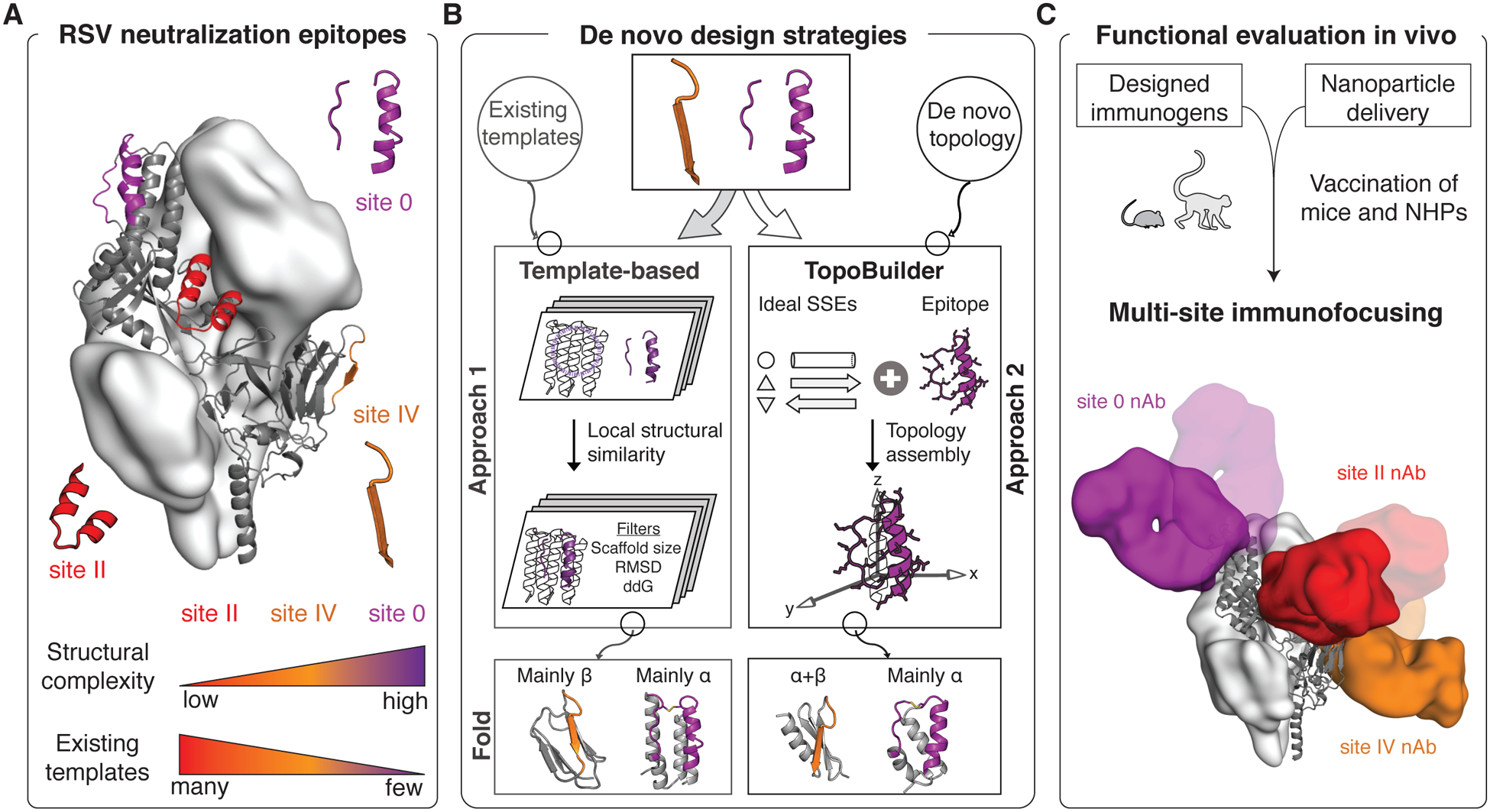
(A) Prefusion RSVF structure (PDB 4JHW) with sites 0, II and IV highlighted. (B) Computational protein design strategies. Approach 1: Design templates were identified in the PDB, followed by in silico folding and design. Approach 2: A template-free design approach was developed (“TopoBuilder”) to tailor the protein topology to the motif’s structural constraints. Bottom: Computational models of designed immunogens. (C) Cocktail formulations of designed immunogens displayed on nanoparticles elicit nAbs focused on three non-overlapping epitopes. SSEs: secondary structure elements; α: alpha-helix; β: beta strand; ddG: computed binding energy.
Results
De novo design of immunogens presenting structurally complex epitopes
The computational design of proteins mimicking structural motifs has been performed previously by first identifying compatible protein scaffolds, which then serve as design templates to graft the motifs (7, 26–28, 31, 32). This approach, referred as template-based design, has been used to transplant functional sites to both structures from the natural repertoire (26–28, 31, 32) or de novo-designed proteins (7). Although most studies focused on linear, regular binding motifs, one study successfully grafted a structurally complex HIV epitope into an existing protein scaffold. However, both the overall structure and sequence of the template remained mostly native (31).
Here, we sought to design accurate mimetics of RSVF neutralization epitopes based on de novo proteins and evaluate their functionality in immunization studies. We chose antigenic sites 0 and IV (Fig 1A), which are both targeted by potent nAbs, and have a high structural complexity: site 0 is a discontinuous epitope consisting of a kinked 17-residue alpha helix and a disordered, seven-residue loop (29, 33), while site IV presents an irregular six-residue bulged β-strand (30).
In a first effort we used a template-based de novo design approach relying on Rosetta FunFolDes (34) to fold and design scaffolds for sites IV and 0. Given the structural complexity of these sites, few structures in the Protein Data Bank (PDB) matched the backbone configuration of the epitopes, even using loose structural criteria (fig S1). Briefly, our best computational design for site IV (S4_1.1), based on a domain excised from prefusion RSVF (preRSVF), bound with weak affinity to the target nAb 101F (KD > 85 μM). After in vitro evolution, we obtained a double mutant (S4_1.5) that bound 101F with a KD of 35 nM and was thermostable up to 65 °C (Fig 2B–D and fig S2–3). For site 0, we used a designed helical repeat protein (PDB 5CWJ (35)) as design template. Our first computational design showed a KD of 1.4 μM to the target D25 nAb, which we improved to a KD of 5 nM upon several truncations, and iterative rounds of computational design and in vitro evolution (Fig 2 and fig S4–5).
Fig 2. Template-based computational design.
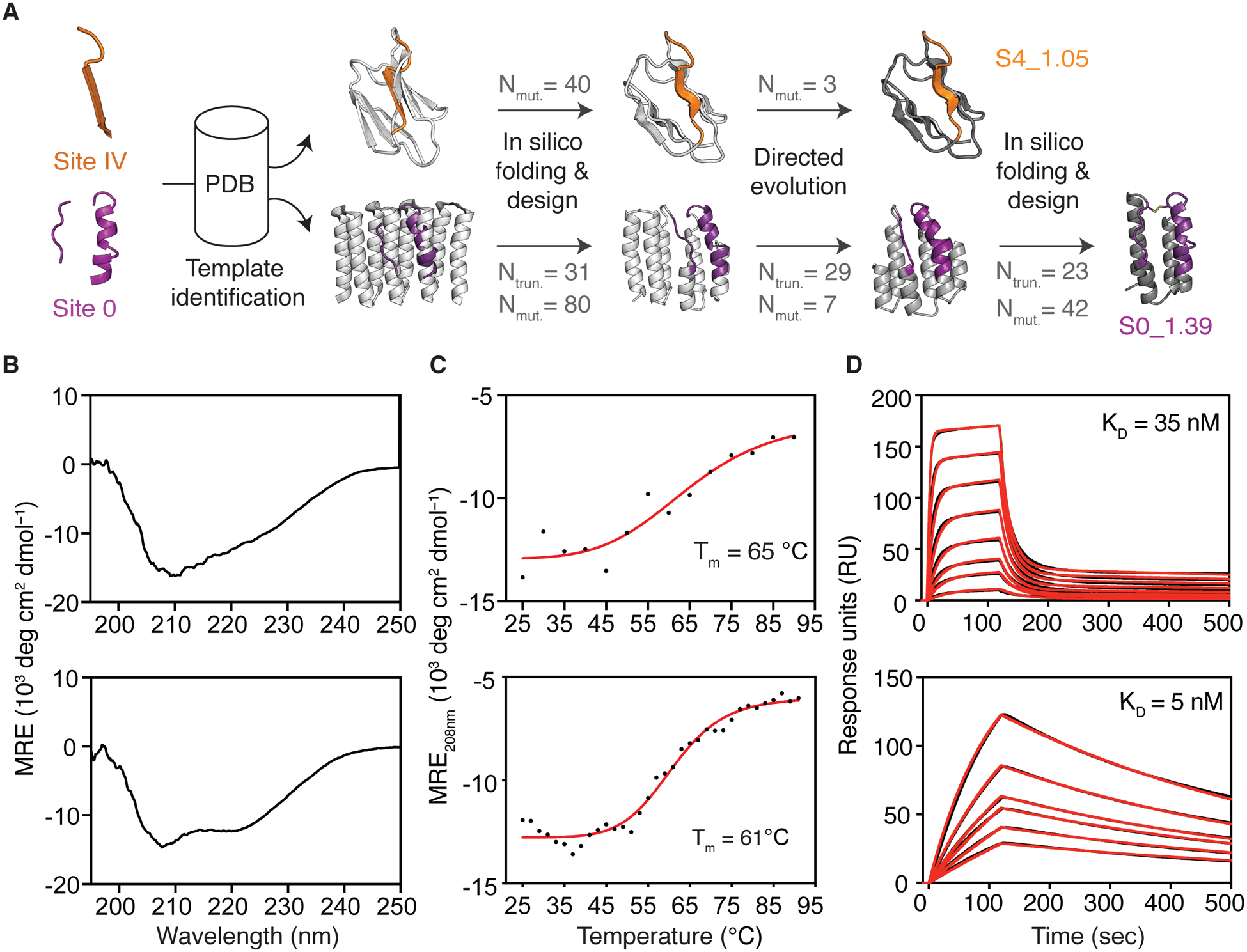
(A) Templates with structural similarity to sites IV and 0 were identified by native domain excision or loose structural matching, followed by in silico folding, design and directed evolution. Computational models of intermediates and final designs (S4_1.5 and S0_1.39) are shown, the number of mutations (Nmut) and truncated residues (Ntrun) are indicated for each step. (B) CD spectra measured at 20 °C of S4_1.5 (top) and S0_1.39 (bottom), are in agreement with the expected secondary structure content of the design models. (C) Thermal melting curves measured by CD in presence of reducing agent. (D) Binding affinity measured by SPR against target antibodies 101F (top) and D25 (bottom). Sensorgrams are shown in black and fits in red. CD: circular dichroism; Tm: melting temperature; SPR: surface plasmon resonance.
The template-based approach led to designs that presented several desired features (e.g. stability and antibody binding), however important limitations emerged during the design process: (1) extensive in vitro evolution optimization was required; (2) binding affinities to target nAbs were one to two orders of magnitude lower than those of the viral protein (preRSVF); (3) suboptimal template topologies constrained the epitope accessibility (fig S6).
In order to address these limitations, we developed a template-free design protocol, TopoBuilder, that generates tailor-made protein topologies to stabilize complex functional motifs. The TopoBuilder consists of three stages (Fig 3A). (1) Topological sampling in 2D space: to quickly define the fold space compatible with the target structural motif, we use the αβα-Form topology definition scheme, a string-based descriptor that allows the extensive enumeration of multilayer protein topologies with alternating secondary structure elements and all possible connections between the secondary structural elements (36, 37). Putative folds are then selected according to basic topological rules (e.g. lack of crossover loops and chain directionality of the functional motif). Together, this allowed the definition of the fold space for a given design task, thereby overcoming a main hurdle in de novo design approaches. (2) 3D projection and parametric sampling: the selected 2D topologies are projected into the 3D space by assembling idealized secondary structures (SSE) around the fixed functional motif. These 3D structures, referred to as “sketches”, are further refined by coarsely sampling structural features of the fold (e.g., distances and orientations between SSEs) using parametric sampling. (3) Flexible backbone sampling and sequence design: to refine the structural features of the sketches at the all-atom level and design sequences that stabilize these structures, we use Rosetta FunFolDes as described before (9, 34).
Fig 3. Template-free de novo design strategy.
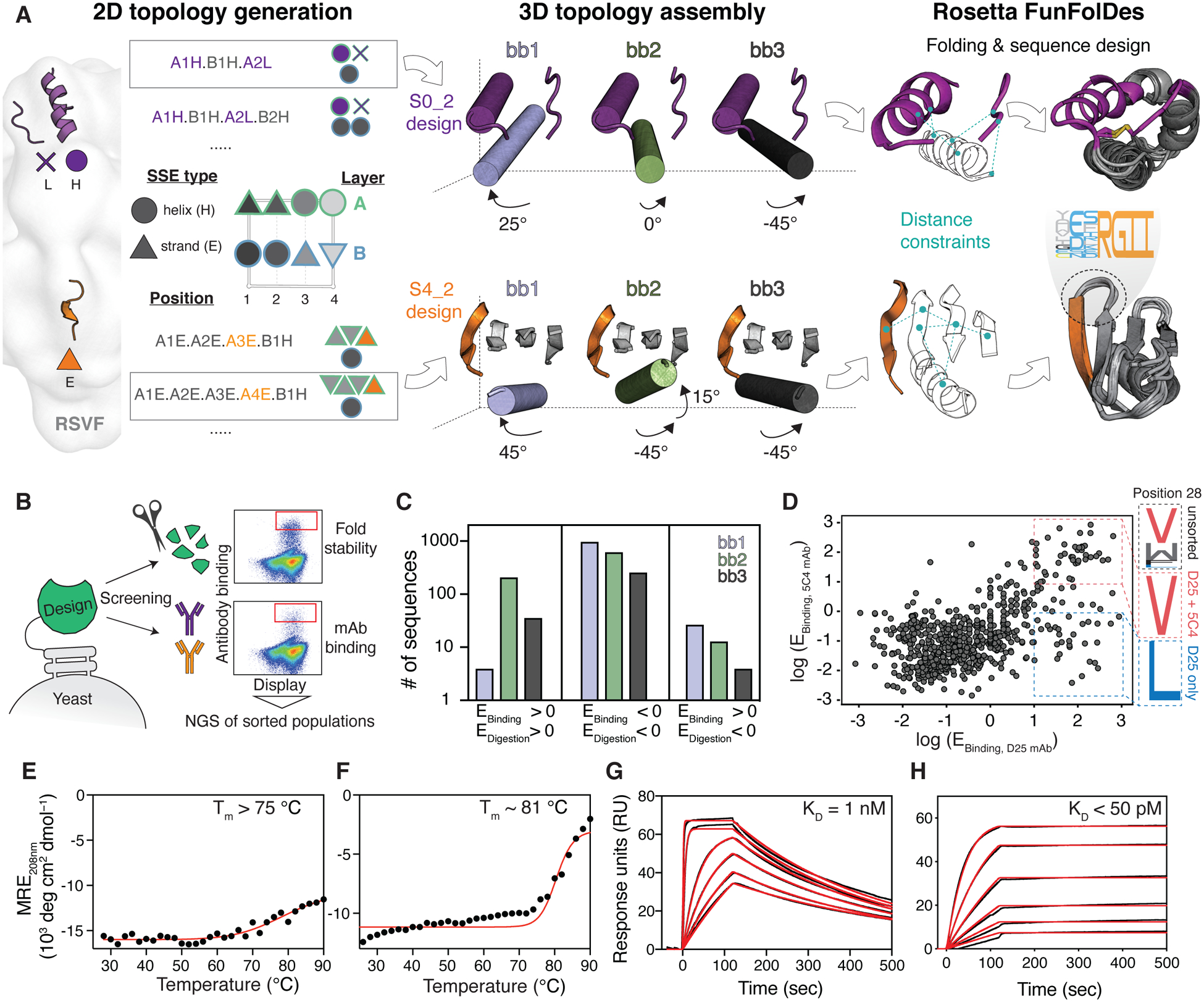
(A) Protein topologies that are compatible with each motif are enumerated in the 2D space. Selected topologies are then projected into the 3D space using idealized SSEs, and their relative orientation is sampled parametrically. Distance constraints are derived from selected topologies to guide in silico folding and sequence design using Rosetta. (B) Designed sequences were screened for high-affinity binding and resistance to chymotrypsin to select stably folded proteins, as revealed by next-generation sequencing (NGS). (C) For the S4_2 design series, enrichment analysis revealed a strong preference for one of the designed helical orientations (S4_2_bb2, green) to resist protease digestion and to bind with high affinity to 101F. (D) To ensure epitope integrity, S0_2_bb3 was screened for binding to both D25 and 5C4. Sequences highly enriched for both D25 and 5C4 binding show convergent sequence features in the critical core position 28 of the site 0 scaffold. (E-F) Thermal melting curves measured by CD for best designs (S4_2.45 (E) and S0_2.126 (F)) showing high thermostability. (G-H) Dissociation constants (KD) of S4_2.45 to 101F (G) and S0_2.126 to D25 (H) as measured by SPR. E: enrichment.
To present antigenic site IV, we designed a fold composed of a β-sheet with four antiparallel strands and one helix (Fig 3A), referred to as S4_2 fold. Within the S4_2 topology, we generated three structural variants (S4_2_bb1–3), by sampling three distinct orientations of the helical element, varying both orientations and lengths to optimize the packing interactions with the β-sheet. Sequences generated from two structural variants (S4_2_bb2 and S4_2_bb3) showed a strong propensity to recover the designed structures in Rosetta ab initio simulations (fig S7).
To evaluate our design approach, we screened a library of designed sequences using yeast display and applied two selective pressures: binding to the 101F antibody and resistance to the nonspecific protease chymotrypsin (Fig 3B), an effective method to digest partially unfolded proteins (7, 38). To reveal structural and sequence determinants of designs that led to stable folds and high-affinity binding to 101F, we performed next-generation sequencing of populations sorted under different conditions, retrieving stability and binding scores for each design. We found that S4_2_bb2-based designs were preferentially enriched over the bb1 and bb3 design series, showing that subtle topological differences in the design template can have substantial impact on function and stability (Fig 3C). Thirteen out of the 14 best-scoring S4_2 variants, bearing between 1 and 38 mutations compared to each other, were successfully purified and biochemically characterized (fig S8). The designs showed mixed alpha/beta CD spectra and bound to 101F with affinities ranging from 1 nM to 200 nM (fig S8). The best variant, S4_2.45 was well folded according to CD and NMR, and only showed partial unfolding even at 90 °C (Fig 3E and fig S9). S4_2.45 showed a KD = 1 nM to the target antibody 101F (Fig 3G), in line with the preRSVF-101F interaction (KD = 4 nM).
Similarly, we built a minimal de novo topology to present the tertiary structure of the site 0 epitope. The choice for this topology was motivated by the native environment of site 0 in preRSVF, where it is accessible for antibodies with diverse angles of approach (33) (fig S6). We explored the topological space within the shape constraints of preRSVF and built three different helical orientations (S0_2_bb1–3) that supported the epitope segments. Rosetta ab initio folding predictions showed that only designs based on one topology (S0_2_bb3) presented funnel-shaped energy landscapes (fig S10). A set of computationally designed sequences based on the S0_2_bb3 template was screened in yeast under the selective pressure of two site 0-specific antibodies (D25 and 5C4), to ensure the presentation of the native epitope conformation. Deep sequencing of the double-enriched clones revealed that subtle sequence variants (e.g., position 28) are sufficient to change the antibody-binding properties of the designs, highlighting the challenges of designing functional proteins (Fig 3D). From the high-throughput screening, we biochemically characterized five sequences, bearing between 3 and 21 mutations compared to each other in a protein of 58 residues (fig S11). The design with best solution behaviour (S0_2.126) showed a CD spectrum of a predominantly helical protein, with extremely high thermostability (Tm = 81 °C, Fig 3F) and a well-dispersed HSQC NMR spectrum (fig S9). Strikingly, S0_2.126 bound with KDs of ~50 pM and 4 nM to D25 and 5C4, respectively, which is in line with the affinities of the nAbs to preRSVF (~150 pM and 13 nM for D25 and 5C4, respectively) (Fig 3H and fig S12).
Across a panel of site-specific human nAbs (39), S4_2.45 and S0_2.126 showed large affinity improvements compared to the first-generation designs, exhibiting a geometric mean affinity closely resembling that of the antibodies to preRSVF (fig S12). These results suggest that the immunogens designed using TopoBuilder were superior mimetics of sites IV and 0 as compared to the template-based designs.
De novo-designed topologies adopt the predicted structures with high accuracy
To evaluate the structural accuracy of the computational design approach, we solved the crystal structure of S4_2.45 in complex with 101F at 2.6 Å resolution. The structure closely resembled our design model, with a backbone RMSD of 1.5 Å (Fig 4A). The epitope was mimicked with an RMSD of 0.23 Å, and retained all essential interactions with 101F (Fig 4D–E and fig S13). Together, the structural data confirmed that we accurately presented an irregular beta strand, a common motif found in many protein–protein interactions (40), in a fully de novo-designed protein with sub-ångström accuracy.
Fig 4. Structural characterization of de novo-designed immunogens.
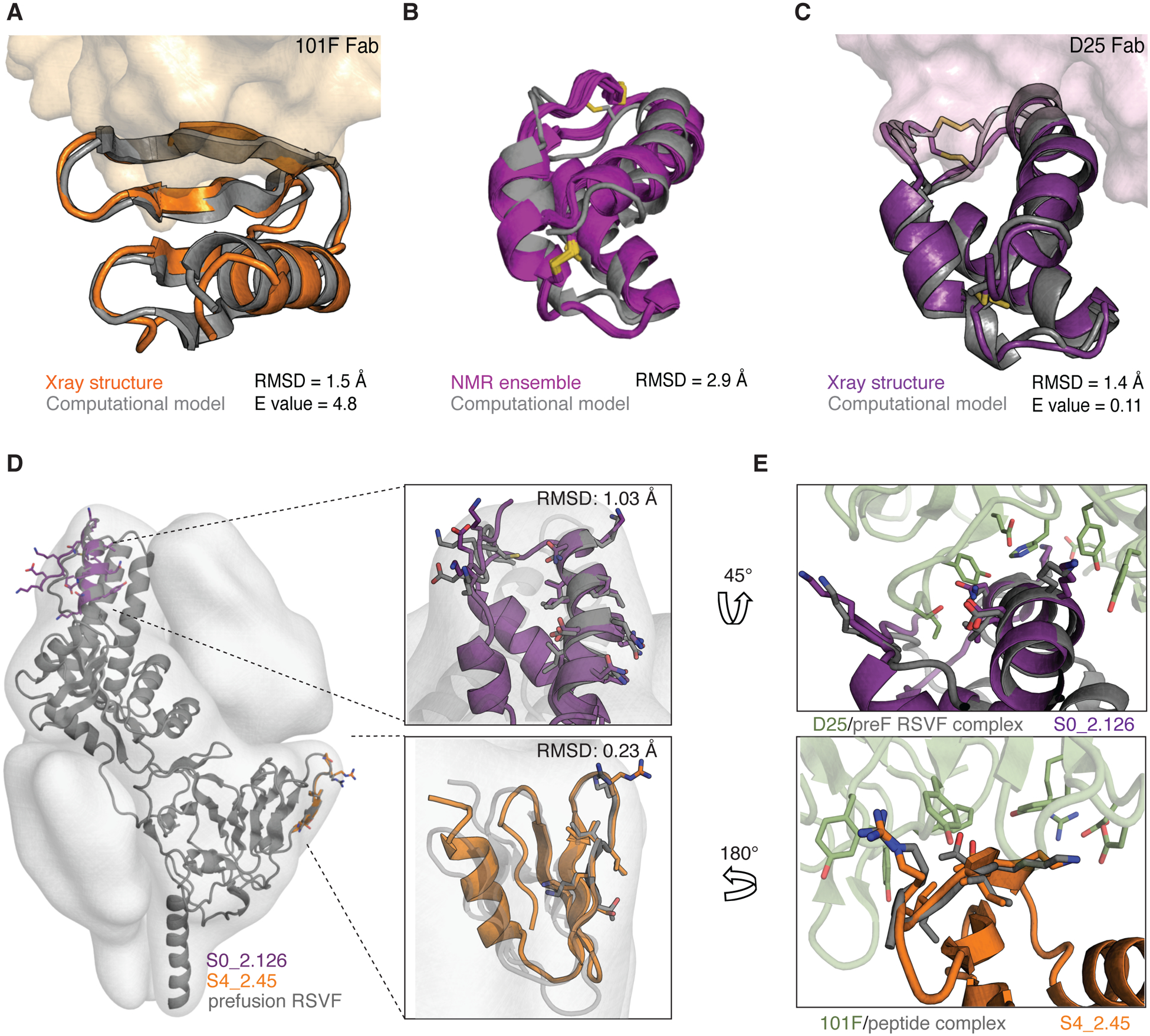
(A) Crystal structure of S4_2.45 (orange) bound to 101F Fab closely matches the design model (gray, RMSD = 1.5 Å). (B) NMR structural ensemble of S0_2.126 (purple) superimposed to the computational model (gray). The NMR structure shows overall agreement with the design model (backbone RMSD of 2.9 Å). (C) Crystal structure of S0_2.126 (purple) bound to D25 Fab closely resembles the design model (gray, RMSD = 1.4 Å). (D) Superposition of the preRSVF sites 0/IV and designed immunogens. Designed scaffolds are compatible with the shape constraints of preRSVF (surface representation). (E) Close-up view of the interfacial side-chain interactions between D25 (top) and 101F (bottom) with designed immunogens as compared to the starting epitope structures.
Next, we solved an unbound structure of S0_2.126 by NMR, confirming the accuracy of the designed fold with a backbone RMSD of 2.9 Å between the average structure and the computational model (Fig 4B). Additionally, we solved a crystal structure of S0_2.126 bound to D25 at a resolution of 3.0 Å. The structure showed backbone RMSDs of 1.4 Å to the design model and 1.03 Å over the discontinuous epitope compared to preRSVF (Fig 4C–E and fig S13). In comparison with native proteins, S0_2.126 showed exceptionally low core packing due to a large cavity (fig S14), but retained a very high thermal stability. The core cavity was essential for antibody binding and highlights the potential of de novo approaches to design small proteins hosting structurally challenging motifs and preserving cavities required for function (2).
Cocktails of designed immunogens elicit nAbs in vivo and reshape pre-existing immunity
Finally, we evaluated the ability of the designed antigens to elicit targeted nAb responses in vivo. Our rationale for combining site 0, II and IV immunogens in a cocktail formulation is that all three sites are non-overlapping in the preRSVF structure (fig S15), and thus might induce a more potent and consistent nAb response in vivo. To increase immunogenicity, each immunogen was multimerized on self-assembling protein nanoparticles. We chose the RSV nucleoprotein (RSVN), a self-assembling ring-like structure of 10–11 subunits, previously shown to be an effective carrier for the site II immunogen (S2_1.2) (13) and formulated a trivalent immunogen cocktail containing equimolar amounts of S0_1.39, S4_1.5 and S2_1.2 immunogen nanoparticles (“Trivax1”, fig S16). The fusion of S0_2.126 and S4_2.45 to RSVN yielded poorly soluble nanoparticles, prompting us to use ferritin particles for multimerization, with a 50% occupancy (~12 copies), creating a second cocktail comprising S2_1.2 in RSVN and the remaining immunogens in ferritin (“Trivax2”, fig S17).
In mice, Trivax1 elicited low levels of preRSVF cross-reactive antibodies, and sera did not show RSV neutralizing activity in most animals (fig S18). By contrast, immunization with Trivax2 (Fig 5A) induced robust levels of preRSVF cross-reactive serum levels (Fig 5B) and six of ten mice showed neutralizing activity (Fig 5C). In these mice, the serum antibody binding responses were equally directed against all three sites (site 0: 32% ± 6%, site II: 38% ± 7%, site IV: 30% ± 6%) (Fig 5D). This is notable, as in previous studies mice have been a difficult model to induce serum neutralization with scaffold-based immunogens (9, 13, 28). Furthermore, Trivax2 only presented ~14% of the preRSVF surface area to be targeted by the immune system. Although serum neutralization titers in mice remained substantially lower compared to the titers induced by preRSVF (Fig 5C), these results demonstrate that vaccine candidates composed of multiple de novo proteins can induce physiologically relevant neutralizing serum levels (defined as similar or higher in vitro neutralizing activity compared to clinically protective serum concentrations of palivizumab (41)).
Fig 5. Immunogenicity of Trivax2 in mice.
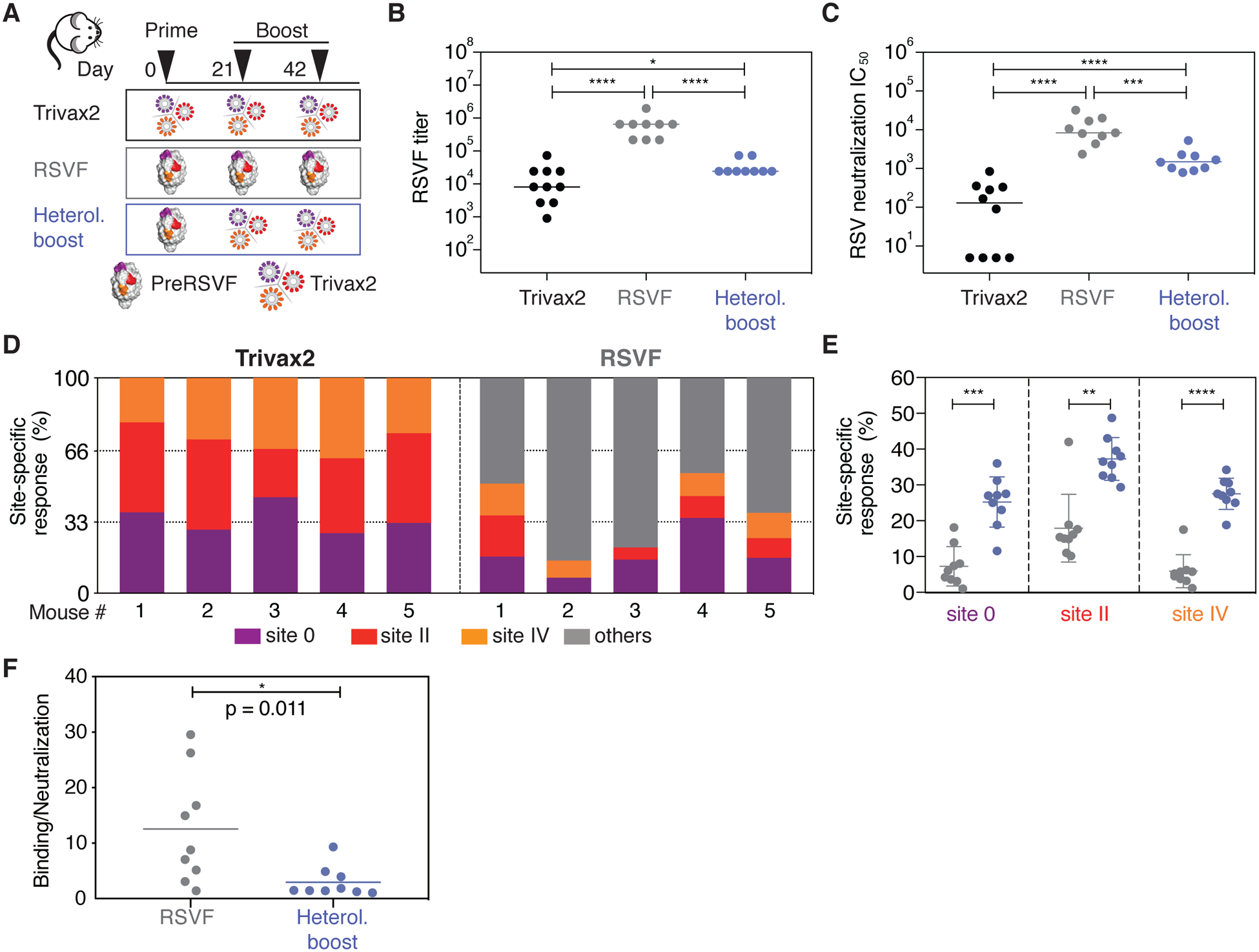
(A) Three groups of mice (n = 9–10) were immunized with Trivax2, preRSVF, or as heterologous prime-boost as indicated. (B) Binding titers measured against preRSVF using ELISA. (C) RSV serum neutralizing titers. (D) Serum composition following three immunizations with Trivax2 or preRSVF. Site-specific responses were dissected from serum at day 56 using competition ELISA. (E) Site-specific responses following preRSVF immunization compared to a heterologous prime-boost cohort, as measured by a competition SPR assay. On average, comparing Trivax2 with preRSVF, the response against sites 0, II, and IV increased 4.4-, 2.3-, and 5.3-fold, respectively. (F) Ratio of preRSVF binding to neutralizing antibody titers. Each symbol represents the ratio of binding IC50 to neutralization IC50 for an individual animal, the line represents the mean for each group. Data are representative from at least two independent experiments. Statistics were computed using Mann–Whitney U test, where * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Given the well-defined epitope specificities induced by the de novo-designed immunogens, we tested the potential of Trivax2 to boost site-specific responses following a priming immunization with preRSVF (Fig 5A). Strikingly, we found that Trivax2 boosting yielded significantly higher levels of site 0, II, and IV antibodies (4.4-, 2.3- and 5.3-fold, respectively) compared to boosting immunizations with preRSVF (Fig 5E). Although overall serum neutralization titers remained inferior to preRSVF boosting (Fig 5C), Trivax2 boosting resulted in a 4.2-fold lower ratio of binding to neutralizing antibodies, a common measure to assess antibody quality (42, 43) (Fig 5F). Thus, de novo-designed immunogens have unique potential to boost site-specific antibodies and to increase the quality of the antibody response compared to repeated boosting immunizations with a viral fusion protein.
In parallel, we performed an immunogenicity study in NHPs to test the trivalent cocktail in a closer-to-human antibody repertoire (Fig 6A). This experiment was designed to test the activity of Trivax1 in both RSV-naïve and -experienced animals to provide further insights into the ability of computationally designed immunogens to elicit focused antibody responses. The previously designed site II immunogen showed promise in NHPs, but the induced neutralizing titers were low and inconsistent across animals even after repeated immunizations (9). In contrast to mice, NHPs immunized with Trivax1 developed robust levels of RSVF cross-reactive serum titers in all animals (Fig 6B), and again antibodies induced were directed against all three epitopes (site 0: 23% ± 5%, site II: 51% ± 6%, site IV: 25% ± 5%) (Fig 6C). Strikingly, six of seven NHPs showed RSV neutralizing serum levels after a single boosting immunization (median IC50 = 312) (Fig 6D). Neutralization titers were maximal at day 84 (median IC50 = 408), and measurements were confirmed by an independent laboratory (fig S19).
Fig 6. Immunogenicity of Trivax1 in NHPs.
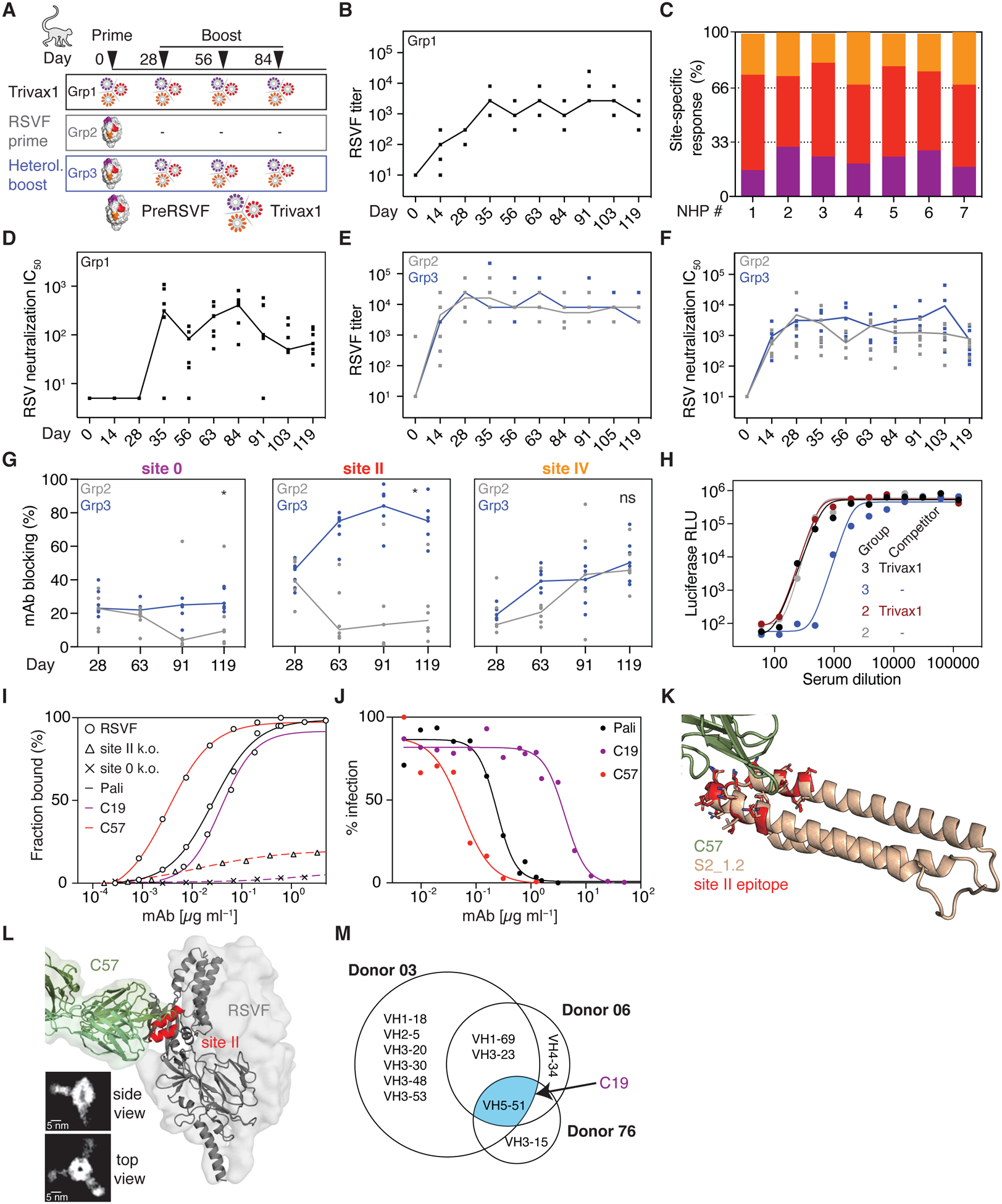
(A) NHP immunization scheme. (B) Trivax1 immunized NHPs developed robust titers cross-reacting with preRSVF. (C) Site-specific antibody responses as dissected by competition SPR. (D) RSV neutralization titers of group 1. (E) PreRSVF titers and (F) RSV neutralization titers in groups 2 (gray) and 3 (blue). (G) Dynamics of site-specific antibody levels. Site 0- and site II-specific titers were significantly higher in group 3 compared to 2 following Trivax1 boosting (* p < 0.05, Mann–Whitney U test). (H) RSV neutralization curves upon depletion of day 91 sera with site 0-, II-, and IV-specific scaffolds. (I) ELISA binding curves of isolated monoclonal antibodies C19 and C57 to preRSVF and site-specific knockouts, in comparison to palivizumab. (J) In vitro RSV neutralization of C19, C57, and palivizumab. (K) X-ray structure of C57 Fab fragment in complex with S2_1.2. (L) Model of C57 bound to preRSVF, as confirmed by negative-stain electron microscopy. Scale bar: 5 nm. (M) Lineage analysis (Venn diagram) of previously identified site 0 nAbs from three different human donors (39). The elicited site 0 nAb C19 is a close homolog of the human VH5–51 lineage (blue). Data are representative from three independent experiments.
Beyond naïve subjects, an overarching challenge for vaccine development against pathogens such as influenza virus, dengue virus and others, is to focus pre-existing immunity onto defined neutralization epitopes that can confer long-lasting protection (13, 22, 44). To mimic a serum response of broad specificity towards RSV, we immunized 13 NHPs with preRSVF. All animals developed strong preRSVF-specific titers (Fig 6E) and cross-reactivity with all the epitope-focused immunogens, indicating that epitope-specific antibodies were primed against all three epitopes (fig S20). Six of those preRSVF-primed animals did not receive boosting immunizations in order to follow the dynamics of epitope-specific antibodies over time (group 2). Seven of the preRSVF-primed animals were boosted three times with Trivax1 (group 3) (Fig 6A). Although preRSVF-specific antibody and neutralization titers remained statistically comparable in both groups (Fig 6E–F), we found that Trivax1 boosting significantly increased antibodies targeting site II and site 0, but not site IV (Fig 6G). In the non-boosted control group, site II and site 0 responses dropped from 37% and 17% at day 28 to 13% and 4% at day 91, respectively (Fig 6G). Compared to the control group, Trivax1 boosting resulted in 6.5-fold higher site II specific responses on day 91 (84% vs 13%), and 6.3-fold higher site 0-specific titers (25% vs 4%) (Fig 6G). By contrast, site IV-specific responses increased to similar levels in both groups, 43% and 40% in groups 2 and 3, respectively.
To evaluate the functional relevance of reshaping the serum antibody specificities, we depleted site 0-, II-, and IV-specific antibodies from pooled sera. In the Trivax1-boosted group, we observed a 60% drop in neutralizing activity as compared to only a 7% drop in the non-boosted control group (Fig 6H). Thus, epitope-focused immunogens can reshape a serum response of broad specificity towards a focused response that predominantly relies on site 0-, II-, and IV-specific antibodies for RSV neutralization.
Looking further into the molecular basis of the neutralizing activity triggered by the epitope-focused immunogens, we isolated two epitope-specific monoclonal Abs (mAbs) from Trivax1-immunized (group 1) animals using single B cell sorting. Through a panel of binding probes, we confirmed that one mAb targeted antigenic site II (C57) and the other site 0 (C19) (Fig 6I and fig S21). Importantly, both C57 and C19 neutralized RSV in vitro (IC50 = 0.03 μg ml−1 and IC50 = 4.2 μg ml−1, respectively). While C19 was less potent, antibody C57 neutralized RSV with approximately one order of magnitude higher potency compared to the clinically used antibody palivizumab, which is similar to the potency of motavizumab and previously reported antibodies induced by a site II epitope-scaffold (9) (Fig 6J).
To elucidate the molecular basis for the potent site II-mediated neutralization, we solved a crystal structure of C57 in complex with S2_1.2 at a resolution of 2.2 Å (Fig 6K and fig S13). C57 recognizes the site II epitope in its native conformation, with a full-atom RMSD of 1.21 Å between the epitope in S2_1.2 and site II in preRSVF. Negative stain EM of the complex C57-preRSVF further confirmed binding to site II, allowing binding of three Fabs per preRSVF trimer (Fig 6L).
RSV nAbs are not known for a strictly constrained VH usage as described for certain influenza and HIV broadly neutralizing antibodies (45, 46). However, an important milestone for computationally designed, epitope-focused immunogens is to elicit nAbs similar to those found in humans upon natural infection or vaccination. In one of the largest RSV antibody isolation campaigns, 30 site 0-specific RSV nAbs were isolated from three human donors (39), derived from 11 different VH genes (Fig 6M). Among those, the VH5–51 was the only lineage shared among all three donors, suggesting that it is a common precursor for site 0 nAbs in humans. We found that the closest human VH gene to C19 is indeed the VH5–51 lineage (89% sequence identity). Thus, computationally designed immunogens can elicit nAbs with similar properties to those found in humans after viral infection (fig S21).
In summary, the computational design strategies developed enabled the design of scaffolds presenting epitopes of unprecedented structural complexity with atomic level accuracy. Upon cocktail formulation, the de novo-designed proteins consistently induced RSV neutralization in naïve animals, mediated through three defined epitopes. In addition, the designed antigens were functional in a heterologous prime-boost immunization regimen, inducing more focused antibody responses towards selected, bona fide neutralization epitopes, and an overall increased quality of the antibody response.
Discussion & Conclusions
Here, we showcased two de novo design strategies to engineer protein scaffolds to present epitopes with high structural complexity. Using template-based de novo design, irregular and discontinuous epitopes were successfully stabilized in heterologous scaffolds. However, this design strategy required extensive optimization by in vitro evolution and the designs remained suboptimal regarding their biochemical and biophysical properties. Moreover, this approach lacks the ability to control the topological features of the designed proteins, constituting an important limitation for functional protein design.
To overcome these limitations, we developed the TopoBuilder. Compared to other approaches, the TopoBuilder has distinctive features and significant advantages to design de novo proteins with structurally complex motifs. First, it assembles topologies tailored to the structural requirements of the functional motif from the start of the design process, rather than through the adaptation (and often destabilization) of a protein structure to accommodate the functional site. Second, the topology assembly resulted in designed sequences that stably folded and bound with high affinity without requiring iterative rounds of optimization through directed evolution, as often necessary in computational protein design efforts (8, 47, 48).
As to the functional aspect of our design work, we have shown that computationally designed immunogens targeting multiple epitopes can induce physiologically relevant levels of functional antibodies in vivo. The elicitation of antibodies targeting conserved epitopes that can mediate broad and potent neutralization remains a central goal for vaccines against pathogens that have frustrated conventional vaccine development efforts (15). For RSV, the development of a prefusion-stabilized version of RSVF has yielded a superior antigen compared to its postfusion counterpart (42, 49), largely attributed to the fact that most preRSVF-specific antibodies are neutralizing (39, 50). Given that Trivax only presents a small fraction (14%) of the antigenic surface of preRSVF, the substantially lower serum titers, and consequently lower bulk serum neutralization elicited by Trivax may be expected, and will likely require significant optimization in terms of delivery and formulation to increase the magnitude of the response.
Nevertheless, we show that a cocktail of de novo-designed proteins induced relevant levels of serum neutralization in the majority of naïve mice and NHPs. Beyond bulk serum titers, Trivax offers an unprecedented level of control over antibody specificities to the single epitope level. In heterologous prime-boost immunization schemes, Trivax profoundly reshaped the serum composition, leading to increased levels of desirable antibody specificities and an overall improved quality of the antibody response. The ability to selectively boost subdominant nAbs targeting defined, broadly protective epitopes could overcome long-standing obstacles in vaccine development against pathogens such as influenza, where the challenge is to overcome established immunodominance hierarchies (51).
Altogether, this study provides a blueprint for the computational design of epitope-focused vaccines. Besides antigens for viral epitopes, the ability to stabilize structurally complex epitopes in a de novo protein with a defined protein topology may prove useful to elicit and isolate mAbs against tertiary epitopes with potentially unique allosteric or therapeutic properties. Beyond immunogens, our work presents a new approach for the design of de novo functional proteins, enabling the assembly of customized protein topologies tailored to structural and functional requirements of the motif. The ability to design de novo proteins presenting functional sites with high structural complexity will be broadly applicable to expand the structural and sequence repertoires, and above all, the functional landscape of natural proteins.
Supplementary Material
Acknowledgements:
We thank W.R. Schief, P. Gainza, S.T. Reddy and B. Lemaitre for helpful discussions and comments on the manuscript. We thank J.E. Crowe, Jr. for providing site IV antibodies. We thank several EPFL facilities: PTPSP (K. Lau, A. Reynaud, F. Pojer, D. Hacker, L. Durrer and S. Quinche) for protein expression and crystallography support; Phenogenomics center (C. Waldvogel, R. Doenlen) for support with mouse experiments; CIME and PTBIOEM (D. Demurtas and S. Nazarov) for electron microscopy support; Flow cytometry core facility; Gene expression core facility for support with next generation sequencing; SCITAS for support in high performance computing. We thank A. McCarthy at ESRF for beam line support. We thank the Behavioural Science Foundation (H. Hotchin, A. Beierschmitt, and R. Palmour) for the NHP immunization and PBMC isolation. Lastly, we thank ExcellGene (Monthey, Switzerland) for help with mammalian protein expression. The computational simulations were also partially facilitated by the CSCS Swiss National Supercomputing Centre.
Funding: This work was supported by the swiss initiative for systems biology (SystemsX.ch), the European Research Council (Starting grant - 716058), the Swiss National Science Foundation (310030_163139) and the EPFL’s Catalyze4Life initiative. F.S. was supported by an SNF/Innosuisse BRIDGE Proof-of-Concept grant. J.B. was supported by the EPFL Fellows postdoctoral fellowship. T.K. received funding from the German Center of Infection Research (DZIF) and the Cluster of Excellence RESIST (EXC 2155) of the German Research foundation. J.T.C. was funded by the ERA-Net PrionImmunity project 01GM1503 (Federal Ministry of Education and Research, Germany). V.M. received funding from “AESI-18” (Instituto de Salud Carlos III), grant MPY 375/18. J.PJ. was funded by the Canada Research Chairs program (J.P.J.), T.J. and X.W. were funded by the NIH NIAID (R01 AI137523). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Competing interests: B.E.C., F.S., C.Y. and J.B. have filed a patent covering the designed antigens.
Data and materials availability: All code used for this study is available through a public github repository: https://github.com/lpdi-epfl/trivalent_cocktail and stored in Zenodo under https://doi.org/10.5281/zenodo.3724330. Structures have been deposited in the Protein Data Bank under accession codes 6S3D (S0_2.126 in complex with D25), 6XWI (S0_2.126 solution NMR structure), 6XXV (S2_1.2 in complex with the Fab fragment of the elicited antibody C57) and 6VTW (S4_2.45 in complex with 101F). Chemical shift data for S0_2.126 were deposited in BMRB under accession code 34481.
References and Notes
- 1.Koga N et al. , Principles for designing ideal protein structures. Nature 491, 222–227 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Marcos E et al. , Principles for designing proteins with cavities formed by curved beta sheets. Science 355, 201–206 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huang PS et al. , De novo design of a four-fold symmetric TIM-barrel protein with atomic-level accuracy. Nat Chem Biol 12, 29–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mravic M et al. , Packing of apolar side chains enables accurate design of highly stable membrane proteins. Science 363, 1418–1423 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang PS, Boyken SE, Baker D, The coming of age of de novo protein design. Nature 537, 320–327 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Dawson WM, Rhys GG, Woolfson DN, Towards functional de novo designed proteins. Curr Opin Chem Biol 52, 102–111 (2019). [DOI] [PubMed] [Google Scholar]
- 7.Chevalier A et al. , Massively parallel de novo protein design for targeted therapeutics. Nature 550, 74–79 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Procko E et al. , A computationally designed inhibitor of an Epstein-Barr viral Bcl-2 protein induces apoptosis in infected cells. Cell 157, 1644–1656 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Correia BE et al. , Proof of principle for epitope-focused vaccine design. Nature 507, 201–206 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones S, Thornton JM, Principles of protein-protein interactions. Proc Natl Acad Sci U S A 93, 13–20 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubinstein ND et al. , Computational characterization of B-cell epitopes. Mol Immunol 45, 3477–3489 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Holliday GL, Fischer JD, Mitchell JB, Thornton JM, Characterizing the complexity of enzymes on the basis of their mechanisms and structures with a bio-computational analysis. FEBS J 278, 3835–3845 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sesterhenn F et al. , Boosting subdominant neutralizing antibody responses with a computationally designed epitope-focused immunogen. PLoS Biol 17, e3000164 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burton DR, What Are the Most Powerful Immunogen Design Vaccine Strategies? Reverse Vaccinology 2.0 Shows Great Promise. Cold Spring Harb Perspect Biol 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rappuoli R, Bottomley MJ, D’Oro U, Finco O, De Gregorio E, Reverse vaccinology 2.0: Human immunology instructs vaccine antigen design. J Exp Med 213, 469–481 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sesterhenn F, Bonet J, Correia BE, Structure-based immunogen design-leading the way to the new age of precision vaccines. Curr Opin Struct Biol 51, 163–169 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Neu KE, Henry Dunand CJ, Wilson PC, Heads, stalks and everything else: how can antibodies eradicate influenza as a human disease? Curr Opin Immunol 42, 48–55 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sok D, Burton DR, Recent progress in broadly neutralizing antibodies to HIV. Nat Immunol 19, 1179–1188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dejnirattisai W et al. , Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat Immunol 17, 1102–1108 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jardine JG et al. , HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science 351, 1458–1463 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Avnir Y et al. , Molecular signatures of hemagglutinin stem-directed heterosubtypic human neutralizing antibodies against influenza A viruses. PLoS Pathog 10, e1004103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barba-Spaeth G et al. , Structural basis of potent Zika-dengue virus antibody cross-neutralization. Nature 536, 48–53 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Corti D et al. , Cross-neutralization of four paramyxoviruses by a human monoclonal antibody. Nature 501, 439–443 (2013). [DOI] [PubMed] [Google Scholar]
- 24.Corti D et al. , A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333, 850–856 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Xu K et al. , Epitope-based vaccine design yields fusion peptide-directed antibodies that neutralize diverse strains of HIV-1. Nat Med 24, 857–867 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ofek G et al. , Elicitation of structure-specific antibodies by epitope scaffolds. Proc Natl Acad Sci U S A 107, 17880–17887 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Correia BE et al. , Computational design of epitope-scaffolds allows induction of antibodies specific for a poorly immunogenic HIV vaccine epitope. Structure 18, 1116–1126 (2010). [DOI] [PubMed] [Google Scholar]
- 28.McLellan JS et al. , Design and characterization of epitope-scaffold immunogens that present the motavizumab epitope from respiratory syncytial virus. J Mol Biol 409, 853–866 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLellan JS et al. , Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340, 1113–1117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McLellan JS et al. , Structure of a major antigenic site on the respiratory syncytial virus fusion glycoprotein in complex with neutralizing antibody 101F. J Virol 84, 12236–12244 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Azoitei ML et al. , Computation-guided backbone grafting of a discontinuous motif onto a protein scaffold. Science 334, 373–376 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Fleishman SJ et al. , Computational design of proteins targeting the conserved stem region of influenza hemagglutinin. Science 332, 816–821 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tian D et al. , Structural basis of respiratory syncytial virus subtype-dependent neutralization by an antibody targeting the fusion glycoprotein. Nat Commun 8, 1877 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bonet J et al. , Rosetta FunFolDes - A general framework for the computational design of functional proteins. PLoS Comput Biol 14, e1006623 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunette TJ et al. , Exploring the repeat protein universe through computational protein design. Nature 528, 580–584 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Taylor WR, A ‘periodic table’ for protein structures. Nature 416, 657–660 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Woolfson DN et al. , De novo protein design: how do we expand into the universe of possible protein structures? Curr Opin Struct Biol 33, 16–26 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Finucane MD, Tuna M, Lees JH, Woolfson DN, Core-directed protein design. I. An experimental method for selecting stable proteins from combinatorial libraries. Biochemistry 38, 11604–11612 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Gilman MS et al. , Rapid profiling of RSV antibody repertoires from the memory B cells of naturally infected adult donors. Sci Immunol 1, 6, eaaj1879 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Watkins AM, Arora PS, Anatomy of beta-strands at protein-protein interfaces. ACS Chem Biol 9, 1747–1754 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fenton C, Scott LJ, Plosker GL, Palivizumab: a review of its use as prophylaxis for serious respiratory syncytial virus infection. Paediatr Drugs 6, 177–197 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Crank MC et al. , A proof of concept for structure-based vaccine design targeting RSV in humans. Science 365, 505–509 (2019). [DOI] [PubMed] [Google Scholar]
- 43.Marcandalli J et al. , Induction of Potent Neutralizing Antibody Responses by a Designed Protein Nanoparticle Vaccine for Respiratory Syncytial Virus. Cell 176, 1420–1431 e1417 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrews SF et al. , Immune history profoundly affects broadly protective B cell responses to influenza. Sci Transl Med 7, 316ra192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lingwood D et al. , Structural and genetic basis for development of broadly neutralizing influenza antibodies. Nature 489, 566–570 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.West AP Jr., Diskin R, Nussenzweig MC, Bjorkman PJ, Structural basis for germ-line gene usage of a potent class of antibodies targeting the CD4-binding site of HIV-1 gp120. Proc Natl Acad Sci U S A 109, E2083–2090 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silva DA et al. , De novo design of potent and selective mimics of IL-2 and IL-15. Nature 565, 186–191 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strauch EM et al. , Computational design of trimeric influenza-neutralizing proteins targeting the hemagglutinin receptor binding site. Nat Biotechnol 35, 667–671 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McLellan JS et al. , Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 342, 592–598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ngwuta JO et al. , Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci Transl Med 7, 309ra162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Angeletti D et al. , Defining B cell immunodominance to viruses. Nat Immunol 18, 456–463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhou J, Grigoryan G, Rapid search for tertiary fragments reveals protein sequence-structure relationships. Protein Sci 24, 508–524 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu X, Wang H, Ke H, Kuhlman B, High-resolution design of a protein loop. Proc Natl Acad Sci U S A 104, 17668–17673 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D, Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci 23, 47–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mas V et al. , Engineering, Structure and Immunogenicity of the Human Metapneumovirus F Protein in the Postfusion Conformation. PLoS Pathog 12, e1005859 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson S et al. , Development of a humanized monoclonal antibody (MEDI-493) with potent in vitro and in vivo activity against respiratory syncytial virus. J Infect Dis 176, 1215–1224 (1997). [DOI] [PubMed] [Google Scholar]
- 57.Wang Y et al. , High-Resolution Longitudinal Study of HIV-1 Env Vaccine-Elicited B Cell Responses to the Virus Primary Receptor Binding Site Reveals Affinity Maturation and Clonal Persistence. J Immunol 196, 3729–3743 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sundling C, Phad G, Douagi I, Navis M, Karlsson Hedestam GB, Isolation of antibody V(D)J sequences from single cell sorted rhesus macaque B cells. Journal of immunological methods 386, 85–93 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Wang Y et al. , HIV-1 Cross-Reactive Primary Virus Neutralizing Antibody Response Elicited by Immunization in Nonhuman Primates. J Virol 91, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tiller T et al. , Efficient generation of monoclonal antibodies from single human B cells by single cell RT-PCR and expression vector cloning. J Immunol Methods 329, 112–124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chao G et al. , Isolating and engineering human antibodies using yeast surface display. Nat Protoc 1, 755–768 (2006). [DOI] [PubMed] [Google Scholar]
- 62.Joyce MG et al. , Iterative structure-based improvement of a fusion-glycoprotein vaccine against RSV. Nat Struct Mol Biol 23, 811–820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Briney B et al. , Tailored Immunogens Direct Affinity Maturation toward HIV Neutralizing Antibodies. Cell 166, 1459–1470 e1411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rohou A, Grigorieff N, CTFFIND4: Fast and accurate defocus estimation from electron micrographs. J Struct Biol 192, 216–221 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.de la Rosa-Trevin JM et al. , Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J Struct Biol 195, 93–99 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Scheres SH, RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Punjani A, Rubinstein JL, Fleet DJ, Brubaker MA, cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 68.Sattler M, Schleucher J, Griesinger C, Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Prog Nucl Mag Res Sp 34, 93–158 (1999). [Google Scholar]
- 69.Herrmann T, Guntert P, Wuthrich K, Protein NMR structure determination with automated NOE-identification in the NOESY spectra using the new software ATNOS. J Biomol NMR 24, 171–189 (2002). [DOI] [PubMed] [Google Scholar]
- 70.Herrmann T, Guntert P, Wuthrich K, Protein NMR structure determination with automated NOE assignment using the new software CANDID and the torsion angle dynamics algorithm DYANA. J Mol Biol 319, 209–227 (2002). [DOI] [PubMed] [Google Scholar]
- 71.Gottstein D, Kirchner DK, Guntert P, Simultaneous single-structure and bundle representation of protein NMR structures in torsion angle space. J Biomol NMR 52, 351–364 (2012). [DOI] [PubMed] [Google Scholar]
- 72.Shen Y, Bax A, Protein backbone and sidechain torsion angles predicted from NMR chemical shifts using artificial neural networks. J Biomol NMR 56, 227–241 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kabsch W, Xds. Acta Crystallogr D 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mccoy AJ et al. , Phaser crystallographic software. J Appl Crystallogr 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot. Acta Crystallogr D 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Adams PD et al. , PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D 66, 213–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Karplus PA, Diederichs K, Linking crystallographic model and data quality. Science 336, 1030–1033 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bonet J, Segura J, Planas-Iglesias J, Oliva B, Fernandez-Fuentes N, Frag’r’Us: knowledge-based sampling of protein backbone conformations for de novo structure-based protein design. Bioinformatics 30, 1935–1936 (2014). [DOI] [PubMed] [Google Scholar]
- 79.Mousa JJ et al. , Human antibody recognition of antigenic site IV on Pneumovirus fusion proteins. PLoS Pathog 14, e1006837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bonet J, Harteveld Z, Sesterhenn F, Scheck A, Correia BE, rstoolbox - a Python library for large-scale analysis of computational protein design data and structural bioinformatics. BMC Bioinformatics 20, 240 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Olmedillas E et al. , Chimeric Pneumoviridae fusion proteins as immunogens to induce cross-neutralizing antibody responses. EMBO Mol Med 10, 175–187 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Zhang R et al. , Envelope-specific B-cell populations in African green monkeys chronically infected with simian immunodeficiency virus. Nat Commun 7, 12131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.