

Graphical abstract
Cellular rigidity-sensing mechanisms to discrete areas of increased or decreased rigidity are not understood. Here we describe a new class of biomimetic surfaces comprising micro and nano-patterned heterogeneous rigidity by focused electron beam exposure. Mesenchymal stem cells exhibited dose dependent focal adhesion co-localization to the exposed regions, a response that was lost on feature dimensions > 500 nm. Concurrently cell function was significantly modulated by both spot rigidity and spot size.
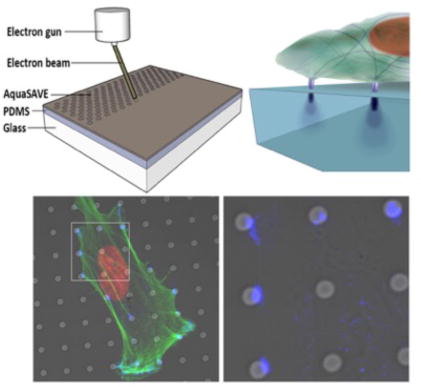
Keywords: electron beam, rigidity, stem cells, mechanotransduction, focal adhesions, polydimethylsiloxane
1. Introduction
As well as responding to biochemical signals, cells directly probe and respond to the physicomechanical properties of their extracellular environment through adhesion complexes and tractive mediated matrix deformation.[1] Increasingly, it is evident that matrix or tissue elasticity has a key role in regulating multiple cell processes[2], including adhesion,[3] migration[4, 5] and differential function[6, 7] through cell-generated actomyosin interactive forces regulated by cell-substrate adhesion and dynamic feedback mechanisms. [5]
The sensitivity of cells to the mechanical properties of the extracellular matrix (ECM) is attributable to the mechanosensitive nature of the molecules involved in the structures of cell adhesion.[8] Among adhesive structures, focal adhesions (FAs) appear to be the most critical, as shown by the reported correlation between FA size and a sustained force exhibiting a constant stress.[9, 10] This mechanosensitivity is thought to be regulated by a conserved local mechanism in which subcellular forces induce an elastic deformation of transmembrane integrin regions, triggering conformational and organizational changes and resulting in integrin activation, which in turn can uncover cryptic binding sites for additional protein binding, enabling FA reinforcement[11] These processes should set a dimensional scale for cellular rigidity sensing.
Modulation to FA formation has been implicated in the onset of differential cell function[12] and recent studies indicate that stem cells show lineage-specific differentiation when cultured on substrates matching the stiffness corresponding to native tissues. Although tissues are associated with ranging Young's modulus values for bulk rigidity, at the sub-cellular level, and particularly at the micro- and nanoscales, tissues are composed of heterogeneous distributions of cellular elements and extracellular particles, and fibers of varying rigidity. Specifically, skeletal stem cells reside in a specialized biophysical and biochemical niche environment which is thought to present physicomechanical cues critical to phenotype maintenance and preventing loss of stemness.[12]
ECM architectures encountered by skeletal stem cells in vivo range from the microscale (insoluble fibrillar ECM proteins), to the nanoscale (apatite crystals), with elastic moduli ranging from 2-7 kPa (plasma membrane)[13] to 5 GPa (collagen type I)[14] to 150 GPa (hydroxyapatite).[15] Critically, how cells sense and respond to the mechanical properties of their surroundings in a heterogeneous environment and the role of mechanical heterogeneity in mediating skeletal stem cell function remains poorly understood.[4, 16]
Elastomeric substrates and hydrogels have been used to present cells with surfaces of specific stiffness, approximating the range of rigidities encountered in physiological Environments.[7] Poly(dimethylsiloxane) (PDMS), in particular, has recently found widespread use in cell adhesion/migration assays[17] microfluidic and MEMS technologies[18] due to its favorable optical, biocompatible and mechanical properties. Indeed, PDMS substrates have been used to study the role of extracellular rigidity on cellular adhesion[19] and differentiation,[20] due to the ease by which its rigidity may be precisely controlled by simply varying the base:accelerator ratio.
Within the realms of micro and nanofabrication, lithographic processes have been widely explored in cell biology for the generation of analytical substrates for probing physicochemical responses at the cellular and sub-cellular level. In particular PDMS has been immensely useful for nanoscale patterning of proteins (soft lithography) and for the generation of pillar substrates with differential rigidity.[9] Significantly, unlike traditional photo-crosslinkable materials and photoresists PDMS is transparent in the visible and ultraviolet wavelengths and cannot, in general, be directly patterned by standard photolithography,[21] without the addition of a photoactive compound.[22] However, PDMS has been shown to be sensitive to deep-UV and e-beam irradiation,[22, 23, 24] which induces cross-linking of the elastomer.
In this work, we show that direct e-beam exposure can significantly alter the rigidity of PDMS. This has enabled us to develop a new class of two-dimensional elastomeric substrates with geometrically patterned heterogeneous rigidity. Specifically, we studied the mechano-response of hMSCs cultured on ∼35 kPa elastomer substrates, modified to present patterns of micro- and nanoscale spots with discrete elastic moduli from ∼50 – ∼350 MPa. Critically, substrates possessing similar bulk rigidity values have been previously shown to induce stem cell differentiation to cartilage and bone specific lineages in vitro.[7, 25] We observed a differential co-localization of FAs to the patterned rigid regions in human skeletal stem cells (hMSCs), and that this response is maintained on micron and submicron features, revealing the existence of a submicron machinery in hMSCs that may be important in the cellular response to local rigidity. We further assessed the influence of heterogeneous rigidity on differential hMSC function through Ingenuity Pathway analysis of osteochondral differentiation. Crucially we noted significant modulation to functional signaling in osteochondral lineage commitment pathways as a function of both spot rigidity and spot size relative to cells cultured under control conditions (unexposed PDMS coupled with chondrogenc or osteogenic growth media). Interestingly, the onset of osteochnodral differentiation was induced on heterogeneous rigidity substrates after only 12 hours in culture. Elucidating the geometrical and mechanical limits of the cellular mechanoresponse to discrete rigidity will enhances current understanding of in vivo cell behavior in processes such as embryogenesis, healing and cellular metastasis.[26] In addition, an understanding of the geometrical basis for rigidity sensing will be essential for the design of implants with artificial smart surfaces for optimal cellular interfacial interaction.
2. Results
2.1. Modulation of PDMS rigidity
PDMS was prepared at a 50:1 ratio of base polymer:accelerating agent and was spin-coated onto standard microscope cover-glasses. Samples were cured for 12h at 70°C to form optically transparent viscoelastic films, ∼ 120 nm thick, which were rendered hydrophilic via a 1 min oxygen plasma treatment to facilitate a further spin-coating application of an electrically conducting layer (AquaSAVE, Mitsubishi Rayon) to suppress charging during e-beam exposure. PDMS substrates were patterned by e-beam exposure using a scanning electron microscope equipped with a Nabity NPGS pattern generator. Patterns were generated on the elastomer surface with e-beam exposure doses ranging from 500-3,000 μC/cm2, using an accelerating voltage of 30 kV and a beam current of ∼ 2.5 nA. The absorbed electron energy within the PDMS at the sub-surface rigidity gradient is determined by two factors: (i) the incident electron energy (30 keV in this study); and (ii) the scattering of electrons within the elastomer, which depends on the density of the material (schematised in Figure 1A). Analysis of Monte Carlo simulations[27] indicated that over 90% of the e-beam energy was absorbed within approximately the top 3 μm of the PDMS (Figure 1B), resulting in a scattering profile having a columnar shape with a broad spreading base that diminishes in intensity with increasing depth. Lateral scattering within the top layer was confined to ∼ 30 nm at 30 keV.
Figure 1. Electron-beam interaction with PDMS thin-films.
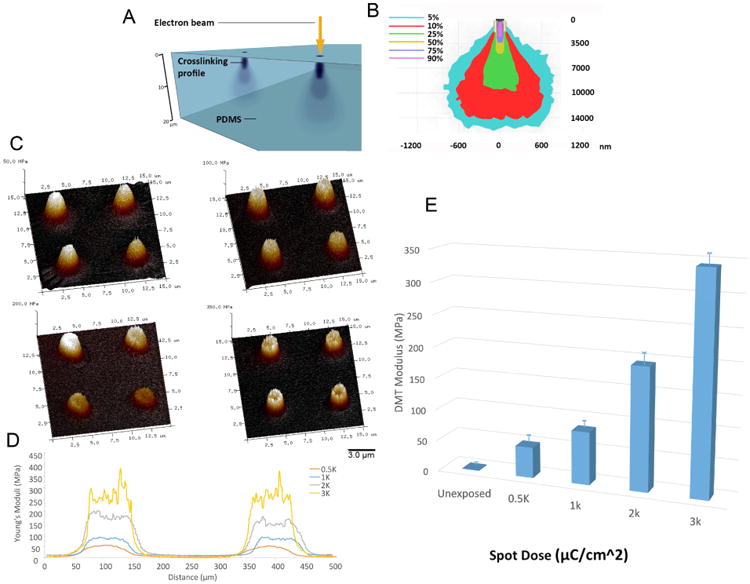
(A) A 120 μm layer of PDMS was deposited onto 22 mm square microscopy cover-glasses by a spin-coating process. Substrates were treated with an oxygen plasma process and coated with a final polymeric discharge layer (AquaSAVE) prior to e-beam patterning. A focused e-beam was rastered over the substrate surface to create arrays of defined surface features (spots) possessing a sub-surface rigidity gradient. (B) Monte Carlo simulations identified the electron trajectory and scatter profile in PDMS substrates. (C,D) Peak-force quantitative AFM nanomechanical mapping (PF-QNM) of 2 μm spots indicated the e-beam exposure of the PDMS film causes an increase in the elastic modulus of the polymer as a function of e-beam dose. (E) shows the function relating Young's modulus changes due to the e-beam exposure dose.
Peak-force quantitative AFM nanomechanical mapping (PF-QNM) was employed to characterize the change in the elastic properties of the PDMS as a function of e-beam exposure. The PF-QNM experiments were carried out on a Dimension Icon AFM (Bruker-Nano Inc., Santa Barbara, CA) operating in peak-force tapping mode under ambient conditions at a scan rate of 2 Hz and a constant impact force of 5 nN with DNP-10 cantilevers, precisely calibrated through a thermal tune process with resulting spring constants of 0.295 and 0.072 N/m. Samples with known elastic moduli were used to validate the tip calibration process (low-density polyethylene 10 MPa and 14 MPa and PDMS 1 MPa).[27] The analysis of the Derjaguin–Mueller–Toporov (DMT) modulus was performed via Nanoscope Analysis software.
As mentioned above, the Monte Carlo simulations indicate that most of the electron energy is deposited in a ∼ 3 μm thick layer at the surface of the PDMS film. In PF-QNM the probe and sample are intermittently brought together (similar to tapping mode) to map and distinguish nanomechanical properties of a material—including modulus, adhesion, dissipation, and deformation. As a force curve is recorded for each pixel of the image, the resolution obtained for all the channels was identical to topographical AFM imaging and quantitative data could directly be obtained without post-processing. The loading force was carefully adjusted so that the tip can effectively indent into the sample and thus give reliable elastic and deformation response and was sufficiently gentle not to wear the tip or damage the sample. The results of the PF-QNM resulting elastic moduli for the electron beam patterned PDMS films are shown in Figure 1C and Figure 1D. PDMS substrates were exposed to e-beam energies from 500 μC/cm2 to 3,000 μC/cm2 which resulted in a significant increase in the stiffness of the ∼35 kPa unexposed polymer – up to four orders of magnitude (∼350 MPa) with 3,000 μC/cm2 doses (Figure 1E).
Concurrent AFM topographical analysis indicated that e-beam exposure of the PDMS surface resulted in the formation of subtle nanometer undulations (Supplementary Figure 1A&B), which fell below the limits of cellular topographical sensing.[36] Nevertheless, in order to isolate the possible involvement of the topographical modulation (arising from e-beam mediated substrate contraction) on cellular function, control PDMS substrates were fabricated by imprinting with a negative resin stamp to replicate topographical undulations in PDMS substrates possessing homogeneous rigidity (Supplementary Figure 1C&D). Cellular responses to these control substrates as assessed through focal adhesion co-localization were not observed, indicating a cellular mecahno-response to these subtle features was not initiated (Supplementary Figure 2 and Supplementary Figure 5).
Samples were cleared of aquaSAVE prior to physicomechanical analysis and the chemical effects of PDMS e-beam irradiation were assessed prior to in vitro cell experiments to ensure that the modulation of cellular function was exclusively rigidity dependent and not as a result of altered surface hydrophobicity and/or protein adsorption. All analysis was conducted with maximum e-beam exposures (3,000 μC/cm2). The wettability of experimental PDMS substrates was analyzed by contact angle measurements (Figure 2A), which confirmed no significant differences between all the materials, used in the study. High resolution XPS of the C 1s, O 1s and Si 2p revealed that the e-beam exposure modified the carbon and oxygen content on the PDMS surface. Decreasing carbon content from 44.7% to 36.5 % and increasing the oxygen content from 30.5 % to 41% (Figure 2B). Furthermore, the O 1s values were associated with peak energy shifts from 530.75 eV for unexposed PDMS to 533.4 eV. C 1s binding energy for untreated PDMS of 282.1 shifted to 284.85 following e-beam exposure and the Si 2p binding energy from 102.8 eV to 103.85 eV after e-beam exposure to O 1s values the corresponding peak shifts from 530.75 eV for unexposed PDMS to 533.4 eV to O 1s values the corresponding peak shifts from 530.75 eV for unexposed PDMS to 533.4 eV (Figure 2C&D&E). A full XPS spectra of exposed and unexposed PDMS is presented in Supplementary Figure 3A.
Figure 2. Chemical modulation of PDMS substrates by focused electron-beam patterning.
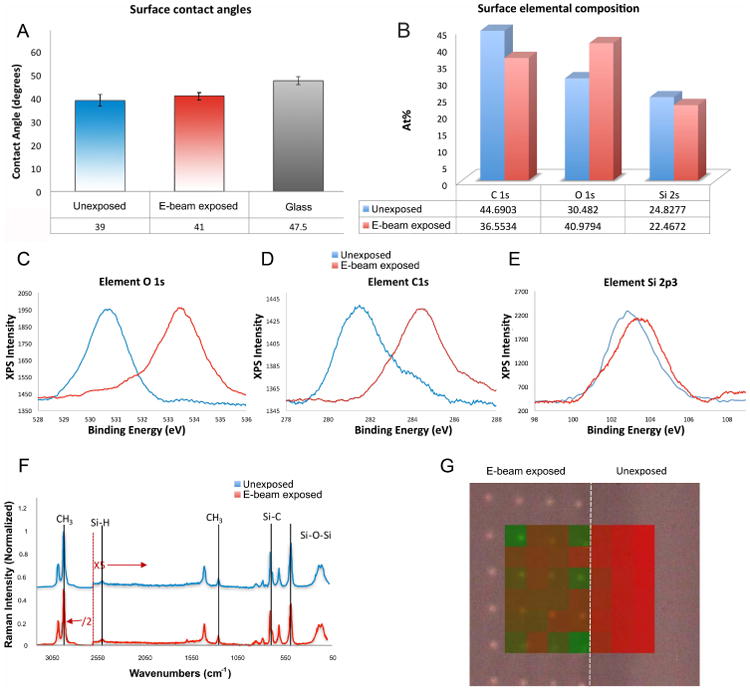
(A) Surface wettability analysis following PDMS treatment with an oxygen plasma. (B)High resolution X-ray photoelectron spectroscopy of (C) O 1s (D) C 1s and (E) Si 2p3. (F) Raman spectra of (F) Raman spectra of e-beam exposed and non-exposed PDMS regions. The spectra below 2600 cm-1 was enhanced five-fold, while spectra above 2600 cm-1 was reduced two-fold, for better visibility of the peaks. (G) Component Discriminant least squares analysis, using peaks from e-beam exposed (green) and unexposed (red), as the spectra of the components, shows spectral differences on PDMS patterned with 1μm spots. The figure shows that the peaks that define the control region are also found in the inter-spot region (red), while the e-beam exposed 1 μm spots are associated with significant peaks (green) that are not present in non-exposed regions.
In order to probe the mechanism of e-beam induced increased rigidity in PDMS, Raman spectroscopy analysis was performed to assess the effects of 3,000 μC/cm2 dose e-beam exposure on polymer chain cross-linking. PDMS has been well characterized via Raman spectroscopy and characteristic spectral peaks have previously been assigned to specific inter- and intra-molecular bonds[28]. Binding energies corresponding to C-Si, C-C sp2 C in phenyl rings, sp3 C, and adventitious C-O and C=O were found in both unexposed and exposed surfaces. (Figure 2F). Component Discriminant least squares analysis, using peaks from e-beam exposed (green) and unexposed (red), as the spectra of the components, indicated that the peaks that define the control region were also found in the inter-spot region (red), while the e-beam exposed 1 μm spots were associated with significant peaks (green) that were not present in non-exposed regions (Figure 2G).
Following e-beam exposure with doses of 3,000 μC/cm2 significant changes were observed in the intensities of Si-O-Si stretches at 490 cm-1, Si-C stretches at 705 cm-1, Si-H stretch at 2500 cm-1, -CH2-CH2- bending at 745 cm-1, CH3 bending at 858 cm-1 and CH stretching at 2905 cm-1. A full assignment of peaks and the spectra can be found in the supplementary information (Supplementary Figure 3B). When analyzing subtle differences between spectra, as observed between e-beam exposed and non-exposed PDMS samples, multivariate analysis like principal component analysis (PCA) and component discriminant least squares (component DLS) are very useful in identifying differences. Raman mapping of e-beam exposed samples patterned with doses of 3,000 μC/cm2 to form arrays of 1μm spots and pristine PDMS samples was carried out using a 3 μm step, with a 534 nm laser. The maps were then post-processed via PCA and DLS analysis to create a ratio of the peak intensities at 490 cm-1 /705 cm-1 and 2500 cm-1 / 2905 cm-1. Data suggest a 9.4% reduction in the presence of Si-O-Si bonds and a 116% increase in the presence of Si-H bonds on e-beam exposed PDMS. A comparison of the peak at 490 /745 cm-1 gives a similar result to that of the 490/705 cm-1, indicating a significant increase in chain crosslinking through CH2-CH2 linkages or Si-H-Si bridges.[29]
To assess protein adsorption to heterogeneously patterned rigidities, ∼35 kPa PDMS substrates were patterned with an array of 2 μm spots possessing an elastic modulus of ∼350 MPa (formed with an e-beam dose of 3,000 μC/cm2). The protein adsorption distribution was determined from the fluorescence intensity profile of labeled fibronectin vs. the bright field DIC intensity on the e-beam exposed materials and was analyzed with ImageJ. The bright field intensity increased sharply at the irradiated regions, demonstrating that e-beam exposed PDMS causes minimal lateral scattering during irradiation and indicating the presence of diffractive modification, consistent with intense crosslinking of the elastomer, as discussed above. On the other hand, the fluorescence intensity profile was unchanged at the sites of e-beam exposure, indicating a uniform distribution of protein adsorption on the patterned substrates (Figure 3C). We note that the surfaces were not subjected to a protein adsorption process prior to cell seeding.
Figure 3. Focal adhesion formation on 2 μm spots of modulated rigidity.
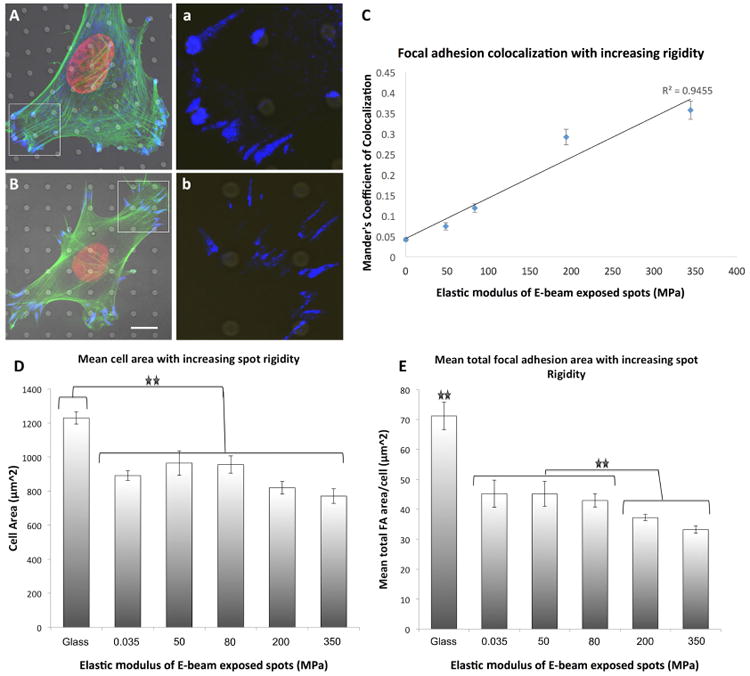
(A) e-beam spots of ∼350 MPa induced differential focal adhesion co-localization in hMSCs. (B) This effect was lost on f 50 MPa spots. High magnification insert of paxillin staining within the boarded area indicated in (a,b). (C) E-beam exposure induced a linear increase in focal adhesion co-localization to spots of altered rigidity. (D) Cellular spreading was not affected in MSCs cultured on 2 μm diameter spots of modulated rigidity. (E) Significant changes in mean FA area were induced by increasing the elastic modulus of 2 μm diameter spots. For statistical analysis of significance see Supplementary Table 1 and Supplementary Table 2. Results are SEM, green = actin, blue = paxillin, red = nucleus, bar = 10 μm.
2.2. Analysis of Focal adhesion formation on heterogeneous rigidities
To study the cellular response to heterogeneously patterned rigidities, PDMS substrates were patterned with an array of spots with diameters ranging from 100 nm – 2000 nm and with elastic moduli ranging from ∼50 KPa (control) to ∼350 MPa. The inter-spot distance was modulated proportionally with spot diameter (edge-edge spacing was maintained at 3) in order to ensure that the cells were exposed to a constant rigid/soft ratio. Substrates were sterilized in 70% ethanol and washed in PBS before seeding of human mesenchymal stem cells (hMSCs) derived from human bone marrow aspirates. Cells were cultured on experimental and control substrates for 12 h before fixing and preparing for immunocytochemistry. Cells were stained for the FA protein paxillin (anti-paxillin) and for filamentous actin (rhodamine conjugated phalloidin) and analysis of FA co-localization on the electron-beam patterned samples was performed with ImageJ (Supplementary Figure 4).
Analysis of FA formation on exposed spot regions showed that FA co-localized to the e-beam exposed spots, and the degree of co-localization increased with applied electron beam dose (Figure 3A-3C). On substrates patterned with ∼350 MPa spots measuring 2 μm in diameter, MSCs formed punctate FAs that co-localized significantly with the underlying exposed region. This effect was observed to diminish with decreasing spot rigidity (Figure 3A-3C). On less stiff spots, elongated FAs were observed to initiate at the irradiated regions yet extended onto the “soft” unexposed inter-spot regions, and co-localization was eliminated with decreasing spot rigidity (Supplementary Figure 2). This was observed as a decrease in the fluorescence signal intensity of paxillin staining on spots of decreasing rigidity (Supplementary Figure 5A) Further, varying the spot rigidity was not observed to significantly modulate cellular spreading (Figure 3D) yet spot with elastic moduli greater than 100 MPa induced significant reductions in the mean cellular total FA area (Figure 3E). For a statistical significance of focal adhesion co-localization as a function of spot elastic modulus see Supplementary Table 1.
Additional analysis of FA co-localization to rigid spots revealed that punctate FA co-localization was dependent on spot size. On the ∼350 MPa spots (a dose of 3,000 C/cm2), reducing the exposed spot diameter from 2 μm to 100 nm significantly decreased FA co-localization (Figure 4A-4C). By decreasing the spot diameter and inter-spot spacing, co-localized, punctate FAs became less frequent (Supplementary Figure 6); rather, FAs were observed to bridge between multiple spots indicating FA sensing machinery can initiate discrete protein reinforcement along the FA plaque. This was observed as an increase in the fluorescence signal intensity of paxillin staining on spots of increased diameter (Supplementary Figure 5B). Again, varying the spot diameter was observed to significantly modulate total FA area, yet not cellular spreading. However, varying the spot size was observed to affect cellular motility (Supplemental movie M1, M2, M3). Specifically, cell velocity and mean migration distance were significantly reduced on substrates patterned with dots > 500 nm in diameter (Supplementary Figure 6). For a statistical significance of focal adhesion co-localization as a function of spot diameter see Supplementary Table 2.
Figure 4. Focal adhesion formation on ∼350 MPa spots of varying diameter.
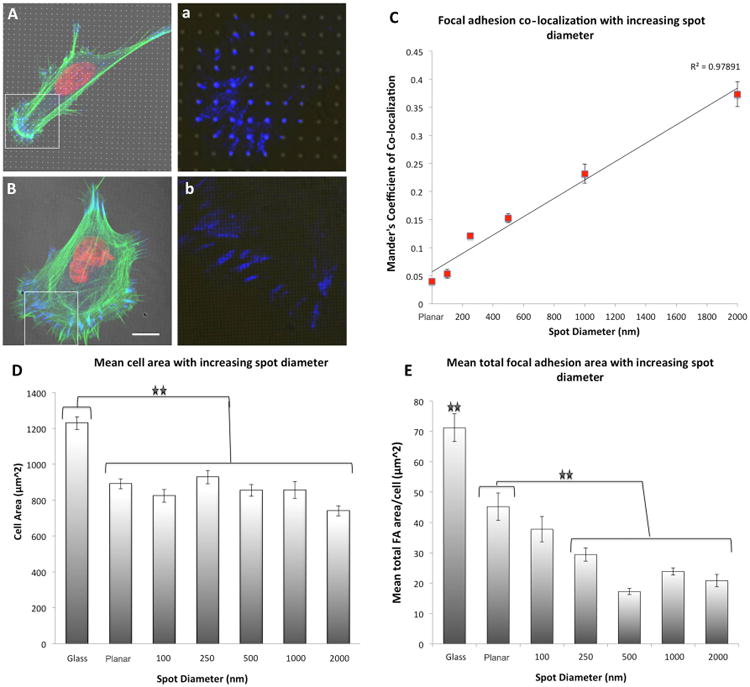
(A) 1 μm spots of ∼350 MPa (formed with doses of 3,000 μC/cm2) induced differential focal adhesion co-localization in hMSCs as a function of spot diameter. This effect was lost on 100 nm spots, high magnification insert of paxillin staining within the boarded area indicated in (a,b). (B) Mander's coefficient of co-localization indicated a linear increase in FA co-localization to the e-beam exposed regions with increasing spot diameter. (D) Cellular spreading was not significantly different in MSCs cultured on spots of modulated rigidity as a function of spot diameter relative to unexposed PDMS, yet significant reductions in cell spreading were noted relative to hMSCs cultured on glass control substrates. (E) Significant reductions in mean FA area were also induced by reducing the spot diameter. For statistical analysis of significance see Supplementary Table 2 and Supplementary Table 3. Results are SEM, green = actin, blue = paxillin, red = nucleus, bar = 10 μm.
2.3. Analysis of Differential Gene Expression on heterogeneous rigidities
The effects of heterogeneous rigidity as a function of both spot rigidity and diameter on differential hMSC function were investigated, with a focus on the regulation of early events in chondrogenesis and osteogenesis. Cells were seeded onto experimental substrates for 12 hours preceding RNA isolation. To perform high-throughput quantitative genomic analysis on 1 mm2 electron-beam patterned samples, real-time PCR was conducted using Fluidigm integrated microfluidic circuit analysis, capable of performing 9,216 simultaneous real-time PCR experiments in nanoliter quantities. Fold changes were expressed relative to cells cultured either in chondrogenic or osteogenic media on unexposed PDMS substrates. Ingenuity Pathway Analysis (IPA) identified 78 genes, which underwent significant modulation in response to the heterogeneous rigidity patterns. These genes were attributed to specific pathways and top functional pathways were assigned an activation z score to infer signaling pathway activation (Figure 5 & 6). Analysis of FA associated signaling pathways in hMSCs cultured on 2 μm diameter spots of increasing rigidity revealed that FA formation was deactivated on unexposed substrates and substrates possessing ∼350 MPa spots. FA deactivation was predominantly due to deactivation of vinculin relative to control chondrogenic and osteogenic conditions (Supplementary Figure 7 & 8)
Figure 5. Functional analysis of hMSCs cultured on PDMS substrates patterned with 2 μm spots of increasing rigidity for 12 hours.
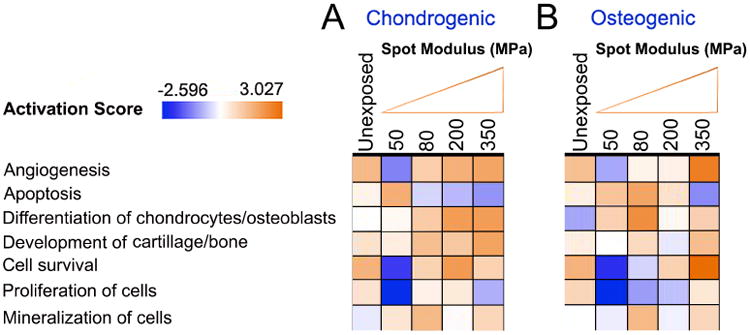
(A) Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation of signaling pathways as a function of spot dose relative to cells cultured on non-exposed homogenous rigidity substrates in (A) chondrogenic and (B) Osteogenic media. Red indicates an increase in pathway activation and green indicates a decrease in pathway activation relative to controls as shown in the Activation Score bar. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Figure 6. Functional analysis of hMSCs cultured on PDMS substrates patterned with ∼350 MPa spots of increasing spot diameter for 12 hours.
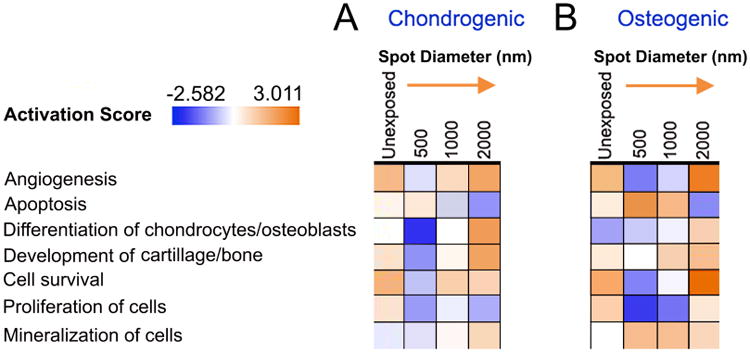
Functional pathway analysis of hMSCs cultured on e-beam patterned spots with diameters ranging from 500-2000 nm revealed significant activation of signaling pathways as a function of spot size relative to cells cultured on control homogeneous rigidity substrates in (A) chondrogenic and (B) Osteogenic media. Red indicates a increase in pathway activation and green indicates a decrease in pathway activation relative to controls as shown in the Activation Score bar. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Functional pathway analysis of hMSCs cultured on 2 μm diameter spots of increasing rigidity indicated dose dependent activation of signaling pathways involved in cell survival, angiogenesis differentiation of chondrocytes and the development of cartilage tissue relative to cells cultured on non-expose substrates in the presence of chondrogenic induction media. Conversely significant deactivations of pathways attributed to cellular apoptosis and cell mineralization were observed. Interestingly deactivation was noted predominantly in hMSCs cultured on 2 μm diameter spots possessing an elastic modulus of ∼50 MPa (500 C/cm2 exposure dose) (Figure 5A). Similar changes were noted in MSC populations relative to cells cultured on non-exposed substrates in the presence of osteogenic induction media (Figure 5B). Pathways associated with chondrocyte and osteoblast development appeared to be dose sensitive, and a linear increase in the prediction of chondrocyte differentiation was observed as a function of spot dose associated with up-regulation of cartilage development genes, BMP2, BMP4, RUNX2, SOX9 and BMPR2 (Supplementary Figure 9). A non-linear response was observed in the prediction of osteogenis however with significant increases occurring with cells culture on ∼80 MPa spots, Here, up-regulation of osteocyte development genes BMP2, RUNX2, coupled with down-regulation in HIFA, SMURF1 and TWIST1 were noted (Supplementary Figure 10). (Fold-change values; Supplementary Table 3).
As the observed influence of heterogeneous rigidity on FA co-localization was lost with spot sizes <500 nm modulations to differential cell function as a function of spot diameter were investigated with spot diameters ranging from 500 nm – 2 μm. Analysis of FA associated signaling pathways in hMSCs cultured on ∼350 MPa spots (3,000 μC/cm2) revealed that FA signaling pathways were deactivated on unexposed substrates and substrates possessing all investigated spot diameters. FA deactivation was predominantly due to down-regulations in the expression of vinculin, paxillin, SRC and integrin 1 relative to control chondrogenic and osteogenic conditions (Supplementary Figure 11 & 12)
Substrates patterned with ∼350 MPa spot with a diameter <2 μm demonstrated significant deactivation of pathways associated with differential cell function. Relative to hMSCs cultured in the presence of chondrogenic induction media, cells cultured on 2 μm diameter spots demonstrates enhanced angiogenic signaling, enhanced chondrocytic differentiation, enhanced cartilage development and enhanced cell survival. These effects were also noted in cells cultured in 1 μm spots, yet to a smaller degree, however these pathways were significantly deactivated in hMSCs cultured on 500 nm diameter spots (Figure 6A). Similarly, relative to hMSCs cultured in the presence of osteogenic induction media, cells cultured on 2 μm diameter spots demonstrates enhanced angiogenic signaling, enhanced osteospecific differentiation, enhanced bone development and enhanced cell survival (Figure 6B). Pathways associated with chondrocyte and osteoblast development also appeared to be sensitive, to the spot diameter and a linear increase in the prediction of chondrocyte differentiation was observed as a function of spot size associated with up-regulation of cartilage development genes, BMP2, BMP4, RUNX2, SOX9 and BMPR2 (Supplementary Figure 13). A non-linear response was observed in the prediction of osteogenis however with significant increases in the prediction of osteogenis noted only with spot sizes of 2000 nm, Here, up-regulation of osteocyte development genes BMP4, BMP2, RUNX2, ACVR2B, NOTCH1, SMURF1 and JAG1 were noted (Supplementary Figure 14). (Fold-change values; Supplementary Table 4).
3. Discussion
For the first time, we report on the effects of e-beam exposure on PDMS crosslinking and subsequent localized increases in surface rigidity at the nanoscale. A similar increase in PDMS cross-linking, accompanied by increased stiffness, as a result of exposure to a Ga+ ion beam, was recently reported by Liu and Fu.[30] Ion beams are typically used to modify the structure of materials (i.e., by sputtering or by ion implantation), and in that work, the resultant mechanical change could be attributed to both modification of the PDMS polymer structure as well as the incorporation of Ga in the polymer. Conversely, we employed direct write focused e-beam patterning, which has not previously been explored to produce geometrical patterns of increased rigidity on viscoelastic PDMS substrates. Significant increases to the elastomer modulus scaled with increasing the e-beam dose, effectively increasing the materials elastic modulus by four orders of magnitude, from ∼35 kPa to ∼350 MPa.
Similarly, a previous study by Bowen et al. explored the effect of e-beam exposure on the mechanical properties of non-cross linked PDMS with an aim to validating PDMS as a potential e-beam resist.[31] Interestingly, the study reports very similar mechanical responses of the pre-polymer to e-beam exposure to those reported herein. Conversely, we show that elastomeric PDMS, widely employed in the medical device industry, may be modified through e-beam patterning, to produce arrays of differential rigidity with feature dimensions ranging from the micron scale to the nanoscale with a range of rigidities, regulated by varying the applied e-beam exposure dose
The analysis of e-beam induced modulation of elasticity was accomplished via Peak-force quantitative AFM, enabling mapping of the substrate mechanical properties with nanoscale resolution. Using lithography to control the size and geometric arrangement of the spots allowed us to engineer substrata with defined distributions of heterogeneously increased rigidity with dimensions ranging from the micron- to the nanoscale. Although spot features were explored in this study, ongoing studies are investigating the effects of anisotropic rigidity features on cell function (Supplementary Figure 15) and it is important to indicate the versatility of e-beam patterning in generating complex and arbitrary feature shapes.[32], in conformations ranging from ordered to non-ordered.[33]
We note that in this study, in contrast to the observations of Russell et al[24] that the PDMS chemistry was affected by focused e-beam exposure; in particular the measured surface carbon content was reduced and the creation of SiO2 was confirmed by the shift in the peak positions of the SI 2p and the O 1s signals (Figure 2F). This was also reported in a previous study by Schnyder et al. which described a very similar reduction in carbon and increase in oxygen composition in PDMS following exposure to UV light.[34] Subsequent degradation of the polymer was confirmed through X-ray photoelectron spectroscopy, and corresponding binding energy shifts reached values corresponding to Si02. The C 1s binding energy for untreated PDMS of 282.1 shifted to 284.85 following e-beam exposure and the Si 2p binding energy from 102.8 eV to 103.85 eV after e-beam exposure. This peak position corresponds exactly with literature values of 102.1 – 103.4 following UV exposure. With respect to O 1s values the corresponding peak shifts from 530.75 eV for unexposed PDMS to 533.4 Ev following e-beam exposure. Again, these values were identical to previously reported studies with UV exposure of PDMS.
We hypothesized that the observed increase in PDMS rigidity as a function of e-beam exposure dose was due to a dose dependent increase in PDMS chain crosslinking. Indeed, Raman spectroscopy suggested a 9.4% reduction in the presence of Si-O-Si bonds and a 116% increase in the presence of Si-H bonds on e-beam exposed PDMS indicating a significant increase in chain crosslinking through CH2-CH2 linkages or Si-H-Si bridges.[29] Critically, we did not observe differential fibronectin adsorption on heterogeneous rigidity substrates a process also verified in a similar study employing photo-crosslinking of micro-domains in a hydrogel system to create heterogeneous rigidity substrata.[35] This is important as it indicates that the observed FA co-localization effects could not be attributed to differences in protein adsorption between the irradiated and non-irradiated regions which has been shown previously to modulate the reinforcement of FAs,[36] dynamic turnover of FA associated proteins [37], and integrin mediated signalling activity.[38]
Focal adhesions play dual physiological functions — as physical structures that direct and regulate tissue and organ morphogenesis through mechanical cellular coupling to the ECM, and as bi-directional mechanosensors that modulate cellular signaling events. The mechanisms of adhesion-mediated sensing of the physical properties of the matrix are yet to be resolved, however cells are exquisitely sensitive to the physical state of the local environment. Matrix rigidity is conveyed through macromolecular FAs in a bidirectional manner, modulating cellular function and FA morphology. Stiffer matrices generate larger FAs through increased intracellular tension, and studies with bulk systems of rigidity have shown that cells respond to rigidity gradients, and migrate from regions of lower to higher rigidity, in a process termed durotaxis.[17, 39]
Major differences in FA reinforcement and density are reported in cells cultured on surfaces with bulk compliance (sub-kPa to the kPa range), relative to cells cultured on surfaces with bulk rigidity (hundreds of kPa to a few MPa).[5, 40] However conflicting hypotheses exist on the mechanisms of bulk-rigidity mediated changes to FA reinforcement and subsequent cell function. It is known that FA reinforcement and induction of actin organization require certain threshold densities of adhesion ligands,[41, 42] however the existence of a minimum length scale at which cells can sense rigidity has not yet been established. Our results demonstrate clearly that the formation of FAs in hMSCs cultured on substrates with heterogeneous rigidity is dependent both on feature stiffness and size. The formation of discrete punctate FAs coupled with an increase in paxillin recruitment on spots 1 μm in diameter, indicate that this length scale lies between 500 nm and 1 μm. Critically, paxillin recruitment to FAs and subsequent phosphorylation has been identified as essential for high FA traction over a broad range of ECM rigidity.[43]
In agreement with a recent study by Monge et al. it was observed that FAs did not extend from the center of the rigidity patterned spots but were rather located at the spot boundary.[35] We also observed that on spots ≤ 1 μm in diameter, FAs had a tendency to extend paxillin domains, and single FAs were associated with multiple rigid spots. This behavior is similar to that observed by Arnold et al. with cells plated on adhesive patches of RGD peptide.[44] That study concluded that adhesive areas 1 μm2 can support the formation of sufficiently mature FAs to withstand the applied load per patch necessary for cell spreading. In contrast, cells were observed to couple to adjacent paxillin domains through a single actin bundle if adhesive patches were ≤ 500 nm, in order to mechanically stabilize adhesion and facilitate cell spreading.[44] Our results suggest that rigidity mediated adhesion is regulated by the same machinery that governs FA assembly and reinforcement and that this machinery is capable of recognizing localized discrepancies in matrix rigidity.
A recent study by Yang et al. explored the influence of heterogeneous rigidity on hMSC adhesion through a photo-reactive hydrogel system. by copolymerizing PEG monoacrylate (PEGA) with a photodegradable PEG diacrylate (PEGdiPDA). Using photolithographic masks, cell culture substrates were synthesized through a UV photo-degradation process at 365 nm to yield hydrogels possessing discrete regions of reduced E. UV exposure induced a decrease in E of the hydrogel from 9.6 - 2.3 kPa, however owing to the resolution limits of photolithography the smallest future size obtainable with this approach was limited to 2μm2. Interestingly, it was also noted in this study that paxillin intensity was increase in FAs formed on regions of increased rigidity.[45] Further studies employed stiff islands of (approx. 36 μm2) concluded that although the island were too large to address the minimal adhesion area required to trigger FA formation, as long as adhesion sites are well-anchored to resist traction forces, the area of adhesion is limited only by the minimal area required to support focal complex initiation,[46] however, the minimum scale for FA initiation and early integrin clustering events in response to discrete rigidity structures is unknown. Recently Meacci et al. reported that it is local FA contraction mechanics mediated by myosin II and α -actinin, and not intracellular tension that plays the central role in FA reinforcement in response to local rigidity sensing.[47] We suggest that this localized contractile unit is a prime candidate for this role and our results set the size scale for this unit.
Direct mechanical cues have been shown to play a significant role in regulating Osteochondral differentiation[48] and it was interesting to note that substrates possessing heterogeneous rigidity with spot rigidities > 50 MPa were able to increase focal adhesion co-localization and initiate the activation of differential functional pathways in hMSCs following only 12h of culture relative to cells cultured in oseospecific or chondrospecific induction media. In this study, it was noted that heterogeneous rigidity induced significant activation of defined pathways involved in the processes of chondrogenic, osteogenic and angiogenic function relative to cells cultured in osteochondrogenic conditions on homogenous substrates. IPA pathway analysis revealed several functional pathways linked to osteochondral growth and differentiation induced by up-regulation of BMP-2/-4, Rac1, RhoA and ROCKII which influence RUNX2 and SOX9 expression.[49] Critically pathways associated with osteochondral differentiation pathways were noted predominantly on 2 μm patterned heterogeneous arrays formed with e-beam doses of 3000 C/cm2 which induced a patterned rigidity increase of the ∼35 kPa elastomer to ∼350 MPa, and that this effect was eliminated on substrate with spots diameters < 500 nm.
It can be argued that substrates possessing high density arrays of spots with a diameter < 500 nm effectively present a bulk rigidity of ∼350 MPa yet did not promote differential function, pointing to the roles of heterogeneous rigidity in mediating stem cell function in vitro. Critically, the effects of heterogeneous rigidity on hMSC focal adhesion formation and differential function reveal the potential of microscale rigidity in engineering next generation biomaterial interfaces to control cell function. Ongoing studies will hence explore the role of the density of heterogeneous rigidity spots on the maintenance of hMSC stemness in vitro, with an aim to elucidating clean, protein free culture conditions.
Conclusions
Tissues do not represent bulk rigidity systems, but rather are composed of heterogeneous distributions of particles, and fibers of varying rigidity. We have developed a new type of biomimetic surface comprising regions of heterogeneous rigidity on the micro- and nanoscale by writing on PDMS films with an electron beam. Peak-force quantitative AFM nanomechanical mapping of these surfaces reveals a substantial increase in the Young's modulus of the elastomer as a function of the e-beam exposure. By monitoring cellular response to these surfaces, we have demonstrated in planar system that the cellular rigidity sensing apparatus is capable of sensing discrete submicron discrepancies in the matrix rigidity, and that FAs demonstrate intrinsic “local” reinforcement in response to rigid features measuring ≳ 500 nm in diameter. At ≲ 500 nm, the ability to sense the rigid features is completely lost. This contrasts with cellular response to other physical cues, such as topography[12, 38] and geometry,[42, 44] where cells respond to features well into the nanoscale. Different cell types respond differently to rigidity and may have different spatial and rigidity requirements to elicit differnetial responses. The versatility of the patterning system presented here can be applied to a broad range of cellular systems in order to elucidate the specific requirements for each. Understanding these requirements can be used to guide the design of new types of tissue scaffolds and may have implications in the treatment of cancer and other diseases.
4. Experimental Section
4.1. Substrate fabrication
Microscope cover-glasses (Corning, NJ, USA) (22 mm2 No. 0) were cleaned for 12 h in a 1 per cent v/v solution of the detergent MICRO-90 (International Products, NJ, USA), rinsed in reverse osmosis water (ROH2O) and blown-dry in a stream of filtered nitrogen. Sylgard 184 PDMS (Dow Corning, MI, USA) was mixed with the supplied accelerating agent at a ratio of 50:1 for 5 min and degassed under vacuum for 10 min at 5 Torr. PDMS (0.5 ml) was applied to the microscope cover-glasses and spin-coated for 45 s at 1000 r.p.m and an acceleration of 400 r.p.m. s−1 to form a uniform film. PDMS-coated cover-glasses were cured for 17 h at 70°C before further processing. Substrates were subjected to an oxygen plasma in a tabletop Harrick PDC32G plasma cleaner for 10 sec at a RF power of 18 W to induce surface hydrophilicity. Samples were next coated with a conductive discharge layer to facilitate e-beam exposure. A 5 nm thick discharge layer was applied to the substrates by spin coating 100 μl of Aquasave (Rayon, Mitsubishi) for 45 s at 4000 rpm and an acceleration of 400 rpm2. Samples were stored at RT until e-beam exposure.
4.1.1. Electron-beam direct-write patterning
The PDMS substrates were patterned by e-beam exposure using a scanning electron microscope (FEI XL 30 Sirion) equipped with a Nabity NPGS pattern generator. A 1 mm2 area consisting of an arrays of spots with diameters ranging from 100 nm to 2 μm were written onto the substrate surface at doses from 500-3,000 μC/cm2, an accelerating voltage of 30 kV and a beam current of ∼ 2.5 nA. Substrates were cleared of Aquasave in deionized water for 3×5 min. and allowed to air dry for 30 min.
Topographical control substrates were fabricated by casting PDMS onto directly e-beam written samples to create negative template. Briefly, e-beam written samples were prepared with doses of 3,000 μC/cm2 and the patterned area isolated with a glass cloning-ring. PDMS with a base:accelerator ratio of 5:1 was introduced into the cloning ring and allowed to cure overnight at RT. The inverse cloning-ring/PDMS shim was subsequently removed AT -80°C from the direct e-beam written pattern and used for the casting of topographical replicas. Topographical PDMS substrates prepared as above were cast onto the PDMS template overnight to yield topographically modified, yet mechanically homogeneous PDMS substrates.
4.2. Surface Characterization
Monte Carlo simulations of electron trajectory in PDMS were conducted with Casino software.[50] Surface physical modification was characterized by nanoindentation, optical profilometry and scanning electron microscopy measurements. Chemical modification was analyzed by water contact angle, angle, X-ray photoelectron spectroscopy (XPS) and confocal laser scanning microscopy measurements. Planar control materials were also subjected to a plasma treatment as described in section 4.1.
4.2.1. Quantitative AFM Nanomechanical Mapping
The PF-QNM experiments were carried out on a Dimension Icon AFM (Bruker-Nano Inc., Santa Barbara, CA) operating in peak-force tapping mode under ambient conditions at a scan rate of 2 Hz and a constant impact force of 5 nN with DNP-10 cantilevers, precisely calibrated through a thermal tune process with resulting spring constants of 0.295 and 0.072 N/m. Samples with known elastic moduli were used to validate the tip calibration process (low-density polyethylene 10 MPa and 14 MPa and PDMS 1 MPa).[27] The analysis of the Derjaguin–Mueller–Toporov (DMT) modulus was performed via Nanoscope Analysis software.
4.2.2. Raman microscopy
Chemical characterization of the substrates was carried out using the Renishaw inVia Raman microscope. Spectra of the control and the etched regions were collected using a 534 nm laser (5% laser power, 10s exposure time, 1 acquisition, ×50 objective), with high confocality and with the pinhole in to reduce the spot size to under a micron, so as to collect data from an etched spot efficiently, and preventing interference patterns from the etching. Spectra in the range of 50 – 3200 cm-1 were obtained using extended use of the grating, or in static mode, centering the grating at 950 cm-1 and 2500 cm-1 to cover the entire range. Raman maps of the samples were obtained by scanning a region with 3 micron steps. The measurements were made under focused tracking to get the best possible Raman scattering possible. This also acts as measurement of the surface depth/ profile. Component DLS analysis and principal component analysis (PCA) of the maps obtained were carried out to differentiate between subtly different Raman spectra.
4.2.3. Contact angle measurements
Surface wettability assessment was carried out at room temperature using 8 μl water droplets with a model 100_00 contact angle goniometer (Rame-Hart, Inc.). Values were averages of measurements on more than three different samples at more than three different locations on each sample.
4.2.4. X-ray photoelectron spectroscopy (XPS)
XPS spectra were recorded with PHI 5500 model spectrometer equipped with a Al K monochromator X-ray source run at 15 kV and 23.3 mA, a hemispherical electron energy analyzer and a multichannel detector. The test chamber pressure was maintained below 2 × 10–9 Torr during the spectrum acquisition. Low energy electron flood gun was used to neutralize possible surface charging. The XPS binding energy (BE) was internally referenced to aliphatic main C 1s peak (BE = 284.6 eV). Survey spectrum was acquired at an analyzer pass energy of 93.9 eV and BE resolution of 0.8 eV, while the high-resolution spectrum was acquired with a pass energy of 23.5 eV and BE resolution 0.05 eV. Angle-dependent XPS was performed by rotating the sample holder to the desired take-off angle (the angle between the surface normal and the detector) through a motor. Spectrum was fitted by a Guassian–Lorentz (BE) was internally referenced to aliphatic main C 1s peak function after subtracting a striped background using the PHI data processing software package under the constraint of setting reasonable BE shift and characteristic full width at high maximum (FWHM) range. Atomic concentration was calculated by normalization of the peak area to the elemental sensitivity factor data provided by PHI database.
4.2.5. Protein adsorption Assay
Surface adsorption of fibronectin was analyzed by fluorescence microscopy. Human fibronectin (Sigma Aldrich) was conjugated directly to Alexa Fluor 488 (Invitrogen) by protein dialysis according to manufacturer instructions (Thermo Scientific). Exposed substrates were prepared as above and immersed in PBS containing 0.5 ug/ml fluorescent fibronectin. Samples were coated for 18 hours before being washed in PBS (3×5 mins) and mounted for microscopy.
4.3. Cell culture
Substrates were sterilized by successive rinsing in 70% ethanol (3×5 seconds) followed by PBS (3×5 seconds). Human mesenchymal stem cells (hMSCs) derived from human bone marrow aspirates were isolated using a protocol previously described[51] hMSCs were cultured in complete medium (MEM alpha, GlutaMAX™ supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin) and maintained at 37°C in a humidified atmosphere containing 5% CO2. Cells were expanded to passage 2 following 1 weeks of culture and subsequently trypsinized in TrypLE™ Express dissociation medium (Invitrogen) and seeded onto untreated experimental, and planar control tissue culture plates at a density of 1 × 104 cells per sample in 1 mL of complete medium. Cells were maintained at 37°C with a 5% CO2 atmosphere in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (Gibco), 1% l-glutamine and 100 IU mg−1 penicillin/streptomycin (Invitrogen). To induce chondrogenesis, cells were subjected to chondrogenic induction media (Lonza, Switzerland) containing dexamethasone, ascorbate, ITS+supplement, GA-1000, sodium pyruvate, proline and L-glutamine, supplemented with 10ng ml−1 TGF-β3 (Lonza). For osteogenic induction, cells were cultured in osteogenic basal media supplemented with L-glutamine, ascorbate, dexamethasone, penicillin/streptomycin, MCGS and β-glycerophosphate (Lonza, Switzerland).
4.4. Fluorescent labeling
Following 12 hours of culture on experimental and control substrates hMSCs cultures were fixed in 4% paraformaldehyde in phosphate-buffered saline solution (PBS), with 1% sucrose at 37°C for 5 min. Once fixed, the samples were washed with PBS. Samples were permeabilized with buffered 0.5% Triton X-100 (10.3 g sucrose, 0.292 g NaCl, 0.06 g MgCl2, 0.476 g [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] (HEPES), 0.5 mL Triton X-100, in 100 mL water, pH 7.2) at 4°C for 5 min. Nonspecific binding sites were blocked with 1% bovine serum albumin (BSA) in PBS at 37°C for 15 min and subsequently incubated for 2 h with anti-paxillin monoclonal anti-human IgG raised in mouse, (1:200, (B.D Biosciences, Sparks, MD). Nonspecific charges (e.g. remaining aldehyde) were neutralized with 0.5% Tween 20/PBS (5 min × 3) to minimize background labeling. A secondary, Fluorescein isothiocyanate-conjugated antibody was added, in 1% BSA/PBS, (1:50, Vector Laboratories, Burlingame, CA) 4°C for 1 h and simultaneously, rhodamine-conjugated phalloidin was added for the duration of this incubation (1:50, Molecular Probes, OR). Substrates were given a final wash in PBS (5 min × 3. Samples were mounted in Vectorshield mounting medium for fluorescence (Vector Laboratories, Burlingame, CA). Cell–substrate and cell–cell interactions were examined by scanning confocal microscopy on a stage maintained at 37°C (live cell imaging). Imaging was performed on an LSM 700 scanning laser confocal microscope with an argon-ion laser (wavelengths 405; 488; 555; 639 nm) fitted with a Zeiss 100× -PLAN Apochromat objective with a numerical aperture of 1.57 and with ZEN software (Carl Zeiss).
4.5. Assessment of gene expression in MSC populations using Fluidigm Biomark
In order to perform high-throughput quantitative genomics on 1 mm2 electon-beam patterned samples, real-time PCR was conducted using integrated microfluidic circuit analysis (Fluidigm Biomark HD system, UK). Approximately 600 hMSCs in 5μl were seeded onto PDMS substrates at second passage (P2) and a cell density of 1.2 × 104 cells/ml for 12 hours.
4.5.1. RNA Isolation
RNA was isolated from hMSCs using an ARCTURUS®PicoPure™ RNA isolation kit according to manufacturer instructions (Applied Biosystems, UK). RNA quality and quantity was measured with 2100 Bioanalyser and RNA 6000 Pico Kit, again according to manufacturer's protocol (Agilent Technologies, USA). Samples with a RIN value >7 was further processed for cDNA conversion. Briefly, total RNA (∼20 ng) was amplified with High Capacity RNA-to-cDNA Kit (Applied Biosystems) using MJ Research PTC 200 Thermal Cycler (Thermo Scientific, Ireland).
4.5.2. Fluidigm Genomic Analysis
The RNA was subjected to a reverse transcription using the SuperScript III Reverse Transcriptase (Invitrogen, U.K.). At all stages of the process, reactions were performed at 4 °C unless stated. Gene analysis was performed using the Fluidigm Fastgene expression Analysis using EvaGreen® on the Biomark HD system protocol (PN 100-3488 C1). In brief, all 96 primers were pooled together (1 μL from each primer set pooled in 104 μL of DNA suspension buffer). Pre-amplification was prepared using 1.25 μL of the cDNA from each sample, 2.5 μL 2× Multiplex Master Mix (Qiagen), 0.5 μL pooled primer mix and 0.75 μL water. This was vortexed, centrifuged and subjected to 22 thermal cycles with the following programme:
| Condition | Hold | 18 cycles | Extension | Hold | |
| Temperature | 95°C | 94°C | 60°C | 72°C | 4°C |
| Time | 15 mins | 30 secs | 90 secs | 10 min | ∞ |
After the 18 thermal cycles, 1.4 μL water, 0.2 μL Exonuclease I Reaction Buffer and 0.4 μL Exonuclease was added to each sample and vortexed, centrifuged and incubated at 37°C for 30 minutes followed by 80°C for 15 minutes. Following heat inactivation, 18 μL of TE buffer was added to each sample. 2.7 μL of the Exonuclease I treated sample was added to 3.0 μL 2× SsoFast EvaGreen Supermix (Bio-Rad) and 0.3 μL 20× DNA Binding Dye sample loading reagent (Fluidigm). Each mixture was vortexed and centrifuged ready to be loaded onto the chip. Additionally, 0.3 μL of each individual primer set was added to 3 μL 2× assay loading reagent and 2.7 μL 1× DNA suspension buffer, vortexed and centrifuged ready for loading on the chip and run on the Fluidigm Biomark™ HD system. A 96.96 Dynamic array integrated fluidic circuit was used during this analysis.
4.5.3. Analysis of signaling pathways
A total of 87 target genes were probed and the gene targets were expressed as relative fold-change to the differentiation controls (Supplementary Table 1). Ingenuity Pathway Analysis (IPA) (Ingenuity Systems, Qiagen) was used to identity canonical signaling and functional pathways. The plots are normalized relative to the control samples on plain PDMS treated with chondrogenic media (Poietics PT-4124; Lonza, Walkersville, MD) or osteogenic media (Poietics PT-3924; Lonza, Walkersville, MD). Comparison analysis on differential gene expression between the samples were performed with a false discovery rate <0.05 (Fischer's exact test) and fold change >1.3.
4.6. Time-lapse videomicroscopy
Time-lapse studies were performed as described elsewhere.[52] Briefly, MSCs were seeded onto patterned and control PDMS substrata and incubated for 1 hour to allow cells to adhere. Cell media as subsequently removed and cells cultured in CO2 independent medium (Gibco) supplemented with 10% fetal bovine serum, 1% l-glutamine and 100 IU mg−1 penicillin/streptomycin (Invitrogen). The substrates were sandwiched to an aluminum microscope slide with vacuum grease. Time-lapse micrographs were recorded with a 20×, 0.7 NA air objective (Olympus) through a cooled CCD camera CoolSNAP HQ (Roper Scientific Inc.) using Simple PCI software (Compix Inc.). Images were captured via Differential interference contrast (DIC) microscopy every 5 mins.
4.7. Image analysis
All images were analyzed using ImageJ (National Institutes of Health). Image stacks consisted of 2-3 planes spaced by 0.40 μm, which were rendered using standard deviation image intensity to produce a single image of the ventral cell surface. Focal adhesions were analyzed in cells from three separate experiments (20 cells each). FA/exposed spot colocalization was analysed by Manders' method with the JACoP plugin.[53] Mander's overlap coefficient is based on the Pearson's correlation coefficient with average intensity values being taken out of the mathematical expression.[54] This coefficient will vary from 0 to 1, the former corresponding to non-overlapping images and the latter reflecting 100% co-localization between both images. Therefore, M1 (or M2) determined the proportion of the fluorescent paxillin signal coincident with the DIC signal of the substrate over its total intensity, given as the following: k1=Si(Ai, coloc))/ (Si Ai) & k2=(Si (Bi, coloc))/ (Si Bi) With Ai, coloc being Ai if Bi>0 and 0 if Bi=0; and Bi, coloc being Bi if Ai>0 and 0 if Ai=0. Live-cell analysis of cell motility was performed with the ImageJ plugin MTrackJ.[55]
4.8. Statistical analysis
All statistical analysis was performed with SPSS Statistics software 20 (IBM, USA) unless otherwise noted. Data are expressed as mean ± SEM with * and ** indicating a 95% and 99.5% confidence interval respectively. ANOVA was used to determine statistical significance followed by post hoc Bonferoni's multiple test correction to determine which groups were statistically different. A right tailed Fisher's exact test was applied to identify significance in activation pathways extracted from individual changes at the genetic level through IPA analysis.
Supplementary Material
Supplementary figure 1. Topographical modification of E-beam exposed PDMS and analysis of topographical control substrates (A) Elastomer contraction was observed to be dose dependent. Low doses of 50 μC/cm2 resulted in undulations of 4 nm depth. PDMS contraction and undulation depth was observed to plateau with doses of 2,200 μC/cm2, resulting undulations were 140 nm in depth with a radius of curvature of 20 μm. (B) Maximum E-beam exposure initiated nanoscale contractions of PDMS resulting in an undulating topography. (C) AFM Phase analysis indicated significance modification to PDMS substrate mechanical properties following E-beam exposure of 3,000 μC/cm2. (D) Typical phase profile of PDMS following E-beam exposure of 3,000 μC/cm2. (E) Exposed substrates we employed to generate negative topographical master stamps. (F,G) Undulating topographies were replicated into into 500 μm thick PDMS films of 35 KPa via direct casting to produce topographical controls with a homogenous rigidity profile. Significant reduction in phase angle was observed following casting onto a topographical master.
Supplementary figure 2. Focal adhesion Co-localization to spots of increasing diameter and rigidity (A) Focal adhesion co-localization to 2 μm spots of modulated rigidity increased as a function of spot rigidity. (B) FA co-localization was unaffected by spots <250 nm in diameter irrespective of spot dose. Data obtained from image analysis of immunofluorescent staining of paxillin indicated that increases in rigidity dependent co-localization are more significant on larger spot diameters. Results are SEM, green = actin, blue = paxillin, red = nucleus, bar = 10 μm.
Supplementary figure 3. XPS, RAMAN and fluorescence absorption Spectra of e-beam exposed PDMS (A) XPS survey of non-exposed and E-beam exposed PDMS indicated a reduction in C content. (B) Raman map of e-beam exposed PDMS and adjacent control, acquired with a 3 μm step shows the ratio of the intensities of the peak at 490 cm-1 and 706 cm-1, suggesting a 9.4% increase in the presence of Si-O-Si bonds on non-exposed regions relative to e-beam exposed regions. The ratio of the intensities of the peak at 2500 cm-1 and 2905 cm-1, suggests a 116% increase in the presence of Si-H bonds in e-beam patterned regions relative to the un-exposed region. Also shown is the principal component analysis of Raman mapping acquired using a static method, with the peak centered at 950 cm-1 and at 2500 cm-1. Both clearly show the difference in the chemical signatures between e-beam exposed and control non-exposed PDMS. (C) Confocal microscopy revealed that differential fibronectin adsorption did not occur on patterned substrates.
Supplementary figure 4. Focal adhesion analysis on e-beam exposed PDMS (A&B) Fluorescent paxillin and DIC images are analyzed in imageJ via the JACoP co-localization plugin, with a returned value of 1 indicating 100% pixel co-localization and a value of 0 indicating no co-localization. Green = actin, blue = paxillin, red = nucleus, bar = 50 μm.
Supplementary figure 5. Relative fluorescence intensity of paxillin staining on e-beam exposed spots as a function of (A) spot rigidity and (B) spot diameter. Fluorescence intensity profiles are expressed as normalized signals to paxillin intensity of FA formed on inter-spots regions. Intensity was observed to increases on spots of increasing elastics modulus and on spots of increased diameter. With maximum intensity occurring on 350 MPa spots of 2 μm diameter.
Supplementary figure 6. Focal adhesion Co-localization to ∼350 MPa spots of increasing diameter and the influence of spot diameter on cellular motility (A) Paxillin staining revealed a significant increase in FA colocalization with increasing spot diameter. Bottom panels represent high magnification of paxillin staining within the boarded insert area. (B) Significant changes in mean cellular velocity were induced by increasing the spot diameter on patterns fabricated with elastic moduli of ∼350 MPa. Mean cell velocity was greatest on glass control substrates but significant reductions were noted in cells were cultured on spots >250 nm in diameter. (C) Greatest migration distances were observed in cells cultured on 100 nm diameter spots. Migration distance was observed to decrease linearly with increasing spot diameter. Results are SEM, green = actin, blue = paxillin, red = nucleus, bar = 10 μm.
Supplementary figure 7. Genomic analysis of the focal adhesion signaling pathway in hMSCs cultured on PDMS substrates patterned with 2 μm spots of increasing electron-beam dose for 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation and deactivation of the FA signaling pathway as a function of spot dose relative to cells cultured on non-exposed homogenous rigidity substrates in chondrogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates were associated with an increased confidence in the prediction of de-activation of FA signaling, with measured down-regulations in the expression of vinculin (VCL) integrin 1 (ITGB1) the JAG1 protein (JAG1) and paxillin (PAX). A linear increase in predicted activation of FA signaling was noted in cells cultured on 50 – 200 MPa and spots of 200 MPa induced up-regulation in expression of integrin 1 and integrin 2, vinculin transforming growth factor 1 (TGFB1), cyclin D1 (CCND1) and CD44. Predicted activation of FA signaling was lost however on PDMS substrates possessing 2 μm spots of ∼350 MPa. Cells cultured on these substrates exhibited down-regulations in VCL, PXN and TGFB1. Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 8. Genomic analysis of the focal adhesion signaling pathway in hMSCs cultured on PDMS substrates patterned with 2 μm spots of increasing electron-beam dose for 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation and deactivation of the FA signaling pathway as a function of spot dose relative to cells cultured on non-exposed homogenous rigidity substrates in osteogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates were associated with a significant increased confidence in the prediction of de-activation with measured down-regulations in the expression of vinculin (VCL) integrin 1 (ITGB1) the JAG1 protein (JAG1) and paxillin (PAX). A linear increase in predicted activation of FA signaling was noted in cell cultured on 2 μm spots with E values raging from 50 – 200 MPa and spots of 200 MPa further induced up-regulation in expression of integrin 1 and integrin 2, vinculin transforming growth factor 1 (TGFB1), cyclin D1 (CCND1) and CD44. Predicted activation of FA signaling was lost however on PDMS substrates possessing spots of 350 MPa. Cells cultured on these substrates exhibited down-regulations in VCL, PXN and TGFB1. Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 9. Genomic analysis of chondrogenesis in hMSCs cultured on PDMS substrates patterned with 2 μm spots of increasing electron-beam dose for 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation and deactivation of the FA signaling pathway as a function of spot dose relative to cells cultured on non-exposed homogenous rigidity substrates in chondrogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates exhibited no changes in the predicted activation of chondrogenesis associated pathways relative to hMSCs cultured on identical substrates in complete (non-chondrogenic) media. A linear increase in the predicted activation of chondrogenesis was noted in cells cultured on 2 μm spots with E values raging from 50 – 200 MPa and in particular, PDMS substrates e-beam patterned with spot of 350 MPa induced up-regulation in expression of bone morphogenetic protein 4 (BMP4), bone morphogenetic protein 2 (BMP2), bone morphogenetic protein receptor 2 (BMPR2), the transcription factors SOX-9 (SOX9) and RUNX2 (RUNX2), integrin 1 (ITGB1) and integrin α1 (ITGA5). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 10. Genomic analysis of osteogenesis in hMSCs cultured on PDMS substrates patterned with 2 μm spots of increasing electron-beam dose for 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation and deactivation of the FA signaling pathway as a function of spot dose relative to cells cultured on non-exposed homogenous rigidity substrates in osteogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates were associated with a significant increased confidence in the prediction of de-activation of osteogenesis associated pathways relative to hMSCs cultured on identical substrates in complete (non-osteogenic) media. A linear increase in the predicted activation of osteogenesis was noted in cells cultured on 2 μm spots with E values raging from 50 – 200 MPa and in particular PDMS substrates e-beam patterned with 2μm spot doses 350 MPa induced up-regulation in expression of osteogenesis associated genes - bone morphogenetic protein 4 (BMP4), bone morphogenetic protein 2 (BMP2), the JAG1 protein (JAG1), The neurogenic locus notch homolog protein 1 (NOTCH1), SMAD Specific E3 Ubiquitin Protein Ligase 1 (SMURF1), activin A receptor type 2B (ACVR2B) and the transcription factor RUNX2 (RUNX2). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway centre represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 11. Genomic analysis of the focal adhesion signaling pathway in hMSCs cultured on PDMS substrates patterned with 350 MPa spots of increasing diameter 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation and deactivation of the FA signaling pathway as a function of spot diameter relative to cells cultured on non-exposed homogenous rigidity substrates in chondrogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates were associated with a significant An increased confidence in the prediction of de-activation of FA signalling with measured down-regulations in the expression of vinculin (VCL) integrin 1 (ITGB1) the JAG1 protein (JAG1) and paxillin (PAX). The predicted de-activation of FA signaling was significantly increased on spot diameters ranging from 500 – 2000 nm in hMSCs cultured on spots of all diameters through down-regulations in the expression of integrin 1 (ITGB1) and integrin 2 (ITGB2), vinculin (VCL) transforming growth factor 1 (TGFB1), cyclin D1 (CCND1), CD44 and paxillin (PXN). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 12. Genomic analysis of the focal adhesion signaling pathway in hMSCs cultured on PDMS substrates patterned with 350 MPa spots of increasing diameter 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned PDMS substrates with spots of increasing diameter revealed significant activation and deactivation of the FA signaling pathway as a function of spot diameter relative to cells cultured on non-exposed homogenous rigidity substrates in osteogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates were associated with a significant reduction in FA activation with measured down-regulations in the expression of vinculin (VCL) integrin 1 (ITGB1) the JAG1 protein (JAG1) and paxillin (PAX). The predicted activation of FA signaling was significantly reduced on spot diameters ranging from 500 – 2000 nm and hMSCs cultured on spots of all diameters were associated with an increased confidence in the prediction of de-activation of activation of FA signaling through down-regulations in the expression of integrin 1 (ITGB1) and integrin 2 (ITGB2), vinculin (VCL) transforming growth factor 1 (TGFB1), cyclin D1 (CCND1), CD44 and paxillin (PXN). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 13. Genomic analysis of chondrogenesis in hMSCs cultured on PDMS substrates patterned with 350 MPa spots of increasing diameter 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned PDMS substrates with spots of increasing diameter revealed revealed significant activation and deactivation of the chondrogenic pathway as a function of spot diameter relative to cells cultured on non-exposed homogenous rigidity substrates in chondrogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates exhibited no changes in the activation of chondrogenesis associated pathways relative to hMSCs cultured on identical substrates in complete (non-chondrogenic) media. An increased confidence in the prediction of de-activation of chondrogenesis was observed in hMSCs cultured on 500 nm spots with down-regulation noted in BMP2, BMP4 BMPR2, ITGA5 and ITGB1. A linear increase in the predicted activation of chondrogenesis was noted with e-beam spot sizes from 1000 – 2000 nm and in particular PDMS substrates e-beam patterned with spot sizes of 2000 nm induced up-regulation in expression of bone morphogenetic protein 4 (BMP4), bone morphogenetic protein 2 (BMP2), bone morphogenetic protein receptor 2 (BMPR2), the transcription factors SOX-9 (SOX9) and RUNX2 (RUNX2), integrin 1 (ITGB1) and integrin α1 (ITGA5). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 14. Genomic analysis of osteogenesis in hMSCs cultured on PDMS substrates patterned with 350 MPa spots of increasing diameter 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned PDMS substrates with spots of increasing diameter revealed revealed significant activation and deactivation of the osteogenic pathway as a function of spot diameter relative to cells cultured on non-exposed homogenous rigidity substrates in osteogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates exhibited an increased confidence in the prediction of de-activation of osteogenesis associated pathways relative to hMSCs cultured on identical substrates in complete (non-osteogenic) media. An increased confidence in the prediction of de-activation of chondrogenesis was also observed in hMSCs cultured on 500 nm and 1000 nm spots with down-regulation noted in BMPR2, SMURF1, activin A receptor type 2A (ACVR2A), activin A receptor type 2B (ACVR2B) and Hypoxia-inducible factor 1-alpha (HIF1A). An increase in the predicted activation of osteogenesis was noted in hMSCs cultured on e-beam spot sizes of 2000 nm which induced up-regulation in the expression of bone morphogenetic protein 4 (BMP4), bone morphogenetic protein 2 (BMP2), RUNX2 (RUNX2), (SMURF1), (NOTCH1), (ACVR2B) and RAC-alpha serine/threonine-protein kinase (AKT1). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary Figure 15. Stripes of alternating rigidity in e-beam exposed PDMS. Example anisotropic pattern fabricated with the maximum e-beam dose (3000 μC/cm2) in PDMS. Green = paxillin, red = actin, line width = 2 μm.
Supplementary Table 1: Statistical analysis of focal adhesion co-localization on heterogeneous rigidity substrates as a function of spot rigidity.
Supplementary Table 2: Statistical analysis of focal adhesion co-localization on heterogeneous rigidity substrates as a function of spot size.
Supplementary Table 3: Statistical analysis of Relative changes in gene expression on heterogeneous rigidity substrates as a function of spot rigidity.
Supplementary Table 4: Statistical analysis of Relative changes in gene expression on heterogeneous rigidity substrates as a function of spot size.
Supplementary Movie 1: Cellular motility on planar PDMS substrates, Cells exhibited rapid polarized migration on unexposed PDMS substrates.
Supplementary Movie 2: Cellular motility on 500 nm spot PDMS substrates, Cellular migration was unaffected on spots measuring 500 nm. Here cells exhibited rapid polarized migration as on unexposed PDMS substrates.
Supplementary Movie 3: Cellular motility on 1 μm spot PDMS substrates, Cellular motility is significantly modulated on spots measuring 1 μm. Here cells were observed to migrate in non-polarized trajectories, with decreased velocity.
Acknowledgments
MB is a Science Foundation Ireland fellow and is supported by the grant: 11/SIRG/B2135. This work carried out by MB, JL and SW was funded principally by the National Institutes of Health (NIH) Common Fund Nanomedicine program (PN2EY016586). The authors would like to thank the following people: Ms. Jessica Brann, for microscopy support, Ms. Gemma Orpella for primer design, Mr. Geoff Goold for technical assistance, Profs. Michael Sheetz, and Michael Dustin for their help, advice and useful discussion and Mr. Maciej Doczyk (http://doczykdesign.com) for graphic design.
Footnotes
Supporting Information: Supporting Information is available from the Wiley Online Library or from the author.
Bibliography
- 1.Pompe T, Glorius S, Bischoff T, Uhlmann I, Kaufmann M, Brenner S, Werner C. Biophysical Journal. 2009;97:2154. doi: 10.1016/j.bpj.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mason BN, Califano JP, Reinhart-King CA. Engineering and Biomaterials for Regenerative Medicine. 2012;Chapter 2:19. [Google Scholar]
- 3.Giannone G, Dubin-Thaler BJ, Dobereiner HG, Kieffer N, Bresnick AR, Sheetz MP. Cell. 2004;116:431. doi: 10.1016/s0092-8674(04)00058-3. [DOI] [PubMed] [Google Scholar]
- 4.Trichet L, Le Digabel J, Hawkins RJ, Vedula SR, Gupta M, Ribrault C, Hersen P, Voituriez R, Ladoux B. Proc Natl Acad Sci U S A. 2012;109:6933. doi: 10.1073/pnas.1117810109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Discher DE, Janmey P, Wang YL. Science. 2005;310:1139. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 6.Vogel V, Sheetz M. Nat Rev Mol Cell Biol. 2006;7:265. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- 7.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 8.Moore SW, Roca-Cusachs P, Sheetz MP. Dev Cell. 2010;19:194. doi: 10.1016/j.devcel.2010.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]; Marshall KL, Lumpkin EA. Adv Exp Med Biol. 2012;739:142. doi: 10.1007/978-1-4614-1704-0_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghassemi S, Meacci G, Liu S, Gondarenko AA, Mathur A, Roca-Cusachs P, Sheetz MP, Hone J. Proc Natl Acad Sci U S A. 2012;109:5328. doi: 10.1073/pnas.1119886109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shemesh T, Geiger B, Bershadsky AD, Kozlov MM. Proc Natl Acad Sci U S A. 2005;102:12383. doi: 10.1073/pnas.0500254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Science. 2009;323:638. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM. Nature. 2010;468:580. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biggs MJ, Richards RG, Gadegaard N, Wilkinson CD, Oreffo RO, Dalby MJ. Biomaterials. 2009;30:5094. doi: 10.1016/j.biomaterials.2009.05.049. [DOI] [PubMed] [Google Scholar]; Biggs MJ, Richards RG, Dalby MJ. Nanomedicine. 2010;6:619. doi: 10.1016/j.nano.2010.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanchez D, Johnson N, Li C, Novak P, Rheinlaender J, Zhang YJ, Anand U, Anand P, Gorelik J, Frolenkov GI, Benham C, Lab M, Ostanin VP, Schaffer TE, Klenerman D, Korchev YE. Biophysical Journal. 2008;95:3017. doi: 10.1529/biophysj.108.129551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wenger MPE, Bozec L, Horton MA, Mesquida P. Biophysical Journal. 2007;93:1255. doi: 10.1529/biophysj.106.103192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saber-Samandari S, Gross KA. Acta Biomaterialia. 2009;5:2206. doi: 10.1016/j.actbio.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 16.Crow A, Webster KD, Hohlfeld E, Ng WP, Geissler P, Fletcher DA. Biophysical Journal. 2012;102:443. doi: 10.1016/j.bpj.2011.11.4020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palama IE, D'Amone S, Coluccia AM, Biasiucci M, Gigli G. Integr Biol (Camb) 2012;4:228. doi: 10.1039/c2ib00116k. [DOI] [PubMed] [Google Scholar]
- 18.Tropmann A, Tanguy L, Koltay P, Zengerle R, Riegger L. Langmuir. 2012;28:8292. doi: 10.1021/la301283m. [DOI] [PubMed] [Google Scholar]; Lin G, Wu VI, Hainley RE, Flanagan LA, Monuki ES, Tang WC. Conf Proc IEEE Eng Med Biol Soc. 2004;4:2607. doi: 10.1109/IEMBS.2004.1403749. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y, Jiang LT, Okada R, Fu J. Langmuir. 2012;28:10789. doi: 10.1021/la300978x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trappmann B, Gautrot JE, Connelly JT, Strange DG, Li Y, Oyen ML, Cohen Stuart MA, Boehm H, Li B, Vogel V, Spatz JP, Watt FM, Huck WT. Nat Mater. 2012;11:642. doi: 10.1038/nmat3339. [DOI] [PubMed] [Google Scholar]
- 21.Xia YN, Whitesides GM. Annual Review of Materials Science. 1998;28:153. [Google Scholar]
- 22.Tsougeni K, Tserepi A, Gogolides E. Microelectron Eng. 2007;84:1104. doi: 10.1016/j.mee.2012.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Desimone JM, York GA, Mcgrath JE, Gozdz AS, Bowden MJ. Macromolecules. 1991;24:5330. [Google Scholar]; Giri R, Sureshkumar MS, Naskar K, Bharadwaj YK, Sarma KSS, Sabharwal S, Nando GB. Advances in Polymer Technology. 2008;27:98. [Google Scholar]
- 24.Russell MT, Pingree LSC, Hersam MC, Marks TJ. Langmuir. 2006;22:6712. doi: 10.1021/la060319i. [DOI] [PubMed] [Google Scholar]
- 25.Moeinzadeh S, Pajoum Shariati SR, Jabbari E. Biomaterials. 2016;92:57. doi: 10.1016/j.biomaterials.2016.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, Hammer DA, Weaver VM. Cancer Cell. 2005;8:241. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]; Liu J, Tan Y, Zhang H, Zhang Y, Xu P, Chen J, Poh YC, Tang K, Wang N, Huang B. Nat Mater. 2012;11:734. doi: 10.1038/nmat3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young TJ, Monclus MA, Burnett TL, Broughton WR, Ogin SL, Smith PA. Meas Sci Technol. 2011;22 [Google Scholar]
- 28.Cai DK, Neyer A, Kuckuk R, Heise HM. J Mol Struct. 2010;976:274. [Google Scholar]
- 29.Albers L, Baumgartner J, Marschner C, Muller T. Chem-Eur J. 2016;22:7970. doi: 10.1002/chem.201600116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu BY, Fu J. J Micromech Microeng. 2015;25 doi: 10.1088/0960-1317/25/7/075010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bowen J, Cheneler D, Robinson APG. Microelectron Eng. 2012;97:34. [Google Scholar]
- 32.Zhu SL, Cheng HH, Blakey I, Stokes N, Ostrikov K, Cortie M. Microelectron Eng. 2015;139:13. [Google Scholar]; Gamboa JR, Mohandes S, Tran PL, Slepian MJ, Yoon JY. Colloid Surface B. 2013;104:318. doi: 10.1016/j.colsurfb.2012.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dalby MJ, Gadegaard N, Tare R, Andar A, Riehle MO, Herzyk P, Wilkinson CDW, Oreffo ROC. Nat Mater. 2007;6:997. doi: 10.1038/nmat2013. [DOI] [PubMed] [Google Scholar]
- 34.Schnyder B, Lippert T, Kotz R, Wokaun A, Graubner VM, Nuyken O. Surface Science. 2003;532:1067. doi: 10.1021/la051086i. [DOI] [PubMed] [Google Scholar]
- 35.Monge C, Saha N, Boudou T, Pozos-Vasquez C, Dulong V, Glinel K, Picart C. Advanced functional materials. 2013;23:3432. doi: 10.1002/adfm.201203580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cukierman E, Pankov R, Stevens DR, Yamada KM. Science. 2001;294:1708. doi: 10.1126/science.1064829. [DOI] [PubMed] [Google Scholar]; Biggs MJ, Richards RG, Gadegaard N, Wilkinson CD, Dalby MJ. J Mater Sci Mater Med. 2007;18:399. doi: 10.1007/s10856-006-0705-6. [DOI] [PubMed] [Google Scholar]
- 37.Kulangara K, Yang Y, Yang J, Leong KW. Biomaterials. 2012;33:4998. doi: 10.1016/j.biomaterials.2012.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dalby MJ, Gadegaard N, Tare R, Andar A, Riehle MO, Herzyk P, Wilkinson CD, Oreffo RO. Nat Mater. 2007;6:997. doi: 10.1038/nmat2013. [DOI] [PubMed] [Google Scholar]
- 39.Pelham RJ, Wang YL. P Natl Acad Sci USA. 1997;94:13661. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nicolas A, Besser A, Safran SA. Biophysical Journal. 2008;95:527. doi: 10.1529/biophysj.107.127399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Massia SP, Hubbell JA. Journal of Cell Biology. 1991;114:1089. doi: 10.1083/jcb.114.5.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]; Geiger B, Spatz JP, Bershadsky AD. Nature Reviews Molecular Cell Biology. 2009;10:21. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 42.Cavalcanti-Adam EA, Volberg T, Micoulet A, Kessler H, Geiger B, Spatz JP. Biophysical Journal. 2007;92:2964. doi: 10.1529/biophysj.106.089730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plotnikov SV, Pasapera AM, Sabass B, Waterman CM. Cell. 2012;151:1513. doi: 10.1016/j.cell.2012.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arnold M, Schwieder M, Blummel J, Cavalcanti-Adam EA, Lopez-Garcia M, Kessler H, Geiger B, Spatz JP. Soft Matter. 2009;5:72. doi: 10.1039/B815634D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang C, DelRio FW, Ma H, Killaars AR, Basta LP, Kyburz KA, Anseth KS. P Natl Acad Sci USA. 2016;113:E4439. doi: 10.1073/pnas.1609731113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoffecker IT, Guo WH, Wang YL. Lab on a Chip. 2011;11:3538. doi: 10.1039/c1lc20504h. [DOI] [PubMed] [Google Scholar]
- 47.Meacci G, Stachowiak M, Liu SM, Iskratsch T, Mathur A, Wolfenson H, Ghassemi S, Roca-Cusachs P, Tabdanov E, Gauthier N, Gondarenko A, O'Shaughnessy B, Hone J, Sheetz M. Biophysical Journal. 2013;104:477A. [Google Scholar]
- 48.O'Reilly A, Kelly DJ. Ann Biomed Eng. 2016;44:3446. doi: 10.1007/s10439-016-1664-9. [DOI] [PubMed] [Google Scholar]
- 49.Gao L, McBeath R, Chen CS. Stem Cells. 2010;28:564. doi: 10.1002/stem.308. [DOI] [PMC free article] [PubMed] [Google Scholar]; Woods A, Wang GY, Beier F. J Biol Chem. 2005;280:11626. doi: 10.1074/jbc.M409158200. [DOI] [PubMed] [Google Scholar]
- 50.http://www.gel.usherbrooke.ca/casino/.
- 51.Thomas D, Fontana G, Chen XZ, Sanz-Nogues C, Zeugolis DI, Dockery P, O'Brien T, Pandit A. Biomaterials. 2014;35:8757. doi: 10.1016/j.biomaterials.2014.06.053. [DOI] [PubMed] [Google Scholar]
- 52.Biggs MJ, Milone MC, Santos LC, Gondarenko A, Wind SJ. Journal of the Royal Society Interface. 2011;8:1462. doi: 10.1098/rsif.2011.0025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolte S, Cordelieres FP. J Microsc-Oxford. 2006;224:213. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 54.Dunn KW, Kamocka MM, McDonald JH. Am J Physiol-Cell Ph. 2011;300:C723. doi: 10.1152/ajpcell.00462.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meijering E, Dzyubachyk O, Smal I. Methods Enzymol. 2012;504:183. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. Topographical modification of E-beam exposed PDMS and analysis of topographical control substrates (A) Elastomer contraction was observed to be dose dependent. Low doses of 50 μC/cm2 resulted in undulations of 4 nm depth. PDMS contraction and undulation depth was observed to plateau with doses of 2,200 μC/cm2, resulting undulations were 140 nm in depth with a radius of curvature of 20 μm. (B) Maximum E-beam exposure initiated nanoscale contractions of PDMS resulting in an undulating topography. (C) AFM Phase analysis indicated significance modification to PDMS substrate mechanical properties following E-beam exposure of 3,000 μC/cm2. (D) Typical phase profile of PDMS following E-beam exposure of 3,000 μC/cm2. (E) Exposed substrates we employed to generate negative topographical master stamps. (F,G) Undulating topographies were replicated into into 500 μm thick PDMS films of 35 KPa via direct casting to produce topographical controls with a homogenous rigidity profile. Significant reduction in phase angle was observed following casting onto a topographical master.
Supplementary figure 2. Focal adhesion Co-localization to spots of increasing diameter and rigidity (A) Focal adhesion co-localization to 2 μm spots of modulated rigidity increased as a function of spot rigidity. (B) FA co-localization was unaffected by spots <250 nm in diameter irrespective of spot dose. Data obtained from image analysis of immunofluorescent staining of paxillin indicated that increases in rigidity dependent co-localization are more significant on larger spot diameters. Results are SEM, green = actin, blue = paxillin, red = nucleus, bar = 10 μm.
Supplementary figure 3. XPS, RAMAN and fluorescence absorption Spectra of e-beam exposed PDMS (A) XPS survey of non-exposed and E-beam exposed PDMS indicated a reduction in C content. (B) Raman map of e-beam exposed PDMS and adjacent control, acquired with a 3 μm step shows the ratio of the intensities of the peak at 490 cm-1 and 706 cm-1, suggesting a 9.4% increase in the presence of Si-O-Si bonds on non-exposed regions relative to e-beam exposed regions. The ratio of the intensities of the peak at 2500 cm-1 and 2905 cm-1, suggests a 116% increase in the presence of Si-H bonds in e-beam patterned regions relative to the un-exposed region. Also shown is the principal component analysis of Raman mapping acquired using a static method, with the peak centered at 950 cm-1 and at 2500 cm-1. Both clearly show the difference in the chemical signatures between e-beam exposed and control non-exposed PDMS. (C) Confocal microscopy revealed that differential fibronectin adsorption did not occur on patterned substrates.
Supplementary figure 4. Focal adhesion analysis on e-beam exposed PDMS (A&B) Fluorescent paxillin and DIC images are analyzed in imageJ via the JACoP co-localization plugin, with a returned value of 1 indicating 100% pixel co-localization and a value of 0 indicating no co-localization. Green = actin, blue = paxillin, red = nucleus, bar = 50 μm.
Supplementary figure 5. Relative fluorescence intensity of paxillin staining on e-beam exposed spots as a function of (A) spot rigidity and (B) spot diameter. Fluorescence intensity profiles are expressed as normalized signals to paxillin intensity of FA formed on inter-spots regions. Intensity was observed to increases on spots of increasing elastics modulus and on spots of increased diameter. With maximum intensity occurring on 350 MPa spots of 2 μm diameter.
Supplementary figure 6. Focal adhesion Co-localization to ∼350 MPa spots of increasing diameter and the influence of spot diameter on cellular motility (A) Paxillin staining revealed a significant increase in FA colocalization with increasing spot diameter. Bottom panels represent high magnification of paxillin staining within the boarded insert area. (B) Significant changes in mean cellular velocity were induced by increasing the spot diameter on patterns fabricated with elastic moduli of ∼350 MPa. Mean cell velocity was greatest on glass control substrates but significant reductions were noted in cells were cultured on spots >250 nm in diameter. (C) Greatest migration distances were observed in cells cultured on 100 nm diameter spots. Migration distance was observed to decrease linearly with increasing spot diameter. Results are SEM, green = actin, blue = paxillin, red = nucleus, bar = 10 μm.
Supplementary figure 7. Genomic analysis of the focal adhesion signaling pathway in hMSCs cultured on PDMS substrates patterned with 2 μm spots of increasing electron-beam dose for 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation and deactivation of the FA signaling pathway as a function of spot dose relative to cells cultured on non-exposed homogenous rigidity substrates in chondrogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates were associated with an increased confidence in the prediction of de-activation of FA signaling, with measured down-regulations in the expression of vinculin (VCL) integrin 1 (ITGB1) the JAG1 protein (JAG1) and paxillin (PAX). A linear increase in predicted activation of FA signaling was noted in cells cultured on 50 – 200 MPa and spots of 200 MPa induced up-regulation in expression of integrin 1 and integrin 2, vinculin transforming growth factor 1 (TGFB1), cyclin D1 (CCND1) and CD44. Predicted activation of FA signaling was lost however on PDMS substrates possessing 2 μm spots of ∼350 MPa. Cells cultured on these substrates exhibited down-regulations in VCL, PXN and TGFB1. Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 8. Genomic analysis of the focal adhesion signaling pathway in hMSCs cultured on PDMS substrates patterned with 2 μm spots of increasing electron-beam dose for 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation and deactivation of the FA signaling pathway as a function of spot dose relative to cells cultured on non-exposed homogenous rigidity substrates in osteogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates were associated with a significant increased confidence in the prediction of de-activation with measured down-regulations in the expression of vinculin (VCL) integrin 1 (ITGB1) the JAG1 protein (JAG1) and paxillin (PAX). A linear increase in predicted activation of FA signaling was noted in cell cultured on 2 μm spots with E values raging from 50 – 200 MPa and spots of 200 MPa further induced up-regulation in expression of integrin 1 and integrin 2, vinculin transforming growth factor 1 (TGFB1), cyclin D1 (CCND1) and CD44. Predicted activation of FA signaling was lost however on PDMS substrates possessing spots of 350 MPa. Cells cultured on these substrates exhibited down-regulations in VCL, PXN and TGFB1. Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 9. Genomic analysis of chondrogenesis in hMSCs cultured on PDMS substrates patterned with 2 μm spots of increasing electron-beam dose for 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation and deactivation of the FA signaling pathway as a function of spot dose relative to cells cultured on non-exposed homogenous rigidity substrates in chondrogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates exhibited no changes in the predicted activation of chondrogenesis associated pathways relative to hMSCs cultured on identical substrates in complete (non-chondrogenic) media. A linear increase in the predicted activation of chondrogenesis was noted in cells cultured on 2 μm spots with E values raging from 50 – 200 MPa and in particular, PDMS substrates e-beam patterned with spot of 350 MPa induced up-regulation in expression of bone morphogenetic protein 4 (BMP4), bone morphogenetic protein 2 (BMP2), bone morphogenetic protein receptor 2 (BMPR2), the transcription factors SOX-9 (SOX9) and RUNX2 (RUNX2), integrin 1 (ITGB1) and integrin α1 (ITGA5). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 10. Genomic analysis of osteogenesis in hMSCs cultured on PDMS substrates patterned with 2 μm spots of increasing electron-beam dose for 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation and deactivation of the FA signaling pathway as a function of spot dose relative to cells cultured on non-exposed homogenous rigidity substrates in osteogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates were associated with a significant increased confidence in the prediction of de-activation of osteogenesis associated pathways relative to hMSCs cultured on identical substrates in complete (non-osteogenic) media. A linear increase in the predicted activation of osteogenesis was noted in cells cultured on 2 μm spots with E values raging from 50 – 200 MPa and in particular PDMS substrates e-beam patterned with 2μm spot doses 350 MPa induced up-regulation in expression of osteogenesis associated genes - bone morphogenetic protein 4 (BMP4), bone morphogenetic protein 2 (BMP2), the JAG1 protein (JAG1), The neurogenic locus notch homolog protein 1 (NOTCH1), SMAD Specific E3 Ubiquitin Protein Ligase 1 (SMURF1), activin A receptor type 2B (ACVR2B) and the transcription factor RUNX2 (RUNX2). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway centre represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 11. Genomic analysis of the focal adhesion signaling pathway in hMSCs cultured on PDMS substrates patterned with 350 MPa spots of increasing diameter 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned 2 μm spot substrates with different rigidities revealed significant activation and deactivation of the FA signaling pathway as a function of spot diameter relative to cells cultured on non-exposed homogenous rigidity substrates in chondrogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates were associated with a significant An increased confidence in the prediction of de-activation of FA signalling with measured down-regulations in the expression of vinculin (VCL) integrin 1 (ITGB1) the JAG1 protein (JAG1) and paxillin (PAX). The predicted de-activation of FA signaling was significantly increased on spot diameters ranging from 500 – 2000 nm in hMSCs cultured on spots of all diameters through down-regulations in the expression of integrin 1 (ITGB1) and integrin 2 (ITGB2), vinculin (VCL) transforming growth factor 1 (TGFB1), cyclin D1 (CCND1), CD44 and paxillin (PXN). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 12. Genomic analysis of the focal adhesion signaling pathway in hMSCs cultured on PDMS substrates patterned with 350 MPa spots of increasing diameter 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned PDMS substrates with spots of increasing diameter revealed significant activation and deactivation of the FA signaling pathway as a function of spot diameter relative to cells cultured on non-exposed homogenous rigidity substrates in osteogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates were associated with a significant reduction in FA activation with measured down-regulations in the expression of vinculin (VCL) integrin 1 (ITGB1) the JAG1 protein (JAG1) and paxillin (PAX). The predicted activation of FA signaling was significantly reduced on spot diameters ranging from 500 – 2000 nm and hMSCs cultured on spots of all diameters were associated with an increased confidence in the prediction of de-activation of activation of FA signaling through down-regulations in the expression of integrin 1 (ITGB1) and integrin 2 (ITGB2), vinculin (VCL) transforming growth factor 1 (TGFB1), cyclin D1 (CCND1), CD44 and paxillin (PXN). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 13. Genomic analysis of chondrogenesis in hMSCs cultured on PDMS substrates patterned with 350 MPa spots of increasing diameter 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned PDMS substrates with spots of increasing diameter revealed revealed significant activation and deactivation of the chondrogenic pathway as a function of spot diameter relative to cells cultured on non-exposed homogenous rigidity substrates in chondrogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates exhibited no changes in the activation of chondrogenesis associated pathways relative to hMSCs cultured on identical substrates in complete (non-chondrogenic) media. An increased confidence in the prediction of de-activation of chondrogenesis was observed in hMSCs cultured on 500 nm spots with down-regulation noted in BMP2, BMP4 BMPR2, ITGA5 and ITGB1. A linear increase in the predicted activation of chondrogenesis was noted with e-beam spot sizes from 1000 – 2000 nm and in particular PDMS substrates e-beam patterned with spot sizes of 2000 nm induced up-regulation in expression of bone morphogenetic protein 4 (BMP4), bone morphogenetic protein 2 (BMP2), bone morphogenetic protein receptor 2 (BMPR2), the transcription factors SOX-9 (SOX9) and RUNX2 (RUNX2), integrin 1 (ITGB1) and integrin α1 (ITGA5). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary figure 14. Genomic analysis of osteogenesis in hMSCs cultured on PDMS substrates patterned with 350 MPa spots of increasing diameter 12 hours Functional pathway analysis of hMSCs cultured on e-beam patterned PDMS substrates with spots of increasing diameter revealed revealed significant activation and deactivation of the osteogenic pathway as a function of spot diameter relative to cells cultured on non-exposed homogenous rigidity substrates in osteogenic media. hMSCs cultured on homogenous, unexposed PDMS substrates exhibited an increased confidence in the prediction of de-activation of osteogenesis associated pathways relative to hMSCs cultured on identical substrates in complete (non-osteogenic) media. An increased confidence in the prediction of de-activation of chondrogenesis was also observed in hMSCs cultured on 500 nm and 1000 nm spots with down-regulation noted in BMPR2, SMURF1, activin A receptor type 2A (ACVR2A), activin A receptor type 2B (ACVR2B) and Hypoxia-inducible factor 1-alpha (HIF1A). An increase in the predicted activation of osteogenesis was noted in hMSCs cultured on e-beam spot sizes of 2000 nm which induced up-regulation in the expression of bone morphogenetic protein 4 (BMP4), bone morphogenetic protein 2 (BMP2), RUNX2 (RUNX2), (SMURF1), (NOTCH1), (ACVR2B) and RAC-alpha serine/threonine-protein kinase (AKT1). Red indicates high relative expression and green indicates low expression of genes. Brown shading of the pathway center represents and increased confidence in the prediction of activation and blue, a decreased confidence as shown in the prediction legend. Statistical significance of pathway modulation was calculated via a right-tailed Fisher's exact test.
Supplementary Figure 15. Stripes of alternating rigidity in e-beam exposed PDMS. Example anisotropic pattern fabricated with the maximum e-beam dose (3000 μC/cm2) in PDMS. Green = paxillin, red = actin, line width = 2 μm.
Supplementary Table 1: Statistical analysis of focal adhesion co-localization on heterogeneous rigidity substrates as a function of spot rigidity.
Supplementary Table 2: Statistical analysis of focal adhesion co-localization on heterogeneous rigidity substrates as a function of spot size.
Supplementary Table 3: Statistical analysis of Relative changes in gene expression on heterogeneous rigidity substrates as a function of spot rigidity.
Supplementary Table 4: Statistical analysis of Relative changes in gene expression on heterogeneous rigidity substrates as a function of spot size.
Supplementary Movie 1: Cellular motility on planar PDMS substrates, Cells exhibited rapid polarized migration on unexposed PDMS substrates.
Supplementary Movie 2: Cellular motility on 500 nm spot PDMS substrates, Cellular migration was unaffected on spots measuring 500 nm. Here cells exhibited rapid polarized migration as on unexposed PDMS substrates.
Supplementary Movie 3: Cellular motility on 1 μm spot PDMS substrates, Cellular motility is significantly modulated on spots measuring 1 μm. Here cells were observed to migrate in non-polarized trajectories, with decreased velocity.