

Abstract
The STING pathway has been proposed as a key regulator of gastrointestinal homeostasis and inflammatory responses. Although STING reportedly protects against gut barrier damage and graft-versus-host disease (GVHD) after MHC-mismatched aHSCT, its effect in clinically relevant MHC-matched allogeneic hematopoietic stem cell transplantation (aHSCT) is unknown. Studies here demonstrate that STING signaling in non-hematopoietic cells promoted MHC-matched aHSCT-induced GVHD and STING agonists increased type I interferon and MHC I expression in non-hematopoietic mouse intestinal organoid cultures. Moreover, mice expressing a human STING allele containing three SNPs associated with decreased STING activity also developed reduced MHC-matched GVHD, demonstrating STING’s potential clinical importance. STING−/− recipients experienced reduced GVHD with transplant of purified donor CD8+ T cells in both MHC-matched as well as mismatched models, reconciling the seemingly disparate results. Further examination revealed that STING deficiency reduced the activation of donor CD8+ T cells early post-transplant and promoted recipient MHC class II+ antigen presenting cell (APC) survival. Therefore, APC persistence in STING pathway absence may account for the increased GVHD mediated by CD4+ T cells in completely mismatched recipients. In total, our findings have important implications for regulating clinical GVHD by targeting STING early post-aHSCT and demonstrate that an innate immune pathway has opposing effects on the outcome of aHSCT depending on the donor/recipient MHC disparity.
One Sentence Summary:
STING differentially regulates experimental GVHD mediated by distinct T cell subsets
Introduction
Allogeneic hematopoietic stem cell transplantation (aHSCT) is a potentially curative therapy used to treat hematologic cancers (1). Despite recent advances, including the use of post-transplant cyclophosphamide, GVHD remains a substantial cause of morbidity and mortality in patients receiving aHSCTs (2–5). Pre-transplant chemotherapy / irradiation for conditioning and tumor treatment initiates widespread cell death and release of endogenous danger signals, as well as bacterial translocation due to epithelial barrier dysfunction, promoting the induction of a pro-inflammatory cytokine storm (6). These signals drive the activation of antigen presenting cells (APCs) and the differentiation of allo-reactive donor T cells, leading to damage of particular host tissues characteristic of GVHD (6). However, the innate immune receptors that initiate this process are not well understood.
We posit that stimulator of interferon genes (STING), an innate immune sensor, can contribute to the inflammatory response following conditioning and transplant. Cyclic dinucleotides (CDNs) produced by bacteria or by cyclic GMP-AMP synthase (cGAS) sensing of dsDNA activate STING (7, 8). STING signaling has been implicated in several diseases due to its role in cytoplasmic DNA sensing from both intracellular and other sources, including from the phagocytosis of dying cells (9–12). STING activation leads to the activation of the transcription factors Interferon Regulatory Factor 3 (IRF3) and Nuclear Factor-Kappa B (NF-κB) and the production of type I interferons (IFN) and other pro-inflammatory cytokines including TNF-α and IL-6, respectively (13). Notably, type I IFN can act as a third signal for CD8+ T cells, induce cross-priming by dendritic cells, and increase the expression of major histocompatibility complex (MHC) class I (14–16).
Although STING has been well studied in the context of viral infection and cancer, its role in aHSCT is largely not understood (17, 18). Recently, STING was reported to protect against gastrointestinal GVHD after MHC-mismatched aHSCT (19), however the role of STING in MHC-matched GVHD remains unexplored. Since type I IFN is known to regulate GVHD dependent on the T cell subsets involved (20), we wanted to investigate if STING could control / influence CD8 vs. CD4 mediated GVHD and if subset effects were dependent on the donor / recipient MHC-matched vs mismatched genetic disparity.
Results
The absence of STING in recipient mice reduces GVHD after MHC-matched aHSCT
To investigate if the absence of STING could affect development of GVHD, aHSCTs were performed using two MHC-matched donor/recipient strain combinations. Because lethality is minimal and not characteristic of these MHC-matched models, we do not include overall survival curves. B6-STING−/− (H2b) mice transplanted with bone marrow (BM) and T cells from either LP/J (H2b, unseparated splenocytes containing 0.8×106 T cells) or C3H.SW (H2b, unseparated and pooled splenocytes and peripheral lymph node cells containing 2×106 CD8+ T cells) donor mice exhibited markedly decreased weight loss and reduced clinical scores compared to wild type (WT) B6 recipients (Fig. 1A–C). Notably, the differences observed in clinical scores in the MHC-matched aHSCTs were maintained for the duration of the transplants extending over a 6–9 week period. To assess for a difference in molecules associated with suppressive function, serum was collected from recipients at 6 weeks following transplant. At this time, we found that serum concentrations of IL-27 and IL-10 were consistently and markedly elevated in STING−/− recipients of C3H.SW donor BM and unseparated and pooled splenocytes and peripheral lymph node cells (2×106 CD8+ T cells) compared to WT recipients (Fig. 1D). Elevated concentrations of these suppressive cytokines were concurrent with the markedly diminished clinical GVHD scores in STING−/− recipients at this time post-HSCT.
Fig. 1. Recipient stimulator of interferon genes (STING) deficiency ameliorates graft-versus-host disease (GVHD) after major histocompatibility complex (MHC)-matched allogeneic hematopoietic stem cell transplant (aHSCT).
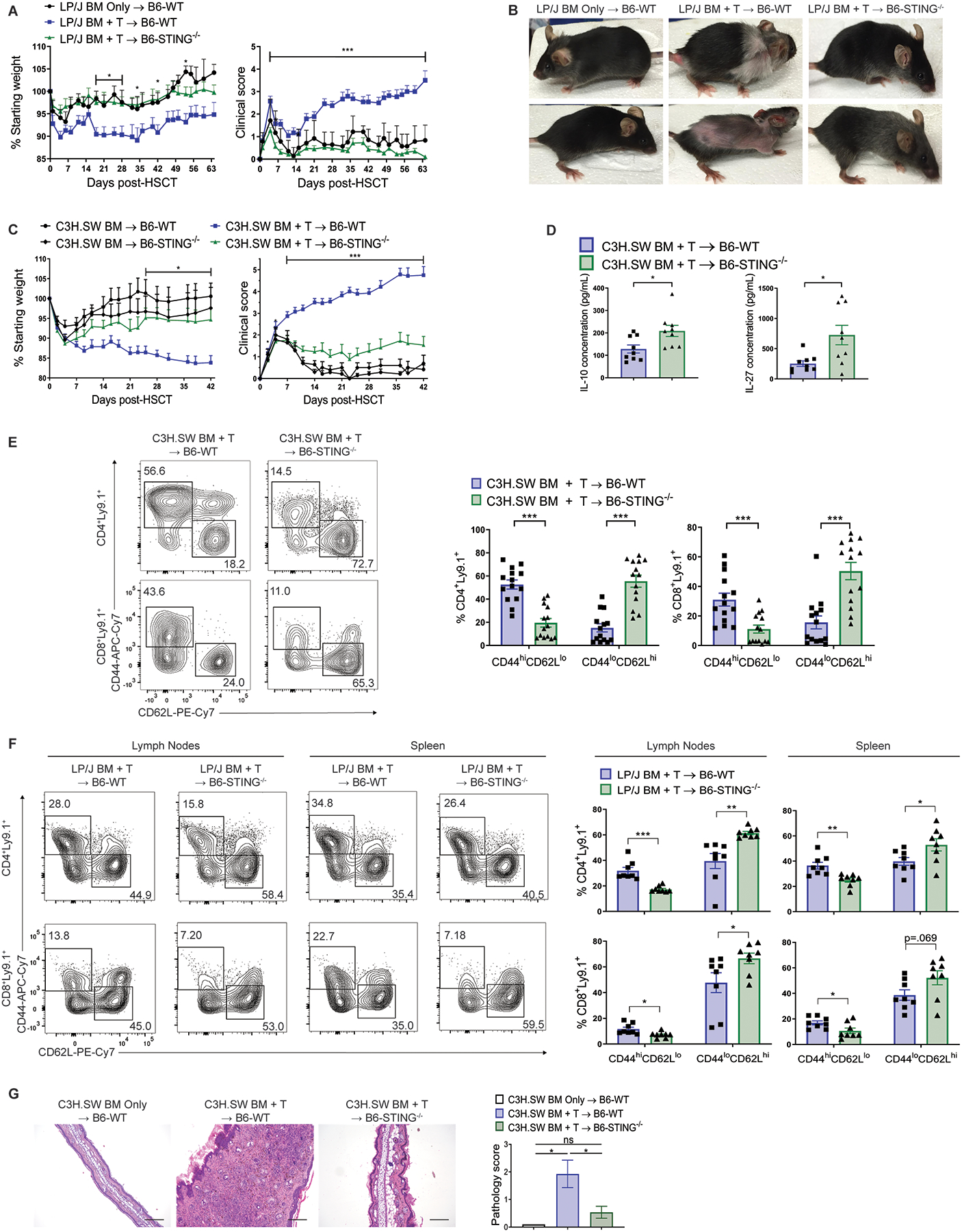
(A) Weight loss and clinical score (B) representative photographs of B6-wild type (WT) and B6-STING−/− recipients after transplant of LP/J bone marrow (BM) and unseparated splenocytes (0.8×106 T cells). Data pooled: 3 experiments (n=7 for WT BM Only, n=14 for WT BM+T, n=15 for STING−/− BM+T). (C) Weight loss and GVHD clinical score of B6-WT or B6-STING−/− recipients after transplant of C3H.SW BM and unseparated and pooled splenocytes and peripheral lymph node cells (2×106 CD8+ T cells). Data pooled: 3 experiments (n=7 for WT BM Only, n=5 for STING−/− BM Only, n=15 each for WT BM+T and STING−/− BM+T). (D) Serum IL-10 and IL-27 concentration from B6-WT or B6-STING−/− recipients 6 weeks after transplant as in (C). Data pooled: 2 experiments (n=9 per group). (E) Representative flow cytometry plots and frequency of donor T cells expressing an effector memory (CD44hiCD62Llo) or naïve (CD44loCD62Lhi) phenotype in B6-WT or B6-STING−/− pooled cervical, axillary, inguinal and mesenteric lymph nodes 6 weeks after transplant as in (C). Data pooled: 3 experiments (n=15 per group). (F) Representative flow cytometry plots and frequency of donor T cells expressing an effector memory or naïve phenotype in B6-WT or B6-STING−/− lymphoid tissues 9 weeks after transplant as in (A). Pooled data: 3 experiments (n=8 per group, representing a total of 15 pooled mice per group). (G) Representative histology and pathology score of ear skin from B6-WT or B6-STING−/− recipients 6 weeks after transplant as in (C). Data pooled: 3 experiments (n=5 for WT BM Only, n=14 for WT BM+T, n=13 for STING−/− BM+T). Scale bars=200μm. GVHD groups compared using two-tailed unpaired t test or one-way ANOVA with Tukey’s multiple comparisons test for multiple groups. * p<0.05, ** p<0.01, *** p<0.001. ns=not significant. Data are means ± SEM.
Donor T cells were examined at different time-points after aHSCT to determine if the absence of recipient STING altered the donor immune phenotype. Peripheral lymphoid tissues from B6-STING−/− recipients 6 weeks after transplant from either donor strain contained greater cellularity, a lower percentage of donor T cells expressing an activated phenotype (CD44hiCD62Llo) and higher frequency of donor T cells with a naïve phenotype (CD44loCD62Lhi) compared to WT recipients, consistent with differences in GVHD between the groups (Fig. 1E and F, fig. S1). Histological analyses of target tissues at that time indicated that skin from STING-deficient recipients had diminished cellular infiltrate, thickening, and overall pathology scores (Fig. 1G). When B6-WT and STING−/− recipients were co-housed for two weeks prior to transplant with C3H.SW BM and T cells, STING-deficient recipients still exhibited reduced weight loss and clinical scores, as well as similar lymphoid phenotypes five weeks after aHSCT as recipients that were not co-housed. These findings suggest that potential differences in the microbiomes of these mouse strains do not account for the amelioration of GVHD in B6-STING−/− mice (fig. S2A, S2B). Examination of B6-WT and STING−/− thymuses - a sensitive GVHD target tissue - 5 weeks after aHSCT with C3H.SW BM and T cells revealed that STING−/− recipients had greater overall cellularity and a higher frequency of donor-derived CD4+CD8+ double positive T cells, consistent with reduced GVHD in these mice (fig. S2C).
Because STING−/− recipients began to exhibit less weight loss and lower clinical GVHD scoring early post-HSCT, pro-inflammatory cytokines were examined within 48 hours after conditioning and transplant. Following transplant with C3H.SW BM and unseparated and pooled splenocytes and peripheral lymph node cells (2×106 CD8+ T cells), analysis of mRNA from colon tissue demonstrated markedly reduced expression of Ifnb1, Tnf, and Il6 in STING-deficient versus WT recipients (Fig. 2A). These findings that the absence of recipient STING resulted in early reduction of cytokines in the colon, a key site of early acute GVHD responses, were corroborated after transplant with LP/J donors (Fig. 2B). Whereas serum IL-10 concentrations were found to be elevated later post-HSCT, Il10 expression was not elevated immediately post-transplant and we also did not detect any elevation of Cxcl10 (Fig. 2A and B). Consistent with the early reduction of pro-inflammatory cytokines in the colon, on day 10 post-transplant we found that STING−/− recipients had lower colon pathology scores and diminished frequencies of donor T cells in the colon lamina propria versus WT on day 10 (Fig. 2C, fig. S2D). STING−/− recipients also had increased frequencies of CD11b+CD11c+Kb+I-Ab+ and CD11c+Kb+I-Ab+ APCs in this tissue 10 days post-aHSCT and these cells had reduced expression of MHC I protein, potentially indicative of reduced inflammatory signaling in these recipients (fig. S2E). Interestingly, despite these early differences in the colon and increased T conventional (CD4+FoxP3−) to T regulatory (CD4+FoxP3+) cell ratios in WT versus STING−/− colons 5 weeks post-aHSCT, we did not observe substantial colon pathology 6 weeks after transplant with C3H.SW BM and unseparated and pooled splenocytes and peripheral lymph node cells (2×106 T cells), consistent with the primarily sclerodermatous nature of GVHD in this model (fig. S3A, S3B). In total, these findings show that the absence of the STING pathway in recipients results in diminished GVHD after MHC-matched aHSCT accompanied by lower inflammatory cytokine expression early post-aHSCT. The elevated suppressive cytokines 1–2 months following aHSCT may not directly affect GVHD induction and instead be a consequence of less inflammation and tissue injury.
Fig. 2. Recipient STING reduces the inflammatory response to pre-transplant conditioning and MHC-matched aHSCT.
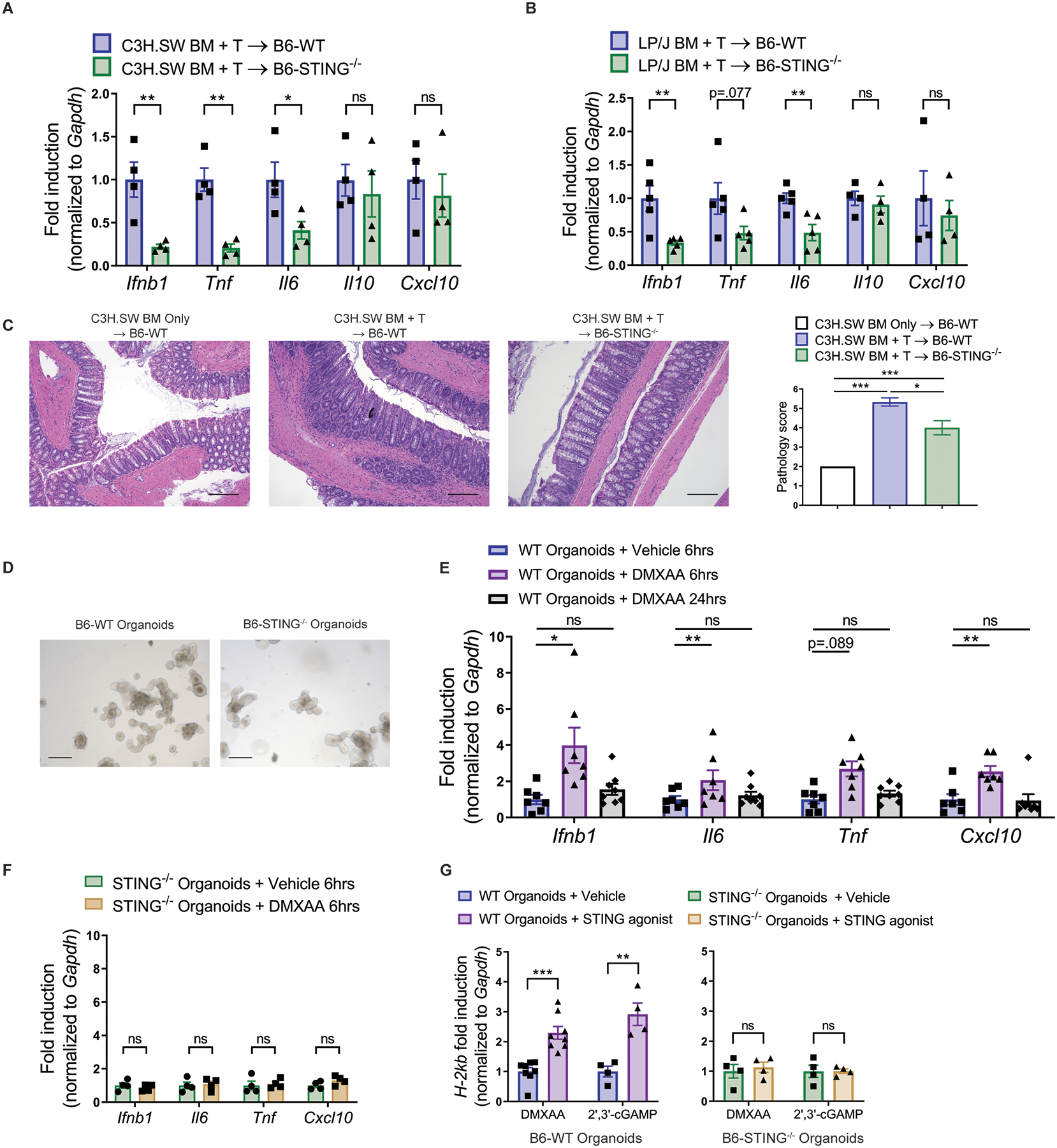
(A) Fold induction of Ifnb1, Tnf, Il6, Il10 and Cxcl10 mRNA from colon tissue 48 hours after transplant with BM and unseparated and pooled splenocytes and peripheral lymph node cells (2×106 CD8+ T cells)from C3H.SW mice. Data pooled: 2 experiments (n=4 per group). (B) Fold induction of Ifnb1, Tnf, Il6, Il10 and Cxcl10 mRNA from colon tissue 48 hours after transplant with BM and unseparated splenocytes (0.8×106 T cells) from LP/J mice. Data pooled: 2 experiments (n=4–5 per group). (C) Representative histology and pathology score of colon tissue from B6-WT or B6-STING−/− recipients 10 days after transplant as in (A). Data pooled: two experiments (n=4 for WT BM Only, n=6 each for WT BM+T and STING−/− BM+T). (D) Representative growth of intestinal organoids derived from B6-WT or B6-STING−/− mice. Scale bars=200μm. (E and F) Fold induction of Ifnb1, Tnf, Il6 and Cxcl10 mRNA by intestinal organoids derived from B6-WT (E) or B6-STING−/− (F) mice and stimulated for 6 or 24 hours with DMXAA (100 μg/mL) or vehicle. (E) Data from two experiments (n=7 each for WT+vehicle and WT+DMXAA at 6hrs, n=8 for WT+DMXAA at 24hrs). (F) Data from one experiment (n=4 per group). (G) Fold induction of H-2kb mRNA by intestinal organoids derived from B6-WT or STING−/− mice and stimulated for 24 hours with DMXAA, 2’3’-cGAMP, or vehicle. Data pooled: 2 experiments (n=8 each for WT organoids + DMXAA, n=4 each for WT organoids + 2’3’-cGAMP and STING−/− organoids). Groups compared using two-tailed unpaired t test or one-way ANOVA with Tukey’s multiple comparisons test for multiple groups. * p<0.05, ** p<0.01, *** p<0.001, ns=non-significant. Data are means±SEM.
Since we observed reduced expression of inflammatory cytokines in the colons of STING−/− recipients early post-aHSCT, we next generated intestinal organoid cultures from B6-WT and B6-STING−/− intestinal tissue to examine their capacity for STING-induced gene regulation (Fig. 2D). STING agonists have been identified which bind exclusively to mouse STING (21, 22). Specifically, 5,6–dimethylxanthenone-4-acetic acid (DMXAA) is known to engage STING molecules at the natural binding site for CDNs in mice and was shown to induce a functional type I IFN response following in vivo administration into B6 mice (22–24). Although cultures from STING−/− mice grow more slowly (19), WT but not STING−/− intestinal organoids upregulated Ifnb1, Tnf, Il6, and Cxcl10 mRNA 6 hours after stimulation with DMXAA, supporting the notion that STING in intestinal tissues can contribute to inflammation in vivo. However, this upregulation was transient, and expression of these cytokines returned to baseline 24 hours after stimulation (Fig. 2E, F). Moreover, type I IFN is known as a potent inducer of MHC expression which is critical for donor T cell recognition of allo-antigen (15). Therefore, we wanted to determine if STING activation - and potentially type I IFN signaling - regulated intestinal organoid MHC class I expression. Indeed, following 24 hours of exposure to DMXAA, an increase in MHC class I H-2kb mRNA expression was detected in WT organoid cultures, but not in organoids lacking STING (Fig. 2G). These results were corroborated using the CDN 2’,3’-cGAMP, the natural ligand for STING (Fig. 2G). Thus, cultures which exhibited elevation of Ifnb1 and other cytokine mRNA at earlier time-points subsequently displayed enhanced MHC class I expression.
STING deficiency in non-hematopoietic recipient cells is sufficient for the amelioration of GVHD
Next, since non-hematopoietic intestinal organoids were able to respond to STING agonists, we considered that STING expression in non-hematopoietic compartments may be important for the regulation of GVHD following MHC-matched HSCT. Therefore, we generated hematopoietic chimeras using congenic B6-WT (CD45.1) and B6-STING−/− (CD45.2) mice as BM donors and recipients. Following complete immune reconstitution and >90% multi-lineage donor chimerism (fig. S4), the chimeras received a second, allogeneic HSCT using MHC-matched LP/J donor BM and unseparated splenocytes (0.8×106 T cells) to initiate GVHD. A striking difference in GVHD severity was observed between chimeras with non-hematopoietic compartments that lacked or expressed STING, paralleling results in non-chimeric STING−/− and WT recipients. Specifically, chimeric recipients lacking STING expression in non-hematopoietic cells experienced decreased clinical scores versus recipients who expressed STING in non-hematopoietic cells, regardless of STING expression in the hematopoietic compartment (Fig. 3A). Similar to our results with B6-STING−/− recipients after MHC-matched aHSCT, mice lacking STING expression in the non-hematopoietic compartment also had lower frequencies of donor CD4+ and CD8+ T cells expressing an activated phenotype in lymphoid tissues 8 weeks post-aHSCT (Fig. 3B). Consistent with their overall GVHD phenotype at this time, histological examination of skin showed reduced inflammation and focal ulceration in chimeric recipients that lacked STING expression in non-hematopoietic cells (Fig. 3C). These findings strongly support the notion that the STING pathway in non-hematopoietic cells promotes pro-inflammatory responses and GVHD after MHC-matched aHSCT.
Fig. 3. STING in non-hematopoietic tissues is important for the induction of GVHD after MHC-matched aHSCT.
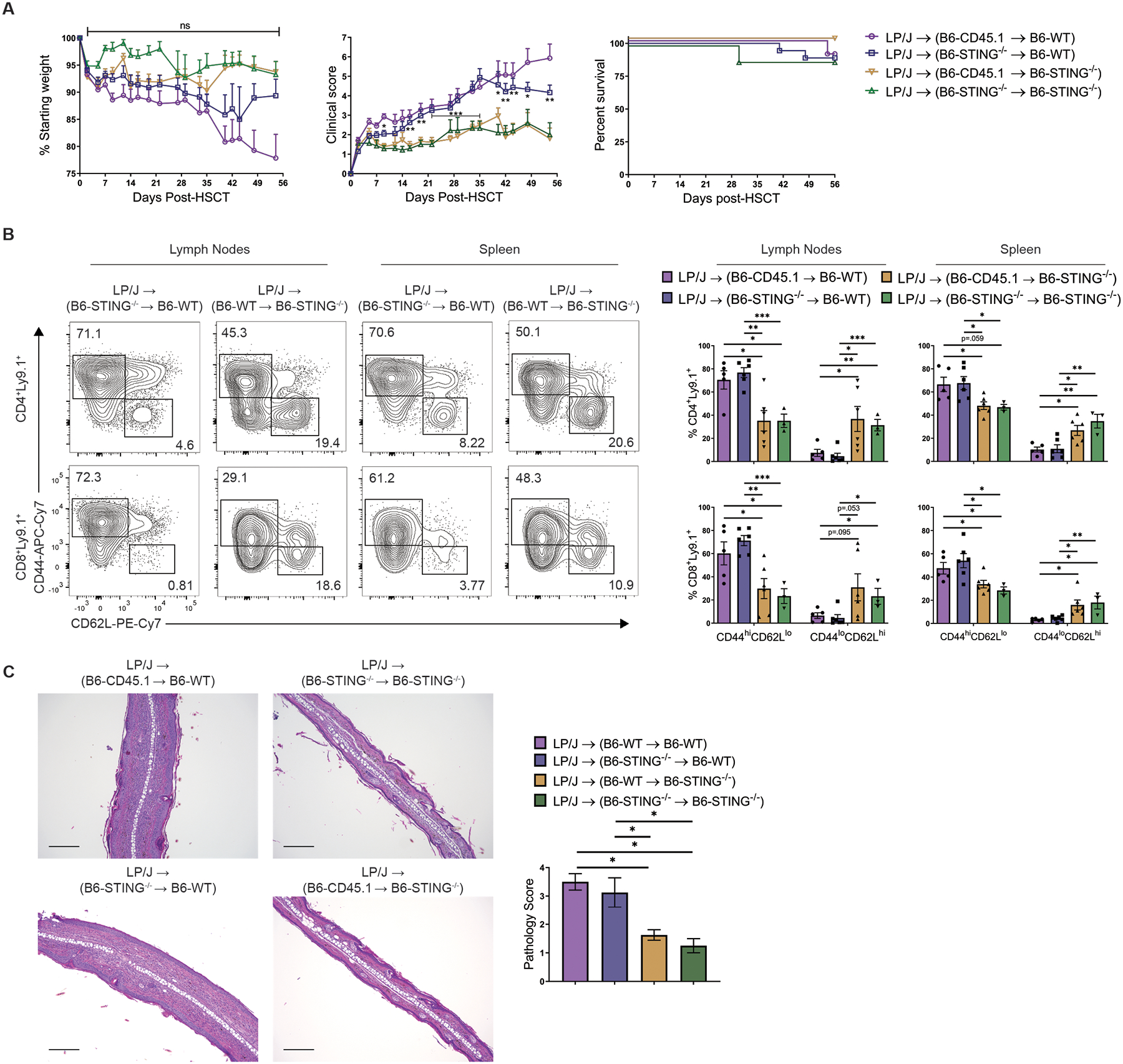
(A) Weight loss, GVHD clinical score, and survival of chimeric (B6-WT donors/B6-STING−/− recipients and B6-STING−/− donors/B6-WT recipients) recipients after second transplant of LP/J BM and unseparated splenocytes (0.8×106 T cells). Data pooled: 2 experiments (n=10 for CD45.1→WT, n=18 for STING−/−→WT, n=15 for CD45.1→STING−/−, n=8 for STING−/−→STING−/−). (B) Representative flow cytometry plots and frequency of donor T cells expressing an effector memory (CD44hiCD62Llo) or naïve (CD44loCD62Lhi) phenotype in lymphoid tissues from chimeric recipients 8 weeks after second transplant with LP/J BM and unseparated splenocytes (0.8×106 T cells). Data pooled: 2 experiments (n=6 per group, representing a total of 14 mice per group). (C) Representative histology and pathology score of ear skin from chimeric recipients 8 weeks after second aHSCT as in (A). Data representative of 2 experiments (n=8 per group). Scale bars=200μm. Chimeric groups compared using one-way ANOVA with Tukey’s multiple comparisons test for multiple groups. * p<0.05, ** p<0.01, *** p<0.001, ns=non-significant. Data are means ± SEM.
Altering the kinetics of STING activation but not T cell numbers abrogates the pathway’s ability to reduce GVHD
Since our MHC-matched transplants were performed using same day (day 0) total body irradiation (TBI) conditioning, we asked if the timing of conditioning in relation to the transplant of BM and T cells might alter the influence of STING on aHSCT outcome. To test this, B6-WT and B6-STING−/− recipients were conditioned at either day −1 or on day 0 prior to transplant with C3H.SW donor cells and GVHD was assessed. As expected, the STING−/− recipients of MHC-matched C3H.SW donor cells conditioned on the day of transplant (day 0) again exhibited markedly diminished GVHD compared to WT (fig. S5A). However, no difference was observed between B6-WT and STING−/− recipients after transplant of MHC-matched donor unseparated and pooled splenocytes and peripheral lymph node cells (2×106 CD8+ T cells) following conditioning on day −1 (fig. S5A). To test the effect of STING on early inflammatory responses to day −1 and day 0 irradiation, we conditioned chimeras (B6-WT donors / B6-STING−/− recipients and B6-STING−/− donors / B6-WT recipients) on day −1 or day 0 and subsequently transplanted LP/J BM and unseparated splenocytes (0.8×106 T cells) on day 0. Analysis of mRNA from chimeric colon tissue 48 hours after irradiation again demonstrated markedly reduced expression of Ifnb1, Tnf, and Il6 in mice lacking STING in their non-hematopoietic compartment compared to those with non-hematopoietic STING. This was observed regardless of STING expression in the hematopoietic compartment and consistent with our findings that STING in the non-hematopoietic compartment promoted MHC-matched GVHD (fig. S5B). Importantly, these results were similar in recipients receiving irradiation on day −1 or day 0, suggesting that the cytokine milieu at the time of transplant was the critical factor in the reduction of GVHD in STING−/− recipients. These findings demonstrate that the effect of STING on GVHD in these models is critically dependent on the timing of conditioning in regard to the time of transplant.
Next, we sought to determine if the strength of the allo-reactive anti-host response might override the ability of STING deficiency to reduce GVHD. Therefore, we performed an MHC-matched aHSCT using unseparated splenocytes containing 10x greater numbers of donor T cells than transplanted in the studies presented thus far. Overall GVHD was heightened in both B6-WT and STING−/− recipients receiving 10x greater numbers of donor T cells. However, at each T cell dose STING−/− recipients again exhibited diminished GVHD compared to WT recipients receiving the same number of donor T cells (fig. S6). Therefore, despite a more vigorous allo-reactive anti-host response, the absence of STING expression in MHC-matched recipients again resulted in diminished GVHD.
Administration of a STING agonist exacerbates GVHD in MHC-matched WT recipients
Based on diminished GVHD in STING-deficient mice following transplant with MHC-matched donors, we hypothesized that further stimulation of STING in WT transplants would result in heightened GVHD. Because we hypothesized that STING is activated by pre-transplant conditioning, DMXAA was administered into B6-WT and STING−/− recipient mice immediately following TBI and prior to MHC-matched transplant with C3H.SW BM and unseparated and pooled splenocytes and peripheral lymph node cells (2×106 CD8+ T cells). B6-WT recipients treated with DMXAA developed clearly heightened GVHD compared to untreated WT recipients as assessed by weight loss, elevated clinical scores and decreased survival (Fig. 4A and B, fig. S7). Analysis of cytokine mRNA production in colon tissue from WT mice indicated that DMXAA treatment elevated Ifnb1, Tnf, and Il6 expression 48 hours post-transplant compared to untreated recipients (Fig. 4C). In contrast, DMXAA administration into B6-STING−/− recipients did not affect GVHD score or survival compared to untreated B6-STING−/− recipients (Fig. 4D). WT recipients receiving DMXAA also had increased frequencies of activated donor T cells in their lymph nodes 7 weeks post-aHSCT versus vehicle control, while no differences were observed in STING−/− recipients receiving DMXAA or vehicle (Fig. 4E). These findings are consistent with diminished expression of these inflammatory cytokines in B6-STING−/− recipients and support the notion that the STING pathway can affect aHSCT outcomes very early post-transplant.
Fig. 4. Further activation of STING with a small molecule agonist at the time of MHC-matched aHSCT exacerbates GVHD.
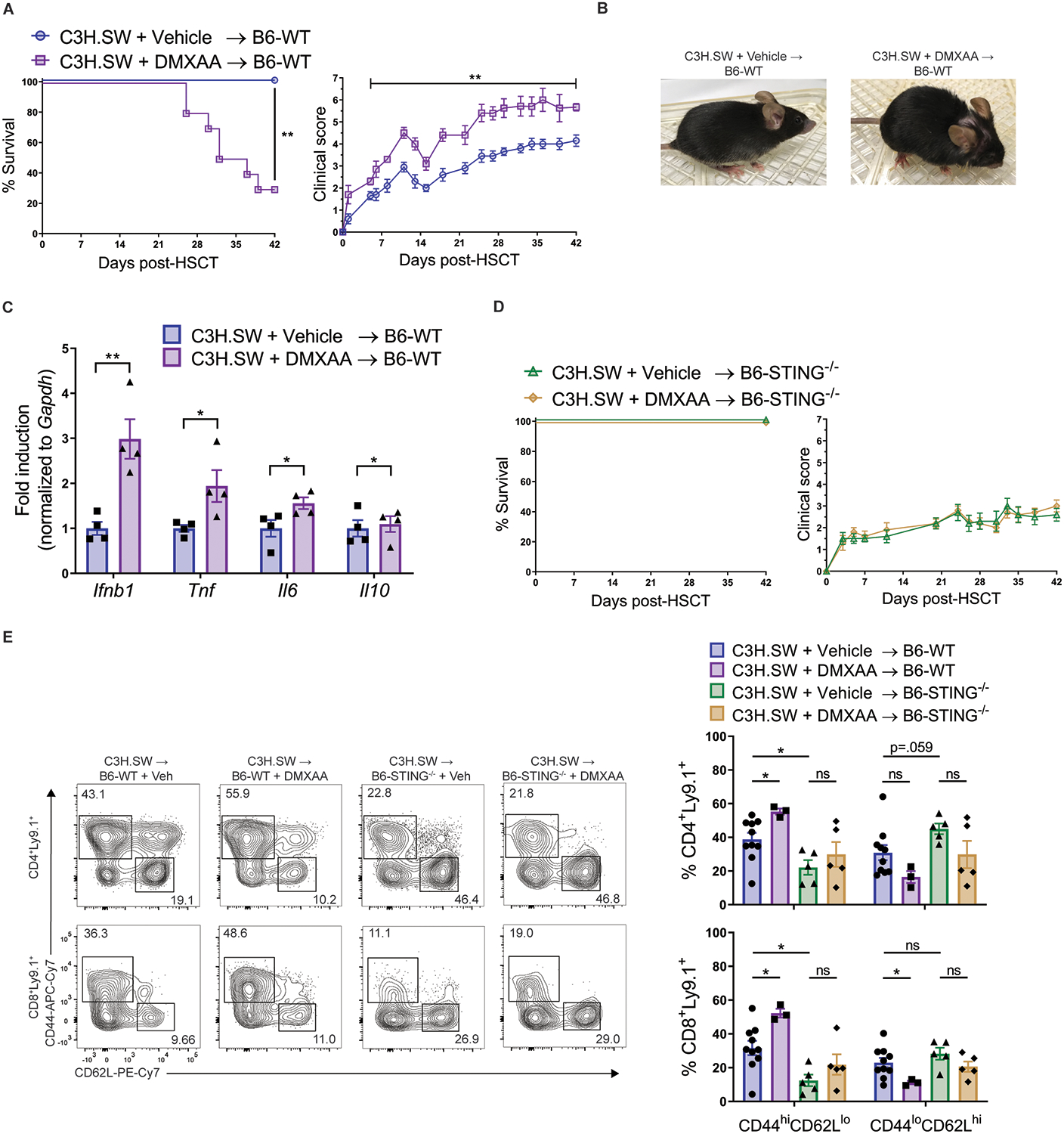
(A) Survival, GVHD clinical score, and (B) representative photographs of B6-WT recipients after transplant with C3H.SW BM and unseparated and pooled splenocytes and peripheral lymph node cells (2×106 CD8+ T cells) and given day 0 intraperitoneal injections of vehicle or DMXAA (25mg/kg). Data pooled: 2 experiments (n=10 per group). (C) Fold induction of Ifnb1, Tnf, Il6 and Il10 mRNA from B6-WT colon tissue 48 hours after transplant and DMXAA administration as in (A). Data from one experiment (n=4 per group). (D) Survival and GVHD clinical score of B6-STING−/− recipients after transplant and DMXAA administration as was performed using B6-WT mice in (A). Data from one experiment (n=5 per group). (E) Representative flow cytometry plots and frequency of donor T cells expressing an effector memory (CD44hiCD62Llo) or naïve (CD44loCD62Lhi) phenotype in B6-WT or B6-STING−/− lymph nodes 6 weeks after transplant as in (A). Pooled data: 2 experiments (n=10 each for WT, n=5 each for STING−/−). Groups compared using two-tailed unpaired t test, one-way ANOVA with Tukey’s multiple comparisons test for multiple groups or log-rank test for survival analyses. * p<0.05, ** p<0.01, ns=non-significant. Data are means ± SEM.
Diminished STING activity in knock-in mice expressing the human STINGHAQ allele is associated with decreased GVHD in recipients after MHC-matched HSCT
Four non-synonymous single nucleotide polymorphisms (SNPs) have been identified in the human STING gene (TMEM173), three of which are present within a human STING allele (HAQ = R71H, G230A, R293Q, Fig. 5A) which has been associated with diminished STING function (25, 26). To corroborate this deficit in mice homozygous for these three SNPs (B6N-STINGHAQ/HAQ), peritoneal exudate cells (PECs) were obtained from B6-WT and knock-in B6N-STINGHAQ/HAQ and stimulated with the CDN 2’,3’-cGAMP and DMXAA. qPCR analysis from these cultures demonstrated that B6N-STINGHAQ/HAQ PECs exhibited markedly lower expression of Ifnb1, Il6 and Tnf in response to both 2’,3’-cGAMP transfection and DMXAA stimulation (Fig. 5B, fig. S8). Notably, while there was statistically significant induction of Ifnb1 mRNA in the STINGHAQ/HAQ PEC cultures exposed to both molecules, this increase was much lower than that observed from WT cultures (Fig. 5B). Next, to begin to evaluate the potential impact of STING in the clinical aHSCT setting, we assessed GVHD in B6N-WT and B6N-STINGHAQ/HAQ recipients after transplant of MHC-matched C3H.SW BM and unseparated and pooled splenocytes and peripheral lymph node cells (2×106 CD8+ T cells). B6N-STINGHAQ/HAQ recipients developed diminished GVHD clinical scores and were found to contain a lower frequency of donor T cells expressing an activated phenotype (CD44hiCD62Llo) versus B6N-WT recipients (Fig. 5C and D), although not as reduced as in recipients lacking STING (see above, Fig. 1). These results indicate that the expression of the human STINGHAQ allele results in decreased GVHD, consistent with our findings in STING-deficient recipients.
Fig. 5. B6N-STINGHAQ/HAQ mice are less responsive to STING ligands and develop reduced GVHD after MHC-matched aHSCT.
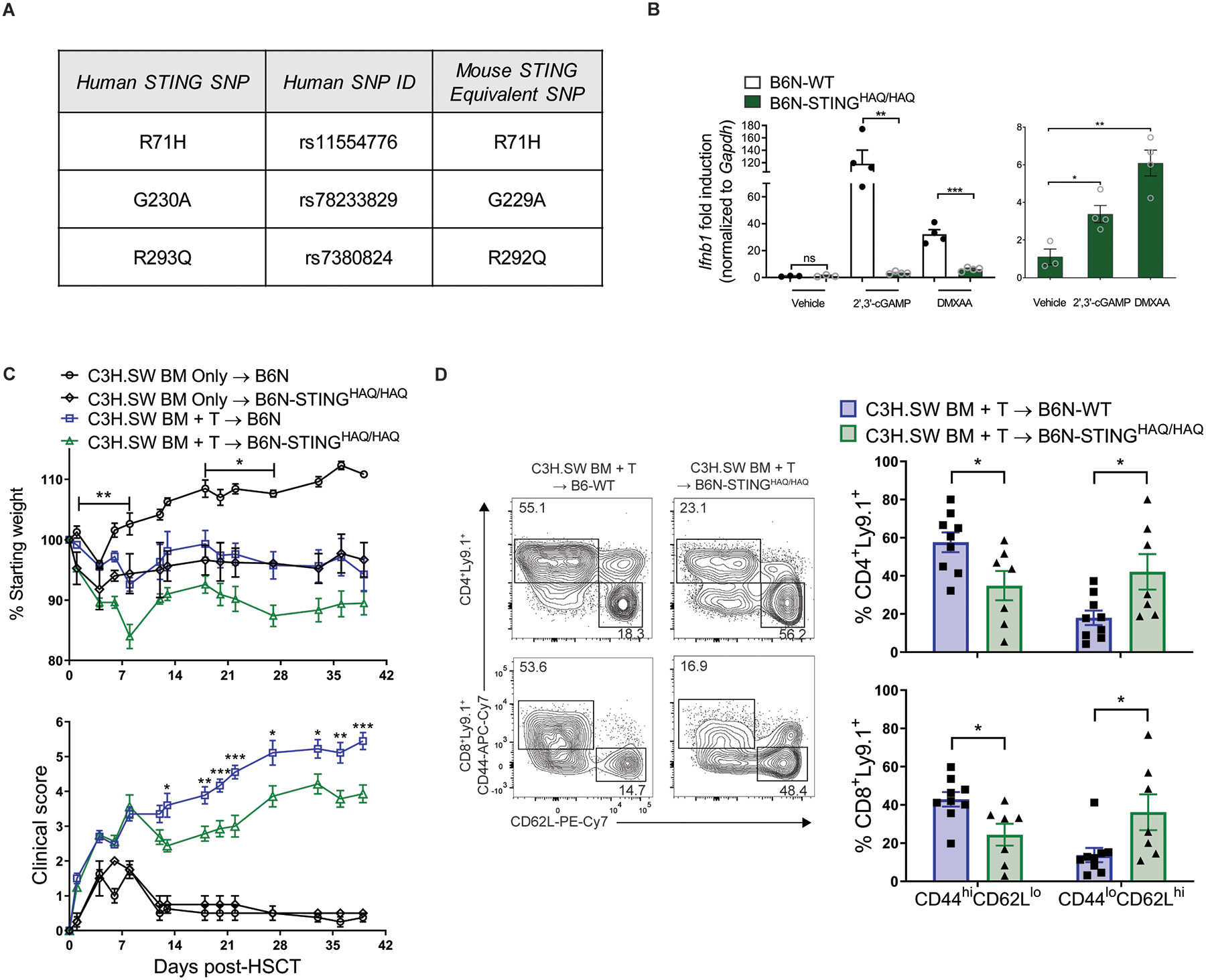
(A) Table describing 3 human STING SNPs replicated in mouse STING and knocked into B6N mice to generate the B6N-STINGHAQ/HAQ mice. (B) Fold induction of Ifnb1 mRNA by peritoneal exudate cells isolated from either B6N-WT or B6N-STINGHAQ/HAQ mice after stimulation for 6 hours with vehicle, 2’,3’-cGAMP (6.7μg/mL) or DMXAA (100μg/mL). Graph on right shows the same B6N-STINGHAQ/HAQ data plotted alone. Data from one experiment (n=3 each for WT and STINGHAQ/HAQ + Vehicle, n=4 each for WT and STINGHAQ/HAQ + cGAMP or DMXAA). (C) Weight loss and GVHD clinical score of B6N-WT or B6N-STINGHAQ recipients after transplant with C3H.SW BM and unseparated and pooled splenocytes and peripheral lymph node cells (2×106 CD8+ T cells). Data from one experiment (n=4 for WT BM Only, n=2 for STINGHAQ/HAQ BM Only, n=10 for WT BM+T, n=8 for STINGHAQ/HAQ). (D) Representative flow cytometry plots and frequency of donor T cells expressing an effector memory (CD44hiCD62Llo) or naïve (CD44loCD62Lhi) phenotype in lymph nodes 6 weeks after transplant as in (C). Data from one experiment (n=9 for WT BM+T, n=7 for STINGHAQ/HAQ). GVHD groups compared using two-tailed unpaired t test or one-way ANOVA with Tukey’s multiple comparisons test for multiple groups. * p<0.05, ** p<0.01, *** p<0.001. Data are means ± SEM.
STING promotes CD8+ T cell-mediated GVHD following aHSCT across MHC-matched or MHC-mismatched transplants
Based on the predominance of CD8+ T cells associated with GVHD induction following MHC-matched minor antigen mismatched C3H.SW BM into B6 recipient aHSCT, we next transplanted highly purified C3H.SW CD8+ donor T cells to test if CD4+ T cells were dispensable for reduced GVHD in B6-STING−/− recipients (27, 28). Following transplant of C3H.SW CD8+ T cells, GVHD weight loss, clinical score, and frequencies of activated donor T cells were again diminished in B6-STING−/− versus WT recipients (Fig. 6A–C).
Fig. 6. Donor CD8+ T cells are sufficient for STING to promote MHC-matched GVHD.
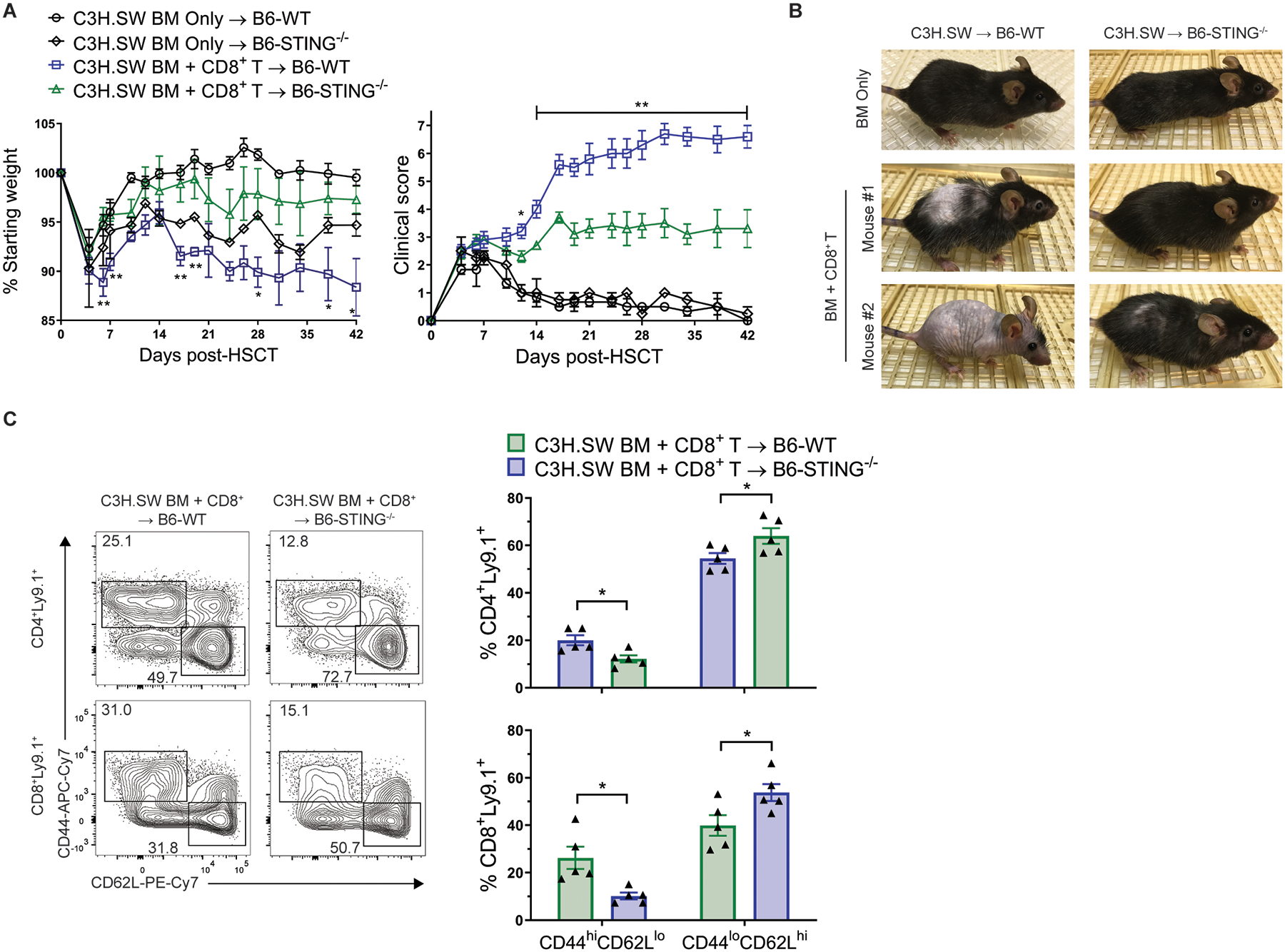
(A) Weight loss, GVHD clinical score and (B) representative photographs, and (C) Representative flow cytometry plots and frequency of donor T cells expressing an effector memory (CD44hiCD62Llo) or naïve (CD44loCD62Lhi) phenotype in lymph nodes from B6-WT and B6-STING−/− recipients 6 weeks after transplant with C3H.SW donor BM and purified CD8+ T cells. Data from 1 experiment (n=3 for WT BM Only, n=2 for STING−/− BM Only, n=5 each for WT BM+T and STING−/− BM+T). GVHD groups compared using two-tailed unpaired t test. * p<0.05, ** p<0.01. Data are means ± SEM.
We also wanted to assess the role of STING after MHC-mismatched aHSCT since GVHD after these pre-clinical transplants is primarily mediated by CD4+ T cells and a minority of clinical transplants are performed in which donor and recipient differ at one or more human leukocyte antigen (HLA) alleles. The results shown here with MHC-matched aHSCT contrast a prior study which reported that the STING pathway protects against GVHD after MHC-mismatched aHSCT using B6-STINGgt/gt (C57BL/6J-Tmem173gt) recipients which lack STING expression (19). Therefore, we first tested if our distinct STING knock-out strain was responsible for our opposing findings by transplanting MHC-mismatched BALB/c BM and unseparated lymph node cells (1.7×106 T cells) into B6-WT or STING−/− recipients. After this transplant, B6-STING−/− recipients exhibited increased clinical scores and decreased survival compared to WT, corroborating the previously reported findings (20) and confirming an opposing effect of STING deficiency after MHC-matched versus mismatched aHSCT (Fig. 7A).
Fig. 7. The absence of STING in recipients ameliorates CD8-dependent, but not CD4-dependent, GVHD after MHC-mismatched aHSCT.
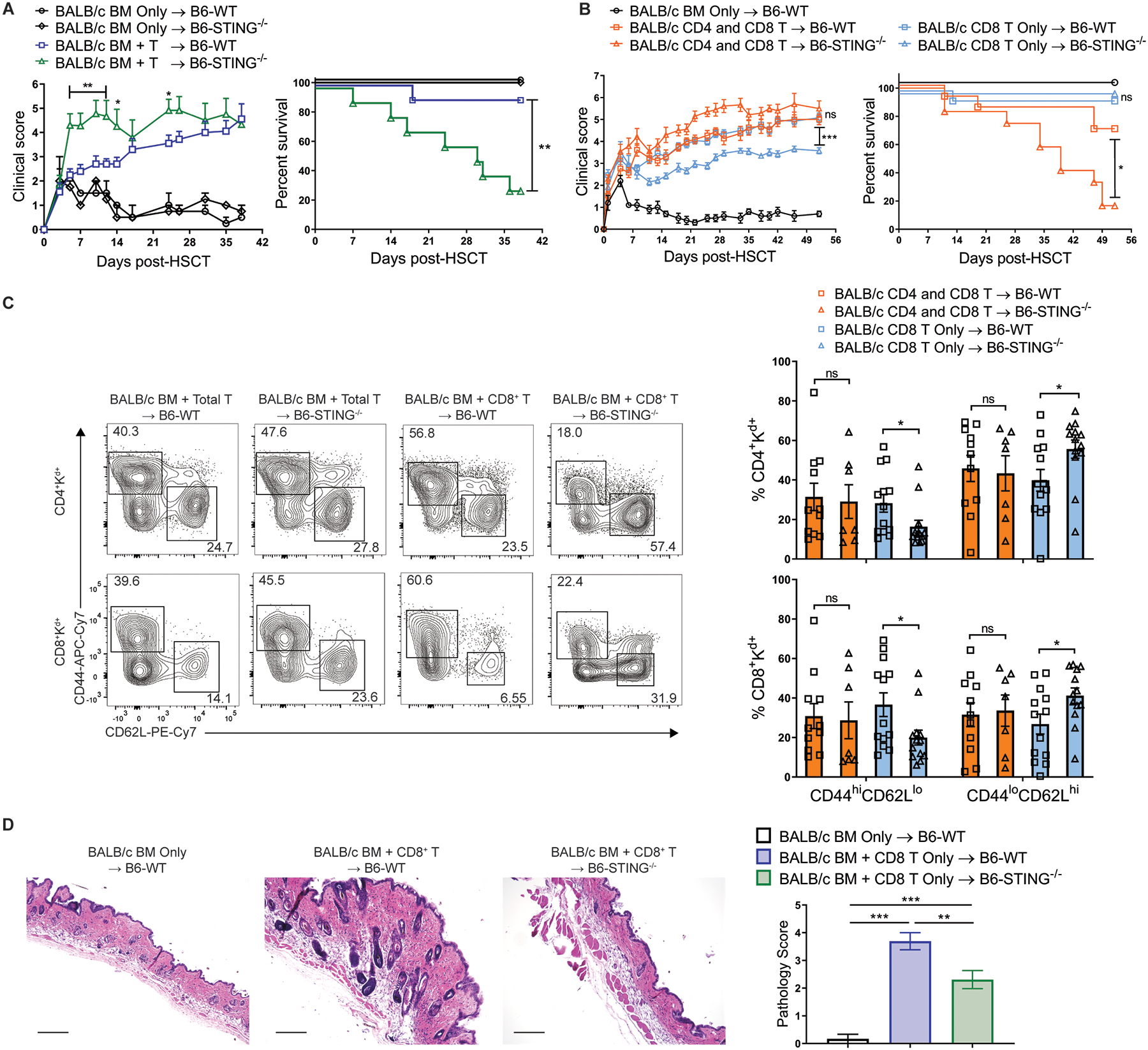
(A) GVHD clinical score and survival of B6-WT or B6-STING−/− recipients after transplant of BALB/c BM and unseparated lymph node cells (1.7×106 T cells). Data pooled: 2 experiments (n=2 each for WT and STING−/− BM Only, n=10 each for WT and STING−/− BM+T). (B) GVHD clinical score and survival of B6-WT or B6-STING−/− recipients after transplant of BALB/c BM and either total T cells or CD8+ T cells alone. Data pooled: 2 experiments (n=5 for WT BM Only, n=13 for WT BM+Total T, n=12 for STING−/− BM+Total T, n=14 for WT BM+CD8+ T, n=13 for STING−/− BM+CD8+ T). (C) Frequency of donor T cells expressing an effector memory (CD44hiCD62Llo) or naïve (CD44loCD62Lhi) phenotype in lymph nodes from B6-WT or B6-STING−/− recipients 8 weeks after transplant as in (B). Data pooled: 2 experiments (n=11 for WT BM+Total T, n=7 for STING−/− BM+Total T, n=13 each for WT BM+CD8+ and STING−/− BM+CD8+ T). (D) Representative histology and pathology score of interscapular skin from B6-WT or B6-STING−/− recipients 8 weeks after transplant as in (B). Scale bars=200μm. Data pooled: 2 experiments (n=5 for WT BM Only, n=13 each for WT BM+CD8+ T and STING−/− BM+CD8+ T). GVHD groups compared using one-way ANOVA with Tukey’s multiple comparisons test for multiple groups or log-rank test for survival analyses. * p<0.05, ** p<0.01, *** p<0.001, ns=non-significant. Data are means ± SEM.
We next hypothesized that the dominant T cell-subset (i.e. CD4+ in MHC-mismatched versus CD8+ in MHC-matched) in each model may explain these opposing results in STING−/− versus WT recipients. We therefore proposed that STING may promote CD8+ T cell-mediated GVHD. CD8+ T cells can mediate GVHD across MHC-mismatched donor-recipient combinations (20, 29, 30). Therefore, an aHSCT was performed using highly purified donor BALB/c CD8+ T cells transplanted into MHC-mismatched B6-WT and STING−/− recipients. In support of our hypothesis, STING−/− recipients receiving purified MHC-mismatched CD8+ T cells exhibited reduced GVHD as assessed by clinical score, donor T cell phenotype and skin pathology, similar to what we observed in MHC-matched recipients (Fig. 7B–D). In transplants utilizing fractionated T cells into B6-WT and B6-STING−/−recipients, there were no differences detected in the serum with regard to concentrations of several cytokines (i.e. IL-10, IL-27). Interestingly, when we transplanted BALB/c BM and CD4+ donor T cells only, we were unable to detect a difference in GVHD between B6-WT and STING−/− as assessed by weight loss, clinical score, survival and donor T cell phenotype (fig. S9A and B), suggesting that CD8+ T cells are necessary for the differences in GVHD we observed between WT and STING−/− recipients.
STING regulates recipient APC survival and donor CD8+ T cell activation
We next sought to reconcile the contrasting results comparing STING−/− versus WT recipients after MHC-matched and mismatched HSCT. We first considered that recipient APCs are critical for the induction of GVHD after aHSCT (31). Furthermore, our data indicate that STING promotes CD8+ T cell-mediated GVHD. Therefore, we hypothesized that STING may contribute to the early activation of donor CD8+ T cells which then eliminate recipient APCs. This would then deprive donor CD4+ T cells of allo-antigen presentation via MHC class II, providing one potential mechanism by which STING might promote CD8+ T cell-mediated GVHD and also protect against CD4+-mediated GVHD. To test this, we utilized the well-characterized B6 BM with or without unseparated lymph node cells (1.2×106 T cells) into BALB/c recipient MHC-mismatched aHSCT model comparing BALB/c-WT or BALB/c-STING−/− recipients. One day post-aHSCT, although there was no detectable difference in the recipient spleen cell numbers between BALB/c-STING−/− versus WT (Fig. 8A), BALB/c-STING−/− mice had greater frequencies and numbers of recipient splenic CD11b+CD11c+Kd+I-Ad+ APCs and CD11b+Kd+ cells as well as increased frequencies of CD11c+Kd+ cells in their colon lamina proprias (Fig. 8B, fig. S12A). These CD11b+CD11c+Kd+IAd+ APCs lacking STING also expressed reduced MHC class I protein and fewer exhibited annexin V binding (Fig. 8C, fig. S10). Moreover, spleens from STING-deficient recipients also contained lower frequencies and numbers of donor CD8+ T cells and lower numbers of donor CD8+ T cells producing IFNγ and TNFα (Fig. 8D–F).
Fig. 8. STING deficiency promotes recipient antigen presenting cell (APC) survival and diminishes donor CD8+ T cell activation early after MHC-mismatched aHSCT.
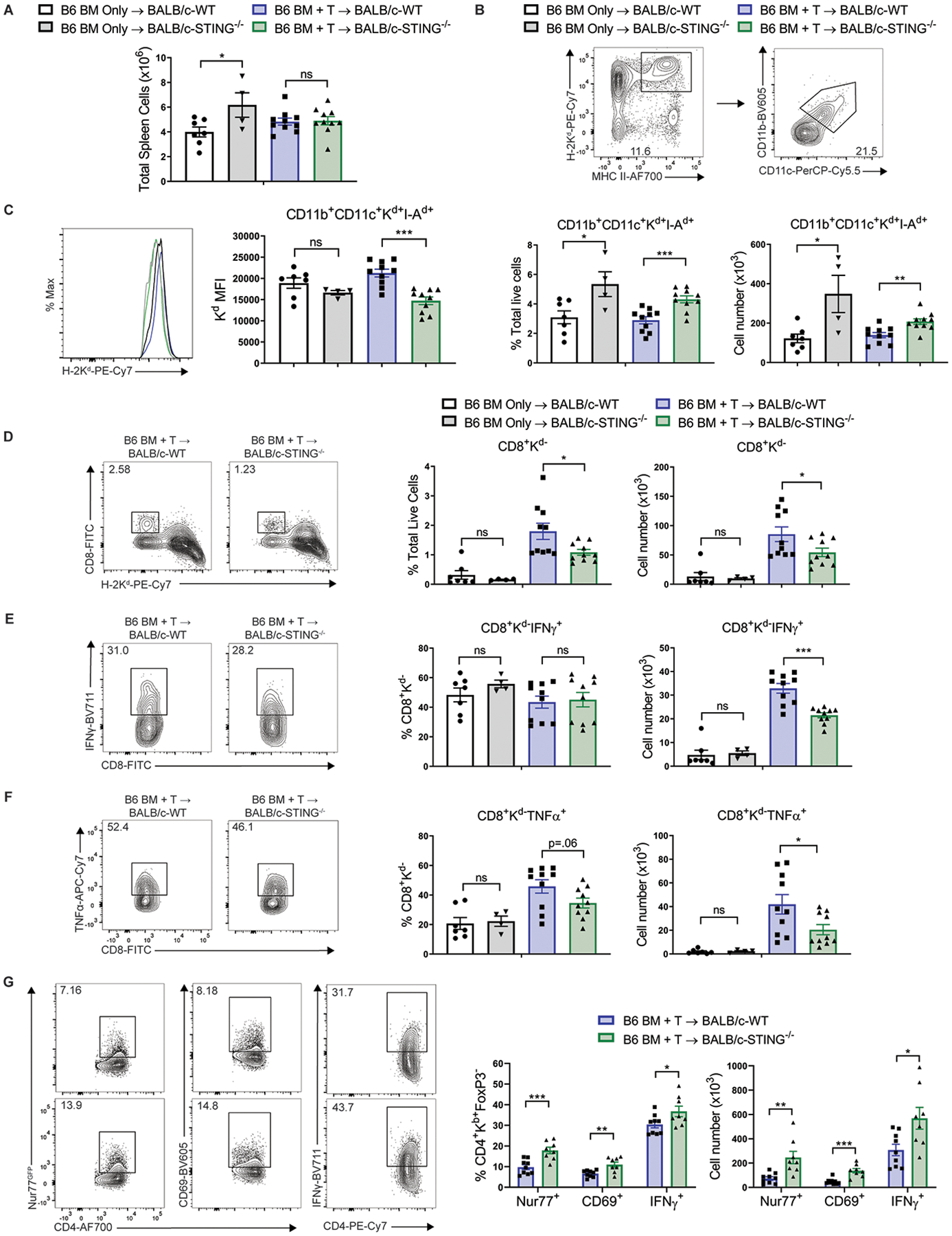
(A) Total spleen cells from BALB/c-WT or BALB/c-STING−/− recipients 24 hours after transplant with B6-WT BM and unseparated lymph node cells (1.2×106 T cells). (B) Gating strategy, frequency and total cell number of recipient APCs in spleens of BALB/c-WT or STING−/− recipients 24 hours after transplant as in (A). (C) H-2Kd median fluorescence intensity on recipient APCs in spleens of BALB/c-WT or STING−/− recipients 24 hours after transplant as in (A) and gated as in (B). (D) Gating strategy, frequency and total cell number of donor CD8+ T cells in spleens of BALB/c-WT or STING−/− recipients 24 hours after transplant as in (A). (E and F) Gating strategy, frequency and total cell number of donor CD8+ T cells producing (E) IFNγ or (F) TNFα in spleens of BALB/c-WT or STING−/− recipients 24 hours after transplant as in (A). Data pooled: 2 experiments (n=7 for WT BM Only, n=4 for STING−/− BM Only, n=10 each for WT BM+T and STING−/− BM+T). (G) Gating strategy, frequency and total cell number of donor CD4+FoxP3− T cells expressing Nur77GFP, CD69, and IFNγ in spleens of BALB/c-WT or STING−/− recipients 6 days after transplant with B6-CD45.1 BM and B6-Nur77GFPFoxP3RFP unseparated lymph node cells (1.2×106 T cells). Data pooled: 2 experiments (n=9 for WT BM+T and n=8 for STING−/− BM+T). GVHD or BM Only groups compared using two-tailed unpaired t test. * p<0.05, ** p<0.01, *** p<0.001. Data are means ± SEM.
To identify if the loss of CD11b+CD11c+Kd+I-Ad+ APCs affected donor CD4 T cell receptor (TCR) engagement and activation, the Nur77GFP transgenic mouse containing a GFP insert in the Nr4a1 (Nur77) locus was used (32), which indicates T cell activation via the TCR but not by inflammatory cytokines (33). Consistent with increased numbers of MHCII+ APCs in the spleens of STING-deficient recipients 24 hours post-aHSCT, examination of splenocytes from STING−/− recipients 6 days after transplant found greater frequencies and numbers of donor Nur77GFP CD4+ T cells expressing GFP, CD69 and IFNγ (Fig. 8G, fig. S11). Chimeric recipients (B6-WT donors / B6-STING−/− recipients and B6-STING−/− donors / B6-WT recipients) that lacked STING in their non-hematopoietic compartments also had increased frequencies and cell numbers of CD11b+Kb+ cells in their spleens 48 hours after transplant with LP/J BM and unseparated splenocytes (0.8×106 T cells), consistent with the notion that STING in the non-hematopoietic compartment can regulate the survival of hematopoietic myeloid cells early after aHSCT (fig. S12B). These data support the hypothesis that recipient STING deficiency leads to reduced donor alloreactive CD8+ T cell activation, greater recipient APC survival, and stronger CD4+ T cell responses compared to WT after aHSCT.
Discussion
Here, we show that recipient STING deficiency reduces pro-inflammatory responses to pre-aHSCT conditioning and transplant and decreases the activation of donor CD8+ T cells early post-transplant in mice. Our data also demonstrate that this reduction in early CD8+ T cell activation dampens subsequent CD8+ T cell-mediated GVHD after both MHC-matched and mismatched aHSCT, dependent on the relative timing of conditioning and transplant. In contrast, we hypothesize that STING deficiency could promote CD4+ T cell-mediated GVHD as a result of the greater numbers of APCs in mice lacking STING thereby providing MHC II and antigen signaling to donor CD4+ T cells. Overall, these findings demonstrate that an innate immune sensor can differentially regulate GVHD mediated by distinct T cell subsets, underscoring the concept that successful therapeutic targeting will require positive or negative pathway regulation dependent on the context of the disease.
STING is expressed in virtually all compartments including tissues important in development of GVHD (ex. GI) (11, 13, 34),. The importance of the STING pathway in the GI tract has recently been appreciated. It has been reported that STING is important in gut homeostasis and control of inflammation as evidenced by the finding that STING−/− mice were more susceptible to DSS induced intestinal inflammation and colitis (35). Notably, the STING pathway was also found to be important in relation to irradiation and GVHD damage (19). While the mechanism of STING activation during aHSCT has not been established, activation of this pathway can occur as a result of danger signals released from damaged cells (i.e. DNA) and the leakage of bacterial products (i.e. DNA and CDNs) through the GI epithelium following pre-transplant conditioning (5). Furthermore, treatment of WT mice with the highly specific mouse STING agonist DMXAA further increased cytokine production post-transplant, suggesting that the magnitude of STING activation under the conditions of our MHC-matched transplant model was not maximal. As a result, we posit that the type and severity of individual conditioning protocols, such as reduced intensity versus myeloablative, is likely to determine the extent of and individual tissues involved in STING regulation of GVHD.
Our studies also specifically found that the absence of STING expression in non-hematopoietic tissues is important for the reduction of GVHD after MHC-matched aHSCT, although we cannot formally rule out a contribution from STING in the hematopoietic compartment. We have not yet identified the precise cell population where STING is activated during aHSCT, but it is notable that recipient non-hematopoietic APC in the intestine - an important GVHD target tissue - have been shown to be capable of inducing lethal GVHD (36). The accessibility of non-hematopoietic and local hematopoietic APCs, together with other types of non-hematopoietic stromal and epithelial cells - to intestinal bacteria, their products, as well as self-DNA following transplant-related intestinal damage likely accounts for local activation of STING in the intestine (35). Notably, we observed upregulated cytokine mRNAs in WT vs STING−/− colon tissue post-aHSCT. A parallel result was observed in DMXAA treated / untreated WT recipients and again identified in DMXAA-treated intestinal organoids. Accordingly, it is conceivable that epithelial cells may be an important site of STING activation post-aHSCT. After STING activation in non-hematopoietic tissues, type I IFN and other pro-inflammatory signals may promote hematopoietic APC activation and migration to lymphoid tissues where allo-reactive donor T cells are activated and GVHD is initiated. In accordance with this idea, others have previously reported that expression of the type I IFN receptor on recipient hematopoietic cells was important for regulating GVHD, however, the underlying type I IFN pathway was not examined (20).
Importantly, our initial data that STING deficiency reduced GVHD after MHC-matched aHSCT contrasted observations reported in a recent study which found that the absence of STING exacerbated GVHD following transplant across fully-mismatched donors and recipients (19). Utilizing a STING-deficient strain distinct from the B6-STINGgt/gt mice, we obtained similar data using our B6-STING−/− recipients (19, 34, 37). Of note, multiple recent studies have demonstrated that the administration of type I IFN or type I IFN-inducing agonists can protect against MHC-mismatched GVHD, but only if given prior to aHSCT (19, 20, 38, 39). These findings are consistent with our data indicating that the timing of irradiation was critical for STING deficiency to protect against MHC-matched GVHD as well as the kinetics of cytokine upregulation in intestinal organoids. To reconcile the differences between MHC-matched and mismatched recipients, we investigated the role of T cell subsets. We found that GVHD was reduced in STING-deficient recipients after CD8+ T cell-mediated GVHD, regardless of the genetic disparity between donors and recipients, and therefore contrasting previous findings using unfractionated MHC-mismatched donor T cells. Since type I IFNs are known as potent inducers of MHC class I expression and MHC class I is critical for CD8+ T cell activation and effector function, we then asked if STING signaling could regulate MHC class I. Since results demonstrated the absence of STING in non-hematopoietic cells is important for its regulation of GVHD, we examined DMXAA-stimulated intestinal organoid cultures and found an upregulation of H-2Kb mRNA in WT but not STING-deficient organoids, supporting the significance of this pathway in elevating CD8+ T cell-mediated GVHD.
Although we did not observe a difference in the number of recipient APCs when T cells were included in the graft versus BM alone in either WT or STING−/− recipients, we consistently noted that WT vs STING−/− recipients harbored reduced numbers of CD11b+CD11c+Kd+IAd+ APCs at several time points and tissues in multiple models which may be a consequence of STING activation in the non-hematopoietic compartment. Further experiments are required to determine if activation of donor CD8+ T cells in WT recipients following T cell replete aHSCT further alters the APC compartment. We have not eliminated this possibility since others have reported that donor T cells can contribute to the elimination of recipient APCs (28). In the current study we consistently noted reduced APC numbers, greater donor CD8+ T cell numbers as well as increased TNFα and IFNγ expression in WT recipients. Overall, the lower numbers of recipient APCs in STING-sufficient recipients following aHSCT may reduce the availability of allo-antigen presented in the context of MHC, i.e. including class II, protecting against allo-reactive CD4+ T cell activation. Therefore, while APC are reduced following both MHC-matched and mismatched HSCT when STING is present, less GVHD occurs in the mismatched transplant since CD4+ T cells are not effectively stimulated. Conversely, when CD4+ T cells are not key GVHD inducers, the STING pathway promotes CD8+ T cell activation and MHC-matched GVHD through cytokine / MHC class I upregulation. It is notable that two other studies have reported that type I IFN and IL-18 signaling can differentially regulate CD4+ versus CD8+ T cell-mediated GVHD (20, 40). STING induces IFNβ production and has been reported to increase IL-18 expression (17, 41). Therefore, in STING−/− recipients where both cytokines may be reduced, our observations could reflect those in prior aHSCT studies which found that type I IFN receptor and IL-18 protected against CD4 mediated GVHD and worsened CD8 mediated GVHD (20, 40). Lastly, although we have not examined the microbiome in WT and STING−/− mice, the composition of gut microbiota were reported similar in co-housed B6-WT and STING-deficient B6-STINGgt/gt mice, therefore a differential impact of bacteria on CD4 and CD8 T cells would need to be investigated to determine if this was a contributing mechanism (19).
In the human population, two common STING alleles have been identified that are associated with reduced STING functionality (25). We have reported that cells from individuals that are homozygous for one of these, the STING-HAQ allele, have a substantially reduced ability to respond to STING agonists (25). Due to the relatively common prevalence of this allele in the human population - estimated to be as frequent as 16.1% among individuals that are ethnically East Asian - considerable translational import might be expected. Peritoneal cells from mice knocked-in for murine STING containing the mouse equivalent of these three SNPs (R71H, G229A, R292Q, B6-STINGHAQ/HAQ), were also markedly less responsive to STING agonists. Of note, Ifnb1 mRNA production was still upregulated, albeit weakly in these cells after stimulation, suggesting that some STING functionality remained (25). Accordingly, STINGHAQ/HAQ mice transplanted with MHC-matched C3H.SW BM and T cells developed reduced GVHD compared to WT. Together with our results from STING knockout mice, these findings illustrate that a reduction in STING activity is capable of altering the severity of GVHD and that STING alleles that have reduced functionality may affect clinical aHSCT. However, human studies are required to confirm a role for these STING alleles in clinical aHSCT.
Since the major cause of death post-HSCT is the recurrence of primary disease, a central benefit of receiving an aHSCT is the accompanying “graft-versus-tumor / leukemia” (GVL) response to target and eradicate residual disease (42). Notably, STING signaling is known to promote the generation of anti-tumor T cell responses and has recently been found to be deregulated in human colorectal carcinomas (18, 43, 44). In fact, a recent study found that in vivo DMXAA administration promoted tumor-specific CD8+ T cells and potent immunity against systemic AML in a pre-clinical model (45). Therefore, this pathway could potentially promote the activation of anti-tumor reactive CD8+ T cells in patients with hematopoietic malignancies receiving aHSCT (44). Although experimental GVL responses in STING-deficient recipients have yet to be examined, we speculate that transient inhibition of this pathway at the time of transplant to regulate GVHD would subsequently allow for tumor-derived dsDNA to activate STING and promote the activation of donor CD8+ anti-tumor T cells in matched related recipients. While there are currently no inhibitors of STING that are in clinical use, several small biomolecules and compounds have been recently identified as potential mouse and human STING inhibitors (46, 47). Overall, the results presented here indicate that the generation of clinically effective agonists and inhibitors of this pathway are likely to result in protocols which will improve outcomes of patients receiving aHSCT.
Materials and Methods
Study design
The objectives of this research study were to investigate the role of STING in GVHD development. This study was performed using mouse models of MHC-matched and mismatched aHSCT designed to induce GVHD in B6-WT and B6-STING−/− recipients. GVHD was monitored by survival, weight loss, clinical scoring, flow cytometry and histopathology. Frequencies of donor T cells expressing an effector memory phenotype were used to examine GVHD on the cellular level. Quantification of tissue RNA and serum cytokines were also used to evaluate STING’s effect on GVHD. Hematopoietic chimeras were generated to examine within which tissues STING affected GVHD. Group sizes and number of independent experiments are indicated in figure legends and were chosen to give sufficient power to the statistical analyses that were to be used. WT and transgenic mice (age and sex matched) were randomized into different cages prior to the beginning of experiments. For experiments utilizing multiple groups with the same mouse strain, recipient mice were randomized into experimental groups. Histology slides were read in a blinded fashion. No data points were excluded as outliers in this study. Primary data are reported in data file S1.
Mice
C3H.SW (Stock: 000438), C57BL/6J (B6, Stock: 000664), C57BL/6N (B6N, Stock: 005304), BALB/c (Stock: 000651), B6-CD45.1 breeder (Stock: 002014) and LP/J breeder (Stock: 000676) mice were purchased from The Jackson Laboratory and maintained in our animal facility. B6-STING−/− mice were generated as previously described (34). B6N-STINGHAQ/HAQ mice were generated as previously described (25). BALB/c-STING−/− mice were generated by backcrossing B6-STING−/− mice onto a BALB/c background for 9 generations. B6-FoxP3RFPNur77GFP mice were generated by crossing B6-Nur77GFP mice (Stock: 016617) purchased from The Jackson Laboratory with B6-FoxP3RFP mice (provided by R. Flavell, Yale University) (32, 48). All mice were maintained in specific pathogen-free housing at the University of Miami and given autoclaved food and water ad libitum. Mice (females and males) were used at 8–16 weeks of age. All animal use procedures were approved by the University of Miami institutional animal care and use committee (IACUC protocol numbers 19–114-VVC ad01 and 18–036).
Bone marrow transplantation
For MHC-matched transplants, B6-WT, B6N-WT, B6-STING−/− or B6N-STINGHAQ/HAQ mice received 8.5Gy (LP/J donors) or 10.5Gy (C3H.SW donors) total body irradiation (30 cGy/min TBI from a Cs137 source) on day 0 (MHC-matched) or day −1 (MHC-mismatched), unless otherwise stated. Three hours later, irradiated mice were injected intravenously (IV) with T cell- depleted BM with or without T cells from sex and age-matched LP/J (10×106 BM cells and unseparated splenocytes containing 8×105 T cells) or C3H.SW (7×106 BM cells and pooled splenocytes and peripheral lymph node cells containing 2×106 CD8+ T cells) donors. For MHC-mismatched transplants, B6-WT or B6-STING−/− mice received 11.5Gy TBI and BALB/c-WT or BALB/c-STING−/− mice received 8.5Gy on day −1 and BM was injected 24 hours later with or without T cells from sex and age-matched BALB/c (7×106 BM cells and lymph node cells containing 1.7×106 T cells) or B6 (5×106 BM cells and lymph node cells containing 1.2×106 T cells) donors. For experiments utilizing positively selected T cell subsets, B6-WT or B6-STING−/− mice received 11.5Gy TBI on day −1 and BM was injected 24 hours later with or without positively selected T cells (see T cell enrichment) from sex and age-matched BALB/c (7×106 BM cells and 1.2×106 CD4+ T cells recombined with 0.6×106 CD8+ T cells, 1.8×106 CD4+ T cells alone, or 1.8×106 CD8+ T cells alone) donors. T cell depletion was performed using HO134 hybridoma supernatant (αThy1.2, 40% of final volume at 25×106 cells/ml) and rabbit complement (Cedarlane Labs) immediately prior to transplant. Mice were monitored 3x per week for weight loss and clinical score as previously described (49, 50). In brief, clinical signs of GVHD were scored on a scale from 0 to 2 for 5 parameters: weight loss, diarrhea, fur texture, posture, and alopecia. Mice exhibiting a clinical score greater than 6 were sacrificed and their death was recorded as the next day, in accordance with our animal protocols. For transplants utilizing the STING agonist DMXAA (InvivoGen), mice were injected once intraperitoneally with 25mg/kg of DMXAA dissolved in 5% NaHCO3 immediately following TBI. For transplants involving co-housing, WT and STING−/− recipients were co-housed for two weeks prior to transplant.
Generation of bone marrow chimeras
To generate bone marrow chimeras, B6-WT or STING−/− mice were irradiated with 12.5Gy split dose (6.25Gy x 2) on day −1 and day 0 and injected IV with 15×106 BM cells from congenic B6-CD45.1 or STING−/− mice. 3 months after congenic BMT, chimeric recipients were conditioned with 8.5Gy and given a second allogeneic BMT using LP/J donor cells.
T cell enrichment
CD4+ and CD8+ T cells were positively selected from lymph nodes of BALB/c donor mice using EasySep PE Positive Selection Kit (STEMCELL Technologies) according to the manufacturer’s instructions along with commercially purchased PE-conjugated antibodies targeting CD4 and CD8 described in table S1. Purity of enriched populations was routinely >90% as assessed by flow cytometry. Less than 1% of the non-desired T cell subsets were detected in the positively selected fraction.
Flow cytometry
At the indicated time points post-HSCT, peripheral blood, spleen, or pooled lymph nodes (mesenteric, inguinal, brachial, axillary, and cervical) were collected from transplant recipients. Lymphoid organs were prepared into single-cell suspensions and filtered through 200μm nylon mesh and PBMCs were separated using a standard Ficoll-Paque (GE Healthcare) density separation. 1×106 cells were stained in PBS containing 2% FBS and 0.1% NaN3 with commercial fluorescently-labeled antibodies purchased from BD Biosciences or Biolegend. For intracellular cytokine staining, splenic single cell suspensions were stimulated in complete media containing 10% FBS, PMA (20ng/mL), ionomycin (1μg/mL) and brefeldin A for 6 hours. After surface staining, cells were fixed using IC Fixation Buffer and permeabilized according to the manufacturer’s instructions (eBioscience). Permeabilized samples were then incubated with commercial fluorescently-labeled antibodies purchased from Biolegend for 1 hour. Flow cytometry data was analyzed using FlowJo version 10 software (TreeStar). All samples were run on a BD LSRFortessa Flow Cytometer. Antibodies used in this study are described in table S1.
Histopathology
At the end of experiments, colon, ear skin, or interscapular skin were collected, formalin-fixed, embedded in paraffin, sectioned, and stained with H&E. Stained sections were reviewed by a trained veterinary pathologist and subsequently analyzed in a blinded fashion using an aggregate scoring system adapted from Kaplan, D et al (51). Sections were given a pathology score of 0–2 (0=normal, 1=moderate, 2=severe) based upon amount of inflammation/infiltration, collagen deposition and dermal thickening for skin, and inflammation/infiltration, edema, mucosal thickening and crypt structure for colon. Scores were then aggregated to calculate an overall histopathology score (max score = 6 for skin, 8 for colon).
Intestinal organoid cultures
To generate intestinal organoid cultures, small intestines from B6-WT or B6-STING−/− were harvested, washed 15–20 times with PBS, incubated with Gentle Cell Dissociation Reagent (STEMCELL Technologies) for 15 minutes and then plated in Matrigel Matrix (Corning) with Intesticult Organoid Growth Medium (STEMCELL Technologies) supplemented with Penicillin and Streptomycin (100μg/mL each). For STING stimulation of organoids, DMXAA (100μg/mL) was dissolved in 5% NaHCO3, filter sterilized through a 0.2μm filter and added to organoid cultures along with media change.
Imaging
Slide and organoid imaging were performed using a Keyence BZ-X700 Inverted Microscope. Scale bars were added using ImageJ Software (NIH).
Peritoneal exudate cells
Briefly, cold PBS containing 2% FBS was injected into the peritoneal cavity of mice immediately following sacrifice using a 27-gauge needle and the cavity was gently massaged. The PBS containing cells was subsequently collected using a 25-gauge needle and washed twice before plating in complete media containing 10% FBS. PECs were stimulated with either the addition of DMXAA (100μg/mL) in 5% NaHCO3 or transfection of 2’,3’-cGAMP (6.7μg/mL, InvivoGen) complexed to Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
qPCR
Colon tissue was collected from mice 48 hours after aHSCT, flushed with PBS, cut longitudinally and disrupted in TRIzol Reagent (Life Technologies). For organoid cultures and PECs, media was removed, and cells were disrupted with cold TRIzol Reagent before collection. RNA was isolated using manufacturer’s recommendations and cDNA was reverse transcribed using qScript cDNA Supermix (QuantaBio). qPCR was run and analyzed using an ABI 7300 (Applied Biosystems). Samples were run in duplicate and normalized to GAPDH expression. Gene fold induction was calculated using the 2−ΔΔCt method. Primers were purchased from Sigma and are described in table S1.
Bead-based immunoassay
Serum was collected from B6-WT and B6-STING−/− recipients 6 weeks post-aHSCT with C3H.SW donors via cardiac puncture. Quantification of serum cytokines was performed using a LEGENDplex 13-panel mouse inflammation kit according to the manufacturer’s instructions (Biolegend). Samples were run on a BD Cytoflex flow cytometer and analyzed using LEGENDplex Software.
Statistical analysis
Numbers of animals per group are described in the figure legends. All measurements were taken from distinct samples. GraphPad Prism 7 was used for all statistical analyses. Significance of differences between two experimental groups were determined using two-tailed unpaired t test. For experiments comparing more than two groups, data was analyzed using a one-way ANOVA with a post-hoc Tukey’s multiple comparisons test. For experiments utilizing multiple t tests over time, p values were adjusted using the two-stage linear step-up procedure of Benjamini, Krieger and Yekutieli. For survival analyses, a Log-rank (Mantel-Cox) test was performed. Statistical tests performed are indicated in the figure legends. Non-significant p values obtained that were between 0.05 and 0.1 are presented to the reader while values >0.10 are presented as non-significant. Significance indicated by * p < 0.05, ** p < 0.01, *** p < 0.001, ns=non-significant. Data shown are means ± SEM.
Supplementary Material
Acknowledgments:
We thank the Sylvester Comprehensive Cancer Center Flow Cytometry Core for technical support, Norman H. Altman and Victor M. Delacruz for interpretation of histology slides, and Thomas R. Malek, Zhibin Chen, Brian Marples, and Natasa Strbo for their helpful discussions regarding the paper.
Funding: These studies were funded by the Sylvester Comprehensive Cancer Center [to R.B.L], NIH R01 [to R.B.L (EY024484-01)] and Kalish Family Foundation [to K.V.K.]. CSB was supported by an NIH F31 (F31CA216999) and an NIH F99 (F99CA245728).
Footnotes
Note: The publisher’s final edited version of this manuscript is available at https://stm.sciencemag.org/content/12/552/eaay5006.short
Supplementary Materials
Fig. S1. STING-deficient recipients exhibit greater lymph node cellularity versus WT recipients.
Fig. S2. Co-housing recipient WT and STING-deficient mice does not alter reduced GVHD in STING−/− recipients after MHC-matched aHSCT.
Fig. S3. C3H.SW donor T cells cause limited intestinal GVHD.
Fig. S4. Donor immune cell reconstitution is similar between B6-WT and B6-STING−/− syngeneic recipients.
Fig. S5. Altering the time and severity of conditioning abrogates the effect of STING deficiency on GVHD following MHC-matched HSCT.
Fig. S6. STING-deficient recipients also develop reduced GVHD after transplant of 10-fold higher numbers of T cells.
Fig. S7. Further activation of STING with a small molecule agonist at the time of MHC-matched aHSCT exacerbates early GVHD-induced weight loss.
Fig. S8. B6N-STINGHAQ/HAQ peritoneal cells have markedly reduced responsiveness to STING ligands.
Fig. S9. CD4+ T cells are not sufficient for exacerbated MHC-mismatched GVHD in STING-deficient recipients.
Fig. S10. Fewer STING-deficient APCs are apoptotic early after aHSCT.
Fig. S11. Donor Tregs are activated more efficiently in STING-deficient recipients.
Fig. S12. Recipient non-hematopoietic STING reduces the survival of recipient APCs early post-aHSCT.
Fig. S13. Gating strategy for the identification of live cells, singlets, and donor T cells to examine activation phenotype.
Table S1. Antibodies and qPCR Primers.
Data File S1. Primary data
Competing interests: B.R.B receives remuneration as an advisor to Kadmon Pharmaceuticals, Inc, Five Prime Therapeutics Inc, Regeneron Pharmaceuticals, Magenta Therapeutics and BlueRock Therapeutics; research support from Fate Therapeutics, RXi Pharmaceuticals, Alpine Immune Sciences, Inc, Abbvie Inc., Leukemia and Lymphoma Society, Childrens’ Cancer Research Fund, KidsFirst Fund and is a co-founder of Tmunity. All other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are available within the paper or supplementary materials. The BALB/c-STING−/− mice are available from R.B.L under a material transfer agreement with the University of Miami and the STINGHAQ/HAQ mice from L.J. under a material transfer agreement with Albany Medical College.
References and Notes:
- 1.Appelbaum FR, Haematopoietic cell transplantation as immunotherapy. Nature 411, 385–389 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Luznik L, O’Donnell PV, Fuchs EJ, Post-transplantation cyclophosphamide for tolerance induction in HLA-haploidentical bone marrow transplantation. Semin Oncol 39, 683–693 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ganguly S, Ross DB, Panoskaltsis-Mortari A, Kanakry CG, Blazar BR, Levy RB, Luznik L, Donor CD4+ Foxp3+ regulatory T cells are necessary for posttransplantation cyclophosphamide-mediated protection against GVHD in mice. Blood 124, 2131–2141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Copelan EA, Hematopoietic stem-cell transplantation. N Engl J Med 354, 1813–1826 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Zeiser R, Blazar BR, Pathophysiology of Chronic Graft-versus-Host Disease and Therapeutic Targets. N Engl J Med 377, 2565–2579 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Heidegger S, van den Brink MR, Haas T, Poeck H, The role of pattern-recognition receptors in graft-versus-host disease and graft-versus-leukemia after allogeneic stem cell transplantation. Front Immunol 5, 337 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Romling U, Galperin MY, Gomelsky M, Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol Mol Biol Rev 77, 1–52 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE, STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rice GI, Rodero MP, Crow YJ, Human disease phenotypes associated with mutations in TREX1. J Clin Immunol 35, 235–243 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S, Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol 6, 49–56 (2005). [DOI] [PubMed] [Google Scholar]
- 11.Barber GN, STING: infection, inflammation and cancer. Nat Rev Immunol 15, 760–770 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, Janssen EM, STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol 193, 6124–6134 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abe T, Barber GN, Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J Virol 88, 5328–5341 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le Bon A, Etchart N, Rossmann C, Ashton M, Hou S, Gewert D, Borrow P, Tough DF, Cross-priming of CD8+ T cells stimulated by virus-induced type I interferon. Nat Immunol 4, 1009–1015 (2003). [DOI] [PubMed] [Google Scholar]
- 15.Lindahl P, Gresser I, Leary P, Tovey M, Interferon treatment of mice: enhanced expression of histocompatibility antigens on lymphoid cells. Proc Natl Acad Sci U S A 73, 1284–1287 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF, Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol 174, 4465–4469 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Ishikawa H, Ma Z, Barber GN, STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia T, Konno H, Ahn J, Barber GN, Deregulation of STING Signaling in Colorectal Carcinoma Constrains DNA Damage Responses and Correlates With Tumorigenesis. Cell Rep 14, 282–297 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer JC, Bscheider M, Eisenkolb G, Lin CC, Wintges A, Otten V, Lindemans CA, Heidegger S, Rudelius M, Monette S, Porosnicu Rodriguez KA, Calafiore M, Liebermann S, Liu C, Lienenklaus S, Weiss S, Kalinke U, Ruland J, Peschel C, Shono Y, Docampo M, Velardi E, Jenq RR, Hanash AM, Dudakov JA, Haas T, van den Brink MRM, Poeck H, RIG-I/MAVS and STING signaling promote gut integrity during irradiation- and immune-mediated tissue injury. Sci Transl Med 9, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robb RJ, Kreijveld E, Kuns RD, Wilson YA, Olver SD, Don AL, Raffelt NC, De Weerd NA, Lineburg KE, Varelias A, Markey KA, Koyama M, Clouston AD, Hertzog PJ, Macdonald KP, Hill GR, Type I-IFNs control GVHD and GVL responses after transplantation. Blood 118, 3399–3409 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, Vogel SN, 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. J Biol Chem 287, 39776–39788 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, Rathinam VA, Monks B, Jin T, Xiao TS, Vogel SN, Vance RE, Fitzgerald KA, Mouse, but not human STING, binds and signals in response to the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid. J Immunol 190, 5216–5225 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ, Metchette K, Dubensky TW Jr., Gajewski TF, Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11, 1018–1030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rewcastle GW, Atwell GJ, Li ZA, Baguley BC, Denny WA, Potential antitumor agents. 61. Structure-activity relationships for in vivo colon 38 activity among disubstituted 9-oxo-9H-xanthene-4-acetic acids. J Med Chem 34, 217–222 (1991). [DOI] [PubMed] [Google Scholar]
- 25.Patel S, Blaauboer SM, Tucker HR, Mansouri S, Ruiz-Moreno JS, Hamann L, Schumann RR, Opitz B, Jin L, The Common R71H-G230A-R293Q Human TMEM173 Is a Null Allele. J Immunol 198, 776–787 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Patel S, Jin L, TMEM173 variants and potential importance to human biology and disease. Genes Immun, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matte CC, Liu J, Cormier J, Anderson BE, Athanasiadis I, Jain D, McNiff J, Shlomchik WD, Donor APCs are required for maximal GVHD but not for GVL. Nat Med 10, 987–992 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Zhang Y, Louboutin JP, Zhu J, Rivera AJ, Emerson SG, Preterminal host dendritic cells in irradiated mice prime CD8+ T cell-mediated acute graft-versus-host disease. J Clin Invest 109, 1335–1344 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Teshima T, Ordemann R, Reddy P, Gagin S, Liu C, Cooke KR, Ferrara JL, Acute graft-versus-host disease does not require alloantigen expression on host epithelium. Nat Med 8, 575–581 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Karimi MA, Bryson JL, Richman LP, Fesnak AD, Leichner TM, Satake A, Vonderheide RH, Raulet DH, Reshef R, Kambayashi T, NKG2D expression by CD8+ T cells contributes to GVHD and GVT effects in a murine model of allogeneic HSCT. Blood 125, 3655–3663 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shlomchik WD, Couzens MS, Tang CB, McNiff J, Robert ME, Liu J, Shlomchik MJ, Emerson SG, Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science 285, 412–415 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Moran AE, Holzapfel KL, Xing Y, Cunningham NR, Maltzman JS, Punt J, Hogquist KA, T cell receptor signal strength in Treg and iNKT cell development demonstrated by a novel fluorescent reporter mouse. J Exp Med 208, 1279–1289 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashouri JF, Weiss A, Endogenous Nur77 Is a Specific Indicator of Antigen Receptor Signaling in Human T and B Cells. J Immunol 198, 657–668 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ishikawa H, Barber GN, STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Canesso MCC, Lemos L, Neves TC, Marim FM, Castro TBR, Veloso ES, Queiroz CP, Ahn J, Santiago HC, Martins FS, Alves-Silva J, Ferreira E, Cara DC, Vieira AT, Barber GN, Oliveira SC, Faria AMC, The cytosolic sensor STING is required for intestinal homeostasis and control of inflammation. Mucosal Immunol 11, 820–834 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Koyama M, Kuns RD, Olver SD, Raffelt NC, Wilson YA, Don AL, Lineburg KE, Cheong M, Robb RJ, Markey KA, Varelias A, Malissen B, Hammerling GJ, Clouston AD, Engwerda CR, Bhat P, MacDonald KP, Hill GR, Recipient nonhematopoietic antigen-presenting cells are sufficient to induce lethal acute graft-versus-host disease. Nat Med 18, 135–142 (2011). [DOI] [PubMed] [Google Scholar]
- 37.Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, Vance RE, The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect Immun 79, 688–694 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swimm A, Giver CR, DeFilipp Z, Rangaraju S, Sharma A, Ulezko Antonova A, Sonowal R, Capaldo C, Powell D, Qayed M, Kalman D, Waller EK, Indoles derived from intestinal microbiota act via type I interferon signaling to limit graft-versus-host disease. Blood 132, 2506–2519 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fischer JC, Bscheider M, Gottert S, Thiele Orberg E, Combs SE, Bassermann F, Heidegger S, Haas T, Poeck H, Type I interferon signaling before hematopoietic stem cell transplantation lowers donor T cell activation via reduced allogenicity of recipient cells. Sci Rep 9, 14955 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Min CK, Maeda Y, Lowler K, Liu C, Clouthier S, Lofthus D, Weisiger E, Ferrara JL, Reddy P, Paradoxical effects of interleukin-18 on the severity of acute graft-versus-host disease mediated by CD4+ and CD8+ T-cell subsets after experimental allogeneic bone marrow transplantation. Blood 104, 3393–3399 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Ahn J, Konno H, Barber GN, Diverse roles of STING-dependent signaling on the development of cancer. Oncogene 34, 5302–5308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Truitt RL, Bortin MM, Rimm AA, Alloimmunization: induction of antileukemic reactivity without modification of anti-host reactivity in H-2-compatible mice. Transplant Proc 12, 143–146 (1980). [PubMed] [Google Scholar]
- 43.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA, Alegre ML, Gajewski TF, STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li T, Cheng H, Yuan H, Xu Q, Shu C, Zhang Y, Xu P, Tan J, Rui Y, Li P, Tan X, Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci Rep 6, 19049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curran E, Chen X, Corrales L, Kline DE, Dubensky TW Jr., Duttagupta P, Kortylewski M, Kline J, STING Pathway Activation Stimulates Potent Immunity against Acute Myeloid Leukemia. Cell Rep 15, 2357–2366 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, van der Goot FG, Turcatti G, Behrendt R, Ablasser A, Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Kwon D, Park E, Sesaki H, Kang SJ, Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) suppresses STING-mediated DNA sensing pathway through inducing mitochondrial fission. Biochem Biophys Res Commun 493, 737–743 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Wan YY, Flavell RA, Identifying Foxp3-expressing suppressor T cells with a bicistronic reporter. Proc Natl Acad Sci U S A 102, 5126–5131 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolf D, Barreras H, Bader CS, Copsel S, Lightbourn CO, Pfeiffer BJ, Altman NH, Podack ER, Komanduri KV, Levy RB, Marked in Vivo Donor Regulatory T Cell Expansion via Interleukin-2 and TL1A-Ig Stimulation Ameliorates Graft-versus-Host Disease but Preserves Graft-versus-Leukemia in Recipients after Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cooke KR, Kobzik L, Martin TR, Brewer J, Delmonte J Jr., Crawford JM, Ferrara JL, An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood 88, 3230–3239 (1996). [PubMed] [Google Scholar]
- 51.Kaplan DH, Anderson BE, McNiff JM, Jain D, Shlomchik MJ, Shlomchik WD, Target antigens determine graft-versus-host disease phenotype. J Immunol 173, 5467–5475 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.