

Abstract

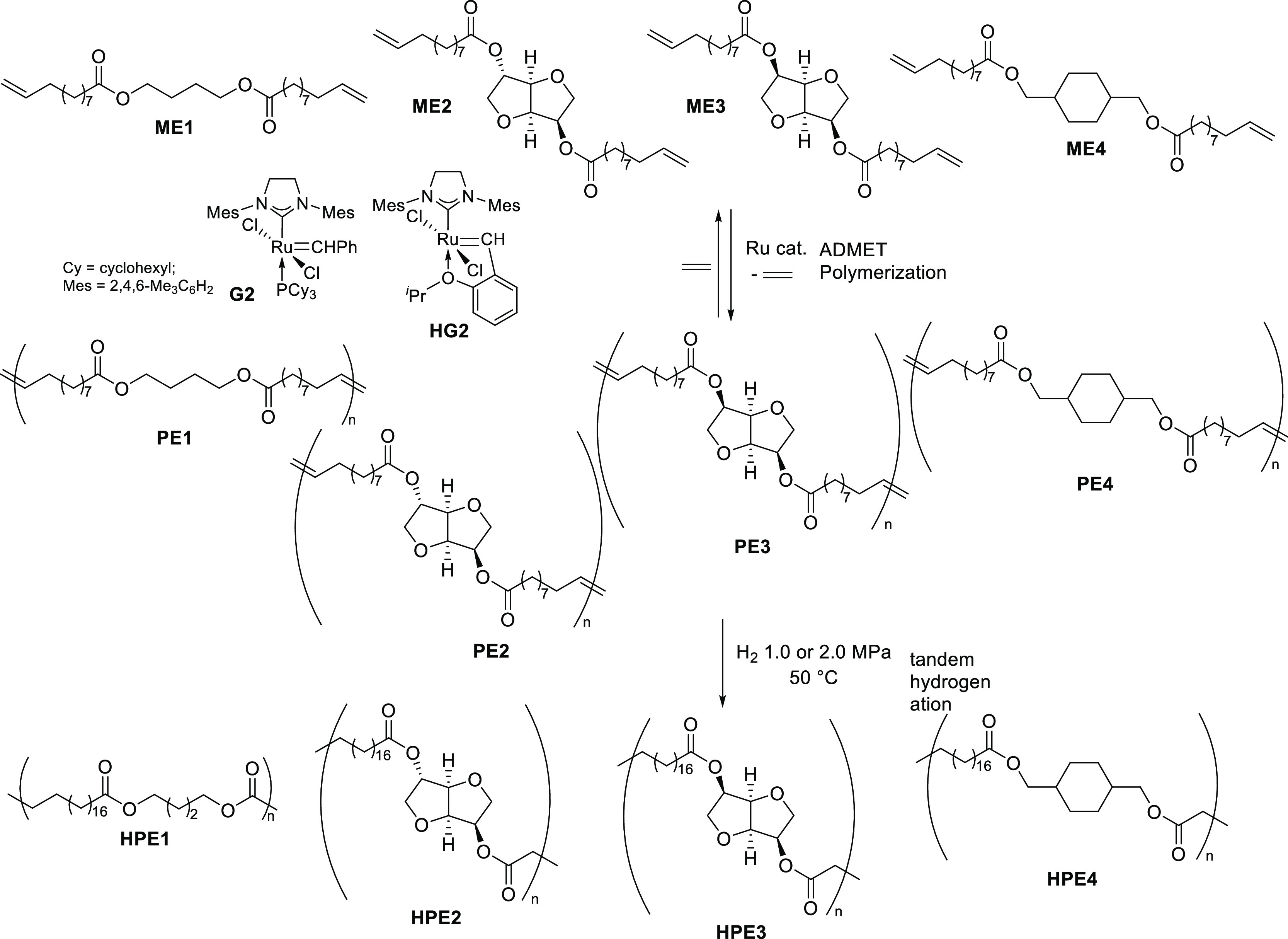
Acyclic diene metathesis (ADMET) polymerization of biobased α,ω-dienes of bis(undec-10-enoate) with diols (1,4-butanediol, isosorbide, isomannide, and 1,4-cyclohexanedimethanol) afforded high-molecular weight unsaturated polyesters, and subsequent tandem hydrogenation (H2 1.0 MPa, 50 °C, 3 h) gave the saturated polymers upon addition of a small amount of Al2O3 (1.0–1.7 wt %). Subsequent reaction of the unsaturated polymers with ethylene afforded the oligomers (by depolymerization and degradation).
Introduction
Synthesis of functional polymers from renewable feedstocks is an important subject in terms of shifting to sustainable alternatives from fossil oil-based polymers.1−7 Development of strategies for synthesis of renewable polymers by controlled polymerization,5−20 including design and development of renewable copolymers with various macromolecular architectures,18,19 has thus become an active field of study. Polyesters generally display tunable mechanical properties and potential biodegradability,12 and study of synthesis and the development of high-performance polymers from bioderived monomers is thus of particular interest.5−20 Various raw materials from renewable resources such as plant oils,8−13,21−37 lignin,14 sugars,15,29,38−41 and terpenes16,17,42 have been studied, and plant oils exemplified as castor oil,5,9−13 which generally convert to fatty acids or fatty acid methyl esters by chemical modifications, are useful feedstock for synthesis of the (aliphatic) polyesters.
Acyclic diene metathesis (ADMET) polymerization has been employed as a useful method for synthesis of linear polymers and polymer architectures.43−47 There have thus been reports for synthesis of biobased polyesters by the polymerization of fatty acids and their derivatives,22−34 and use of a castor oil derivative, ω-undecenoate, containing both the olefinic double bond and carboxylate at the termini is an ideal method for synthesis of high-purity α,ω-dienes (undecenyl-undecenoate) as valuable monomers for the ADMET polymerization.22−34 The polymerization using ruthenium-carbene catalysts, especially called second-generation catalysts, RuCl2(PCy3) (IMesH2)(CHPh) [G2; IMesH2 = 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene, Cy = cyclohexyl] and RuCl2(IMesH2)(CH-2-OiPr-C6H4) (HG2), generally afforded high-molecular weight unsaturated polyesters. Moreover, it was demonstrated that transesterification of the resultant unsaturated polymers (depolymerizations) containing isosorbide was achieved under reflux conditions in methanol in the presence of a small amount of sulfuric acid (85 °C overnight, Scheme 1).23 There was also a report for synthesis of end-functionalized (co)polymers by the ADMET polymerization of monomers derived from castor oil and isosorbide in the presence of terminal olefins (methyl 10-undecenoate).29
Scheme 1. Synthesis of Biobased Polyesters by ADMET Polymerization and Subsequent Degradation (Transesterification)23 and Hydrogenation.27,28.

It was also demonstrated that hydrogenation of olefinic double bonds in the resultant polymers afforded long-chain aliphatic polyesters,27,28,31 and the copolymers of undec-10-en-1-yl undec-10-enoate and undeca-1,10-diene possessed high melting temperatures depending upon the methylene repeat units (ratio of each monomer units).27 These hydrogenations were conducted in the presence of prepared RuCl2(PCy3)2(CHOEt) (4.0 MPa, 110 °C, 2 days, Scheme 1),27 H[(μ2-H)(C5Ph4O)Ru(CO)2]2 (4.0 MPa, 100 °C, overnight),31 or 10 wt % of Pd/C (H2 5.0 MPa, 80 °C, 20 h)28 under rather harsh conditions after isolation of unsaturated polymers by precipitation. In general, the resultant unsaturated polymers containing (long) aliphatic chains prepared by ADMET polymerization were hydrogenated using (i) p-toluenesulfonyl hydrazide and tri-n-propyl amine,48−50 (ii) RhCl(PPh3)3,51,52 RuHCl(CO) (PCy3)2,53 or Pd/C28,54 catalysts under high hydrogen pressure (3.1–13.8 MPa, 80–90 °C, 2–5 days) or (iii) Ir(1,5-cyclooctadiene)(PCy3)-(pyridine) under mild conditions (ca. Ir 2 mol %, ca. 2 MPa at room temperature, 20 h);54 no reports concerning the tandem systems have been studied for synthesis of the saturated polyesters. Moreover, ADMET polymerization has been known as the condensation polymerization by removal of ethylene (or propylene);43−47 however, study of the depolymerization of ADMET-derived polymers in the presence of ethylene and a polymerization catalyst was not carried out.
We recently reported the synthesis of a new biobased polyester by ADMET polymerization of the monomer (α,ω-diene) derived from castor oil and eugenol (obtained from clove oil).33 Because we also reported the synthesis of conjugated polymers by ADMET polymerization including the end modification and grafting,47,56,57 we thus have an interest in the synthesis of biobased polyesters by AMDET polymerization and subsequent (tandem) hydrogenation and in the degradation of the polymers in the presence of ethylene (to confirm the equilibrium in this system). In this paper, we wish to demonstrate a method for the synthesis of biobased saturated polyesters by tandem ADMET polymerization and subsequent hydrogenation without isolation of unsaturated polymers (Scheme 2, HPE1-4), and we also demonstrate the subsequent reaction (depolymerization) with ethylene after the ADMET polymerization.
Scheme 2. Synthesis of a Biobased Polyester by Tandem ADMET Polymerization and Subsequent Hydrogenation in the Presence of the Ruthenium Catalyst.
Results and Discussion
ADMET Polymerization of Biobased α,ω-Diene Monomers
Four α,ω-diene monomers derived from castor oil (ω-undecenoate) and 1,4-butanediol, isosorbide, isomannide, and 1,4-cyclohexanedimethanol have been chosen in this study. These diols can be obtained from sugars (glucose and mannitol) and have been used as renewable building blocks.5,6 These monomers, butane-1,4-diyl bis(undec-10-enoate) (ME1), dianhydro-d-glucityl bis(undec-10-enoate) (ME2),23 dianhydro-d-mannityl bis(undec-10-enoate) (ME3), and cyclohexane-1,4-dimethanol bis(undec-10-enoate) (ME4), were prepared by treating 10-undecenoyl chloride with the corresponding diol in toluene in the presence of NEt3 (Scheme 3), according to the reported procedure.33 The resultant monomers were identified by NMR spectra and were confirmed by atmospheric pressure chemical ionization (APCI) mass spectrometry (see, Experimental Section).a
Scheme 3. Synthesis of Biobased Monomers (ME1–ME4).

ADMET polymerizations of ME1–ME4 were conducted in the presence of two types of ruthenium-carbene catalysts, RuCl2(PCy3)(H2IMes)(CHPh) [G2; IMesH2 = 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene, Cy = cyclohexyl] and RuCl2(IMesH2)(CH-2-OiPr-C6H4) (HG2) because these catalysts afforded high-molecular weight polymers in the polymerization of 4-allyl-2-methoxyphenyl 10-undecenoate, prepared from biorenewable eugenol (clove oil) and undecenoate (castor oil).33 As reported for the synthesis of all-trans poly(9,9-n-alkyl fluorene-2,7-vinylene)s by ADMET polymerization,58,59 the polymerizations were carried out in a small amount of CHCl3 (0.14 mL for monomer 300 mg) using a sealed Schlenk tube equipped with a high-vacuum valve in the presence of a catalyst (Scheme 2). The reactions were conducted in an oil bath heated at 50 °C, and the mixture was then cooled with a liquid nitrogen bath and the headspace of the flask was evacuated to remove the ethylene formed in this condensation polymerization (for details, see the Experimental Section);33,58,59 efficient removal of ethylene is thus beneficial for obtaining high-molecular weight polymers.33,58 The results are summarized in Table 1.
Table 1. ADMET Polymerization of Monomers (ME1–4) Using Ruthenium-Carbene Catalysts (G2, HG2).a.
| run | monomer (mmol) | conc.b/M | cat. (mol %) | time/h | yieldc/% | Mnd | Mw/Mnd |
|---|---|---|---|---|---|---|---|
| 1 | ME1 (0.71) | 5.07 | HG2 (2.0) | 6 | 87 | 7700 | 1.34 |
| 2 | ME1 (0.71) | 5.07 | HG2 (2.0) | 12 | 93 | 8500 | 1.35 |
| 3 | ME1 (0.71) | 5.07 | HG2 (2.0) | 24 | 91 | 15,900 | 1.46 |
| 4 | ME1 (0.71) | 5.07 | HG2 (2.0) | 24 | 92 | 13,300 | 1.39 |
| 5 | ME1 (0.71) | 5.07 | HG2 (1.5) | 24 | 94 | 15,700 | 1.32 |
| 6 | ME1 (0.71) | 5.07 | HG2 (1.0) | 24 | 93 | 12,400 | 1.24 |
| 7 | ME1 (0.71) | 5.07 | G2 (1.5) | 24 | 91 | 12,700 | 1.25 |
| 8 | ME1 (0.71) | 5.07 | G2 (1.0) | 24 | 89 | 8800 | 1.22 |
| 9 | ME1 (0.71)e | 5.07 | HG2 (2.0) | 24 | 87 | 11,800 | 1.43 |
| 10 | ME1 (0.71)f | 5.07 | HG2 (2.0) | 24 | 85 | 9100 | 1.41 |
| 11 | ME1 (2.36) | 6.21 | HG2 (2.0) | 6 | 90 | 8300 | 1.44 |
| 12 | ME1 (2.36) | 6.21 | HG2 (2.0) | 12 | 92 | 10,200 | 1.48 |
| 13 | ME1 (2.36) | 6.21 | HG2 (2.0) | 24 | 93 | 16,400 | 1.51 |
| 14 | ME2 (0.65) | 4.64 | HG2 (2.0) | 12 | 86 | 8700 | 1.39 |
| 15 | ME2 (0.65) | 4.64 | HG2 (2.0) | 24 | 88 | 14,000 | 1.42 |
| 16 | ME2 (0.65) | 4.64 | HG2 (1.0) | 24 | 88 | 11,000 | 1.21 |
| 17 | ME2 (0.65) | 4.64 | G2 (1.0) | 24 | 86 | 9300 | 1.30 |
| 18 | ME2 (2.08) | 6.12 | HG2 (2.0) | 6 | 87 | 7100 | 1.51 |
| 19 | ME2 (2.08) | 6.12 | HG2 (2.0) | 12 | 90 | 10,300 | 1.55 |
| 20 | ME2 (2.08) | 6.12 | HG2 (2.0) | 24 | 94 | 14,700 | 1.58 |
| 21 | ME3 (0.65) | 4.64 | G2 (1.5) | 24 | 89 | 8700 | 1.29 |
| 22 | ME3 (0.65) | 4.64 | HG2 (1.5) | 24 | 91 | 9800 | 1.31 |
| 23 | ME3 (0.65) | 4.64 | HG2 (2.0) | 24 | 92 | 10,100 | 1.35 |
| 24 | ME3 (0.65) | 4.64 | HG2 (2.0) | 24 | 93 | 10,200 | 1.38 |
| 25 | ME3 (2.08) | 6.12 | HG2 (2.0) | 6 | 91 | 6700 | 1.49 |
| 26 | ME3 (2.08) | 6.12 | HG2 (2.0) | 12 | 90 | 8600 | 1.47 |
| 27 | ME3 (2.08) | 6.12 | HG2 (2.0) | 24 | 94 | 10,900 | 1.46 |
| 28 | ME4 (0.63) | 4.50 | G2 (2.0) | 24 | 88 | 8600 | 1.32 |
| 29 | ME4 (0.63) | 4.50 | HG2 (2.0) | 24 | 86 | 9200 | 1.35 |
| 30 | ME4 (2.09) | 6.15 | HG2 (2.0) | 24 | 94 | 9400 | 1.42 |
Conditions: monomer 300 mg in CHCl3 0.14 mL or monomer 1000 mg in CHCl3 0.34 mL (runs 11–13, 0.38 mL), 50 °C.
Initial monomer concentration in mmol/mL.
Isolated yield as MeOH insoluble fraction.
GPC data in THF versus polystyrene standards.
In CH2Cl2.
In toluene.
It was revealed that the polymerization of ME1 using HG2 afforded high-molecular weight polymer (PE1) with unimodal molecular weight distribution (run 3, Mn = 15,900; Mw/Mn = 1.46), and the Mn values in the resultant polymers increased over the time course (runs 1–3). The results conducted under the same conditions were close (runs 3 and 4), although the Mn value was somewhat different probably because of increase in viscosity under these conditions (containing polymer with a high Mn value); it seems very difficult to conduct the polymerization under higher ME1 concentration because of technical difficulty of stirring magnetically. As reported previously,33 the Mn value was also affected by the Ru catalyst (G2vsHG2, runs 6–8) and amount of the catalyst (HG2) loaded (runs 3–6). HG2 seemed to be more suited in terms of synthesis of high-molecular weight PE1; the Mn values in the resultant polymer (PE1) prepared by HG2 were higher than those prepared by G2 [Mn = 15,700 (run 5) versus 12,700 (run 7), 12,400 (run 6) versus 8800 (run 8)] under the same conditions. The Mn value was also affected by the solvent employed (runs 3, 9, 10), although these polymerizations were conducted under rather high-monomer concentration conditions that are generally required in this condensation polymerization.43−47
It turned out that resonances ascribed to protons of terminal olefins (at 4.84, 4.91, and 5.72 ppm) in the 1H NMR spectrum (in CDCl3 at 25 °C, Figure 1a) for ME1 were no longer present and resonances ascribed to protons assigned to the internal olefins (at 5.29–5.38 ppm) were observed in the resultant polymer (PE1, Figure 1b), whereas the other resonances remained [also shown in Figures S1, S9 in the Supporting Information].a Similarly, the disappearance of resonances ascribed to carbon in the terminal olefins along with observation of the resonances in the internal olefins (with remaining the other resonances) was observed in the 13C{1H} NMR spectra (Figures S2, S10, Supporting Information).a The results thus clearly indicate the formation of polymers by ADMET polymerization.22−34,43−59 The resultant polymers (PE1) possess a sole (rather sharp) melting temperature (Tm) at 46.9 °C (run 3, Mn = 15,900), whereas the Tm value was affected by the molecular weight [Figure S39, Supporting Information, Tm value = 46.9 °C (Mn = 15,900, run 3), 40.9 °C (12,400, run 6), 36.6 °C (8500, run 2), and 34.8 °C (7700, run 1)].
Figure 1.

1H NMR spectrum (in CDCl3 at 25 °C) for (a) ME1 (monomer), (b) PE1 (ADMET polymer, run 3), (c) HPE1 (after tandem hydrogenation, run 33), (d) ME2, (e) PE2 (run 15), (f) HPE2 (run 37), (g) ME3, (h) PE3 (run 23), (h) HPE3 (run 41), (i) ME4, (j) PE4 (run 29), and (j) HPE4 (run 43). The full spectra are shown in the Supporting Information.a
The ADMET polymerizations of ME2, ME3, and ME4 by HG2, conducted under the same conditions, also afforded high-molecular weight polymers (PE2, PE3, and PE4, respectively) with unimodal molecular weight distributions [Mn (Mw/Mn) = 14,000 (1.42), 10,100 (1.35), and 9200 (1.35) for PE2 (run 15), PE3 (run 23), and PE4 (run 29), respectively]. The results conducted under the same conditions were reproducible (runs 23 and 24, Mn = 10,100 and 10,200, and Mw/Mn = 1.35 and 1.38). The resultant polymers showed resonances ascribed to protons (carbon) in the internal olefins in their NMR spectra along with disappearance of resonances because of the terminal olefins (shown in Figure 1 and Supporting Information),a strongly indicating that these polymers also possess structures formed by the ADMET polymerization (Scheme 2). It seems that the Mn value in PE2 prepared under the optimized conditions (Mn = 14,000, run 15) was higher than those reported previously (Mn = 4400–8400),23 and the Tm value in the resultant polymer (32.3 °C, run 15) is higher than that in the low-molecular weight polymer (25.9 °C, run 14, Mn = 8700, Figure S41, Supporting Information). The resultant polymers prepared by ME3 and ME4 showed rather broad and low melting temperature at 5.4 °C, according to the DSC thermograms obtained (Figures S42, S43, Supporting Information). The results thus indicate that the polymerization using HG2 gave high-molecular weight polymers (PE1–4) under the optimized conditions.b
It should be noted that the Mn values in the resultant polymers also increased over time in the ADMET polymerizations of ME1 and ME2 under the scaled-up conditions (runs 11–13 and 18–20, monomer 1000 mg scale), as observed under small-scale conditions (run 1–3, 14, 15, monomer 300 mg scale). The resultant polymers possessed Mn values similar to those conducted under small-scale conditions (monomer 300 mg scale), although the initial monomer concentrations were rather high under the scaled-up conditions (monomer 1000 mg, CHCl3 0.34 or 0.38 mL). Therefore, syntheses of high-molecular weight polymers (PE1–PE4) have been demonstrated by the ADMET condensation polymerization in the presence of HG2.
Tandem ADMET Polymerization and Hydrogenation: Synthesis of Biobased Saturated Polyesters
It should be noted that olefinic double bonds in the resultant polymers (PE1–PE4), prepared by the ADMET polymerizations of the monomers (ME1–ME4) by G2 or HG2, could be hydrogenated in a tandem manner (without isolation of unsaturated polymers) upon addition of a small amount of alumina (Al2O3) in situ (Scheme 4), as shown in Table 2 and Figure 1.a
Scheme 4. Synthesis of Biobased Polyesters (HPE1–HPE4) by Tandem ADMET Polymerization and Hydrogenation Using Ruthenium Catalysts.

Table 2. Synthesis of Saturated Polyesters by Tandem ADMET Polymerization and Hydrogenation Using Ruthenium-Carbene Catalystsa.
| PE (ADMET)c |
HPE (after H2)e |
|||||||
|---|---|---|---|---|---|---|---|---|
| run | monomer (mmol) | cat. | H2/MPa | yieldb/% | Mnd | Mw/Mnd | Mnd | Mw/Mnd |
| 31 | ME1 (0.71) | HG2 | 1.0 | 88 | 12,800 | 1.41 | 13,800 | 1.45 |
| 32 | ME1 (2.36) | HG2 | 1.0 | 90 | 16,400 | 1.51 | 16,600 | 1.48 |
| 33 | ME1 (2.36) | HG2 | 1.0f | 94 | 12,300 | 1.46 | 13,200 | 1.48 |
| 34 | ME2 (0.65) | HG2 | 1.0 | 87 | 14,000 | 1.42 | 15,900 | 1.44 |
| 35 | ME2 (0.65) | G2 | 1.0 | 85 | 11,900 | 1.38 | 13,100 | 1.28 |
| 36 | ME2 (0.65) | HG2 | 2.0 | 90 | 13,200 | 1.45 | 16,000 | 1.49 |
| 37 | ME2 (2.08) | HG2 | 1.0 | 93 | 13,700 | 1.48 | 15,800 | 1.53 |
| 38 | ME3 (0.65) | G2 | 1.0 | 93 | 9500 | 1.41 | 10,400 | 1.42 |
| 39 | ME3 (0.65) | HG2 | 1.0 | 92 | 10,200 | 1.38 | 11,100 | 1.39 |
| 40 | ME3 (0.65) | HG2 | 0.5 | 90 | 9700 | 1.37 | 10,900 | 1.34 |
| 41 | ME3 (0.65) | HG2 | 2.0 | 91 | 10,700 | 1.34 | 11,800 | 1.32 |
| 42 | ME3 (2.08) | HG2 | 1.0 | 92 | 10,900 | 1.46 | 11,000 | 1.53 |
| 43 | ME4 (2.09) | HG2 | 1.0 | 92 | 8600 | 1.48 | 9200 | 1.51 |
Conditions: Ru cat. 2.0 mol %, monomer 300 mg in CHCl3 0.14 mL or monomer 1000 mg in CHCl3 0.34 mL (run 32, 0.38 mL), 50 °C. ADMET polymerization 24 h, and hydrogenation 3 h after addition of Al2O3. Al2O3 5 mg (1.7 wt %, monomer 300 mg scale) or 10 mg (1.0 wt %, monomer 1000 mg scale).
Isolated yield as MeOH insoluble fraction.
Sample before hydrogenation.
GPC data in THF versus polystyrene standards.
Sample after hydrogenation.
Hydrogenation at 25 °C.
After the ADMET polymerization, the reaction mixture was moved into an autoclave (20 mL scale) with addition of a small amount of Al2O3 powder (1.7 or 1.0 wt % to monomer charged), and the solution was pressurized with hydrogen (1.0 or 2.0 MPa) and was stirred for 3 h at 50 °C (runs 31, 32, 34–37, 38, 39, 41–43). As shown in Figure 1, resonances ascribed to olefinic protons disappeared with the other resonances remaining in all cases, and no significant differences in the Mn values and Mw/Mn values were observed. Disappearance of resonances due to protons/carbons in the olefinic double bond could be confirmed in the sample treated even at 25 °C (run 33, Figures S17, and S18). These results could demonstrate the successful synthesis of saturated biobased polyesters (HPE1–4) in a tandem manner.
It turned out that in the tandem reaction for ME1, optimization of reaction conditions (hydrogen pressure and reaction time) is necessary for obtainment of saturated polymers (HPE1); the reaction after 1 h (under H2 1.0 MPa, run S2 in Table S1, Supporting Information) or the reaction at 0.5 MPa (3 h, run S4, Table S1) gave the polymers with incomplete hydrogenation (Figures S19, S20, Supporting Information).a Incompletion of the hydrogenation could also be confirmed by DSC thermograms (Figure S40, Supporting Information), as described below.
It should be noted that tandem hydrogenation did not proceed in the absence of Al2O3 under the same conditions (runs S6, S7 in Table S1; on the basis of NMR spectra, Figures S29, S30, Supporting Information). Addition of alumina is thus prerequisite to accomplish the tandem reaction under rather mild conditions compared to the conventional independent hydrogenation of the ADMET polymers using rhodium, ruthenium, or Pd/C catalysts (3.1–13.8 MPa, 80–110 °C).27,28,32,51−55 Interestingly, only 1.0 wt % Al2O3 powder is effective for the tandem synthesis under the scaled-up conditions (monomer 1000 mg scale) for syntheses of HPE1–HPE4 (runs 32, 37, 42, 43), and no significant differences in the Mn values and Mw/Mn values were confirmed before/after hydrogenation.
It was revealed that in the tandem reaction for ME3, the Tm values in the resultant polymers were affected by the hydrogen pressure employed [runs 39–41, DSC thermograms in Figure S42; Tm = 56.1 °C (run 41, H2 2.0 MPa), 50.6 °C (run 39, H2 1.0 MPa), and 50.0 °C (run 40, H2 0.5 MPa)], although no significant differences (disappearance of protons/carbon ascribed to the olefinic double bonds) in these NMR spectra were seen in all cases (Figure S23 through S28, Supporting Information).a Because the property (melting and/or glass transition temperature, crystallinity, etc.) of polyesters is generally influenced by the preciseness of the microstructure,12 it seems likely that the observed difference would be probably due to the presence of unsaturated olefinic double bonds that remained (even though we do observe resonances in the NMR spectra).
As shown in Figure 2 and DSC thermograms in the resultant polymers (HPE1 and HPE2), the Tm values increased by hydrogenation of olefinic double bonds [Tm = 67.8 °C (HPE1, run 32) and 53.2 °C (HPE2, run 37); Tm = 46.9 °C (PE1, run 3) and 32.3 °C (PE2, run 15)] because of increase in the alkyl chain length, and the Tm values in the resultant HPE3 and HPE4 also increased after hydrogenation [Figure S42, S43, Supporting Information; Tm = 41.7 °C (HPE4, run 43) versus 5.4 °C (PE4)].
Figure 2.

DSC thermograms for (a) PE1 (run 13) and HPE1 (run 32) and (b) PE2 (run 15) and HPE2 (run 37). Polymer samples before/after tandem hydrogenation. Detailed data are shown in Table 2.
Tandem ADMET Polymerization and Reaction with Ethylene
ADMET polymerizations of ME1 and ME2 were conducted by G2 (or HG2) at 50 °C for 6 h (or 12 h) and the resultant mixture was then pressurized with ethylene at 0.8 MPa and was stirred for 1 h at 50 °C. The results are summarized in Table 3, and the 1H and 13C{1H} NMR spectra in the resultant sample for the reaction with ME2 (run 49) are shown in Figure 3. Detailed NMR spectra in the reaction with ME1 (run 46) are shown in Figures S33 and S34, and the spectra in the reaction with ME2 (run 49) are shown in Figures S35–S37, Supporting Information.a Selected GPC traces before/after the depolymerization are also shown in Figures S44–S47.
Table 3. Reaction with Ethylene after ADMET Polymerizationa.
| ADMET
polymerizationb |
reaction with ethylened |
|||||||
|---|---|---|---|---|---|---|---|---|
| run | monomer | cat. | time/h | Mnc | Mw/Mnc | Mnc | Mw/Mnc | yielde/% |
| 44 | ME1 | HG2 | 12 | 10,200 | 1.39 | 2000 | 1.51 | 87 |
| 45 | ME1 | G2 | 12 | 9800 | 1.55 | 1800 | 1.46 | 88 |
| 46 | ME1 | G2 | 6 | 8400 | 1.62 | 1500 | 1.54 | 85 |
| 47 | ME2 | HG2 | 12 | 10,800 | 1.46 | 2200 | 1.45 | 89 |
| 48 | ME2 | G2 | 12 | 11,300 | 1.45 | 1600 | 1.45 | 89 |
| 49 | ME2 | G2 | 6 | 8500 | 1.54 | 1400 | 1.48 | 84 |
Conditions: Ru cat. 2.0 mol % and monomer 300 mg in CHCl3 0.14 mL. 50 °C.
ADMET polymerization at 50 C.
GPC data in THF versus polystyrene standards.
Ethylene 0.8 MPa at 50 °C for 1 h.
Isolated yield as MeOH insoluble fraction.
Figure 3.

1H NMR spectrum (in CDCl3 at 25 °C, expanded at 4.3–6.2 ppm) for (a) ME2 (monomer), (b) PE2 (ADMET polymer, run 15), and (c) DPE2 (after reaction with ethylene, run 49). The 13C{1H} NMR spectrum (in CDCl3 at 25 °C, expanded at 80–190 ppm) for (d) ME2 (monomer), (e) PE2 (ADMET polymer, run 15), and (f) DPE2 (after reaction with ethylene, run 49). The full spectra are shown in the Supporting Information (Figures S33–S35).a
It turned out that decreases in the Mn values were observed in all cases when the resultant reaction mixtures after the ADMET polymerization were stirred in ethylene for 1 h at 50 °C. It seems that the Mn values in the reaction with HG2 are rather high compared to those in the reaction with G2, and the values seems rather high when the samples after 12 h (polymerization) were used instead of 6 h. These may be due to rather high-molecular weight samples (by HG2 or 12 h) or due to partial decomposition of the catalyst (especially after 12 h), although the exact reason is unclear at this moment.
Figure 3 shows 1H and 13C{1H} NMR spectra (in CDCl3 at 25 °C) for the sample for the reaction with ME2 (run 49), and the NMR spectra for ME2 and PE2 (run 15) are also placed for comparison. It was revealed that resonances ascribed to protons/carbons in the terminal olefins were observed in the sample after the reaction with ethylene (Figure 3c,f),a whereas the other resonances remained. These results suggest that the resultant polymer was reacted with ethylene to give oligomers by depolymerization (degradation). On the basis of the integration ratio of internal/terminal olefins in the reaction product [terminal/internal = (2.00 + 4.14):2.45, Figure S33], it seems that the major product after the reaction would be a dimer (including a trimer, etc.), which also seems to be appropriate from speculation of the GPC data. The similar trend was also observed in the reaction of ME1, although the resultant materials seem to be a mixture of oligomers. Although we may need to explore in more detail, the results could suggest that the resultant ADMET polymer could be reacted with ethylene to afford the oligomer without further addition of the ruthenium catalyst.
Concluding Remarks
We have demonstrated that ADMET polymerization of biobased α,ω-diene monomers (derived from castor oil and sugars) afforded high-molecular weight polymers with unimodal molecular weight distribution, and subsequent tandem hydrogenation with addition of Al2O3 (1.0–1.7 wt %) afforded saturated polyesters (HPE1-HPE4) efficiently (Scheme 5). The efficient tandem hydrogenation system under rather mild conditions [1.0 or 2.0 MPa at 50 °C, 3 h (this study, tandem) versus 4.0–5.0 MPa at 80–110 °C, >10–48 h (reported, independent hydrogenation)]27,28,31 has thus been introduced through this effort. Resultant polymers are saturated, biorenewable semicrystalline polyesters with the melting temperature at 41.7–67.8 °C. Moreover, once formed ADMET polymers (PE1–PE4) were reacted with ethylene to afford oligomers (by depolymerization and degradation, Scheme 5) because ADMET polymerization is a type of condensation polymerization with an equilibrium. Various middle segment- (circled in red, Scheme 5) and carboxylate-containing terminal olefins derived from plant oil can be considered as the building blocks for the synthesis of symmetric polyesters, which are important for obtainment of materials with high Tm,12 by adopting this approach. Further improvements (such as catalyst efficiency, tandem hydrogenation conditions preferably under low pressure, complete depolymerization, alternatives of chloroform, efficient synthesis from the long-chain esters, etc.) should be considered as the future project; however, the approach adopted here should be beneficial to the development of a green sustainable process and materials which should be promising alternatives to those based on fossil fuels.
Scheme 5. Synthesis of a Biobased Polyester by Ruthenium-Catalyzed ADMET Polymerization and Tandem Hydrogenation and Depolymerization with Ethylene.

Experimental Section
General Procedure
All experiments were carried out under nitrogen atmosphere or using standard Schlenk techniques unless otherwise specified. Anhydrous grade dichloromethane and toluene (>99.5%, Kanto Chemical Co., Inc.) were transferred into a bottle containing molecular sieves (mixture of 3A 1/16, 4A 1/8 and 13× 1/16) in a drybox. RuCl2(PCy3)(IMesH2)(CHPh) [G2; Cy = cyclohexyl, IMesH2 = 1,3-bis(2,4,6-trimethylphenyl)imidazolin-2-ylidene], and RuCl2(IMesH2) (CH-2-OiPr-C6H4) (HG2) were purchased from Aldrich Chemical Co., and were used as received. Chemicals of reagent grades such as 10-undecenoyl chloride (>99%), isosorbide >98.0%(GC), isomannide >98.0%(GC), 1,4-butanediol >99.0%(GC), 1,4-cyclohexanedimethanol >99.0%(GC), and triethylamine (>99%) were purchased from Tokyo Chemical Industry, Co., Ltd., and were used as received. Ethyl vinyl ether (>98%), alumina (activated Al2O3), and Celite were purchased from Fujifilm Wako Pure Chemical Industries, Ltd.
All 1H and 13C{1H} NMR spectra were recorded using a Bruker AV500 spectrometer (500.13 MHz for 1H, 125.77 MHz for 13C). All chemical shifts were reported in parts per million (ppm) with reference to SiMe4 at 0.00 ppm. Obvious multiplicities and routine coupling constants are usually not listed, and all spectra were obtained in the solvent (CDCl3) indicated at 25 °C unless otherwise noted. Molecular weights and the molecular weight distributions of resultant polymers were measured by gel permeation chromatography (GPC). The measurements were performed at 40 °C on a Shimadzu SCL-10A using a RID-10A detector (Shimadzu Co., Ltd.) in THF (containing 0.03 wt % of 2,6-di-tert-butyl-p-cresol, flow rate 1.0 mL/min). HPLC-grade THF (Fujifilm Wako Pure Chemical Ind., Inc.) was used as the eluent with a flow rate of 1.0 mL/min and was degassed prior to use. GPC columns (ShimPAC GPC-806, 804 and 802, 30 cm × 8.0 mm diameter, spherical porous gel made of styrene/divinylbenzene copolymer, ranging from <102 to 2 × 107 MW) were calibrated versus polystyrene standard samples. Differential scanning calorimetric (DSC) data for polymer were measured using a Hitachi DSC 7020 analyzer. Nitrogen was used as the purge gas and all samples (5–7 mg) were placed in standard aluminum pans. Polymer samples were first heated from 25 to 150 °C and then cooled to −100 °C. The glass transition (Tg) and melting (Tm) temperature were determined upon the second heating cycle. All runs were performed at a rate of 10 °C/min. Atmospheric pressure chemical ionization (APCI) mass spectrometry was performed on a Bruker MicroTOF II-SDT1.
Synthesis of Butane-1,4-diyl Bis(undec-10-enoate) (ME1)
10-Undecenoyl chloride (8.00 g, 39.4 mmol) was added dropwise into a toluene solution (50.0 mL) containing 1,4-butanediol (1.77 g, 19.7 mmol, 0.50 equiv) and triethylamine (5.00 g, 49.3 mmol, 1.25 equiv) for over 30 min at 0 °C. The stirred mixture was warmed to room temperature and was stirred for 24 h. After the reaction reached completion, by confirmation of consumption of 10-undecenoyl chloride by TLC, the reaction mixture was then neutralized with 2 M HCl and was washed with 10% NaHCO3 (15 mL × 2), deionized water (15 mL × 2), and brine (15 mL × 2). The solution was then dried over anhydrous MgSO4 and then filtered through a Celite pad. The filtrate was evaporated under reduced pressure, and the crude product was then purified by column chromatography using n-hexane and ethyl acetate (8/2) as an eluent to yield ME1 as a colorless oil (5.25 g, 91% yield). 1H NMR (CDCl3): δ 1.22 (br s, 16H, CH2), 1.30 (t, J = 6.7 Hz, 4H, CH2), 1.54 (t or q, J = 7.3 Hz, 4H, −CH2CH2COO−), 1.63 (4H, −CH2CH2OCO−), 1.96 (q or dt, J = 6.9 Hz, 4H, −CH2CH=CH2), 2.22 (t, J = 7.5 Hz, 4H, −CH2COO−), 4.02 (t, J = 5.9 Hz, 4H, −CH2OCO−), 4.84 (dt, J = 10.3, 1.1 Hz, 2H, CH2=CH−), 4.91 (dd, J = 17.0, 1.8 Hz, 2H, CH2=CH−), 5.72 (ddt, J = 17.0, 10.3, 6.9 Hz, 2H, −CH=CH2). 13C{1H} NMR (CDCl3): δ 24.9, 25.3, 28.8, 29.0, 29.1, 29.1, 29.2, 33.7, 34.2, 63.6 (−CH2OCO−), 114.1 (CH2=CH−), 138.9 (CH2=CH−), 173.5 (−COO−). APCI-MS: calcd for C26H46O4m/z, 423.35 [M + H]+; found, 423.3.
Synthesis of Dianhydro-d-glucityl Bis(undec-10-enoate) (ME2)
10-Undecenoyl chloride (8.00 g, 39.4 mmol) was added dropwise into a toluene solution (50.0 mL) containing isosorbide (2.88 g, 19.7 mmol, 0.50 equiv) and triethylamine (5.00 g, 49.3 mmol, 1.25 equiv) for over 30 min at 0 °C. The stirred mixture was then warmed to room temperature and was stirred for 24 h. After the reaction reached completion, by confirmation of consumption of 10-undecenoyl chloride by TLC, the reaction mixture was then neutralized with 2 M HCl and was washed with 10% NaHCO3 (15 mL × 2), deionized water (15 mL × 2), and brine (15 mL × 2). The solution was then dried over anhydrous MgSO4 and then filtered through a Celite pad. The filtrate was evaporated under reduced pressure, and the crude product was then purified by column chromatography using n-hexane and ethyl acetate (8/2) as an eluent to yield ME2 as a colorless oil (6.46 g, 94% yield). 1H NMR (CDCl3): δ 1.27 (br s, 16H, −CH2−), 1.34 (t, J = 6.7 Hz, 4H, −CH2−), 1.59 (m, 4H, −CH2CH2COO−), 2.00 (q or dt, J = 6.9 Hz, 4H, −CH2CH=CH2), 2.27 (t, J = 7.7 Hz, 2H, −CH2COO−), 2.33 (t, J = 7.1 Hz, 2H, −CH2COO−), 3.76 (dd, J = 9.8, 5.4 Hz, 1H, −CH2–OCH−), 3.91–3.95 (m, 3H, −CH2–OCH–, −CH2–OCH−), 4.44 (d, J = 4.6 Hz, 1H, −CH2–OCH−), 4.80 (t, J = 5.0 Hz, 1H, −CH2–OCH−), 4.89 (d, J = 10.2 Hz, 2H, CH2=CH−), 4.96 (dd, J = 17.1, 1.60 Hz, 2H, CH2=CH−), 5.12 (q, J = 5.6 Hz, 1H, −CHOCO−), 5.16 (d, J = 3.2 Hz, 1H, −CHOCO−), 5.77 (ddt, J = 17.0, 10.3, 6.7 Hz, 2H, −CH=CH2). 13C{1H} NMR (CDCl3): δ 24.8, 28.9, 29.0, 29.1, 29.2, 33.8, 34.0, 34.1, 70.3, 73.7, 74.5, 80.7, 85.9, 114.1 (CH2=CH−), 139.1 (−CH=CH2), 172.8 (−COO−), 173.1 (−COO−). APCI-MS: calcd for C28H46O6m/z, 479.34 [M + H]+; found, 479.3.
Synthesis of Dianhydro-d-mannityl Bis(undec-10-enoate) (ME3)
10-Undecenoyl chloride (8.00 g, 39.4 mmol) was added dropwise into a toluene solution (50.0 mL) containing isomannide (2.88 g, 19.7 mmol, 0.5 equiv) and triethylamine (5.00 g, 49.3 mmol, 1.25 equiv) for over 30 min at 0 °C. The stirred mixture was then warmed to room temperature and stirred for 24 h. After the reaction reached completion, by confirmation of consumption of 10-undecenoyl chloride by TLC, the reaction mixture was then neutralized with 2 M HCl and was washed with 10% NaHCO3 (15 mL × 2), deionized water (15 mL × 2), and brine (15 mL × 2). The solution was dried over anhydrous MgSO4 and then filtered through a Celite pad. The filtrate was evaporated under reduced pressure. The crude product was purified by column chromatography using n-hexane and ethyl acetate (8/2) as an eluent to yield ME3 as a colorless oil (6.33 g, 82% yield). 1H NMR (CDCl3): δ 1.20 (br s, 16H, −CH2−), 1.34 (br, 4H, −CH2−),1.55 (t, J = 6.8 Hz, 4H, −CH2CH2COO−), 1.94 (d, J = 6.3 Hz, 4H, −CH2CH=CH), 2.28 (t, J = 7.3 Hz, 4H, −CH2COO−), 3.70 (t, J = 7.6 Hz, 2H, −CH2–OCH−), 3.93 (t, J = 7.1 Hz, 2H, −CH2–OCH−), 4.60 (s, 2H, −CH2–OCH−), 4.83 (d, J = 10.1 Hz, 2H, −CH=CH2), 4.89 (d, J = 17.1 Hz, 2H, −CH=CH2), 4.99 (d, J = 4.4 Hz, 2H, −CHOCO−), 5.70 (ddt, J = 16.8, 10.0, 6.8 Hz, 2H, −CH=CH2). 13C{1H} NMR (CDCl3): δ 24.8, 28.8, 28.9, 29.1, 29.2, 33.7, 33.8, 70.3, 73.5, 80.3, 114.1 (CH2=CH−), 138.9 (−CH=CH2), 172.9 (−COO−). APCI-MS: calcd for C28H46O6m/z, 479.34 [M + H]+; found, 479.3.
Synthesis of Cyclohexane-1,4-dimethanol Bis(undec-10-enoate) (ME4)
10-Undecenoyl chloride (8.00 g, 39.4 mmol) was added dropwise into a toluene solution (50.0 mL) containing 1,4-cyclohexanedimethanol (2.84 g, 19.7 mmol, 0.5 equiv) and triethylamine (5.00 g, 49.3 mmol, 1.25 equiv) for over 30 min at 0 °C. The stirred mixture was then warmed to room temperature and was stirred for 24 h. After the reaction reached completion, by confirmation of consumption of 10-undecenoyl chloride by TLC, the reaction mixture was then neutralized with 2 M HCl and was washed with 10% NaHCO3 (15 mL × 2), deionized water (15 mL × 2), and brine (15 mL × 2). The solution was dried over anhydrous MgSO4 and was then filtered through a Celite pad, and the filtrate was evaporated under reduced pressure. The crude product was purified by column chromatography using n-hexane and ethyl acetate (8/2) as an eluent to yield ME4 as a colorless oil (6.02 g, 88% yield). 1H NMR (CDCl3): δ 1.29 (br s, 20H, −CH2−), 1.37 (t, J = 6.3 Hz, 4H, −CH2−), 1.61 (t, J = 7.1 Hz, 6H, −CH2−), 1.80 (d, J = 7.1 Hz, 4H, −CH2CH2COO−), 2.03 (q or dt, J = 6.90 Hz, 4H, −CH2CH=CH2), 2.29 (t, J = 7.5 Hz, 4H, −CH2COO−), 3.89 and 3.99 (d, J = 6.6 Hz, 7.2 Hz, 4H, −CH2OCO−), 4.92 (dd, J = 10.2, 1.0 Hz, 2H, −CH=CH2), 4.98 (dd, J = 17.3, 1.7 Hz, 2H, −CH=CH2), 5.80 (ddt, J = 17.3, 10.3, 6.7 Hz, 2H, −CH=CH2). 13C{1H} NMR (CDCl3): δ 25.0, 25.3, 28.9, 29.0, 29.1, 29.2, 29.3, 33.8, 34.3, 34.5, 37.1, 66.9, 69.1, 114.1 (CH2=CH−), 139.1 (−CH=CH2), 173.8 (−COO−). APCI-MS: calcd for C30H52O4m/z, 477.39 [M + H]+; found, 477.40.
ADMET Polymerization Using Ruthenium-Carbene Catalysts
The typical polymerization procedure is as follows. In the drybox, monomers (ME1 0.71 mmol, 300 mg, conc. 5.29 mmol/mL), which were pretreated by passing the mixed solution (n-hexane and toluene, 2:1) through a Celite pad and was removed the volatiles in vacuo in the drybox, and a CHCl3 solution (0.14 mL, anhydrous) containing a prescribed amount of the 2nd generation Hoveyda-Grubbs catalyst (HG2) were added into a 25 mL scale sealed Schlenk tube. The reaction mixture was taken out from the drybox and was magnetically stirred in an oil bath set at 50 °C. The mixture was then placed into a liquid nitrogen bath to remove the ethylene gas from the reaction by opening the valve connected to the vacuum line for a short period (1 min), and the valve was then closed and the tube was placed into the oil bath to continue the reaction.32,57,58 The procedure removing ethylene was repeated for a certain period (30 min for the first time then every 1.0 h until 6 h). The polymerization mixture was then cooled to room temperature and was quenched with excess ethyl vinyl ether (two drops, ca. 100 mg) while stirring for 1.0 h. The resultant solution was then dissolved in chloroform (2.0 mL) for dilution, and the solution was added dropwise into the stirred cold methanol (50 mL). The solution was stirred for 15 min, and the precipitates were then collected by filtration and dried in vacuo to yield PE1 as a white solid (91% yield). Similar polymerization protocol was used for polymerization of ME2 (300 mg, 0.65 mmol, conc. 4.84 mmol/mL), ME3 (300 mg, 0.65 mmol, conc. 4.84 mmol/mL), and ME4 (300 mg, 0.63 mmol, conc. 4.69 mmol/mL) to yield the polymers PE2, PE3, and PE4, respectively.
PE1. 1H NMR (CDCl3): δ 1.26 (br s, 20H, −CH2−), 1.56 (t, J = 7.3 Hz, 4H, −CH2CH2COO−), 1.67 (4H, −CH2CH2OCO−), 1.92 (4H, −CH2CH=CH2−), 2.26 (t, J = 7.5 Hz, 4H, −CH2COO−), 4.06 (s, 4H, −CH2OCO−), 5.29–5.38 (2H, −CH=CH−). 13C{1H} NMR (CDCl3): δ 25.0, 25.3, 29.1, 29.1, 29.2, 29.3, 29.6, 32.6, 34.2, 63.7 (−CH2OCO−), 130.3 (−CH=CH−), 173.8 (−COO−).
PE2. 1H NMR (CDCl3): δ 1.27 (br s, 20H, −CH2−), 1.61 (t, J = 6.6 Hz, 4H, −CH2CH2COO−), 1.94 (4H, −CH2CH=CH2−), 2.30 (t, J = 7.7 Hz, 2H, −CH2COO−), 2.34 (t, J = 7.1 Hz, 2H, −CH2COO−), 3.78 (dd, J = 9.8, 5.4 Hz, 1H, −CH2OCH−), 3.92–3.97 (m, 3H, −CH2–OCH–, −CH2–OCH−), 4.46 (d, J = 4.6 Hz, 1H, −CH2–OCH−), 4.82 (t, J = 5.0 Hz, 1H, −CH2–OCH−), 5.13 (dd, J = 5.6 Hz, 1H, −CHOCO−), 5.18 (d, J = 3.2 Hz, 1H, −CHOCO−), 5.37 (2H, −CH=CH−). 13C{1H} NMR (CDCl3): δ 24.8, 24.9, 29.0, 29.1, 29.2, 29.3, 32.6, 33.9, 34.2, 70.3, 73.5, 73.7, 80.7, 86.0, 130.3 (−CH=CH−), 172.9 (−COO−), 173.2 (−COO−).
PE3. 1H NMR (CDCl3): δ 1.31 (20H −CH2−), 1.65 (t, J = 6.8 Hz, 4H, −CH2CH2COO−), 1.97 (4H, −CH2CH=CH−), 2.39 (t, J = 7.3 Hz, 4H, −CH2COO−), 3.81 (t, J = 7.6 Hz, 2H, −CH2–OCH−), 4.04 (t, J = 7.1 Hz, 2H, −CH2–OCH), 4.70 (s, 2H, −CH2–OCH−), 5.10 (d, J = 4.4 Hz, 2H, −CHOCO−), 5.39 (2H, −CH=CH−). 13C{1H} NMR (CDCl3): δ 24.9, 29.0, 29.1, 29.2, 29.3, 29.6, 33.9, 70.4, 73.5, 80.4, 130.3 (−CH=CH−), 173.2 (−COO−).
PE4. 1H NMR (CDCl3): δ 1.24 (24H, −CH2−), 1.60 (6H, −CH2−), 1.78 (d, J = 7.1 Hz, 4H, −CH2CH2COO−), 2.03 (4H, −CH2CH=CH2), 2.28 (t, J = 7.5 Hz, 4H, −CH2COO−), 3.89 and 3.99 (d, J = 6.6 Hz, 4H, −CH2OCO−), 5.39 (2H, −CH=CH−). 13C{1H} NMR (CDCl3): δ 25.0, 25.3, 28.9, 29.2, 29.3, 29.5, 31.9, 34.4, 37.1, 66.9, 69.1, 130.3 (−CH=CH−), 173.9 (−COO−).
Tandem ADMET Polymerization and Subsequent Hydrogenation Using Ruthenium-Carbene Catalysts
The typical polymerization procedure is the same as that described above. After ADMET polymerization, the solution was transferred into an autoclave with addition of a prescribed amount of alumina powder, and the reactor was then pressurized with hydrogen at 1.0 MPa. The reactor was then set into an oil bath preheated at 50 °C and was stirred for 3 h. The resultant solution was then dissolved in chloroform (2.0 mL) for dilution, and the solution (after filtration through Celite pad) was added dropwise into the stirred cold methanol (50 mL). The solution was stirred for 15 min, and the precipitates were then collected by filtration and dried in vacuo to yield hydrogenated HPE1 as a whit e solid.
HPE1. 1H NMR (CDCl3): δ 1.30 (28H, −CH2−), 1.63 (4H, −CH2CH2COO−), 1.72 (s, 4H, −CH2CH2OCO−), 2.31 (t, J = 7.5 Hz, 4H, −CH2COO−), 4.11 (s, 4H, −CH2OCO−). 13C{1H} NMR (CDCl3): δ 25.0, 25.4, 29.2, 29.3, 29.5, 29.6, 29.7, 34.3, 63.7 (−CH2OCO−), 173.9 (−COO−).
HPE2. 1H NMR (CDCl3): δ 1.27 (28H, −CH2−), 1.64 (m, 4H, −CH2CH2COO−), 2.33 (t, J = 7.7 Hz, 2H, −CH2COO−), 2.39 (t, J = 7.1 Hz, 2H, −CH2COO−), 3.81 (dd, J = 9.8 and 5.4 Hz, 1H, −CH2OCH−), 3.96–4.02 (m, 3H, −CH2–OCH–, −CH2–OCH−), 4.49 (d, J = 4.6 Hz, 1H, −CH2–OCH−), 4.84 (t, J = 5.0 Hz, 1H, −CH2–OCH−), 5.16 (dd, J = 5.6 Hz, 1H, −CHOCO−), 5.20 (d, J = 3.2 Hz, 1H, −CHOCO−). 13C{1H} NMR (CDCl3): δ 24.8, 24.9, 29.1, 29.2, 29.5, 29.6, 29.7, 34.0, 34.2, 70.4, 73.5, 73.7, 80.8, 86.0, 172.9 (−COO−), 173.2 (−COO−).
HPE3. 1H NMR (CDCl3): δ 1.25 (28H), 1.64 (4H), 2.38 (4H), 3.79 (m, 2H), 4.02 (m, 2H), 4.69 (2H), 5.09 (2H). δ 1.25 (28H −CH2−), 1.64 (t, J = 6.8 Hz, 4H, −CH2CH2COO−), 2.38 (t, J = 7.3 Hz, 4H, −CH2COO−), 3.79 (t, J = 7.6 Hz, 2H, −CH2–OCH−), 4.02 (t, J = 7.1 Hz, 2H, −CH2–OCH−), 4.69 (d, J = 4.1 Hz, 2H, −CH2–OCH−), 5.09 (2H, −CHOCO−). 13C{1H} NMR (CDCl3): δ 24.9, 29.1, 29.3, 29.5, 29.6, 29.7, 33.9, 70.4, 73.5, 80.4, 173.3 (−COO−).
HPE4. 1H NMR (CDCl3): δ 1.27 (32H), 1.62 (6H), 1.81 (4H), 2.31 (4H), 3.85 and 3.95 (4H). δ 1.27 (32H, −CH2−), 1.62 (6H, −CH2−), 1.81 (d, J = 7.1 Hz, 4H, −CH2CH2COO−), 2.31 (t, J = 7.5 Hz, 4H, −CH2COO−), 3.85 and 3.95 (d, J = 6.6 Hz, 4H, −CH2OCO−). 13C{1H} NMR (CDCl3): δ 25.0, 25.3, 28.9, 29.2, 29.3, 29.5, 29.6, 29.7, 31.9, 34.4, 37.1, 66.9, 69.1, 174.0 (−COO−).
Reaction with Ethylene in the Resultant Polymer Prepared by ADMET Polymerization Using Ruthenium-Carbene Catalysts. Confirmation of the Reversible Nature in This Catalysis
The typical polymerization procedure is the same as that described above (50 °C, 6 h). After ADMET polymerization, the solution was transferred to an autoclave, and the reactor was then pressurized with ethylene at 0.8 MPa. The reactor was then set into an oil bath preheated at 50 °C and was stirred for 1 h. The resultant solution was then dissolved in chloroform (2.0 mL) for dilution, and the solution was added dropwise into the stirred cold methanol (50 mL). The solution was stirred for 15 min, and the precipitates were then collected by filtration and dried in vacuo to yield (depolymerized) oligomers (DPE1 or DPE2) as white-gray precipitates (solid). NMR spectra in the resultant mixture after the removal of volatiles are shown in the Supporting Information.a
DPE1. 1H NMR (CDCl3): δ 1.28 (s, 22H), 1.61 (t, J = 6.9 Hz, 4H), 1.69 (s, 4H), 1.95 (d, J = 4.2 Hz, 4H), 2.28 (t, J = 7.6 Hz, 4H), 4.09 (s, 4H), 4.90–5.01 (d, J = 9.8 Hz, 4H), 5.37 (t, J = 18.2 Hz, 16H), 5.76–5.85 (m, 2H). 13C{1H} NMR (CDCl3): δ 25.0, 25.4, 29.0, 29.1, 29.2, 29.3, 29.4, 29.6, 32.6, 34.3, 63.7, 130.3, 173.9 (−COO−).
DPE2. 1H NMR (CDCl3): δ 1.24–1.38 (22H), 1.57–1.65 (quint, J = 6.4 and 8.1 Hz, 4H), 1.95 (d, J = 4.2 Hz, 2H), 2.02 (quart, J = 7.2 and 7.2 Hz, 2H), 2.30 (t, J = 7.5 Hz, 2H), 2.36 (t, J = 7.5 Hz, 2H), 3.78 (dd, J = 4.2 and 5.5 Hz, 2H), 3.93–3.99 (4H), 4.46 (d, J = 4.6 Hz, 2H), 4.82 (t, J = 4.9 Hz, 2H), 4.92 and 4.98 (d, J = 10.1 Hz, 4H), 5.14 (quart, J = 5.6 and 5.5 Hz, 2H), 5.18 (d, J = 2.6 Hz, 2H), 5.37 (t, J = 17.7 Hz, 2H), 5.77–5.83 (2H). 13C{1H} NMR (CDCl3): δ 24.8, 24.9, 28.9, 29.0, 29.1, 29.2, 29.3, 29.4, 29.6, 32.5, 32.6, 34.0, 34.2, 70.3, 73.5, 73.7, 80.7, 86.0, 114.2, 130.3, 139.2, 172.9 and 173.2 (−COO−).
Acknowledgments
This project was partly supported by JST SICORP grant number JPMJSC19E2, Japan, and Tokyo Metropolitan Government Advanced Research Grant number R2-1. PC expresses his thanks to the Tokyo Metropolitan government (Tokyo Human Resources Fund for City Diplomacy) for fellowship of the graduate study, and M.M.A. expresses his thanks to The Follow-up Research Fellowship (Tokyo Human Resources Fund for City Diplomacy, Tokyo Metropolitan University) for conducting collaboration research. The authors express their thanks to Profs. S. Komiya and A. Inagaki (Tokyo Metropolitan Univ., TMU) for discussion.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c01965.
Additional polymerization results, selected NMR spectra, DSC thermograms, and GPC trace before/after depolymerization (PDF)
The authors declare no competing financial interest.
Footnotes
Selected NMR spectra for monomers (ME1–4), the resultant unsaturated polymers (PE1–4), polymers after hydrogenation (JPE1–4), and reaction mixture after reaction of PE1 or PE2 with ethylene are shown in the Supporting Information.
These ADMET polymerizations were conducted at 50 °C, according to our previous reports,33,58,59 and the polymerization at high temperature caused decrease in the activity and observation of side reactions (such as addition polymerization) for synthesis of conjugated polymers.58 Unpublished results.
Supplementary Material
References
- Gandini A. Polymers from renewable resources: A challenge for the future of macromolecular materials. Macromolecules 2008, 41, 9491–9504. 10.1021/ma801735u. [DOI] [Google Scholar]
- Coates G. W.; Hillmyer M. A. A Virtual Issue ofMacromolecules: “Polymers from Renewable Resources”. Macromolecules 2009, 42, 7987–7989. 10.1021/ma902107w. [DOI] [Google Scholar]
- Mülhaupt R. Green polymer chemistry and bio-based plastics: Dreams and reality. Macromol. Chem. Phys. 2013, 214, 159–174. 10.1002/macp.201200439. [DOI] [Google Scholar]
- Kawashima N.; Yagi T.; Kojima K. How Do Bioplastics and Fossil-Based Plastics Play in a Circular Economy?. Macromol. Mater. Eng. 2019, 304, 1900383. 10.1002/mame.201900383. [DOI] [Google Scholar]
- Monomers and polymers from chemically modified plant oils and their fatty acids. In Polymers from Plant Oils, 2nd ed.; Gandini A., Lacerda T. M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA: and Scrivener Publishing LLC, Beverly, MA, USA, 2019; pp 33–82. [Google Scholar]
- Yao K.; Tang C. Controlled polymerization of next-generation renewable monomers and beyond. Macromolecules 2013, 46, 1689–1712. 10.1021/ma3019574. [DOI] [Google Scholar]
- Wang Z.; Yuan L.; Tang C. Sustainable elastomers from renewable biomass. Acc. Chem. Res. 2017, 50, 1762–1773. 10.1021/acs.accounts.7b00209. [DOI] [PubMed] [Google Scholar]
- Coulembier O.; Degée P.; Hedrick J. L.; Dubois P. From controlled ring-opening polymerization to biodegradable aliphatic polyester: Especially poly(β-malic acid) derivatives. Prog. Polym. Sci. 2006, 31, 723–747. 10.1016/j.progpolymsci.2006.08.004. [DOI] [Google Scholar]
- Meier M. A. R.; Metzger J. O.; Schubert U. S. Plant oil renewable resources as green alternatives in polymer science. Chem. Soc. Rev. 2007, 36, 1788–1802. 10.1039/b703294c. [DOI] [PubMed] [Google Scholar]
- Mutlu H.; Meier M. A. R. Castor oil as a renewable resource for the chemical industry. Eur. J. Lipid Sci. Technol. 2010, 112, 10–30. 10.1002/ejlt.200900138. [DOI] [Google Scholar]
- Biermann U.; Bornscheuer U.; Meier M. A. R.; Metzger J. O.; Schäfer H. J. Oils and fats as renewable raw materials in chemistry. Angew. Chem., Int. Ed. 2011, 50, 3854–3871. 10.1002/anie.201002767. [DOI] [PubMed] [Google Scholar]
- Stempfle F.; Ortmann P.; Mecking S. Long-chain aliphatic polymers to bridge the gap between semicrystalline polyolefins and traditional polycondensates. Chem. Rev. 2016, 116, 4597–4641. 10.1021/acs.chemrev.5b00705. [DOI] [PubMed] [Google Scholar]
- Metathesis reactions applied to plant oils and polymers derived from the ensuing products. In Polymers from Plant Oils, 2nd ed.; Gandini A., Lacerda T. M., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA: and Scrivener Publishing LLC, Beverly, MA, USA, 2019; pp 83–108. [Google Scholar]
- Zakzeski J.; Bruijnincx P. C. A.; Jongerius A. L.; Weckhuysen B. M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. 10.1021/cr900354u. [DOI] [PubMed] [Google Scholar]
- Fenouillot F.; Rousseau A.; Colomines G.; Saint-Loup R.; Pascault J.-P. Polymers from renewable 1,4:3,6-dianhydrohexitols (isosorbide, isomannide and isoidide): A review. Prog. Polym. Sci. 2010, 35, 578–622. 10.1016/j.progpolymsci.2009.10.001. [DOI] [Google Scholar]
- Wilbon P. A.; Chu F.; Tang C. Progress in renewable polymers from natural terpenes, terpenoids, and rosin. Macromol. Rapid Commun. 2013, 34, 8–37. 10.1002/marc.201200513. [DOI] [PubMed] [Google Scholar]
- Thomsett M. R.; Storr T. E.; Monaghan O. R.; Stockman R. A.; Howdle S. M. Progress in the synthesis of sustainable polymers from terpenes and terpenoids. Green Mater. 2016, 4, 115–134. 10.1680/jgrma.16.00009. [DOI] [Google Scholar]
- Hillmyer M. A.; Tolman W. B. Aliphatic polyester block polymers: Renewable, degradable, and sustainable. Acc. Chem. Res. 2014, 47, 2390–2396. 10.1021/ar500121d. [DOI] [PubMed] [Google Scholar]
- Fortman D. J.; Brutman J. P.; De Hoe G. X.; Snyder R. L.; Dichtel W. R.; Hillmyer M. A. Approaches to sustainable and continually recyclable cross-linked polymers. ACS Sustainable Chem. Eng. 2018, 6, 11145–11159. 10.1021/acssuschemeng.8b02355. [DOI] [Google Scholar]
- Nakajima H.; Dijkstra P.; Loos K. The recent developments in biobased polymers toward general and engineering applications: Polymers that are upgraded from biodegradable polymers, analogous to petroleum-derived polymers, and newly developed. Polymers 2017, 9, 523. 10.3390/polym9100523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples, see refs:21−36Mosnáček J.; Matyjaszewski K. Atom transfer radical polymerization of Tulipalin A: A naturally renewable monomer. Macromolecules 2008, 41, 5509–5510. 10.1021/ma8010813. [DOI] [Google Scholar]
- Rybak A.; Meier M. A. R. Acyclic diene metathesis with a monomer from renewable resources: Control of molecular weight and one-step preparation of block copolymers. ChemSusChem 2008, 1, 542–547. 10.1002/cssc.200800047. [DOI] [PubMed] [Google Scholar]
- Fokou P. A.; Meier M. A. R. Use of a renewable and degradable monomer to study the temperature-dependent olefin isomerization during ADMET polymerizations. J. Am. Chem. Soc. 2009, 131, 1664–1665. 10.1021/ja808679w. [DOI] [PubMed] [Google Scholar]
- Mutlu H.; Meier M. A. R. Unsaturated PA X,20 from renewable resources via metathesis and catalytic amidation. Macromol. Chem. Phys. 2009, 210, 1019–1025. 10.1002/macp.200900045. [DOI] [Google Scholar]
- De Espinosa L. M.; Ronda J. C.; Galià M.; Cádiz V.; Meier M. A. R. Fatty acid derived phosphorus-containing polyesters via acyclic diene metathesis polymerization. J. Polym. Sci., Part A: Polym. Chem. 2009, 47, 5760–5771. 10.1002/pola.23620. [DOI] [Google Scholar]
- Fokou P. A.; Meier M. A. R. Studying and suppressing olefin isomerization side reactions during ADMET polymerizations. Macromol. Rapid Commun. 2010, 31, 368–373. 10.1002/marc.200900678. [DOI] [PubMed] [Google Scholar]
- Ortmann P.; Mecking S. Long-spaced aliphatic polyesters. Macromolecules 2013, 46, 7213–7218. 10.1021/ma401305u. [DOI] [Google Scholar]
- Lebarbé T.; Neqal M.; Grau E.; Alfos C.; Cramail H. Branched polyethylene mimicry by metathesis copolymerization of fatty acid-based α,ω-dienes. Green Chem. 2014, 16, 1755–1758. 10.1039/c3gc42280a. [DOI] [Google Scholar]
- Shearouse W. C.; Lillie L. M.; Reineke T. M.; Tolman W. B. Sustainable polyesters derived from glucose and castor oil: Building block structure impacts properties. ACS Macro Lett. 2015, 4, 284–288. 10.1021/acsmacrolett.5b00099. [DOI] [PubMed] [Google Scholar]
- Llevot A.; Grau E.; Carlotti S.; Grelier S.; Cramail H. ADMET polymerization of bio-based biphenyl compounds. Polym. Chem. 2015, 6, 7693–7700. 10.1039/c5py01232e. [DOI] [Google Scholar]
- Barbara I.; Flourat A. L.; Allais F. Renewable polymers derived from ferulic acid and biobased diols via ADMET. Eur. Polym. J. 2015, 62, 236–243. 10.1016/j.eurpolymj.2014.11.035. [DOI] [Google Scholar]
- Dannecker P.-K.; Biermann U.; Sink A.; Bloesser F. R.; Metzger J. O.; Meier M. A. R. Fatty Acid–Derived Aliphatic Long Chain Polyethers by a Combination of Catalytic Ester Reduction and ADMET or Thiol-Ene Polymerization. Macromol. Chem. Phys. 2019, 220, 1800440. 10.1002/macp.201800440. [DOI] [Google Scholar]
- Le D.; Samart C.; Kongparakul S.; Nomura K. Synthesis of new polyesters by acyclic diene metathesis polymerization of bio-based α,ω-dienes prepared from eugenol and castor oil (undecenoate). RSC Adv. 2019, 9, 10245–10252. 10.1039/c9ra01065c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser B. R.; Vermillion K. E.; Banks B. N.; Doll K. M. Renewable aliphatic polyesters from fatty dienes by acyclic diene metathesis polycondensation. J. Am. Oil Chem. Soc. 2020, 97, 517–530. 10.1002/aocs.12338. [DOI] [Google Scholar]
- Quinzler D.; Mecking S. Linear semicrystalline polyesters from fatty acids by complete feedstock molecule utilization. Angew. Chem., Int. Ed. 2010, 49, 4306–4308. 10.1002/anie.201001510. [DOI] [PubMed] [Google Scholar]
- Lebarbé T.; Maisonneuve L.; Nga Nguyen T. H.; Gadenne B.; Alfos C.; Cramail H. Methyl 10-undecenoate as a raw material for the synthesis of renewable semicrystalline polyesters and poly(ester-amide)s. Polym. Chem. 2012, 3, 2842–2851. 10.1039/c2py20394d. [DOI] [Google Scholar]
- Stempfle F.; Ritter B. S.; Mülhaupt R.; Mecking S. Long-chain aliphatic polyesters from plant oils for injection molding, film extrusion and electrospinning. Green Chem. 2014, 16, 2008–2014. 10.1039/c4gc00114a. [DOI] [Google Scholar]
- For examples, see refs:38−41Nasiri M.; Reineke T. M. Sustainable glucose-based block copolymers exhibit elastomeric and adhesive behavior. Polym. Chem. 2016, 7, 5233–5240. 10.1039/c6py00700g. [DOI] [Google Scholar]
- Lillie L. M.; Tolman W. B.; Reineke T. M. Structure/property relationships in copolymers comprising renewable isosorbide, glucarodilactone, and 2,5-bis(hydroxymethyl)-furan subunits. Polym. Chem. 2017, 8, 3746–3754. 10.1039/c7py00575j. [DOI] [Google Scholar]
- Gallagher J. J.; Hillmyer M. A.; Reineke T. M. Isosorbide-based polymethacrylates. ACS Sustain. Chem. Eng. 2015, 3, 662–667. 10.1021/sc5008362. [DOI] [Google Scholar]
- Wang J.; Mahmud S.; Zhang X.; Zhu J.; Shen Z.; Liu X. Biobased amorphous polyesters with high Tg: Trade-off between rigid and flexible cyclic diols. ACS Sustainable Chem. Eng. 2019, 7, 6401–6411. 10.1021/acssuschemeng.9b00285. [DOI] [Google Scholar]
- Strick B. F.; Delferro M.; Geiger F. M.; Thomson R. J. Investigations into apopinene as a biorenewable monomer for ring-opening metathesis polymerization. ACS Sustainable Chem. Eng. 2015, 3, 1278–1281. 10.1021/acssuschemeng.5b00255. [DOI] [Google Scholar]
- Schwendeman J. E.; Church A. C.; Wagener K. B. Synthesis and catalyst issues associated with ADMET polymerization. Adv. Synth. Catal. 2002, 344, 597–613. . [DOI] [Google Scholar]
- Mutlu H.; de Espinosa L. M.; Meier M. A. R. Acyclic diene metathesis: a versatile tool for the construction of defined polymer architectures. Chem. Soc. Rev. 2011, 40, 1404–1445. 10.1039/b924852h. [DOI] [PubMed] [Google Scholar]
- Atallah P.; Wagener K. B.; Schulz M. D. ADMET: The future revealed. Macromolecules 2013, 46, 4735–4741. 10.1021/ma400067b. [DOI] [Google Scholar]
- Schulz M. D.; Wagener K. B. Precision polymers through ADMET polymerization. Macromol. Chem. Phys. 2014, 215, 1936–1945. 10.1002/macp.201400268. [DOI] [Google Scholar]
- Chen Y.; Abdellatif M. M.; Nomura K. Olefin metathesis polymerization: Some recent developments in the precise polymerizations for synthesis of advanced materials (by ROMP, ADMET). Tetrahedron 2018, 74, 619–643. 10.1016/j.tet.2017.12.041. [DOI] [Google Scholar]
- Hydrogenation of ADMET polymer using p-toluenesulfonyl hydrazide and tri-n-propyl amine. See, refs:48−50Boz E.; Nemeth A. J.; Alamo R. G.; Wagener K. B. Precision ethylene/vinyl bromide polymers. Adv. Synth. Catal. 2007, 349, 137–141. 10.1002/adsc.200600433. [DOI] [Google Scholar]
- Rojas G.; Inci B.; Wei Y.; Wagener K. B. Precision polyethylene: Changes in morphology as a function of alkyl branch size. J. Am. Chem. Soc. 2009, 131, 17376–17386. 10.1021/ja907521p. [DOI] [PubMed] [Google Scholar]
- Boz E.; Nemeth A. J.; Ghiviriga I.; Jeon K.; Alamo R. G.; Wagener K. B. Precision ethylene/vinyl chloride polymers via condensation polymerization. Macromolecules 2007, 40, 6545–6551. 10.1021/ma070933g. [DOI] [Google Scholar]
- Hydrogenation of ADMET polymer using RhCl(PPh3)3, see refs (50) and (51):Inci B.; Lieberwirth I.; Steffen W.; Mezger M.; Graf R.; Landfester K.; Wagener K. B. Decreasing the alkyl branch frequency in precision polyethylene: Effect of alkyl branch size on nanoscale morphology. Macromolecules 2012, 45, 3367–3376. 10.1021/ma3002577. [DOI] [Google Scholar]
- Baughman T. W.; Chan C. D.; Winey K. I.; Wagener K. B. Synthesis and morphology of well-defined poly(ethylene-co-acrylic acid) copolymers. Macromolecules 2007, 40, 6564–6571. 10.1021/ma070841r. [DOI] [Google Scholar]
- Sworen J. C.; Smith J. A.; Berg J. M.; Wagener K. B. Modeling branched polyethylene: copolymers possessing precisely placed ethyl branches. J. Am. Chem. Soc. 2004, 126, 11238–11246. 10.1021/ja047850p. [DOI] [PubMed] [Google Scholar]
- Li H.; Rojas G.; Wagener K. B. Precision long-chain branched polyethylene via acyclic diene metathesis polymerization. ACS Macro Lett. 2015, 4, 1225–1228. 10.1021/acsmacrolett.5b00641. [DOI] [PubMed] [Google Scholar]
- Pesko D. M.; Webb M. A.; Jung Y.; Zheng Q.; Miller T. F. III; Coates G. W.; Balsara N. P. Universal relationship between conductivity and solvation-site connectivity ether-based polymer electrolytes. Macromolecules 2016, 49, 5244–5255. 10.1021/acs.macromol.6b00851. [DOI] [Google Scholar]
- Abdellatif M. M.; Nomura K. Precise synthesis of amphiphilic multiblock copolymers by combination of acyclic diene metathesis (ADMET) polymerization with atom transfer radical polymerization (ATRP) and click chemistry. ACS Macro Lett. 2012, 1, 423–427. 10.1021/mz300061a. [DOI] [PubMed] [Google Scholar]
- Miyashita T.; Kunisawa M.; Sueki S.; Nomura K. Synthesis of poly(arylene vinylene)s containing different end groups by combined acyclic diene metathesis polymerization with Wittig-type coupling. Angew. Chem., Int. Ed. 2017, 56, 5288–5293. 10.1002/anie.201700466. [DOI] [PubMed] [Google Scholar]
- For example, see refs (58) and (59):Nomura K.; Morimoto H.; Imanishi Y.; Ramhani Z.; Geerts Y. Synthesis of high molecular weighttrans-poly(9,9-di-n-octylfluorene-2,7-vinylene) by the acyclic diene metathesis polymerization using molybdenum catalysts. J. Polym. Sci., Part A: Polym. Chem. 2001, 39, 2463–2470. 10.1002/pola.1223. [DOI] [Google Scholar]
- Yamamoto N.; Ito R.; Geerts Y.; Nomura K. Synthesis of All-Trans High Molecular Weight Poly(N-alkylcarbazole-2,7-vinylene)s and Poly(9,9-dialkylfluorene-2,7-vinylene)s by Acyclic Diene Metathesis (ADMET) Polymerization Using Ruthenium–Carbene Complex Catalysts. Macromolecules 2009, 42, 5104–5111. 10.1021/ma900775x. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.