

Abstract
The current COVID-19 outbreak warrants the design and development of novel anti-COVID therapeutics. Using a combination of bioinformatics and computational tools, we modelled the 3D structure of the RdRp (RNA-dependent RNA polymerase) of SARS-CoV2 (severe acute respiratory syndrome coronavirus-2) and predicted its probable GTP binding pocket in the active site. GTP is crucial for the formation of the initiation complex during RNA replication. This site was computationally targeted using a number of small molecule inhibitors of the hepatitis C RNA polymerase reported previously. Further optimizations suggested a lead molecule that may prove fruitful in the development of potent inhibitors against the RdRp of SARS-CoV2.
Introduction
The ongoing COVID-19 (coronavirus disease 2019) global crisis has resulted in an unprecedented level of international turbulence, both in terms of global health and economy. The first ever patient diagnosed to have fallen from an infection caused by the then strain of coronavirus, 2019-nCoV, is traced back to the 1st of December 2019.1 Within a span of over three months, the disease—COVID 19—has been declared a global pandemic and has infected 619533 individuals, resulting in 376,320 deaths as of 2nd June 2020.2 Of the seven strains of coronavirus known to infect humans, SARS-CoV (severe acute respiratory syndrome coronavirus), MERS-CoV (Middle East respiratory syndrome coronavirus), and the current strain SARS-CoV2 (severe acute respiratory syndrome coronavirus-2) are known to cause severe lower respiratory tract dysfunction.3 The primary symptoms of COVID-19 are fever, cough, and discomfort in the upper chest accompanied with labored breathing, similar to those of SARS-CoV and MERS-CoV infections. Severe cases are reported to show sepsis, secondary infections, and organ failure. Reports suggest a possible combination of natural selection and zoonotic transfer events that led to the emergence of this novel strain.4
The ∼29.9 kbp (+) ss-RNA genome of SARS-CoV2 shares 96.2% identity with the bat CoV RaTG13 genome and 79.5% identity with the SARS-CoV genome.5 The amino acid sequence encoded by the genome shares 82.3 and 77.2% identities with the amino acid sequences encoded by the genomes of bat CoV SL-CoVZC45 and SARS-CoV, respectively.6 The first polyprotein coding DNA sequence of the SARS- CoV2 genome (isolate Wuhan-Hu-1, NC_045512)7 is predicted to encode for 14 nonstructural proteins, of which “nonstructural protein 12” (YP_009725307.1) is composed of two domains, RdRp (RNA-dependent RNA polymerase) and NiRAN (nidovirus RdRp-associated nucleotidyl transferase). The RdRp is a key enzyme in RNA viruses which mediates the replication of their RNA genomes.8 The RdRp from nidoviruses in addition to the canonical finger, palm, and thumb subdomains, possesses two extra domains—the interface and the NiRAN domain.9,10 The interface domain primarily acts as a linker between the RdRp and NiRAN domains and is also predicted to have interactions with the accessory proteins of the replicase complex—the Nsp7 and the Nsp8.11,12 The NiRAN domain has been observed to possess GTP (guanosine triphosphate)- and UTP (uridine triphosphate)-binding activities that aid in the replication initiation process.9 RdRp enzymes in general are known to exhibit both primer-dependent and primer-independent modes of replication initiation, while some are exclusive to either mode.8,13−16 Although SARS-CoV RdRp exhibits a primer-dependent activity in vitro,17 the RdRp from its sister species—EAV (equine arteritis virus)—demonstrates a primer-independent activity.18 The RdRp enzymes from SARS-CoV and EAV share a similar structural conservation, suggesting that the nidovirus RdRp may exhibit both modes of initiation in vitro.16 Moreover, the nsp7–nsp8 complex has been observed to have a primase activity, possibly aimed at a primer-dependent initiation.19 Interestingly, the primer-independent activity of RdRp is attributed to the binding of single NTP (nucleotide triphosphate), preferably GTP within or in the vicinity of the active site pocket.20−22
In this report, using computer-aided homology modeling, docking, and molecular simulations, we have predicted the protein structure and probable small-molecule inhibitors against SARS-CoV2 RdRp (CoV2-RdRp).
Results and Discussion
Prediction of the GTP Binding Site and the Initiation Complex for CoV2-RdRp
A BLASTp search with the amino acid sequence for CoV2-RdRp against the PDB database yielded greater than 93% sequence identity with the sequence for RdRp from SARS-CoV (CoV1-RdRp). A homology modelling using the cryo-EM structure of CoV1-RdRp (PDB ID: 6NUR(23)) as the template yields a globular structure of CoV2-RdRp with two distinct structural architectures. The overall structure is constituted of an RdRp-like structure (∼570 amino acids) fused with a NiRAN domain (∼360 amino acids) at the N-terminus. Similar to the RdRps from viruses such as dengue,24 hepatitis C,25−27 polio,28 and others, the 3D structure of CoV2-RdRp exhibits the canonical RNA polymerase fold comprising the finger, palm, and thumb subdomains (Figure 1A). A superimposition of the CoV2-RdRp and CoV1-RdRp structures (Figure S1A) highlights the conservation of the secondary structural elements, which is also evident from the low root mean square deviation (rmsd) value of 0.93 Å. While this study was in progress, an experimentally determined (cryo-EM) structure of CoV2-RdRp (Nsp12) in complex with Nsp7 and Nsp8 was reported (PDB IDs—6M71 and 7BTF(11)). The experimentally determined structure and the model generated in this study superimpose with each other with an rmsd of 0.43 Å, suggesting that the model structure is nearly identical to the experimentally determined structure (Figure 1B). In light of this, the results of our study would remain unaltered largely should it be reproduced using the EM structure.
Figure 1.

Overall architecture of RdRp domains of SARS-CoV2. (A) 3D structure of CoV2-RdRp, displaying the RdRp domain with the finger, palm, and thumb subdomains and the NiRAN domain (finger subdomain in royal blue, palm subdomain in magenta, thumb subdomain in yellow, and NiRAN domain in faded raspberry). The extended structures Δ1 and Δ2 are shown in red and green colors. (B) Comparison of the 3D structures of the CoV2-RdRp model generated in this study and the cryo-EM-determined CoV2-RdRp (PDB ID: 7BTF) shows no significant differences between them (cryo-EM RdRp in salmon and model RdRp in cyan). (C) Superimposition of the highly conserved regions of CoV2-RdRp and HCV-RdRp, displaying the conservation of the overall fold and the secondary structural elements (CoV2-RdRp in dark cyan and HCV-RdRp in orange). (D) Pairwise sequence alignment of the conserved regions of CoV2-RdRp and HCV-RdRp, suggesting the possible Asp-catalytic dyad. The dyad shown within a blue curly bracket.
In order to derive insights into the structural similarities and differences between the structures of CoV2-RdRp and HCV-RdRp (RdRp from the hepatitis C virus), we superimposed them. The overlay resulted in an rmsd value of about 5 Å and showed an overall conservation of the RdRp architecture, with exceptions to a few secondary structural elements and flexible loops. Removal of the nonaligning regions resulted in better superimposition of the three RdRp subdomains—fingers, palm, and thumb (Figure 1C), with an rmsd value of 3.2 Å. Similar to those in the HCV-RdRp polymerase, two extended structures [Δ1 (400–447 aa) and Δ2 (544–556 aa) motifs] protruding from the finger subdomain make interactions with the thumb subdomain. These two loop-helix-loop motifs bridge the finger and thumb subdomains. Similar to other known polymerases, the CoV2-RdRp active site is situated in the palm subdomain with two conserved aspartic acid residues, as evident from the pairwise sequence alignment of the superimposing sequences (Asp760 and Asp761) (Figure 1D). Regardless of the variations in the detailed structures, their general architectures are largely similar. For further analysis, only the RdRp region of the model (amino acids 360–932) was chosen.
Previous reports suggest the binding of a GTP molecule adjacent to the first NTP near the active-site pocket for the de novo initiation of RNA replication by RdRp enzymes.20,29,30 This GTP is added by a nucleotidyl transfer reaction.20 Studies have reported structures of GTP-bound RdRp enzymes from other positive sense single-stranded RNA viruses such as the bovine viral diarrhea virus (BVDV; PDB ID: 1S49(20)), enterovirus D68 (EV-D68; PDB ID: 3N6M(31)), poliovirus (PolV; PDB ID: 1RA7(32)), Mammalian orthoreovirus 3-Dearing (MRV3; PDB ID: 1N1H(33)), and Japanese encephalitis virus (JEV; PDB ID: 4HDG(34)) and in double-stranded RNA viruses such as the cypovirus (CypoV; PDB ID: 6TY9(35)) and Pseudomonas phage Phi 6 (Phi6; PDB ID: 1HI0(36)). A ligand-binding site search suggested the presence of a probable GTP binding pocket at the finger−palm interface of the CoV2-RdRp. Docking analysis at this site exhibited multiple GTP binding conformations (Figure S1B). Further stabilization of the model GTP complex yielded the minimum energy conformations model for GTP binding at the predicted site (Figures 2A and S1C). The α-, β-, and γ-phosphate groups of the GTP molecule form H-bond interactions with Lys798, Pro620, and Arg553, respectively (Figure S1D). The guanosine group makes potential H-bonding interactions with Asp761, Ala762, Glu811, and Ser814 residues of the enzyme. Comparison of the CoV2-RdRp model in complex with GTP to that of the experimentally determined structures of RdRp GTP complexes revealed a significant similarity in the GTP binding sites. The CoV2-RdRp aligned with EV-D68-RdRp, BVDV-RdRp (Figure S2A), PolV-RdRp (Figure S2B), JEV-RdRp (Figure S2C), and CypoV-RdRp with rmsd values of 3.08, 3.02, 3.01, and 3.54 Å with significance values of 1.76 × 10–8, 3.83 × 10–9, 8.45 × 10–5, and 6.29 × 10–3, respectively. Further inspection revealed the presence of similar residues such as Glu, Asp, and Lys in the GTP binding pocket, making H-bond interactions possible with the bound GTP (Figure S3A,B), suggesting that GTP binding is an essential aspect of all RdRp enzymes with possible implications toward the initiation of the RNA replication process.
Figure 2.

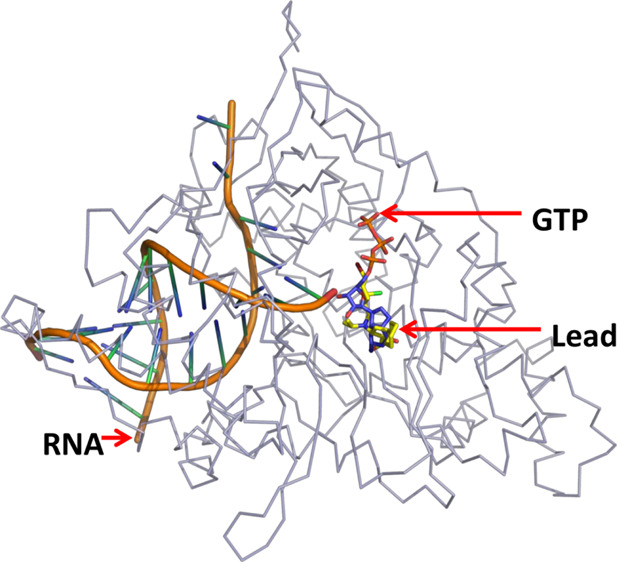
Predictions of the GTP binding site and initiation complex for CoV2-RdRp. (A) Zoomed-in view of the energy-minimized conformation of GTP bound to the predicted binding site (blue indicates positively charged regions, red indicates negatively charged regions, green indicates neutral regions, and gray indicates regions beyond the GTP binding pocket). (B) Superimposition of GTP-bound CoV2-RdRP and the GTP- and RNA-bound Pseudomonas phage φ6-RdRp reveals the conservation of the GTP binding pocket and its close proximity to the template RNA. (GTP bound to CoV2-RdRP and Pseudomonas phage φ6-RdRp in blue and cyan, respectively, CoV2-RdRP ribbon in gray, Pseudomonas phage φ6-RdRp in red, and RNA indicated with a black arrow.) (C). Superimposition of the GTP- and RNA template-bound CoV2-RdRP model and the RNA- and remdesivir (monophosphate)-bound CoV2-RdRP (PDB ID: 7BV2) reveals proximity of the GTP binding pocket to the primer RNA (model CoV2-RdRp in cyan, cryo-EM-determined CoV2-RdRp in red, RNA indicated with a black arrow, GTP in deep blue and orange (phosphate) sticks, and remdesivir in yellow and green (phosphate) sticks). (D) Predicted model of the CoV2-RdRp initiation complex generated following comparison with the cryo-EM-determined RNA-bound CoV2-RdRP, depicting the GTP and RNA molecules (GTP in green and orange sticks and RNA indicated with a black arrow).
Furthermore, we attempted to generate a probable model of the initiation complex of the CoV2-RdRp. A comparison of the reported initiation models for the Pseudomonas phage φ6-RdRp (PDB ID: 1HI0(36)) (Figures 2B and S4A) and the mammalian orthoreovirus 3-Dearing-RdRp (PDB ID: 1N1H(33)) (Figure S4B) with the GTP-bound model of the CoV2-RdRp revealed that the GTP in CoV2-RdRp sits in the vicinity of the template RNA, similar to the GTP of Pseudomonas phage φ6-RdRp and the enterobacterial phage λ-RdRp. This suggests that the predicted GTP binding pocket of CoV2-RdRp possibly plays an important role in the initiation process. A recently published study demonstrates the CoV2-RdRp bound to the template and primer RNA molecules (PDB ID: 7BV2).37 A comparison of the GTP-bound CoV2-RdRp model shows interactions between the primer RNA and the bound GTP. This further suggests that the bound GTP may play a role in primer-independent replication or the GTP binding site may act as the receptor site for incoming nucleotides prior to its addition to the growing RNA chain. The superimposition of the GTP-bound model structure and the RNA- and remdesivir (monophosphate)-bound cryoEM structure of CoV2-RdRp37 is presented in Figure 2C. The GTP in the model structure and the remdesivir molecule in the cryo-EM structure occupy overlapping regions in the active-site region of the CoV2-RdRp; however, the GTP is stabilized via interactions with amino acid residues, remdesivir shows interactions with amino acid residues and the nucleotides from the bound RNA molecule (Figure S5). Taking together the aforementioned interaction and comparison of the model and experimentally determined structures, we propose the probable initiation complex of CoV2-RdRp bound to RNA and GTP molecules in Figure 2D.
Exploration of Inhibitors Targeting the GTP Binding Site of CoV2-RdRp
Over the past several years, a target-based approach has led to the design of a number of small molecule inhibitors against HCV-RdRp.26,27,38−43 Importantly, the current drug (sofosbuvir)44 for HCV treatment targets the HCV-RdRp. The binding of GTP is a key step in the RNA replication process, suggesting the therapeutic importance of the GTP binding site pocket.20,29 Previous reports26,27 involving studies with thiophene scaffold inhibitors against HCV-RdRp suggest that these inhibitors target the thumb site II, nearly 35 Å from the polymerase active site. The inhibitor binding to this site may interfere with the GTP binding function of the RdRp.45 We sought to explore whether these inhibitors and the current anti-HCV drug (sofosbuvir) and a broad spectrum antiviral drug (remdesivir) exhibit any binding affinity with the CoV2 polymerase, specifically targeting the GTP binding site. The 2D structures of six small molecules are presented in Figure 3A–F (left panels). Directed docking of a few proposed HCV-RdRp inhibitors along with the triphosphate/metabolite forms of sofosbuvir and remdesivir (Figure 3, right panels) exhibited binding interactions within the GTP binding pocket (Figure S6). The docking properties for these candidate molecules are listed in Table 1 (rows 1–7). The detailed view of the molecular interactions within the GTP binding site for the three molecules with the most favorable docking properties is presented in Figure S7. A comparison of the remdesivir (triphosphate)-bound model structure and the remdesivir (monophosphate)-bound cryo-EM structure of the RdRp revealed that the molecules occupy similar locations within the active-site region of the molecule (Figure S8A). A comparison of the atomic interactions of these molecules with the amino acids of the RdRp shows common interactions with Lys545, Arg555, Val557, and Cys622 (Figure S8B).
Figure 3.

Molecular docking of probable inhibitory molecules onto the predicted GTP binding site. Panels (A–F) show the 2D structures in the stick model (left sides) and the corresponding computationally directed bindings of sofosbuvir (triphosphate form), SNH, remdesivir (triphosphate form), JPC, CCT, and FIH at the GTP binding sites, respectively (right sides). Positive, negative, and neutral regions of the surface model of the enzyme are indicated by blue, red, and green colors, respectively. Gray indicates regions beyond the GTP binding pocket. The small molecules in the binding cavities are shown in the stick model with carbon, nitrogen, oxygen, fluoride, phosphorous, sulfur, and chloride atoms in yellow, blue, red, sky-blue, orange, tv-yellow, and green colors, respectively.
Table 1. Docking Properties of the Small Molecule Inhibitors at the Predicted GTP Binding Site of CoV2-RdRpa.
| Pubchem/DrugBank ID | ligand name | mol. wt (Da) | docking score (kcal/mol) | binding energy (kcal/mol) | H-bond residues | |
|---|---|---|---|---|---|---|
| 1 | 9543495 | FIH | 391.4 | –5.79 | –30.2 | Asp761, Asp618, Trp617, and Ala762 |
| 2 | DBMET03038 | remdesivir (triphosphate) | 527.2 | –5.66 | –34.7 | Lys545, Arg555, Thr556, Asp623, and Ser682 |
| 3 | 9543494 | CCT | 398.5 | –5.59 | –19.7 | Asp761, Trp617, and Ala762 |
| 4 | 657065 | JPC | 434.3 | –5.38 | –13.4 | Trp617, Ala762, Asp761, and Asp760 |
| 5 | 9543496 | SNH | 435.5 | –4.46 | –30.1 | Asp760, Asp618, His810, and Ser814 |
| 6 | 503535 | IPC | 379.5 | –4.12 | –16.2 | Asp761 and Trp617 |
| 7 | GS-461203 | sofosbuvir (triphosphate) | 500.2 | –4.04 | –29.7 | Ser759, Asp960 and Lys798 |
| 8 | 16074785 | lead (optimized) | 478.8 | –6.5 | –30.7 | Asp761, Ala762 and Trp617 |
Binding energy represented as “ΔG_binding”.
Based on the molecular interactions and docking scores, we attempted to optimize the top three candidate molecules in order to achieve a stronger binding affinity. On computationally mutating the small molecule JPC (“3-[(2,4-dichlorobenzoyl) (isopropyl)amino]-5-phenylthiophene-2-carboxylic acid”) to “3-[(2-bromo,4-chlorobenzoyl)(isopropyl)amino]-5-phenylthiophene-2-carboxylic acid” (2D structure presented in Figure 4A, left panel), a significant rise in the docking affinity was observed (Figure 4A, right panel). The interactions and the docking properties for this lead optimized molecule are presented in Figure 4B and Table 1 (Row 8), respectively. The detailed view of the interactions for this molecule is presented in Figure 4C.
Figure 4.

Molecular docking of the lead-optimized molecule onto the predicted GTP site and its molecular interactions with the RdRp amino acid residues. (A) Computationally directed binding of the lead-optimized molecule (2D structure in the left panel) at the GTP binding sites (right panel) (blue indicates positively charged regions, red indicates negatively charged regions, green indicates neutral regions, and gray indicates regions beyond the GTP binding pocket). (B) Molecular interactions of the lead optimized molecule with the amino acid residues at the GTP binding sites. (C) Detailed view of the interactions between the amino acid residues lining the GTP binding pocket and the lead optimized molecule (please refer figure for details of the representation format).
The ADME/T (absorption, distribution, metabolism, elimination, or toxicity) property values of all the eight molecules satisfied the acceptable ranges in terms of Lipinski’s rule of five46 and other pharmacokinetic parameters such as probable H-bonding atoms, human oral absorption (<25%: poor; >above 80%: good), and the IC50 values for blockage of hERG (human ether-a-go-go-related gene) K+ channels (<−5: satisfactory). Interestingly, the lead optimized molecule is predicted to have 94.7% of oral absorption and its hERG value evidenced the degree of drug likeliness. The detailed results of this analysis are listed in Table 2.
Table 2. Drug Likeliness Properties of the Small Molecules Predicted to Bind the GTP Binding Site of CoV2-RdRp.
| ligand name | H-bond donor | H-bond acceptor | % of human oral absorption | QP log hERG | Lipinski rule of 5 | |
|---|---|---|---|---|---|---|
| 1 | FIH | 3 | 8.4 | 91.496 | –4.195 | 0 |
| 2 | CCT | 3 | 9.9 | 76.146 | –4.316 | 0 |
| 3 | JPC | 3 | 7.6 | 91.313 | –4.431 | 0 |
| 4 | remdesivir | 5 | 16.6 | 35.214 | –5.655 | 2 |
| 5 | SNH | 3 | 10.9 | 77.260 | –4.317 | 0 |
| 6 | sofosbuvir | 3 | 14.9 | 52.851 | –5.422 | 2 |
| 7 | IPC | 2 | 6.9 | 100.000 | –2.968 | 0 |
| 8 | lead (optimized) | 3 | 7.6 | 94.742 | –5 | 0 |
Molecular Dynamics simulation studies of the native and ligand-bound complexes of CoV2-RdRp
MD (Molecular dynamics) simulations were performed for the modelled structure of the RdRp protein and docked complexes for the GTP, lead optimized, and FIH compounds for a 50 ns time period. Here, the GTP molecule was considered for the simulation studies because it plays a vital role in the RdRp functional mechanism; thereby, the structural stability and potential interactions of the GTP in the active-site pocket of the RdRp protein can be understood. Additionally, the complexes of the top scored ligand molecules (lead optimized and FIH) were considered for understanding the structural stability and potential interacting residues for the inhibitory mechanism of the complexes. In MD simulation studies, rmsd and RMSF (root mean square fluctuation) are the indicators for analyzing the stability of the complexes. The rmsd, RMSF, SASA (solvent accessible surface area), and H-bonding analysis results are presented in Figure 5 and Table 3. A comparison of the simulation statistics for the native and GTP-bound RdRp suggests that while the GTP-bound complex initially undergoes higher deviations and fluctuation, the complex gets stabilized over the course of the simulation. Additionally, there is a significant rise in the accessible surface area of the GTP-bound complex, suggesting that the RdRp assumes an open conformation following the binding of GTP, possible assuming a preinitiation state in order to accommodate the incoming template and/or the primer RNA molecule. Interestingly, all the phosphate groups of the GTP molecule displayed conformational changes during the simulation and the first 50 residues at the N-terminal from the palm subdomain were found relaxed. Also, in the first 5 ns, the GTP exhibited approximately 15 H-bond interactions within the binding site, which got stabilized to 8 H-bond interactions throughout the simulation period.
Figure 5.

MD simulations for the native RdRp and its GTP-bound, FIH-bound, and the lead optimized compound-bound complexes. (A) Comparative rmsd plots of the native and ligand-bound complexes for a 50 ns time period. (B) Comparative RMSF plots of the native and ligand-bound complexes for a 50 ns time period. (C) Comparative SASA profiles of the native and ligand-bound complexes. (D) Comparative H-bond interactions of the GTP, lead optimized, and FIH compound complexes for 50 ns. (black curve—FIH; red curve—GTP; green curve—lead compound; and blue curve—native protein).
Table 3. Comparison of the MD Simulation Statistics of the Native CoV2-RdRp and Small Molecule-Bound CoV2-RdRp Complexes for 50 ns.
| NAME | RMSD (Å) | RMSF (Å) | SASA (Å2) | H-bonds |
|---|---|---|---|---|
| native RdRp | 7.5−10 | aa1-aa50: 10–17.5 remaining aa: <10 | 23,000–26,000 | NA |
| RdRp-GTP | 5−12.5 | aa1-aa50: 10–20 aa700-aa800: 5 | 40,000–48,000 | 15 (0–5 ns); 8 (6–50 ns) |
| RdRp-FIH | 5 | aa1-aa50: 5 remaining aa: <2.5 | 42,000–45,000 | 4 (0–50 ns) |
| RdRp-lead | 5−10 | aa1-aa50: 10 remaining aa: <7.5 | 41,000–44,000 | 5 (0–50 ns) |
The complexes of RdRp with both FIH and the lead optimized compound exhibited significantly lesser deviations and fluctuations along with a relative decrease in the solvent accessibility as compared to the GTP complex. This suggests stronger competitive binding of the inhibitor molecules along with the generation of a possible half-closed RdRp conformation, indicating the inhibitory potential of these molecules. Interestingly, the FIH complex showed relatively lesser fluctuations and deviations as those of the lead molecule complex, suggesting that in a physiological state, the FIH molecule may exhibit stronger inhibition. However, the lead molecule exhibited higher H-bond interaction as compared to FIH. Of note, in the rmsd plot of the lead molecule—RdRp complex, an abrupt deviation is observed between the 15 and 25 ns time period and the remaining periods are highly stable. During this sudden deviation, the N-terminal region (50 residues) was found relaxed from the palm subdomain at 15 ns, after which the region again tends toward to the subdomain and stabilizes the complex during the rest of the cycle. These suggest significant conformational changes following the binding of the molecule, possibly causing the RdRp molecule to assume a closed state.
Density Functional Theory Studies of the FIH and Lead Optimized Compound
The DFT (density functional theory) analysis investigates the electronic features of the atoms in a charge transfer reaction, using the HOMO (highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital), and MESP (molecular electrostatic surface potential) analyses. This provides key information about the global and local indices of the compounds that help to understand its chemical reactivity. The electron donor/acceptor properties of the molecules are indicated by the distribution of frontier molecular orbitals. The DFT study was performed for the top two docking scored molecules (FIH and lead optimized compound). These two molecules show low HOMO–LUMO energy gaps, signifying higher stability and chemical reactivity of the molecules. The calculated stereoelectronic properties and profiles of these compounds are presented in Table 4 and the HOMO, LUMO, and MESP contour maps are shown in Figure S9. In FIH, the HOMO regions are localized in the thiophene-2-carboxaldehyde group (energy value −0.22 eV), while the LUMO regions are localized at the sulfonyl amino group (energy value −0.02 eV). Together, the higher stability of the energy value of the HOMO as compared to the LUMO suggests that the compound may favor a nucleophilic reaction. The calculated HOMO–LUMO energy of the gap of the compound is −0.20 eV. The electrostatic charge surface potential analysis revealed that highly negatively charged surfaces are present around the molecule, while positively charged surfaces are found in the sulfonyl and −OH group. For the lead molecule, the HOMO regions are present in the sulfur and hydrogen atom of thiophene group and the LUMO regions localize in the hydroxyl group of the benzoyl ring. The calculated HOMO and LUMO energies of the compound are −0.24 and −0.002 eV, respectively, suggesting that the compound may favor the nucleophilic reaction. The charged surface density of the lead molecule is extremely low with only the negatively charged surface observed in the molecule. The calculated energy gap of the lead molecule (−0.22 eV) is higher than that of the FIH molecule (−0.20 eV), suggesting that the FIH molecule may demonstrate a higher inhibitory reaction potential under physiological conditions.
Table 4. Molecular Electronic Features of the FIH and Lead Optimized Compounds.
| S. no. | compound name | HOMO (eV) | LUMO (eV) | EHOMO – ELUMO (eV) |
|---|---|---|---|---|
| 1 | FIH | –0.22 | 0.02 | –0.20 |
| 2 | lead optimized | –0.24 | –0.00 | –0.24 |
Conclusions
The urgency arising from the current COVID-19 pandemic not only entails major research initiatives on the development of novel antiviral molecules but also drives the repurposing of the available drugs and inhibitors reported in previous studies, particularly on (+) ss-RNA viruses. This study reports the probable GTP binding site in the active site region between the finger and palm subdomains of the CoV2-RdRp. Comparison with the previously reported RdRp structures from RNA viruses shows the conservation of this GTP binding pocket and moreover suggests its close proximity to the RNA template and the RNA primer. The RdRp enzymes are known to function in both primer-dependent and -independent manner. This possibly indicates the dual function of the predicted GTP binding site. In the case of a de novo initiation of the RNA replication process, this site could possibly incorporate the first and the second nucleotide, GTP being the second nucleotide, as experimentally demonstrated in a previous study.20 Alternatively, in the case of a primer-assisted initiation process, this site can potentially function as a nucleotide-binding site for an incoming nucleotide prior to its addition to the growing RNA chain, as evident from the proximity of the GTP to the primer RNA. Thus, theoretically, the inhibition of this site by small molecules could effectively dampen both modes of initiation, thus rendering the RdRp enzyme dysfunctional. In addition, this study suggests that the HCV-RdRp inhibitors reported in previous studies are likely to bind the CoV2-RdRp enzyme at the predicted GTP binding site. Based on the predicted interactions, this study also reports a lead optimized molecule exhibiting a higher binding affinity and drug likeliness. Biochemical experiments need to be carried out to examine the in vitro inhibitory potential of these molecules against CoV2-RdRp.
Computational Methods
Protein Structure Prediction
The primary amino acid sequence of CoV2-RdRp was retrieved from the NCBI database (accession Y_009725307). A BLASTp search was performed against the PDB database to identify the structural homologues of CoV2-RdRp. The protein structure for RdRp-CoV1 (PDB ID: 6NUR) was considered as the template for homology modelling to building the CoV2-RdRp structure using the I-TASSER server.47 The structure generated was used for further studies.
In Silico Protein and Ligand Preparation
The modeled structure of CoV2-RdRp was further improved using protein preparation wizard (OPLS-2015) incorporated in “Schrodinger Suite 2019”48 to improve the quality of the structure by adding hydrogen atoms and hydrogen bonds and removal of the steric clashes. This minimized model of the CoV2-RdRp structure was used in further analysis. The four potential inhibitors of HCV-RdRp along with the known inhibitors of antiviral drugs remdesivir and sofosbuvir were selected for interaction studies with CoV2-RdRp. The HCV-RdRp inhibitors (JPC, CCT, SNH, IPC, and FIH) were retrieved from the reported crystal structures (PDBs ID: 1YVZ, 1YVX, 2D41, 1D3Z), while structures for the triphosphate forms of remdesivir (ID: DBMET03038) and sofosbuvir (ID: GS-461203) were retrieved from DrugBank. All the inhibitors were prepared by assigning appropriate geometries, bond orders, tautomers, and ionization states using the LigPrep module implemented in “Schrodinger Suite 2019”.
GTP Binding Site Prediction and Docking Analysis
The minimized model was used to predict the GTP binding site using the SiteMap module in the “Schrodinger Suite 2019”. In order to verify the accuracy of the site, the GTP binding site was regenerated using the Phyre server,49 CASTp server,50 and the results were compared. The results predicted the near-identical sites in the core region of CoV2-RdRp. This site was then used to generate a grid in order to dock the inhibitors. The prepared inhibitors were docked within the GTP binding site pocket of the CoV2-RdRp protein using the Glide XP (extra Precision) module implemented in “Schrodinger Suite 2019”.
Molecular Mechanics Generalized Born Surface Area Calculation
The binding free energies of the enzyme–inhibitor complexes were analyzed by molecular mechanics generalized Born surface area calculation.51 The calculation was performed on the basis of the following equation
where ΔEMM represents the minimized molecular energy changes in the gas phase, GSGB represents the surface calculation using the GB model, and GSA represents the accessible surface area of the complexes.
Lead Optimization
Lead optimization is an important method in the drug development process to improve the potency and pharmacokinetic properties of the drug molecule.52 Here, lead optimization was performed against the JPC compound using “Schrodinger Suite 2019”. The JPC compound has two chlorine atoms in its benzoyl ring. In order to enhance the potency of the molecule, we modified the molecule with different combinations of fluorine (F), bromine (Br), and chlorine (Cl) atoms at the second and fourth position of its benzoyl ring. The modified lead molecules were analyzed for their docking and ADME/T properties.
ADME/T Property Prediction
The seven inhibitors were analyzed for their drug likeliness and pharmacokinetic properties such as ADME/T.53 The QikProp module implemented in the “Schrodinger Suite 2019” was used to estimate the drug likeliness of the inhibitors using Lipinski’s rule of five46 and other pharmacokinetic parameters such as probable H-bonding atoms, human oral absorption (<25%: poor; >above 80%: good) and the IC50 values for blockage of human ether-a-go-go-related gene (hERG) K+ channels (<−5: satisfactory).54
MD Simulation Studies
The structural stability of native and inhibitor-bound complexes of SARS-CoV2-RdRp was analyzed using the GROMACS (Groningen Machine for Chemical Simulation)55 software for a 50 ns time period. The Gromos56a force field with its default parameters was applied to the protein and inhibitor complex(es). The inhibitor topology was refined and generated using the PRODRG server56 and incorporated into the structure for understanding the dynamics and behavior of the complex. A cubic box of 10 Å dimension was set with SPC216 (simple point charge) water molecules and the periodic boundary condition was applied in the all directions. Further, the system was neutralized by including 12 Na2+ ions in the native and complex system(s). Energy minimization and equilibration are important steps in the generation of the MD. The steepest decent (SD) algorithm was performed for 50,000 steps with a tolerance of 1000 kJ/mol to minimize the system. The thermostat and barostat algorithms such as NVT [canonical ensemble-substance (N), volume (V), and temperature (T)] and NPT [isothermal isobaric-substance (N), pressure (P), and temperature (T)] ensembles were used. The linear constraint solver (LINCS) and particle mesh Ewald (PME) method was used to constraint the covalent bonds and long-range electrostatic interactions of the systems, respectively. The temperature (310 K) and the pressure (1 atm) of the systems were equilibrated by the V-rescale coupling and Parrinello–Rahman (PR) method. The preprocessed systems were used to perform the MD simulations for a 50 ns time period.
DFT Calculation
In quantum mechanical calculation, DFT analysis determines the molecular electronic features such as electron density, frontier molecular orbital (HOMO and LUMO) density, and molecular electrostatic map to predict the chemical reactivity of proposed inhibitors.57,58 The DFT study was carried out for the top two predicted inhibitors in order to understand the inhibitory reaction against the SARS-CoV2-RdRp. Geometry optimization of the compound was performed using a hybrid DFT approach at B3LYP (Becke’s three-parameter exchange potential and the Lee–Yang–Parr correlation functional) with the 6-31G** basis set.59,60 The Poisson–Boltzmann solver was used to calculate the energy under aqueous conditions, simulating physiological conditions, which provides the information about the global and local indices of the molecules to their chemical activity. The spatial distributions of electronic features in the charge transfer mechanism were understood from the HOMO and LUMO molecular orbitals. All DFT calculations were carried out using Jaguar, version 9.1.61,62
Acknowledgments
This work was supported by the core funding from the National Institute of Immunology, New Delhi, India. J.J. gratefully acknowledges the MHRD-RUSA 2.0 [F.24/51/2014-U, Policy (TNMulti-Gen), Dept. of Edn., Govt. of India] for the infrastructure facilities provided to the Department of Bioinformatics, Alagappa University.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02096.
Prediction of small-molecule inhibitors targeting the SARS-CoV2 RdRp (PDF)
Author Contributions
M.A., A.D., and R.M. have contributed equally to the work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Huang C.; Wang Y.; Li X.; Ren L.; Zhao J.; Hu Y.; Zhang L.; Fan G.; Xu J.; Gu X.; Cheng Z.; Yu T.; Xia J.; Wei Y.; Wu W.; Xie X.; Yin W.; Li H.; Liu M.; Xiao Y.; Gao H.; Guo L.; Xie J.; Wang G.; Jiang R.; Gao Z.; Jin Q.; Wang J.; Cao B. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO . Coronavirus Disease 2019 (COVID-19): Situation Report—134, 2020.
- Corman V. M.; Muth D.; Niemeyer D.; Drosten C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Virus Res. 2018, 100, 163–188. 10.1016/bs.aivir.2018.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen K. G.; Rambaut A.; Lipkin W. I.; Holmes E. C.; Garry R. F. The Proximal Origin of SARS-CoV-2. Nat. Med. 2020, 26, 450. 10.1038/s41591-020-0820-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y.-R.; Cao Q.-D.; Hong Z.-S.; Tan Y.-Y.; Chen S.-D.; Jin H.-J.; Tan K.-S.; Wang D.-Y.; Yan Y. The Origin, Transmission and Clinical Therapies on Coronavirus Disease 2019 (COVID-19) Outbreak – an Update on the Status. Mil. Med. Res. 2020, 7, 11. 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah R.; Rodriguez-Morales A. J.; Jha R.; Chu D. K. W.; Gu H.; Peiris M.; Bastola A.; Lal B. K.; Ojha H. C.; Rabaan A. A.; Zambrano L. I.; Costello A.; Morita K.; Pandey B. D.; Poon L. L. M. Complete Genome Sequence of a 2019 Novel Coronavirus (SARS-CoV-2) Strain Isolated in Nepal. Microbiol. Resour. Announce. 2020, 9, e00169–20. 10.1128/MRA.00169-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F.; Deng Y.; Li W. Coronavirus Disease 2019 (COVID-19): What We Know?. J. Med. Virol. 2020, 92, 719. 10.1002/jmv.25766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman S.; Prasad B.; Selvarajan R. RNA Dependent RNA Polymerases: Insights from Structure, Function and Evolution. Viruses 2018, 10, 76. 10.3390/v10020076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann K. C.; Gulyaeva A.; Zevenhoven-Dobbe J. C.; Janssen G. M. C.; Ruben M.; Overkleeft H. S.; van Veelen P. A.; Samborskiy D. V.; Kravchenko A. A.; Leontovich A. M.; Sidorov I. A.; Snijder E. J.; Posthuma C. C.; Gorbalenya A. E. Discovery of an Essential Nucleotidylating Activity Associated with a Newly Delineated Conserved Domain in the RNA Polymerase-Containing Protein of All Nidoviruses. Nucleic Acids Res. 2015, 43, 8416–8434. 10.1093/nar/gkv838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posthuma C. C.; te Velthuis A. J. W.; Snijder E. J. Nidovirus RNA Polymerases: Complex Enzymes Handling Exceptional RNA Genomes. Virus Res. 2017, 234, 58–73. 10.1016/j.virusres.2017.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Yan L.; Huang Y.; Liu F.; Zhao Y.; Cao L.; Wang T.; Sun Q.; Ming Z.; Zhang L.; Ge J.; Zheng L.; Zhang Y.; Wang H.; Zhu Y.; Zhu C.; Hu T.; Hua T.; Zhang B.; Yang X.; Li J.; Yang H.; Liu Z.; Xu W.; Guddat L. W.; Wang Q.; Lou Z.; Rao Z. Structure of the RNA-Dependent RNA Polymerase from COVID-19 Virus. Science 2020, 368, 779. 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subissi L.; Posthuma C. C.; Collet A.; Zevenhoven-Dobbe J. C.; Gorbalenya A. E.; Decroly E.; Snijder E. J.; Canard B.; Imbert I. One Severe Acute Respiratory Syndrome Coronavirus Protein Complex Integrates Processive RNA Polymerase and Exonuclease Activities. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, E3900–E3909. 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng K. K.-S.; Arnold J. J.; Cameron C. E.. Structure–Function Relationships among RNA-Dependent RNA Polymerases. RNA Interference; Current Topics in Microbiology and Immunology; Springer, 2008; pp 137–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C. C.; Singh P.; Ecker D. J. J. De Novo Initiation of Viral RNA-Dependent RNA Synthesis. Virology 2001, 287, 251–260. 10.1006/viro.2001.1039. [DOI] [PubMed] [Google Scholar]
- van Dijk A. A.; Makeyev E. V.; Bamford D. H. Initiation of Viral RNA-Dependent RNA Polymerization. J. Gen. Virol. 2004, 85, 1077–1093. 10.1099/vir.0.19731-0. [DOI] [PubMed] [Google Scholar]
- Snijder E. J.; Decroly E.; Ziebuhr J. The Nonstructural Proteins Directing Coronavirus RNA Synthesis and Processing. Adv. Virus Res. 2016, 96, 59–126. 10.1016/bs.aivir.2016.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Velthuis A. J. W.; Arnold J. J.; Cameron C. E.; van den Worm S. H. E.; Snijder E. J. The RNA Polymerase Activity of SARS-Coronavirus Nsp12 Is Primer Dependent. Nucleic Acids Res. 2009, 38, 203–214. 10.1093/nar/gkp904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerens N.; Selisko B.; Ricagno S.; Imbert I.; van der Zanden L.; Snijder E. J.; Canard B. De Novo Initiation of RNA Synthesis by the Arterivirus RNA-Dependent RNA Polymerase. J. Virol. 2007, 81, 8384–8395. 10.1128/jvi.00564-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Te Velthuis A. J. W.; Van Den Worm S. H. E.; Snijder E. J. The SARS-Coronavirus Nsp7+nsp8 Complex Is a Unique Multimeric RNA Polymerase Capable of Both de Novo Initiation and Primer Extension. Nucleic Acids Res. 2012, 40, 1737–1747. 10.1093/nar/gkr893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi K. H.; Groarke J. M.; Young D. C.; Kuhn R. J.; Smith J. L.; Pevear D. C.; Rossmann M. G. The Structure of the RNA-Dependent RNA Polymerase from Bovine Viral Diarrhea Virus Establishes the Role of GTP in de Novo Initiation. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 4425–4430. 10.1073/pnas.0400660101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y.; Sun F.; Li X.; Pang H.; Xu X.; Bartlam M.; Rao Z. Insights into SARS-CoV Transcription and Replication from the Structure of the Nsp7-Nsp8 Hexadecamer. Nat. Struct. Mol. Biol. 2005, 12, 980–986. 10.1038/nsmb999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y.; Ma Q.; Restle T.; Shang W.; Svergun D. I.; Ponnusamy R.; Sczakiel G.; Hilgenfeld R. Nonstructural Proteins 7 and 8 of Feline Coronavirus Form a 2:1 Heterotrimer That Exhibits Primer-Independent RNA Polymerase Activity. J. Virol. 2012, 86, 4444–4454. 10.1128/jvi.06635-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchdoerfer R. N.; Ward A. B. Structure of the SARS-CoV Nsp12 Polymerase Bound to Nsp7 and Nsp8 Co-Factors. Nat. Commun. 2019, 10, 2342. 10.1038/s41467-019-10280-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap T. L.; Xu T.; Chen Y.-L.; Malet H.; Egloff M.-P.; Canard B.; Vasudevan S. G.; Lescar J. Crystal Structure of the Dengue Virus RNA-Dependent RNA Polymerase Catalytic Domain at 1.85-Angstrom Resolution. J. Virol. 2007, 81, 4753–4765. 10.1128/jvi.02283-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesburg C. A.; Cable M. B.; Ferrari E.; Hong Z.; Mannarino A. F.; Weber P. C. Crystal Structure of the RNA-Dependent RNA Polymerase from Hepatitis C Virus Reveals a Fully Encircled Active Site. Nat. Struct. Biol. 1999, 6, 937–943. 10.1038/13305. [DOI] [PubMed] [Google Scholar]
- Biswal B. K.; Wang M.; Cherney M. M.; Chan L.; Yannopoulos C. G.; Bilimoria D.; Bedard J.; James M. N. G. Non-Nucleoside Inhibitors Binding to Hepatitis C Virus NS5B Polymerase Reveal a Novel Mechanism of Inhibition. J. Mol. Biol. 2006, 361, 33–45. 10.1016/j.jmb.2006.05.074. [DOI] [PubMed] [Google Scholar]
- Biswal B. K.; Cherney M. M.; Wang M.; Chan L.; Yannopoulos C. G.; Bilimoria D.; Nicolas O.; Bedard J.; James M. N. G. Crystal Structures of the RNA-Dependent RNA Polymerase Genotype 2a of Hepatitis C Virus Reveal Two Conformations and Suggest Mechanisms of Inhibition by Non-Nucleoside Inhibitors. J. Biol. Chem. 2005, 280, 18202–18210. 10.1074/jbc.M413410200. [DOI] [PubMed] [Google Scholar]
- Gong P.; Peersen O. B. Structural Basis for Active Site Closure by the Poliovirus RNA-Dependent RNA Polymerase. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 22505–22510. 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjith-Kumar C. T.; Gutshall L.; Kim M.-J.; Sarisky R. T.; Kao C. C. Requirements for De Novo Initiation of RNA Synthesis by Recombinant Flaviviral RNA-Dependent RNA Polymerases. J. Virol. 2002, 76, 12526–12536. 10.1128/jvi.76.24.12526-12536.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G.; Hamatake R. K.; Mathis D. M.; Racela J.; Rigat K. L.; Lemm J.; Colonno R. J. De Novo Initiation of RNA Synthesis by the RNA-Dependent RNA Polymerase (NS5B) of Hepatitis C Virus. J. Virol. 2000, 74, 851–863. 10.1128/jvi.74.2.851-863.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Lou Z.; Miao Y.; Yu Y.; Dong H.; Peng W.; Bartlam M.; Li X.; Rao Z. Structures of EV71 RNA-Dependent RNA Polymerase in Complex with Substrate and Analogue Provide a Drug Target against the Hand-Foot-and-Mouth Disease Pandemic in China. Protein Cell 2010, 1, 491–500. 10.1007/s13238-010-0061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A. A.; Peersen O. B. Structural Basis for Proteolysis-Dependent Activation of the Poliovirus RNA-Dependent RNA Polymerase. EMBO J. 2004, 23, 3462–3471. 10.1038/sj.emboj.7600357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y.; Farsetta D. L.; Nibert M. L.; Harrison S. C. RNA Synthesis in a Cage - Structural Studies of Reovirus Polymerase Λ3. Cell 2002, 111, 733–745. 10.1016/S0092-8674(02)01110-8. [DOI] [PubMed] [Google Scholar]
- Surana P.; Satchidanandam V.; Nair D. T. RNA-Dependent RNA Polymerase of Japanese Encephalitis Virus Binds the Initiator Nucleotide GTP to Form a Mechanistically Important Pre-Initiation State. Nucleic Acids Res. 2014, 42, 2758–2773. 10.1093/nar/gkt1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y.; Zhang Y.; Zhou K.; Sun J.; Zhou Z. H. Conservative Transcription in Three Steps Visualized in a Double-Stranded RNA Virus. Nat. Struct. Mol. Biol. 2019, 26, 1023–1034. 10.1038/s41594-019-0320-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butcher S. J.; Grimes J. M.; Makeyev E. V.; Bamford D. H.; Stuart D. I. A Mechanism for Initiating RNA-Dependent RNA Polymerization. Nature 2001, 410, 235–240. 10.1038/35065653. [DOI] [PubMed] [Google Scholar]
- Yin W.; Mao C.; Luan X.; Shen D.-D.; Shen Q.; Su H.; Wang X.; Zhou F.; Zhao W.; Gao M.; Chang S.; Xie Y.-C.; Tian G.; Jiang H.-W.; Tao S.-C.; Shen J.; Jiang Y.; Jiang H.; Xu Y.; Zhang S.; Zhang Y.; Xu H. E. Structural Basis for the Inhibition of the RNA-Dependent RNA Polymerase from SARS- CoV-2 by Remdesivir. Science 2020, 368, 1499. 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powdrill M. H.; Deval J.; Narjes F.; De Francesco R.; Götte M. Mechanism of Hepatitis C Virus RNA Polymerase Inhibition with Dihydroxypyrimidines. Antimicrob. Agents Chemother. 2010, 54, 977–983. 10.1128/AAC.01216-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Ng K. K.-S.; Cherney M. M.; Chan L.; Yannopoulos C. G.; Bedard J.; Morin N.; Nguyen-Ba N.; Alaoui-Ismaili M. H.; Bethell R. C.; James M. N. G. Non-nucleoside Analogue Inhibitors Bind to an Allosteric Site on HCV NS5B Polymerase. J. Biol. Chem. 2003, 278, 9489–9495. 10.1074/jbc.M209397200. [DOI] [PubMed] [Google Scholar]
- Hirashima S.; Suzuki T.; Ishida T.; Noji S.; Yata S.; Ando I.; Komatsu M.; Ikeda S.; Hashimoto H. Benzimidazole Derivatives Bearing Substituted Biphenyls as Hepatitis C Virus NS5B RNA-Dependent RNA Polymerase Inhibitors: Structure-Activity Relationship Studies and Identification of a Potent and Highly Selective Inhibitor JTK-109. J. Med. Chem. 2006, 49, 4721–4736. 10.1021/jm060269e. [DOI] [PubMed] [Google Scholar]
- Ahmed-Belkacem A.; Guichou J.-F.; Brillet R.; Ahnou N.; Hernandez E.; Pallier C.; Pawlotsky J.-M. Inhibition of RNA Binding to Hepatitis C Virus RNA-Dependent RNA Polymerase: A New Mechanism for Antiviral Intervention. Nucleic Acids Res. 2014, 42, 9399–9409. 10.1093/nar/gku632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Leveque V.; Ma H.; Johnson K. A.; Klumpp K. NTP-Mediated Nucleotide Excision Activity of Hepatitis C Virus RNA-Dependent RNA Polymerase. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, E348. 10.1073/pnas.1214924110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Deore R.; W Chern J. J. NS5B RNA Dependent RNA Polymerase Inhibitors: The Promising Approach to Treat Hepatitis C Virus Infections. Curr. Med. Chem. 2010, 17, 3806–3826. 10.2174/092986710793205471. [DOI] [PubMed] [Google Scholar]
- Stedman C. Sofosbuvir, a NS5B Polymerase Inhibitor in the Treatment of Hepatitis C: A Review of Its Clinical Potential. Ther. Adv. Gastroenterol. 2014, 7, 131–140. 10.1177/1756283X13515825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressanelli S.; Tomei L.; Rey F. A.; De Francesco R. Structural Analysis of the Hepatitis C Virus RNA Polymerase in Complex with Ribonucleotides. J. Virol. 2002, 76, 3482–3492. 10.1128/jvi.76.7.3482-3492.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski C. A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discovery Today: Technol. 2004, 1, 337–341. 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Yang J.; Zhang Y. I-TASSER Server: New Development for Protein Structure and Function Predictions. Nucleic Acids Res. 2015, 43, W174–W181. 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manual User. Schrödinger Release 2019-3: Glide; Schrödinger, LLC: New York, NY, 2019. Schrödinger Release 2018-3 LigPrep, Schrödinger, LLC, New York, NY, 2018. 2018.
- Kelley L. A.; Mezulis S.; Yates C. M.; Wass M. N.; Sternberg M. J. E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian W.; Chen C.; Lei X.; Zhao J.; Liang J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffucci I.; Contini A. Improved Computation of Protein-Protein Relative Binding Energies with the Nwat-MMGBSA Method. J. Chem. Inf. Model. 2016, 56, 1692–1704. 10.1021/acs.jcim.6b00196. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L. Efficient Drug Lead Discovery and Optimization. Acc. Chem. Res. 2009, 42, 724–733. 10.1021/ar800236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butina D.; Segall M. D.; Frankcombe K. Predicting ADME Properties in Silico: Methods and Models. Drug Discovery Today 2002, 7, S83. 10.1016/S1359-6446(02)02288-2. [DOI] [PubMed] [Google Scholar]
- Sato T.; Yuki H.; Ogura K.; Honma T. Construction of an Integrated Database for HERG Blocking Small Molecules. PLoS One 2018, 13, e0199348. 10.1371/journal.pone.0199348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindahl E.Gromacs Workshop Introduction to Molecular Simulation and Gromacs; Springer, 2007; pp 1–25.
- Schüttelkopf A. W.; Van Aalten D. M. F. PRODRG: A Tool for High-Throughput Crystallography of Protein-Ligand Complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- Min M.; Xingjun J.; Xueding W.; Hao Z.; Weiqing Y.; Yuanyuan Z.; Changrong P.; Zicheng L.; Jing Y.; Quan D.; Menglin M. Synthesis and Quantitative Structure-Activity Relationships Study for Arylpropenamide Derivatives as Inhibitors of Hepatitis B Virus Replication. Chem. Biol. Drug Des. 2016, 88, 451–459. 10.1111/cbdd.12774. [DOI] [PubMed] [Google Scholar]
- Mariadasse R.; Choubey S. K.; Jeyakanthan J. Insights into Exogenous Tryptophan-Mediated Allosteric Communication and Helical Transition of TRP Protein for Transcription Regulation. J. Chem. Inf. Model. 2020, 60, 175–191. 10.1021/acs.jcim.9b00755. [DOI] [PubMed] [Google Scholar]
- Bielecki J.; Lipiec E. Basis Set Dependence Using DFT/B3LYP Calculations to Model the Raman Spectrum of Thymine. J. Bioinf. Comput. Biol. 2016, 14, 1650002. 10.1142/S0219720016500025. [DOI] [PubMed] [Google Scholar]
- Choubey S. K.; Mariadasse R.; Rajendran S.; Jeyaraman J. Identification of Novel Histone Deacetylase 1 Inhibitors by Combined Pharmacophore Modeling, 3D-QSAR Analysis, in Silico Screening and Density Functional Theory (DFT) Approaches. J. Mol. Struct. 2016, 1125, 391–404. 10.1016/j.molstruc.2016.06.082. [DOI] [Google Scholar]
- Bochevarov A. D.; Harder E.; Hughes T. F.; Greenwood J. R.; Braden D. A.; Philipp D. M.; Rinaldo D.; Halls M. D.; Zhang J.; Friesner R. A. Jaguar: A High-Performance Quantum Chemistry Software Program with Strengths in Life and Materials Sciences. Int. J. Quantum Chem. 2013, 113, 2110–2142. 10.1002/qua.24481. [DOI] [Google Scholar]
- Maniyazagan M.; Rameshwaran C.; Mariadasse R.; Jeyakanthan J.; Premkumar K.; Stalin T. Fluorescence Sensor for Hg2+ and Fe3+ Ions Using 3,3″–Dihydroxybenzidine: Α–Cyclodextrin Supramolecular Complex: Characterization, in-Silico and Cell Imaging Study. Sens. Actuators, B 2017, 242, 1227–1238. 10.1016/j.snb.2016.09.093. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.