

Abstract
Multiple modes of cell death have been identified, each with a unique function and each induced in a setting-dependent manner. As billions of cells die during mammalian embryogenesis and daily in adult organisms, clearing dead cells and associated cellular debris is important in physiology. In this Review, we present an overview of the phagocytosis of dead and dying cells, a process known as efferocytosis. Efferocytosis is carried out by macrophages and to a lesser extent by other ‘professional’ phagocytes (such as monocytes and dendritic cells) and non-professional phagocytes, such as epithelial cells. Recent discoveries have informed this process and how it functions to maintain tissue homeostasis, tissue repair, and organismal health. Here, we outline the mechanisms of efferocytosis, from the recognition of dying cells through to phagocytic engulfment and homeostatic resolution, and highlight the pathophysiological consequences that can arise when this process is abrogated.
Introduction
Cell death and the effective clearance of dying cells are fundamental processes that maintain homeostasis in multicellular organisms. In nearly all physiological and most pathological scenarios, cells participate in their demise by a programmed cascade of signaling events (“regulated” cell death)1 whereby damaged or obsolete cells die in a controlled manner and are replaced with new cells arising from stem cell progenitors2. Cell death is important for development; billions of cells are eliminated during mammalian embryogenesis and development in order to shape new structures and maintain organ function3,4. Large numbers of cells also die during the resolution of pathological events, including tissue damage and infections. Cell death must be carefully controlled; extensive damage, for example caused by heat, mechanical compression or osmotic pressure, can cause cells to undergo necrosis, releasing their intracellular contents to the surrounding milieu and leading to the activation of inflammatory immune pathways that can damage surrounding healthy cells and tissues.
Removal of cellular corpses is important in both homeostasis and disease. The engulfing of dead cells by professional phagocytes, a multistep process known as efferocytosis [G], allows multicellular organisms to recycle cellular components. When disposal of cell corpses is defective, autoimmune and other pathologies can arise (Fig. 1). Whereas the degradation and the recycling of a cell’s mass are common features in the clearance of any dead cell, some features of cell clearance are unique to a specific mode of cell death (Box 1) Dying cells can expose and secrete signals that attract phagocytes, favour their engulfment, or promote a return to tissue homeostasis depending on their mode of death. Different forms of cell death can also confer pro-inflammatory or anti-inflammatory signals through modulating macrophage activity following efferocytosis.
Figure 1. Efferocytosis is critical for tissue homeostasis.

Efferocytosis can be carried out by professional phagocytes (red boxes), such as macrophages and dendritic cells, or to a lesser extent by non-professional phagocytes (blue boxes) such as epithelial cells. Disruption of normal efferocytosis can contribute to the development of a wide range of pathologies (light grey boxes) across a variety of tissues. (dark grey boxes). COPD, chronic obstructive pulmonary disease; IPD, idiopathic pulmonary disease; SLE, systemic lupus erythematosus.
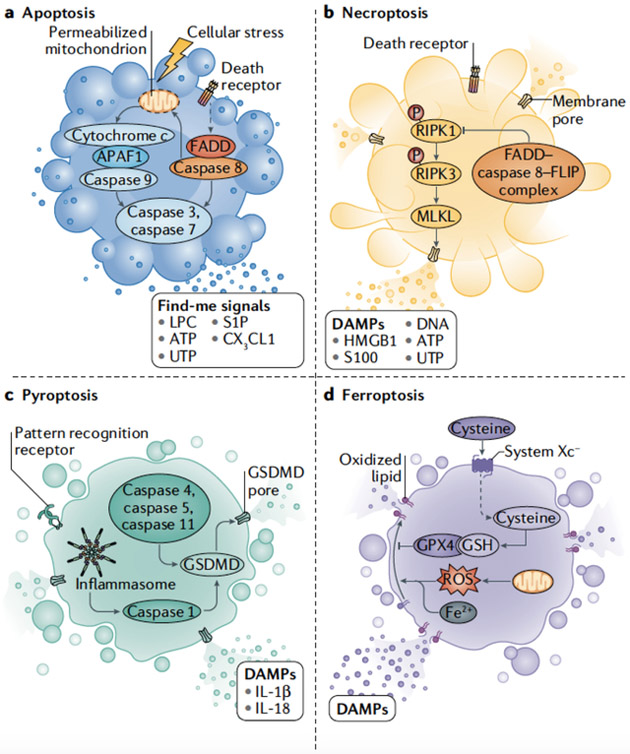
BOX 1: Modes of programmed cell death.
Different modes of cell death have unique activating stimuli and present different signaling moieties to the phagocyte, leading to efferocytosis and a variety of distinct physiological outcomes (see the figure).
a. Apoptosis
Apoptosis can be triggered by the activation of a mitochondrial pathway by cellular stress (intrinsic apoptosis) or through the activation of death receptors at the cell surface (extrinsic apoptosis). The Bcl-2 proteins regulate intrinsic apoptosis; anti-apoptotic Bcl-2 proteins (Bcl-2, Bcl-XL, Bcl-W, Mcl-1 and BFL-1) prevent uncontrolled apoptotic initiation, whereas pro-apoptotic Bcl-2 proteins (Bak, Bax and Bok) trigger mitochondrial outer membrane permeabilization (MOMP). Mitochondrial intermembrane proteins SMAC, Omi, and cytochrome c are released into the cytosol following MOMP. Cytochrome c activates apoptotic protease activating factor-1 (APAF-1), which in turn activates the serine protease caspase-9; active caspase-9 activates the executioner caspases, caspase-3 and caspase-7, which contribute to the archetypal features of apoptotic cells by cleaving cellular proteins246. Death receptors known to mediate extrinsic apoptosis include the tumor necrosis family members, including TNFR1, the Fas receptor (CD95) and the TRAIL receptors. Receptor ligation promotes recruitment of adaptor proteins, including FADD, which bind and activate caspase-8 by oligomerization. Caspase-8 cleaves and activates the executioner caspases, which can be inhibited by X-linked inhibitor of apoptosis (XIAP). Caspase-8 also cleaves the BCL-2 family protein BID, activating it to induce MOMP247 and releasing SMAC and Omi (as above). These proteins antagonize the function of XIAP, permitting executioner caspase activation and apoptosis.
b. Necroptosis
Necroptosis is a regulated form of necrosis that is also activated by extrinsic apoptotic receptors. Necroptosis is initiated through the activation of RIPK1, which binds and activates RIPK3 following autophosphorylation. RIPK3-mediated phosphorylation of the mixed-lineage kinase domain-like pseudokinase (MLKL) promotes its oligomerization and insertion into the plasma membrane, forming a membrane-disrupting pore, leading to death248. This process is inhibited by the activation of caspase-8, together with its apoptotic inhibitor c-FLIPL, and the proteolytic activity of this complex cleaves RIPK1 to prevent necroptosis without engaging apoptosis249.
RIPK3 and necroptosis can also be activated independently of RIPK1 by Toll-like receptors that engage the adapter protein TRIF, or by the activation of the viral sensor ZBP1250,251.
c. Pyroptosis
During bacterial infection, inflammatory caspases (namely caspase 4, caspase 5 and/or caspase 11) directly recognize lipopolysaccharide on intracellular, Gram-negative bacteria and trigger pyroptosis252-255. Alternatively, the inflammatory caspase, caspase-1, is activated by multiprotein inflammasomes. Inflammasomes generally contain a pattern-recognition receptor (PRR) that senses PAMPs or DAMPs, caspase-1, and the bridging molecule apoptosis-associated speck-like protein containing a CARD (ASC). The inflammasomes activate caspase-1 by oligomerization. Caspase-mediated cleavage of the pore-forming protein Gasdermin-D disengages its autoinhibitory domain, leading to insertion of the Gasdermin-D pore into the plasma membrane and subsequent plasma membrane permeabilization255. Active caspase-1 also proteolytically-processes the pro-inflammatory cytokines IL-1β and IL-18 into their active forms, which are subsequently released from pyroptotic cells. Pyroptosis clears pathogens by destroying their replicative niche and engaging the inflammatory response. Although beneficial under most circumstances, uncontrolled pyroptosis can lead to organ failure, sepsis, and death.
d. Ferroptosis
Free cytosolic iron can propagate lipid peroxidation via the Fenton reaction, leading to reactive intermediates that promote ferroptosis, which is characterized by membrane rupture as a result of high levels of lipid peroxides. Glutathione peroxidase 4 (GPX4) functions to reduce lipid peroxides in cellular membranes; GPX4 inhibition or a reduction in the levels of the GPX4 co-factor glutathione can therefore result in uncontrolled lipid peroxidation of poly-unsaturated fatty acids and ferroptosis256,257. Under pathological scenarios ferroptosis often accompanies other cell death routines; however, inhibition of apoptosis or necroptosis is not sufficient to inhibit ferroptosis.
Box 1 Figure.
There are some situations in which cell death and corpse disposal are uncoupled phenomena, meaning disposal does not immediately follow cell death. One such example is the shedding of dead intestinal epithelial cells from the villi tip to the intestinal lumen5, although recent evidence suggests efferocytosis of dead intestinal epithelial cells nevertheless occurs as well6. Likewise, cornification of the outermost layer in the epidermis constitutes an extreme instance where the presence of the cell’s remnants has a biological role, serving as a physical barrier for environmental insults7. Conversely, in the process of entosis [G], cells engulf and kill healthy neighboring cells. Although the responsible mechanisms are typically distinct from those of dead cell removal, shared aspects have been reported8-11. Nevertheless, in the vast majority of cases, dying cells are rapidly cleared by efferocytosis, and such uncoupled events are not considered further herein.
Here, we summarize our current knowledge of the mechanisms of efferocytosis, and how efferocytosis affects the physiology of the organism, including effects on inflammation and the adaptive immune response. Further, we consider the consequences of defects in this critical homeostatic mechanism.
Recognition of dying cells
In early studies, cell shrinkage preceding cell death was linked to a non-inflammatory mechanism of cell clearance, whereas cell swelling prior to death was associated with an inflammatory response12-14. These morphologies were associated with the processes of apoptosis and necrosis, respectively. Over the past decades, more molecularly-diverse programs for regulated cell death have been identified1, with each form greatly influencing the biological consequences of cell death (Box 1). Both apoptotic and non-apoptotic dying cells display and release molecular cues to signal to phagocytes and direct the subsequent phagocytic and immune response.
Find-me signals of apoptotic cells
Apoptosis is the archetypal form of ‘silent’ cell death (Box 1). Processing of the apoptotic cell by the actions of activated caspases and their substrates, encapsulation of the cell into apoptotic bodies, and its subsequent disposal and recycling by surrounding phagocytic cells prevents the release of pro-inflammatory cellular contents and inflammation. During apoptosis, the cell releases soluble signals into the milieu to attract macrophages and stimulate their scavenging potential. These ‘find-me’ signals are diverse, including modified membrane lipids, such as lysophosphatidylcholine (LPC)15 and sphingosine-1-phosphate (S1P); nucleotides including ATP and UTP16; and chemokines such as Fractalkine (encoded by CX3CL1)17. The importance of each find-me signal depends on a number of factors, including the type of phagocyte and dead cell18. Moreover, the variety of find-me signals suggests that there is inherent redundancy that ensures dead cells are recognized by macrophages.
LPC and S1P are apoptosis-specific find-me signals. During apoptosis, caspase-3 cleaves and thereby activates calcium-independent phospholipase A2, which then synthesizes LPC from phosphatidylcholine15. Similarly, some apoptotic cells upregulate the S1P mitogen-activated protein kinases SPK1 and SPK219,20, which phosphorylate the membrane lipid sphingosine to produce S1P. Although the mechanism governing S1P release from apoptotic cells remains obscure, LPC is released by the ATP-binding cassette transporter 1 (ABC-1) following its activation21.
In addition to their roles as chemotactic factors, find-me signals also modify phagocyte behavior when bound to their cognate receptors. As an example, the binding of S1P by S1P receptor (S1PR) on macrophages can protect them from cell death and promotes an autocrine signaling loop comprised of HIF1α, erythropoietin (EPO), EPO receptor, and PPARα, the latter of which upregulates the expression of receptors for dying cells22. Consequently, S1P may cause long-term potentiation of efferocytosis.
Signals from non-apoptotic dying cells
In contrast to apoptosis, plasma membrane integrity is compromised during execution of non-apoptotic cell death, and nearby cells are exposed to inflammatory signals released from the ruptured, dying cells. Cells infected with intracellular pathogens can release pathogen-associated molecular patterns (PAMPs) upon non-apoptotic cell death. PAMPs are pathogen-specific cues that engage pattern recognition receptors (PRR) on or in the phagocyte (Table 1); these have a variety of effects on macrophage function and immune activation that have been reviewed elsewhere23,24.
Table 1. Damage-associated molecular patterns released from necrotic cells.
Data from refs207,208,209,210,211,212,213,214,215,216. BAI1, brain-specific angiogenesis inhibitor 1; cGAS, cyclic GMP–AMP synthase; C1qR, C1q receptor; HMGB1, high-mobility group protein B1; IL-1R, IL-1 receptor; IL-18R, IL-18 receptor; PAMP, pathogen-associated molecular pattern; PRR, pattern recognition receptor; RAGE, receptor for advanced glycosylation end products; TIM, T cell immunoglobulin mucin receptor; TLR, Toll-like receptor.
| Origin | Molecule | Receptor |
|---|---|---|
| Intracellular Pathogens | PAMPs | PRR (cell surface, endosome, cytosol) |
| Nucleus | HMGB1 | TLR2, TLR4, RAGE |
| Histones | TLR2, TLR4, TLR9 | |
| Nucleus and mitochondria | DNA | TLR9, cGAS, AIM2, ZBP1 |
| Cytosol | RNA | TLR3, TLR7, TLR8 |
| ATP, UTP | P2Y2R | |
| Uric acid crystals | NLRP3 | |
| S100 | RAGE | |
| Inflammasome | IL-1β, IL-18 | IL-1R, IL-18R |
| IL-1 | IL-1R | |
| IL-33 | ST2 | |
| Cell surface | Phosphatidylserine exposure | TIM1, TIM4, BAI1, Stabilin 2, CD300f, RAGE, MerTK, Axl, Integrin |
| Endoplasmic Reticulum | Calreticulin | CD91 |
| C1q | C1qR | |
| ICAM3 | CD14 |
Unlike PAMPs, which are derived from microbes, damage-associated molecular patterns (DAMPs) are of cellular origin and can be liberated upon cell death. DAMPs trigger inflammatory responses, and may also serve as chemoattractants for macrophages. DAMPs are metabolically diverse entities, including genomic and mitochondrial DNA, nuclear proteins (HMGB, histones)25, cytoplasmic proteins (S100), cytokines (IL-1α, IL-33, IL-36), and other small molecules (ATP, UTP, uric acid crystals) (Table 1)26. In addition, inflammasome [G] -mediated caspase-1 activation generates inflammatory cytokines IL-1β and IL-18 during pyroptosis (see Box 1) that lead to inflammatory immune activation after cellular demise.27 Below, we review the relevance of DAMPs during efferocytosis, the ability of DAMPs to modulate inflammation, and specific DAMPs and their effects.
DNA as a DAMP.
Several mechanisms ensure low DNA burden following apoptotic death and contribute to its immune-silent phenotype. In healthy cells, caspase-activated DNase (CAD) exists in complex with its inhibitory chaperone ICAD and remains constitutively inactive in the cytosol28,29. Active caspase-3 cleaves ICAD28,30, promoting CAD homodimerization, nuclear translocation, and DNA hydrolysis between nucleosomes. Nuclear pieces are then neatly packaged with cytoplasm into apoptotic bodies that are eventually digested during efferocytosis31. In contrast, nuclear and mitochondrial DNA (mtDNA), as well as pathogen-derived DNA molecules in those cells dying due to an infection, can be released to the extracellular environment from non-apoptotic dying cells. Toll-like receptor 9 (TLR9) is activated by unmethylated CpG sequences such as those found in mtDNA or bacterial DNA (Table 1), and activation of TLR9 triggers downstream inflammatory responses. circulating DNA DAMPs can accumulate in the body in cases where non-apoptotic cell death is widespread; for example, mtDNA was found to be elevated in the plasma of trauma patients32, likely as a result of injury-induced cell death.
DNA in the extracellular environment is processed by DNase-I33, while DNase-III (also known as TREX1) clears cytoplasmic DNA34, and DNase-II processes DNA from dying cells in the phagocyte’s lysosomes to help maintain negligible levels of DNA following efferocytosis. Should DNA escape to the cytosol, it can be recognized by cytosolic DNA sensors35, including cyclic GMP-AMP synthase (cGAS) and the inflammasome component AIM2. cGAS is activated upon cytosolic DNA binding and subsequently catalyzes a reaction between GTP and ATP to form cyclic GMP-AMP (cGAMP)36. The newly synthesized cGAMP binds to and activates the stimulator of interferon genes protein (STING), leading to TANK binding kinase 1 (TBK1)-dependent phosphorylation of the interferon regulatory factor IRF337. These events trigger the IRF3-mediated activation of a Type I interferon response.
DNase-II-deficient or DNase III-deficient mice die during embryogenesis and this embryonic lethality can be prevented if the response to misplaced DNA is abrogated through deletion of cGAS, STING, or the Type-I interferon receptor (IFNAR)38-41. It is possible that these DNases function to limit cytosolic DNA following efferocytosis during development, thereby preventing this lethal interferonopathy, although how the DNA of engulfed corpses might be released from the phagosome or lysosome to become cytosolic, triggering such responses, remains unknown.
Similarly, recognition of cytosolic DNA by AIM2 causes AIM2 to recruit and activate caspase-1, resulting in IL-1β processing and release, contributing to inflammation42,43. Again, when and how defects in the clearance of DNA during efferocytosis may engage AIM2 remains unclear.
Protein DAMPs.
High mobility group protein B1 (HMGB1) is a nuclear protein that binds to DNA and assists replication, repair and transcription44,45,46. Although some HMGB1 can be released to the extracellular milieu under steady state conditions47 or during apoptosis48, it is predominantly released during forms of immunogenic cell death25. Efficient efferocytosis can thus limit the release of HMGB1. Based on its redox status, HMGB1 can function as a chemotactic agent (reduced) or as an inflammatory agent (oxidized)49. In its reduced form, HMGB1 can prevent the induction of immune tolerance [G] to antigens associated with the dying cell48. Reduced HMGB1 may bind to TLRs or the receptor for advanced glycation end products (RAGE) (Table 1)50, leading to immune cell activation and cytokine production. Recently, binding of reduced HMGB1 to the chemokine receptor CXCR4 was observed during tissue regeneration following injury51, highlighting the possible importance of this molecule in the response to dying cells. S100 proteins can also be released from dying cells52, and efferocytosis can limit the release of these proteins from dying cells. Again, TLRs and RAGE appear to be the primary receptors on macrophages that promote inflammatory activation in response to S100 proteins.
IL-1-family cytokines, such as IL-1α, IL-33 and IL-36, are released from non-apoptotic dying cells and lead to activation of the MyD88-NF-κB axis (Table 1)53,54, resulting in a sustained inflammatory response. A compelling case has been made that IL-1 family members represent the ‘true’ DAMPs that signal inflammatory immune responses upon non-apoptotic forms of cell death55. Like other DAMPs, these proteins are released only when the plasma membrane of a cell is compromised. However, unlike HMGB1 and S100 proteins, each IL-1 family cytokine is controlled at several levels, including intracellular processing and the existence of specific receptor antagonists that regulate the extent to which the cytokine acts if released. For example, mice or humans lacking the IL-36 receptor antagonist display a greatly exacerbated inflammatory response to tissue injury (reviewed in55).
Nucleotides.
ATP and UTP can act as find-me signals and can be actively secreted by the dying cell (Table 1)56. This process of ATP and UTP release is governed in apoptotic cells by Pannexin-1 (PANX1), a plasma transmembrane protein that forms constitutively inactive hexameric channels57. During apoptosis, PANX1 is activated by caspase cleavage57,58. Inhibition of PANX1 channel function does not affect cell death but does limit macrophage recruitment to the apoptotic cell58,59. Necroptotic, pyroptotic, and other necrotic cells60 passively release nucleotides60; a PANX1-mediated mechanism for the release of nucleotides was initially proposed in pyroptosis61, although this mechanism is under debate59. These nucleotides are recognized by purinergic G-protein coupled receptors on phagocytes to trigger the find-me response62.
Display and binding of eat-me signals
In addition to generating find-me signals, dying cells display ‘eat-me’ signals on their cell surface (Fig. 2a). These eat-me signals allow phagocytes to discriminate dying cells from their healthy neighbors, which bear ‘don’t-eat-me’ signals (Fig. 2B).
Figure 2. Cell surface signals regulating efferocytosis.

Cells display proteins and lipids to communicate with phagocytes. These protein signals can be “eat-me” signals or “don’t eat me” signals, and interact with phagocytes via receptors on the phagocyte cell membrane. A) Dying cells expose a variety of signals on their surface that serve as “eat-me” signals for surveying phagocytes. A primary signal exposed on dying cells is the externalization of phosphatidylserine (PS). PS can interact with a variety of receptors on the surface of the phagocyte including TIM1, TIM4, BAI1, RAGE, the CD300 family, and the Stabilin proteins. Additionally, signaling can occur through receptors including MerTK, via adaptors including Gas6 and protein S, or through avβ5 integrin via MFGE-8. Calreticulin expressed on the surface of the dying cell can act as an eat-me signal through binding to CD91 expressed on the phagocyte outer membrane. B) Healthy cells decorate their plasma membranes with “don’t-eat-me” signals that protect them from phagocytic targeting. Don’t-eat-me signals include CD47 which is recognized by SIRPα on the phagocyte surface, and CD31, which homodimerizes with CD31 on phagocytes. Similarly, CD24 on viable cells engages Siglec-10 on phagocytes to inhibit engulfment. Signaling in each case occurs through SHP1/2 and suppression of pathways required for phagocytosis, including actin remodeling. Class I MHC (MHC-I) can signal through LILRB1 to mitigate phagocytic activation, although the signaling cascade is not fully delineated. In combination, these signals regulate the initiation of efferocytosis.
Phosphatidylserine.
One feature shared across all forms of cell death is the loss of phospholipid asymmetry in the plasma membrane. In most healthy cell types, phosphatidylethanolamine and phosphatidylserine reside in the inner leaflet of the plasma membrane, whereas phosphatidylcholine and sphingomyelin are restricted to the outer leaflet. The generation and maintenance of membrane lipid asymmetry requires the ATP-dependent action of the flippase [G] ATP11, which confines phosphatidylserine to the inner leaflet of the plasma membrane and restricts its lateral mobility in healthy cells63. During apoptosis, the inactivation of ATP11 by caspase-3 cleavage, together with the caspase-3-dependent activation of the scramblase [G] XK-related protein 8 (XKR8),64 promotes the exposure of phosphatidylserine on the surfaces of apoptotic cells. Additionally, this loss of phospholipid asymmetry can be achieved by the activation of the pore-forming protein MLKL [G] during necroptosis65,66 (see Box 1). In other forms of cell death, the externalization of phosphatidylserine also contributes to the engulfment of the dead cells60, although how it is exposed is less clear. Loss of intracellular ATP upon membrane rupture, such as in ferroptosis65,67, or the activation of pore-forming proteins such as gasdermins [G] in pyroptosis,60 may lead to a loss of ATP-dependent ATP11 activity and passive diffusion of phospholipids to the outer leaflet.
Phosphatidylserine exposure can also occur transiently in healthy cells through the action of the calcium-activated scramblase, TMEM16F68,69. Cells with such transient phosphatidylserine exposure are not engulfed; in contrast, healthy cells lacking functional ATP11 constitutively expose phosphatidylserine and are rapidly engulfed by macrophages10. This finding suggests that ATP11 actively prevents engulfment, perhaps via its ability to restrict the lateral mobility of phosphatidylserine even when it is exposed.
Several biologically redundant systems for phosphatidylserine recognition exist; however, not all of them are typically expressed or engaged on a given phagocytic cell70,71. Surface receptors for phosphatidylserine include T-cell immunoglobulin mucin-1 (TIM1) and TIM4, brain-specific angiogenesis inhibitor 1 (BAI1), stabilin-2, and members of the CD300 family. Alternatively, soluble bridging proteins can mediate the indirect binding of phagocyte surface receptors such as integrins (bridged by milk fat globule-EGF factor 8 (MFG-E8)) and TAM receptors (for example MERTK, which is bridged by dimers of Protein S and GAS6) to phosphatidylserine exposed on the surface of the dying cell72. The CD300 family member, CD300f, appears to have distinct functions depending on the phagocyte; on macrophages it promotes engulfment, while on dendritic cells it inhibits engulfment of dead cells73. The class B scavenger receptor CD36, a well-characterized lipid-binding receptor with preference for anionic phospholipids, likewise recognizes and binds exposed phosphatidylserine on dying cells74,75.
Phosphatidylserine not only functions as a link between the dying cell and the phagocyte, but its binding to these receptors also triggers an anti-inflammatory response mediated by the secretion of IL-10, TGFβ, and prostaglandins, and the active inhibition of the production of pro-inflammatory cytokines, including TNFα and IL-1β76-80.
Other eat-me signals.
While phosphatidylserine is a potent and well-characterized eat-me signal, other signals can have roles in the recognition and engulfment of dying cells. Exposed LPC on the plasma membrane of dying cells can bind to IgM, which in turn binds to Fc receptors on phagocytes, including macrophages81. Hence, LPC appears to function as both a find-me and eat-me signal. Proteins present in the lumen of the endoplasmic reticulum (ER) such as calreticulin can be exposed at the plasma membrane of dying cells and serve as an eat-me signal in the absence of don’t-eat-me signals82 (Fig. 2a). Calreticulin is recognized by phagocytes via the receptor LRP1 (also known as CD91) in coordination with complement component C1q and mannose-binding lectin (MBL)82,83.
Don’t eat me signals.
Healthy cells express don’t-eat-me signals that prevent engulfment even when eat-me signals are exposed. Like eat-me signals and receptors, there appears to be extensive redundancy in this inhibition of engulfment (Fig. 2b). Two examples of don’t-eat-me signals are the cell surface proteins CD47 and CD24, which are recognized by the receptors SIRPα and SIGLEC-10, respectively, on macrophages84,85 (Fig. 2B). Similarly, homotypic interaction of CD31 (on healthy cells and macrophages) appears to also prevent engulfment86. Finally, class I MHC molecules on healthy cells engage the inhibitory receptor, LILRB1, preventing engulfment and inhibiting expression of inflammatory mediators87. In each case, it appears that the inhibition of engulfment upon recognition of these don’t-eat-me singles is mediated by the protein phosphatases, SHP1 and SHP288.
Disruption of this active inhibition of engulfment can promote the uptake of healthy cells, and such disruption is under investigation as potential anti-cancer therapies. Approaches to blockade of the don’t-eat-me signal include antibodies, directed against the signal or receptor as well as small molecules that bind specific receptors89-91. Importantly, upon disruption of don’t-eat-me signals, cancer cells are more likely to be engulfed than normal, healthy cells. Such engulfment appears to be promoted by calreticulin as the eat-me signal92. Why calreticulin might be preferentially exposed on the cell surface in many cancers remains unclear.
Since don’t-eat-me signals prevent engulfment, even when eat-me signals are present, it follows that dying cells must lose this signal if they are to be effectively cleared by efferocytosis. However, very little is known about how this occurs. One study has suggested that during apoptosis, the levels of CD31 and CD47 decline in a caspase-dependent manner, which appears to be a consequence of active shedding of these proteins in released microparticles93. The extent to which this occurs in different cell types undergoing apoptosis, and how the disruption of these and other don’t-eat-me signals occurs during other forms of cell death remain unresolved.
Mechanisms of engulfment
Uptake of dying cells
Once the phagocyte recognizes a dying cell, engulfment of the cell corpse requires rapid plasma membrane reorganization and synthesis to allow for effective phagocytic internalization of the dead cell94. A dynamic mesh of cortical actin fibers lying underneath the plasma membrane allows for phagocyte motility and environmental sampling. Upon recognition of a dying cell, the phagocyte initiates actin remodeling that allows for invagination and localized extravagation of the plasma membrane and ultimately formation of the phagosome. The signaling mechanisms that bridge receptor ligation to activation of actin remodeling and associated pathways, vary depending on the receptor engaged but generally require the orchestrated activation of kinases (such as those from the Srk, Syk and protein kinase C families) and inactivation of phosphatases including SHP-1, processes that have been well reviewed previously95. During efferocytosis, two primary mechanisms leading to the reorganization of actin exist and both converge on a key regulator, the Rho family small GTPase Rac1. In the first system, Rac1 activation is mediated through LDL Receptor Related Protein 1 (LRP1) and the adapter protein GULP96; however, the precise mechanism as to how LRP1 and GULP activate Rac1 is not known. The second system leading to Rac1 activation is dependent on the guanine nucleotide exchange factor (GEF) DOCK180 and the phagocytic regulatory protein engulfment and cell motility protein (ELMO). Following receptor ligation by the dying cell, another GEF called TRIO loads GTP onto the small GTPase RhoG, leading to the recruitment of ELMO18,97-99. ELMO is then able to interact with the SH3 domain of DOCK180100. Together, the DOCK180-ELMO complex serves as a GEF for Rac1, leading to Rac1 activation. Once activated via either system, Rac1 is subsequently able to direct localized actin polymerization necessary to coat or grasp the cargo by activating nucleation-promoting factors of the WASP family, SCAR and WAVE, which recruit the ARP2/3 complex and function together to establish an actin nucleation core95. In addition to forming a nucleation core for de novo actin polymerization, the ARP2/3 complex binds established actin filaments, allowing for new actin synthesis while maintaining actin networking and branching, processes that are essential for phagosome formation101.
While actin polymerization is a critical component of phagosome formation and efficient capturing of the dead cell, actin depolymerization is of equal importance for the scission of the phagosome from the plasma membrane. The processes of phagosome sealing is highly dependent on various phosphoinositides including PtdIns(3,4,5)P3 which activates Rho-family GAPs leading to deactivation of the GTPases including Rac1, resulting in depolymerization102. A coordinated effort between actin depolymerization and dynamin exists to promote scission of the phagosome Dynamin-2 also directs downstream trafficking of the now complete early phagosome103.
Degradation by lysosomes
Following recognition and entrapment of the dying cell, the phagosome and the cell corpse are destined for a well-orchestrated destructive end. The phagosome fuses with lysosomes, which contain a large variety of proteases, nucleases, and lipases that digest the phagosome cargo. This fusion is promoted or impaired by modifications of the phagosome, as discussed below.
The phagosome containing the cell corpse is targeted to lysosomes through a multistep maturation process, which begins immediately following the formation of the phagosome upon dynamin-dependent membrane scission and is marked by multiple biochemical changes at the phagosome membrane. These alterations to the phagosomal membrane and the cargo contained within it are facilitated by vesicular trafficking events to and from the phagosome. Control of these processes is governed by the Rab GTPase family of proteins (Fig. 3a), which alternate between a GTP-bound (active) and a GDP-bound (inactive) state 104 Following activation, Rab proteins associate with effector molecules, the intracellular functions of which include motor-driven trafficking, vesicle fusion, and signaling that can promote ‘Rab conversion,’ and the activation of other downstream Rab GTPase family members105.
Figure 3. Phagocyte processing of a dying cell.

Upon engulfment by a phagocyte, a dying cell progresses through a canonical program leading to the degradation of the dead cell by the lysosome. Additional modifying pathways can be engaged that alter the degradative process and subsequent downstream immune signaling pathways. A) Efferocytosis of dying cells is a multistep process, involving initiation of phagocytosis, maturation of the phagosome, degradation of phagolysosome contents, and the resultant downstream effects on physiological outcomes. B) The process of LC3-associated phagocytosis (LAP) is characterized by recruitment of LC3 to the phagosome membrane. Following engagement of phagocytosis, a PI3-kinase complex containing Rubicon, UVRAG, Beclin1, VPS34, and VPS15, is assembled at the phagophore. Assembly of this PI3-kinase complex leads to the recruitment and activation of the LC3 ligation machinery. Ligation of LC3 to the phagosome lipid membrane promotes rapid maturation and lysosomal fusion. This expeditious process leads to efficient clearance of the dead cell and immune silence. If LAP is abrogated, there is a delay in phagosome maturation and lysosomal fusion. The impaired clearance of the dying cell leads to inflammatory immune activation through processes that are, to date, not fully understood.
Early phagosomes acquire RAB5, which organizes endocytic trafficking and phagosome biogenesis during maturation106. RAB5 is critical to phagosome development as abrogation of its function leads to defects in maturation, preventing the formation of late phagosomes107. The precise mechanisms that initiate the early recruitment of RAB5 to the phagosome are not well understood106. RAB5 stimulates the fusion of nascent phagosomes with early endosomes (also known as sorting endosomes), and recruits and activates multiple effector proteins, including early endosomal antigen 1 (EEA1), and the vacuolar fusion proteins MON1A and MON1B, a process which has been reviewed elsewhere108,109.
The class III PI3 kinase VPS34 is recruited to RAB5-positive early phagosomes, where it catalyzes the formation of the signaling lipid phosphatidylinositol 3-phosphate (PI3P) from phosphatidylinositol110. Both VPS34 and PI3P are required for the optimal progression of phagosome maturation. The catalytic activity and targeting of VPS34 to the membrane of the early phagosome is enhanced by the serine/threonine kinase VPS15 108,110. VPS15 forms a complex with VPS34, and also binds directly to active RAB5111. The function of VPS34 is considered further below, in our consideration of LC3-associated phagocytosis.
Early phagosomes transition to late phagosomes, which are defined by their acquisition of distinct biochemical markers, including the small GTPase RAB7, and the concomitant loss of early markers such as RAB5. Late phagosomes are more acidic than early phagosomes due to increased proton pumping into the phagosome lumen that is mediated by the multimeric protein complex vacuolar ATPase (V-ATPase), which translocates H+ across endosomal and phagosomal membranes (Fig. 3a)112,113.
RAB7 and its effectors are vital for the successful maturation of the phagosome, as inhibition of RAB7 activation blocks phagosome–lysosome fusion and results in failure to acidify the phagosome108,114. Two RAB7 effector proteins, RAB7-interacting lysosomal protein (RILP) and oxysterol-binding protein–related protein 1 (ORPL1), accumulate on late endosomes, phagosomes, and lysosomes114. Through their direct association with the molecular motor dynein–dynactin, these proteins coordinate microtubule-dependent vesicular trafficking of RAB7-positive compartments114. RILP, dynein, intact microtubules, and lysosome-associated membrane protein 1 (LAMP1) and LAMP2 are required for the fusion of late phagosomes with the lysosomal compartment115. LAMP-1 and LAMP-2 are heavily glycosylated membrane proteins that preserve lysosomal membrane integrity and are essential for facilitating phagosome-lysosome fusion through interaction with RAB7 115.
The direct fusion of the mature phagosome with a lysosome is coordinated in part by the formation of a Ca2+-dependent SNARE complex [G], composed of VAMP7 and syntaxin 7 (Fig. 3a)116,117. Both syntaxin 7 and VAMP7 are recruited to phagosomes, and abrogation of syntaxin 7 function inhibits phagosome–lysosome fusion118. After fusion, the newly formed phagolysosome [G] can be defined by its high acidity (pH 4.5–5.0) and the presence of active cathepsins [G]. Acidification, which itself promotes degradation of the internalized cell corpse, is also required for the activation of lysosomal acid hydrolases. These hydrolytic enzymes mediate the potent destructive capacity of the lysosome in concert with other effectors, including oxidants, cationic peptides, and lipases (Fig. 3a)119.
Modifications to phagosome maturation
LC3-associated phagocytosis.
The phagosome can be modified to either facilitate or impair its maturation and thus influence degradation of phagosome contents following its fusion with lysosomes. One such modification occurs during LC3-associated phagocytosis (LAP) [G] (Fig. 3b). In LAP, a subset of proteins required for canonical autophagy, in combination with regulators unique to LAP, enable the conjugation of the microtubule-associated protein 1A/1B light chain 3 (LC3) family [G] to the phagosome membrane upon phagosome sealing (Fig. 3b)120-124. LAP is dependent on the protein Rubicon and reactive oxygen species (ROS) generated by the nicotamide adenine dinucleotide phosphate oxidases (NADPH oxidase-2, also known as NOX2, in macrophages), both of which are completely dispensable for canonical autophagy121. A subset of the autophagy proteins (ATGs) are also required for LAP (Fig. 3b). The roles of autophagy-related proteins in phagocytosis have been reviewed elsewhere123,124.
LAP is engaged in response to certain phagocytic cargo, including dying cells123, although the precise signaling events that trigger this process remain obscure. VPS34 is recruited to the phagosome as part of a complex that contains Rubicon, Beclin-1, UVRAG, and VPS15, all of which appear to be required for PI3K activity of the LAP complex. The generation of PI3P on the phagosome membrane recruits the LC3-conjugation machinery, and abrogation of LC3 lipidation at the membrane impairs phagosome maturation and lysosome-mediated degradation (Fig. 3B)125,126. In autophagy, LC3 family proteins promote autophagosome–lysosome fusion as such fusion is impeded by the depletion of LC3127. The RAB7 effector protein FYCO1 interacts with LC3 and PI3P and facilitates autophagosome–lysosome fusion128. Similarly, LC3 on the phagosome promotes phagosome–lysosome fusion to form the phagolysosome. A Rubicon homolog, PLEKHM1, interacts with both RAB7 and LC3 and putatively facilitates phagosomal and endosomal trafficking129,130. These processes, which are involved in lysosomal targeting and fusion, are likely to be common to autophagy and phagocytosis. To date, it is not believed that the ability to conjugate LC3 to the phagosome directly affects the functional capacity of the lysosome; the impaired maturation and degradation observed in LAP-deficient cells and animals has not been associated with a direct lysosomal defect125,126.
Liver X receptors.
Other pathways with roles in efferocytosis include the liver X receptors (LXR s) and the peroxisome proliferator-activated receptors (PPARs), nuclear receptor families that regulate genes involved in lipid metabolism and transport131-134. Upon engulfing a dying cell, a phagocyte can effectively double its lipid content, requiring an appropriate transcriptional response that is mediated by these nuclear receptors. LXR deficiency in macrophages results in their inability to clear apoptotic thymocytes, but does not alter engulfment of inert beads or other substrates135. Likewise, mice that are deficient in LXRs accumulate dead cells in multiple tissues, due to a failure in efferocytosis in vivo135. Pharmacologic LXR agonists improve efferocytosis and clearance by stimulating the expression of receptors, including MerTK, both in cell culture and in vivo136. In addition, engagement of LXRs during efferocytosis induces the expression of the cholesterol transporter, ABCA1, which effluxes excess cholesterol137, and genes involved in lipid oxidation by mitochondria, permitting efficient catabolism of the excess lipids137,138.
Similar to LXRs, the PPAR family affects efferocytosis. Expression of PPARγ occurs in response to engulfment of apoptotic cells and remains elevated until efferocytosis resolves139. In concert with activation of LXRs, PPARγ activation has been shown to enhance efferocytosis by macrophages135. Indeed, inhibition of PPARγ using a pharmaceutical antagonist resulted in impaired efferocytosis139.
Resolution of phagocytosis
Once the phagosome has fused with lysosomes and its cargo has been degraded, a resolution phase restores homeostasis within the phagocyte, allowing further phagocytosis. Since dying cells are not the only phagocytic cargo, the outcome of phagocytosis can vary dependent upon what cargo is internalized. In the context of efferocytosis, some of the components of the cell corpse can be recaptured and recycled for use by the phagocytic cell following lysosomal degradation. Sugars, amino acids, lipids and nucleotides are recycled to replenish cellular stores and can potentially be used as building blocks and an energy source by the phagocytic cell123. The internalization and degradation of cargo can also influence the activation of signaling pathways; for example, DNA that escapes degradation can activate the DNA sensing cGAS-STING pathway and lead to the production of type I interferons (IFN), as discussed above. These events can lead to the loss of immune tolerance to the apoptotic corpse and can potentially result in autoimmunity125.
Rapid maturation and generation of the phagolysosome and efficient degradation of the cell corpse is facilitated by LAP (as described above) and promotes the production of anti-inflammatory cytokines including IL-10, a process that promotes immune silence (Fig. 3B). In contrast, disruption of LAP decreases phagosome maturation, leading to activation of inflammatory signaling pathways and the production of pro-inflammatory mediators including IL-1β and IL-6 (Fig. 3B) 121.. The exact process leading to this switch in cytokine production in the absence of LAP has not been well defined. Furthermore, the failure to efficiently degrade the cell corpse in LAP-deficient phagocytes may result in leakage of phagosome contents, such as DNA, into the cytosol, which in turn induces type I interferon production via sensing by STING140. STING-dependent IFN production by tumor associated macrophages in LAP-deficient mice promoted T cell-mediated anti-cancer immunity in several cancer models140.
Following efferocytosis, the phagocyte must restore the functions of cytoskeletal components, such as actin and microtubules, to ensure that its phagocytic capacity is maximized for further phagocytic events. This restoration of function has not been well-studied compared to the upstream processes of phagocytosis. However, some details can be inferred from studies evaluating effectors such as RAB27A in the events leading to phagocytosis. RAB27A negatively impacts phagocytosis by prolonging the actin-coating of nascent phagosomes, leading to impaired transition to the phagosome sealing stage141. Thus, RAB27A and similar effectors could be critical for the restoration of actin and other cytoskeletal networks following completion of phagocytosis. However, it is clear that the restoration of the cytoskeleton following phagocytosis is an ATP-dependent process and likely requires multiple components142. An additional aspect of recovery is the recycling or continued expression of membrane receptors to recognize subsequent cargo. Impaired recycling results in decreased membrane expression of receptors such as TREM2 and TLR4, which recognize a variety of amyloids and pathogen products respectively126,143, and may also decrease the membrane expression of receptors that recognize dying cells. This decrease in available surface receptors may impair the endocytic and phagocytic capacity of the phagocytic cell, suggesting that these restorative events are critical for ensuring the phagocyte can continue recognizing and clearing phagocytic cargo.
Efferocytosis and disease
The fact that so much redundancy in the recognition of dying cells is built into efferocytosis highlights its importance in pathophysiology. Indeed, many autoimmune and inflammatory pathologies are associated with defects in this process, with uncleared or improperly cleared cellular corpses resulting in inflammation, exacerbated tissue damage, and organ dysfunction. However, defects in other aspects of the efferocytotic machinery can result in increased protection against some pathologies.
Systemic pathology
The most common autoimmune disorder associated with defective efferocytosis is systemic lupus erythematosus (SLE), a chronic, systemic autoimmune pathology that affects multiple organ systems, such as the lung, kidneys, skin, and central nervous system (CNS). Although it is rare to observe uncleared dead cells when examining healthy tissue under the microscope, dead cells can often be observed in the blood, skin, and lymph nodes of patients with SLE, and disease severity is strongly correlated with defective efferocytosis in vitro and accumulation of dead cells in vivo 144,145. Patients with SLE also display elevated levels of circulating autoantigens, such as extracellular DNA, which bind autoantibodies to form immune complexes that accumulate or are deposited in the glomerular and vessel walls of the kidney146,147.
Defects at nearly every point of the efferocytosis pathway are implicated in the pathogenesis of SLE (Table 2). Mice with genetic deletions that abrogate components of find-me signaling pathways (for example, production of S1P), eat-me signaling pathways (for example, TIM4, MFG-E8, Protein S, MerTK, and C1q), and in components required for the processing of engulfed dead cells (LXRs, PPARs, ABCA1, and Rubicon) all display a progressive SLE-like disease (see Table 2 for references).
Table 2. Diseases associated with aberrations in efferocytosis pathways.
ABCA1, ATP-binding cassette transporter 1; BAI1, brain-specific angiogenesis inhibitor 1; C1qa, complement C1q subcomponent subunit A; EAE, experimental autoimmune encephalomyelitis; ELMO1, engulfment and cell motility protein 1; GVHD, graft-versus-host disease; IBD, inflammatory bowel disease; LRP1, LDL receptor-related protein 1; LXR, liver X receptor; MFGE8, milk fat globule-EGF factor 8; MS, multiple sclerosis; PPAR, peroxisome proliferator-activated receptor; RAGE, receptor for advanced glycosylation end products; SLE, systemic lupus erythematosus; TIM4, T cell immunoglobulin mucin receptor 4.
| Signal | Molecule | Pathology Associated | Ref. |
|---|---|---|---|
| Systemic | |||
| Find-me | G2A | Autoimmunity | 205 |
| Sphingosine-1-phosphate | SLE | 206 | |
| Eat-me | TIM4, MerTK | SLE | 159,207,208 |
| RAGE | Sepsis | 102,209 | |
| CD300f | Autoimmunity | 210 | |
| Integrins | Autoimmunity | 211 | |
| MFG-E8 | SLE; Autoimmunity | 212,213 | |
| Gas6 | GVHD | 190,214 | |
| Protein S | Autoimmunity | 215 | |
| SCARF1 | Autoimmunity | 216 | |
| C1qa | SLE | 217 | |
| Processing | LXRα/β | SLE; autoimmunity | 132 |
| PPAR | SLE | 132 | |
| ABCA1 | SLE | 218 | |
| Rubicon, Beclin-1, VPS34, ATG5, ATG7, ATG16 | SLE, anti-cancer immunity | 125,140 | |
| Central Nervous System | |||
| Find-me | Pannexin-1 | MS/EAE | 219 |
| Eat-me | BAI1 | Learning and memory | 220 |
| TREM2 | Alzheimer's disease | 126 | |
| MerTK | MS/EAE; Parkinson's | 221 | |
| Gas6 | Toxin-induced demyelination | 222 | |
| C1qa | Epilepsy | 223 | |
| Engulfment | GULP | Schizophrenia | 224 |
| Processing | LXRα/β | MS/EAE | 132 |
| Rubicon | Alzheimer's disease | 126 | |
| Eye | |||
| Eat-me | MerTK | Retinal degeneration | 221 |
| Processing | LXRα/β | Autoimmune uveitis | 211 |
| Liver | |||
| Eat-me | TIM4 | Ischemia reperfusion injury | 225 |
| Gas6 | Ischemia reperfusion injury | 226 | |
| Pancreas | |||
| Eat-me | MFG-E8 | Type 1 diabetes mellitus | 167 |
| Testes | |||
| Eat-me | MerTK | Reduced fertility | 221 |
| Engulfment | ELMO1 | Testicular pathology | 227 |
| Lung | |||
| Find-me | P2Y2 | Allergic airway inflammation | 228 |
| Eat-me | RAGE | Fibrosis; Allergic airway inflammation | 102 |
| MerTK | Allergic airway inflammation | 221 | |
| CD36 | Lung injury | 194 | |
| Engulfment | RAC1 | Allergic airway inflammation | 229 |
| Cardiovascular system | |||
| Find-me | Pannexin-1 | Hypertension | 230 |
| CX3CR1 | Atherosclerosis | 231 | |
| Eat-me | BAI1 | Dyslipidemia | 137 |
| MFG-E8 | Atherosclerosis | 167 | |
| CD36 | Diet-induced obesity | 232 | |
| LRP1 | Atherosclerosis | 233 | |
| C1qa | Atherosclerosis | 234 | |
| SLC2A1 | Atherosclerosis | 235 | |
| Processing | DOCK180 | Cardiovascular pathologies | 236 |
| LXRα/β | Atherosclerosis | 237 | |
| PPAR | Atherosclerosis | 238 | |
| DRP1 | Atherosclerosis | 239 | |
| Kidney | |||
| Eat-me | Stabilin-1, Stabilin-2 | Glomerular fibrosis | 240 |
| Gas6 | Nephritis | 214 | |
| Engulfment | ELMO1 | Diabetic nephropathy | 227 |
| Gastrointestinal system | |||
| Find-me | G2A | IBD/Colitis | 241 |
| Eat-me | CD300f | IBD/Colitis | 210 |
| MerTK | Colon cancer | 221 | |
| Integrins | IBD/Colitis | 242 | |
| MFG-E8 | IBD/Colitis | 167 | |
| RAC1 | IBD/Colitis | 243 | |
| Skin, Muscle, Mucosal Tissue and Joints | |||
| Eat-me | RAGE | Muscle regeneration | 244 |
| C1qa | Wound healing | 182 | |
| MerTK | Arthritis | 101 | |
| Gas6 | Melanoma | 245 | |
| Integrins | Carcinoma | 242 | |
| CD36 | Wound healing | 194 | |
| Processing | DNase II | Polyarthritis | 187 |
| Engulfment | RAC1 | Arthritis | 243 |
Neurodegenerative diseases
The central nervous system (CNS) contains phagocytic cells called microglia. These resident phagocytes are phenotypically similar to macrophages and clear dead cells and cellular debris in the CNS148. However, other cells of the CNS, such as oligodendrocytes, astrocytes, and neuronal progenitors can also act as mediators of efferocytosis. Like all organ systems, the CNS requires efficient efferocytosis for homeostasis, but efferocytosis is also fundamental for the reorganization of neuronal circuits and initiating the regenerative response after injury149. Consequently, multiple neurodegenerative pathologies are associated with defects in efferocytosis (Table 2), including Multiple Sclerosis (MS), Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease150,151. Defects at multiple steps of efferocytosis can result in MS, including defects in Pannexin-1 find-me signaling, MerTK eat-me signaling, and LXRα and LXRβ, which are engaged upon processing of the corpse. Decreased expression of the bridging molecule MFG-E8 has also been observed in a mouse model of Alzheimer’s disease, and expression of a dominant negative mutant of MFG-E8 in microglia cells decreased engulfment of apoptotic neurons152,153.
Retinal degeneration
Retinal pigment epithelial (RPE) cells are the primary phagocytes in the eye, and shed photoreceptor outer segments (POS) are their primary phagocytic cargo. Although RPE are post-mitotic cells that must function throughout an organism’s lifetime, POS are shed daily in concert with circadian rhythms, and defects in this process can result in macular degeneration and vision loss. The LAP machinery is required to process POS and to ensure proper chromophore [G] regeneration in RPE cells 154,155. Similarly, defects in MerTK and integrins in RPE cells result in retinal degeneration156. Modulation of efferocytosis could be leveraged as a potential therapy for some eye diseases; for example, activation of LXRα and LXRβ signaling can alleviate inflammation associated with autoimmune uveitis157.
Pulmonary disorders
The lung and associated airway structures are laden with both professional phagocytes (alveolar macrophages and dendritic cells) and non-professional phagocytes (airway epithelial cells). These phagocytic cells primarily clear apoptotic neutrophils, which are rapidly recruited to the lung during damage. Neutrophils are short-lived and their clearance is central to the resolution of inflammation158.
Allergic airway inflammation is associated with an influx of neutrophils and eosinophils to the airway lumen. Deletion of genes encoding protein components of the efferocytotic machinery, such as MerTK or Rac1, in mouse macrophages results in airway hyperresponsiveness (AHR) and fibrosis , caused by unresolved allergic airway inflammation 159. Other pulmonary disorders, such as cystic fibrosis (CF) and chronic obstructive pulmonary disease (COPD), are also characterized by the recruitment of neutrophils and other inflammatory cells to the airways, and alveolar macrophages from patients with CF and COPD display decreased phagocytic capacity for apoptotic cells160,161.
Atherosclerosis
The heart and vasculature contain professional phagocytes, such as monocytes, macrophages, and dendritic cells, which engulf metabolically-derived oxidized lipids that then induce apoptosis in these phagocytes. These apoptotic but lipid-full cells, known as foam cells, must then be cleared via efferocytosis162,163. Failure to effectively clear dead cells in general is a major factor in the progression of atherosclerosis. Mature atherosclerotic plaques contain uncleared foam cells, and it has been suggested that defective efferocytosis contributes to plaque expansion, plaque rupture, acute coronary syndromes and stroke164-166.
Proper processing of engulfed dead cells requires lipid sensing and cholesterol efflux (see above), and mice lacking receptors for dead cells (e.g. MFG-E8167) also display increased atherosclerosis. As discussed above, uptake of apoptotic cells by phagocytes stimulates cholesterol efflux, primarily though upregulation of the cholesterol efflux regulatory protein ABCA1168,169. Decreased expression of ABCA1 promotes inflammation and atherosclerotic lesions, and overexpression of ABCA1 can improve disease progression and decrease inflammation170,171. Synthetic agonists of LXRs or PPARs improve the efferocytotic capacity of phagocytic cells and have been used successfully to treat patients with atherosclerosis. HMG-CoA reductase inhibitors, commonly known as statins, lower cholesterol and inflammation and also inhibit the action of RhoA, a negative regulator of engulfment that is highly expressed in atherosclerotic lesions172. In addition, LysM-Cre Atg5flox/flox mice, in which myeloid cells lack the autophagy protein Atg5, are deficient for canonical autophagy and LAP, and display more atherosclerotic lesions than wild-type animals in murine models of the disease173,174. It is therefore possible that effective pharmacologic treatments of atherosclerosis function, at least in part, by improving efferocytosis.
Liver pathologies
The liver neutralizes exogenous threats that reach this organ via the blood and has been referred to as a “graveyard” for dying lymphocytes, as apoptotic immune cells often traffic via the blood to the liver for their removal. Although much of the required efferocytosis is carried out by liver-resident macrophages called Kupffer cells, other liver cells, including hepatocytes, endothelial cells, and stellate cells can mediate efferocytosis175.
Defective efferocytosis has been implicated in a variety of liver pathologies, such as autoimmune hepatitis, fatty liver disease, and primary biliary cholangitis (Table 2). Molecules such as TIM4 and GAS6 are critical for the resolution of hepatic ischemia reperfusion injury176, suggesting dead cell clearance is essential for regulating hepatic inflammation.
Pathology of diabetes
Normal turnover of pancreatic islet β-cells is crucial for maintaining glucose homeostasis, but the excessive loss of β-cells or β-cell dysfunction results in diabetes mellitus (DM). The clearance of apoptotic islet β-cells is critical for maintaining immune tolerance, and type 1 DM (T1DM) is an autoimmune disease wherein T cells destroy the insulin-producing β-cells in the pancreas. While the etiology of T1DM is not fully understood, it is possible that defects in efferocytosis result in an accumulation of apoptotic β-cells, inflammation, and the generation of autoantibodies and autoreactive T cells that mediate T1DM177.
Phagocytes from non-obese diabetic (NOD) mice, a polygenic model for spontaneous T1DM, display a marked defect in efferocytosis in vitro, as well as high levels of autoantibodies in vivo178. Similarly, mice lacking MFG-E8 exhibit increased insulin sensitivity compared with their wild-type counterparts179. A common complication in patients with diabetes is compromised wound healing; phagocytes isolated from diabetic wounds have been shown to be defective in the clearance of apoptotic neutrophils from the wound site, increased inflammation, and delayed wound healing, suggesting efferocytosis is a general feature of diabetes180,181.
Pathology of the gastrointestinal system
Intestinal epithelial cells (IECs) are continuously renewed to ensure that the intestine maintains optimal barrier and absorptive functions. Due to the high turnover of cells in this tissue, efferocytosis is constantly required to prevent unwanted inflammation. Although IECs themselves are capable of engulfing and clearing apoptotic cells, dendritic cells and macrophages are the primary phagocytes in the small intestinal lamina propria6. Defects in the efferocytotic machinery caused by ablation of the phosphatidylserine receptor CD300f is associated with inflammatory bowel syndrome and colitis, as well as the progression of colon carcinoma73.
Impaired wound healing and regeneration
The skin experiences a high level of turnover of epithelial cells that are shed or cleared by macrophages, dendritic cells, and Langerhans cells [G] to promote an immune-silent response. Wound healing requires that apoptotic neutrophils be efficiently cleared, and mice deficient for C1q or CD36 display delayed wound healing and increased inflammation at the wound site182.
Efferocytosis of apoptotic cells is fundamentally important for normal wound healing. Prevention of efferocytosis by blockade of phosphatidylserine recognition or ablation of MerTK impaired tissue repair in murine models of lung and intestinal damage183. Furthermore, in Drosophila melanogaster, the engulfment of apoptotic cells is required to ‘license’ the repair machinery of macrophages for wound healing184.
Leukocytes are largely absent from skeletal muscle, but injury induces necrotic death and the release of DAMPs to quickly recruit immune cells to facilitate debris clearance. Inflammatory cytokines also promote the proliferation of muscle stem cells known as satellite cells. RAGE-deficient mice display delayed regeneration after acute injury of skeletal muscles due to decreased macrophage infiltration. Further, satellite cells can also engulf and clear dying cells, and RAGE expression is increased in satellite cells upon damage. In the absence of RAGE, damaged skeletal muscle displayed delayed differentiation of activated satellite cells185.
Upon ischemia-reperfusion injury in the heart, efferocytosis of dead cells drives fatty acid oxidation and increased electron transport in the mitochondria, which inhibit inflammation186. Animals in which macrophages have a genetic defect in electron transport display inflammation and reduced wound repair upon heart injury.
Rheumatoid arthritis
Rheumatoid arthritis is a chronic autoimmune disease characterized by increased circulating autoantibodies, which results in progressive joint destruction. Studies have highlighted a prominent role for an immune suppression by efferocytosis in the prevention of arthritic pathologies. Specifically, MerTK-deficient mice develop more severe collagen-induced arthritis, inflammation, and increased secondary necrosis than wild type mice as a consequence of defective efferocytosis101. Mice deficient in both DNAse-II and the type I IFN receptor, IFNAR (while DNAse-II deficiency is lethal, this is prevented by co-ablation of IFNAR, as discussed above) are unable to properly process engulfed cellular cargo and develop spontaneous polyarthritis with increased inflammation reminiscent of human rheumatoid arthritis187. Finally, LXR agonists, which mimic aspects of efferocytotic signaling, have therapeutic benefits in mouse models of inflammatory arthritis188.
Reduced male fertility
During spermatogenesis, vast numbers of germ cells are generated with the majority undergoing apoptosis and clearance. Sertoli cells are large phagocytes that line the seminiferous tubules of the testes and are responsible for clearing apoptotic cells as well as providing support during germ cell differentiation. ELMO and MerTK are both required for Sertoli cell-mediated efferocytosis, and ablation of the genes encoding either of these molecules results in testicular pathology and reduced fertility99,189. Overexpression of BAI1 can rescue the fertility defects, but not the vision defects found in MerTK-knockout mice, suggesting that the phosphatidylserine receptor function of BAI1 can function in place of MerTK in Sertoli cells but not in retinal pigment epithelial cells.
Beneficial effects
In some cases, defects in efferocytosis are associated with beneficial effects. Mice lacking GAS6 show delayed graft versus host disease [G] (GVHD) compared with their wild-type counterparts190. Ablation of the LAP machinery (Rubicon, Beclin-1, UVRAG, ATG5, and ATG7) in myeloid cells display increased anti-tumor immunity140. Similarly, anti-tumor immunity is enhanced in mice lacking Tim4123, or animals treated with annexin V to block recognition of phosphatidylserine on dead cells191,192. In melanoma, interfering with efferocytic clearance may be directly beneficial, as pharmacological inhibition of MerTK can block the growth of melanoma both in culture and in human melanoma xenograft models193. Finally, genetic ablation of CD36 protected animals from ischemia-reperfusion injury of the lung, although whether this relates to efferocytosis is unclear194.
Conclusions and Perspectives
Efferocytosis is governed by a plethora of factors, including unique membrane lipids and lipid reorganization, multiple effector proteins that serve functions such as recognition, the activation of phagocytosis and, ultimately, degradation of the cell corpse. As discussed, the multistep process from cell death to cell clearance is a delicate process with multiple points of redundancy, indicating the importance of efferocytosis in development, homeostasis, and pathophysiology.
The homeostatic function of nearly every major physiological system is dependent, at least in part, on the ability of phagocytes to clear cellular debris. Dysfunction of efferocytosis leads to severe disease and activation of efferocytosis, in the context of clearing apoptotic cells and he engagement of the LAP machinery, promotes immune silence through the production of anti-inflammatory signals. As current evidence suggests that efferocytosis and LAP engagement are beneficial in many settings, promoting an inflammatory response by targeting LAP and efferocytosis may prove beneficial in the treatment of various malignancies. The generation of targeted therapeutics against components of these pathways may facilitate the treatment of cancer, including checkpoint blockade, by increasing the immunogenicity of apoptotic tumor cells. As the study of efferocytosis and associated molecular pathways is an expanding field, new insights into the activation and regulation in response to dying cells is likely to lead to the discovery of new treatment paradigms for pathologies including cancer, autoimmunity, neurodegeneration, and beyond.
Acknowledgements
This work was supported by grants from the US National Institutes of Health; AI40646 and CA231620 to D.R.G., AI138492 and CA231423 to B.L.H, ALSAC, and the John H. Sununu Endowed Fellowship to B.L.H.
Glossary
- Efferocytosis
Engulfment and clearance of dead and dying cells, usually (although not exclusively) by myeloid cells such as macrophages.
- Entosis
A process by which a living cell invades or is engulfed by another. This process differs from the clearance of apoptotic cells by phagocytosis as the internalized cell is still alive.
- Inflammasome
Any molecular complex capable of activating caspase-1.
- Immune Tolerance
Lack or inhibition of an adaptive immune response to specific antigens.
- Flippase
A membrane protein that transports phosphatidylserine from the outer leaflet to the inner leaflet of the plasma membrane.
- Scramblase
A membrane protein that, upon activation, equilibrates (“scrambles”) the distribution of phospholipids between the inner and outer leaflets of the plasma membrane.
- MLKL
Mixed lineage kinase-like protein, MLKL is the executioner in necroptosis, disrupting the plasma membrane following its phosphorylation.
- Gasdermins
Proteins that, upon activation, creates pores in the plasma membrane; Gasdermin D and Gasdermin E function in pyroptosis.
- Dynamin
A member of the protein GTPase family, mainly involved in the scission of newly-formed vesicles from a membrane and their fusion with another membrane.
- Guanine nucleotide exchange factors (GEFs)
Enzymes that activate monomeric GTPases including Ras, Rac, and Rho. GEFs stimulate the release of guanosine diphosphate (GDP) to allow binding of guanosine triphosphate (GTP).
- SNARE complex
SNAP receptor complex involved in vesicle membrane fusion to other membranes.
- Phagolysosome
The intracellular vesicle that results from the fusion of the phagosome with lysosomes.
- Cathepsins
A class of proteases mostly present in lysosomes.
- LC3-Associated Phagocytosis (LAP)
A non-apoptotic function of several proteins of the autophagy pathway, resulting in lipidation of LC3 family proteins on the phagosome membrane, enhancing fusion of the phagosome with lysosomes.
- Microtubule-associated protein 1A/1B light chain 3 (LC3) family
The ATG8 protein family, including Microtubule-associated proteins 1A/1B light chain 3 (LC3) proteins, Gamma-aminobutyric acid receptor-associated protein (GABARAP), GEC-1, and GATE-16.
- Langerhans Cell
A specialized dendritic cell residing in the epidermis of the skin.
- Graft versus host disease
Pathology resulting from the introduction and action of allogeneic T lymphocytes, which can occur following transplant of bone marrow from which T cells have not been removed.
Footnotes
Competing interests
DRG is on the scientific advisory board of Inzen. The other authors declare no competing interests.
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Peer review information
Nature Reviews Molecular Cell Biology thanks Edward Thorp, Martin Herrman and the other, anonymous, reviewer for their contribution to the peer review of this work.
References
- 1.Galluzzi L et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541, doi: 10.1038/s41418-017-0012-4 (2018).A useful set of definitions of diverse modes of cell death based on mechanisms rather than morphology.
- 2.Bergmann A & Steller H Apoptosis, stem cells, and tissue regeneration. Sci Signal 3, re8, doi: 10.1126/scisignal.3145re8 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Suzanne M & Steller H Shaping organisms with apoptosis. Cell Death Differ 20, 669–675, doi: 10.1038/cdd.2013.11 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuchs Y & Steller H Programmed cell death in animal development and disease. Cell 147, 742–758, doi: 10.1016/j.cell.2011.10.033 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blander JM Death in the intestinal epithelium-basic biology and implications for inflammatory bowel disease. FEBS J 283, 2720–2730, doi: 10.1111/febs.13771 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cummings RJ et al. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature 539, 565–569, doi: 10.1038/nature20138 (2016).A demonstration of the role of clearance of dead intestinal epithelial cells by intestinal macrophages and dendritic cells in controlling inflammation of the tissue.
- 7.Eckhart L, Lippens S, Tschachler E & Declercq W Cell death by cornification. Biochim Biophys Acta 1833, 3471–3480, doi: 10.1016/j.bbamcr.2013.06.010 (2013). [DOI] [PubMed] [Google Scholar]
- 8.Florey O, Krajcovic M, Sun Q & Overholtzer M Entosis. Curr Biol 20, R88–89, doi: 10.1016/j.cub.2009.11.020 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Overholtzer M et al. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131, 966–979, doi: 10.1016/j.cell.2007.10.040 (2007). [DOI] [PubMed] [Google Scholar]
- 10.Segawa K et al. Phospholipid flippases enable precursor B cells to flee engulfment by macrophages. Proc Natl Acad Sci U S A 115, 12212–12217, doi: 10.1073/pnas.1814323115 (2018).Genetic ablation of the phospholipid flippase, ATP11C, results in loss of developing B cells due to their engulfment.
- 11.Metayer LE, Vilalta A, Burke GAA & Brown GC Anti-CD47 antibodies induce phagocytosis of live, malignant B cells by macrophages via the Fc domain, resulting in cell death by phagoptosis. Oncotarget 8, 60892–60903, doi: 10.18632/oncotarget.18492 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerr JF, Wyllie AH & Currie AR Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 26, 239–257, doi: 10.1038/bjc.1972.33 (1972). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Majno G & Joris I Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol 146, 3–15 (1995). [PMC free article] [PubMed] [Google Scholar]
- 14.Wyllie AH, Kerr JF & Currie AR Cell death: the significance of apoptosis. Int Rev Cytol 68, 251–306 (1980). [DOI] [PubMed] [Google Scholar]
- 15.Lauber K et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell 113, 717–730 (2003). [DOI] [PubMed] [Google Scholar]
- 16.Elliott MR et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 461, 282–286, doi: 10.1038/nature08296 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Truman LA et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood 112, 5026–5036, doi: 10.1182/blood-2008-06-162404 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Elliott MR, Koster KM & Murphy PS Efferocytosis Signaling in the Regulation of Macrophage Inflammatory Responses. J Immunol 198, 1387–1394, doi: 10.4049/jimmunol.1601520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gude DR et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a "come-and-get-me" signal. FASEB J 22, 2629–2638, doi: 10.1096/fj.08-107169 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weigert A et al. Apoptotic cells promote macrophage survival by releasing the antiapoptotic mediator sphingosine-1-phosphate. Blood 108, 1635–1642, doi: 10.1182/blood-2006-04-014852 (2006). [DOI] [PubMed] [Google Scholar]
- 21.Peter C et al. Release of lysophospholipid 'find-me' signals during apoptosis requires the ATP-binding cassette transporter A1. Autoimmunity 45, 568–573, doi: 10.3109/08916934.2012.719947 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Luo B et al. Erythropoeitin Signaling in Macrophages Promotes Dying Cell Clearance and Immune Tolerance. Immunity 44, 287–302, doi: 10.1016/j.immuni.2016.01.002 (2016). [DOI] [PubMed] [Google Scholar]
- 23.Kumar H, Kawai T & Akira S Pathogen recognition by the innate immune system. Int Rev Immunol 30, 16–34, doi: 10.3109/08830185.2010.529976 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Akira S, Uematsu S & Takeuchi O Pathogen recognition and innate immunity. Cell 124, 783–801, doi: 10.1016/j.cell.2006.02.015 (2006). [DOI] [PubMed] [Google Scholar]
- 25.Scaffidi P, Misteli T & Bianchi ME Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418, 191–195, doi: 10.1038/nature00858 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Shi Y, Evans JE & Rock KL Molecular identification of a danger signal that alerts the immune system to dying cells. Nature 425, 516–521, doi: 10.1038/nature01991 (2003). [DOI] [PubMed] [Google Scholar]
- 27.Shi J, Gao W & Shao F Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci 42, 245–254, doi: 10.1016/j.tibs.2016.10.004 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Liu X, Zou H, Slaughter C & Wang X DFF, a heterodimeric protein that functions downstream of caspase-3 to trigger DNA fragmentation during apoptosis. Cell 89, 175–184, doi: 10.1016/s0092-8674(00)80197-x (1997). [DOI] [PubMed] [Google Scholar]
- 29.Enari M et al. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391, 43–50, doi: 10.1038/34112 (1998). [DOI] [PubMed] [Google Scholar]
- 30.Sakahira H, Enari M & Nagata S Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis. Nature 391, 96–99, doi: 10.1038/34214 (1998). [DOI] [PubMed] [Google Scholar]
- 31.Arandjelovic S & Ravichandran KS Phagocytosis of apoptotic cells in homeostasis. Nat Immunol 16, 907–917, doi: 10.1038/ni.3253 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Q et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107, doi: 10.1038/nature08780 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hawes MC, Wen F & Elquza E Extracellular DNA: A Bridge to Cancer. Cancer Res 75, 4260–4264, doi: 10.1158/0008-5472.CAN-15-1546 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Yang YG, Lindahl T & Barnes DE Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886, doi: 10.1016/j.cell.2007.10.017 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Paludan SR & Bowie AG Immune sensing of DNA. Immunity 38, 870–880, doi: 10.1016/j.immuni.2013.05.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Motwani M, Pesiridis S & Fitzgerald KA DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet, doi: 10.1038/s41576-019-0151-1 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Tanaka Y & Chen ZJ STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal 5, ra20, doi: 10.1126/scisignal.2002521 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawane K et al. Requirement of DNase II for definitive erythropoiesis in the mouse fetal liver. Science 292, 1546–1549, doi: 10.1126/science.292.5521.1546 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Gao D et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A 112, E5699–5705, doi: 10.1073/pnas.1516465112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baum R et al. STING Contributes to Abnormal Bone Formation Induced by Deficiency of DNase II in Mice. Arthritis Rheumatol 69, 460–471, doi: 10.1002/art.39863 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ahn J, Gutman D, Saijo S & Barber GN STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A 109, 19386–19391, doi: 10.1073/pnas.1215006109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cunha LD et al. AIM2 Engages Active but Unprocessed Caspase-1 to Induce Noncanonical Activation of the NLRP3 Inflammasome. Cell Rep 20, 794–805, doi: 10.1016/j.celrep.2017.06.086 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Lugrin J & Martinon F The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol Rev 281, 99–114, doi: 10.1111/imr.12618 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Stros M HMGB proteins: interactions with DNA and chromatin. Biochim Biophys Acta 1799, 101–113, doi: 10.1016/j.bbagrm.2009.09.008 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Yang H, Wang H, Chavan SS & Andersson U High Mobility Group Box Protein 1 (HMGB1): The Prototypical Endogenous Danger Molecule. Mol Med 21 Suppl 1, S6–S12, doi: 10.2119/molmed.2015.00087 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertheloot D & Latz E HMGB1, IL-1alpha, IL-33 and S100 proteins: dual-function alarmins. Cell Mol Immunol 14, 43–64, doi: 10.1038/cmi.2016.34 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bonaldi T et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 22, 5551–5560, doi: 10.1093/emboj/cdg516 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kazama H et al. Induction of immunological tolerance by apoptotic cells requires caspase-dependent oxidation of high-mobility group box-1 protein. Immunity 29, 21–32, doi: 10.1016/j.immuni.2008.05.013 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Venereau E et al. Mutually exclusive redox forms of HMGB1 promote cell recruitment or proinflammatory cytokine release. J Exp Med 209, 1519–1528, doi: 10.1084/jem.20120189 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wen Q, Liu J, Kang R, Zhou B & Tang D The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun 510, 278–283, doi: 10.1016/j.bbrc.2019.01.090 (2019). [DOI] [PubMed] [Google Scholar]
- 51.Tirone M et al. High mobility group box 1 orchestrates tissue regeneration via CXCR4. J Exp Med 215, 303–318, doi: 10.1084/jem.20160217 (2018).HMGB1, released upon tissue injury, promotes the repair of liver and muscle via interaction with the chemokine receptor, CXCR4.
- 52.Rock KL & Kono H The inflammatory response to cell death. Annu Rev Pathol 3, 99–126, doi: 10.1146/annurev.pathmechdis.3.121806.151456 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen CJ et al. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat Med 13, 851–856, doi: 10.1038/nm1603 (2007). [DOI] [PubMed] [Google Scholar]
- 54.Eigenbrod T, Park JH, Harder J, Iwakura Y & Nunez G Cutting edge: critical role for mesothelial cells in necrosis-induced inflammation through the recognition of IL-1 alpha released from dying cells. J Immunol 181, 8194–8198, doi: 10.4049/jimmunol.181.12.8194 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin SJ Cell death and inflammation: the case for IL-1 family cytokines as the canonical DAMPs of the immune system. FEBS J 283, 2599–2615, doi: 10.1111/febs.13775 (2016).In this useful commentary, the idea that IL-1 family cytokines function as the "true" DAMPs, released upon cell death, is convincingly argued.
- 56.Idzko M, Ferrari D & Eltzschig HK Nucleotide signalling during inflammation. Nature 509, 310–317, doi: 10.1038/nature13085 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chiu YH et al. A quantized mechanism for activation of pannexin channels. Nat Commun 8, 14324, doi: 10.1038/ncomms14324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chekeni FB et al. Pannexin 1 channels mediate 'find-me' signal release and membrane permeability during apoptosis. Nature 467, 863–867, doi: 10.1038/nature09413 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Qu Y et al. Pannexin-1 is required for ATP release during apoptosis but not for inflammasome activation. J Immunol 186, 6553–6561, doi: 10.4049/jimmunol.1100478 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Wang Q et al. Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int Immunol 25, 363–372, doi: 10.1093/intimm/dxs161 (2013). [DOI] [PubMed] [Google Scholar]
- 61.Yang D, He Y, Munoz-Planillo R, Liu Q & Nunez G Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 43, 923–932, doi: 10.1016/j.immuni.2015.10.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen J, Zhao Y & Liu Y The role of nucleotides and purinergic signaling in apoptotic cell clearance - implications for chronic inflammatory diseases. Front Immunol 5, 656, doi: 10.3389/fimmu.2014.00656 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Segawa K & Nagata S An Apoptotic 'Eat Me' Signal: Phosphatidylserine Exposure. Trends Cell Biol 25, 639–650, doi: 10.1016/j.tcb.2015.08.003 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Suzuki J, Denning DP, Imanishi E, Horvitz HR & Nagata S Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406, doi: 10.1126/science.1236758 (2013). [DOI] [PubMed] [Google Scholar]
- 65.Kloditz K & Fadeel B Three cell deaths and a funeral: macrophage clearance of cells undergoing distinct modes of cell death. Cell Death Discov 5, 65, doi: 10.1038/s41420-019-0146-x (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zargarian S et al. Phosphatidylserine externalization, "necroptotic bodies" release, and phagocytosis during necroptosis. PLoS Biol 15, e2002711, doi: 10.1371/journal.pbio.2002711 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ousingsawat J, Schreiber R & Kunzelmann K TMEM16F/Anoctamin 6 in Ferroptotic Cell Death. Cancers (Basel) 11, doi: 10.3390/cancers11050625 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fujii T, Sakata A, Nishimura S, Eto K & Nagata S TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc Natl Acad Sci U S A 112, 12800–12805, doi: 10.1073/pnas.1516594112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Suzuki J, Umeda M, Sims PJ & Nagata S Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838, doi: 10.1038/nature09583 (2010). [DOI] [PubMed] [Google Scholar]
- 70.Hochreiter-Hufford A & Ravichandran KS Clearing the dead: apoptotic cell sensing, recognition, engulfment, and digestion. Cold Spring Harb Perspect Biol 5, a008748, doi: 10.1101/cshperspect.a008748 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wu Y, Tibrewal N & Birge RB Phosphatidylserine recognition by phagocytes: a view to a kill. Trends Cell Biol 16, 189–197, doi: 10.1016/j.tcb.2006.02.003 (2006). [DOI] [PubMed] [Google Scholar]
- 72.Geng K et al. Requirement of Gamma-Carboxyglutamic Acid Modification and Phosphatidylserine Binding for the Activation of Tyro3, Axl, and Mertk Receptors by Growth Arrest-Specific 6. Front Immunol 8, 1521, doi: 10.3389/fimmu.2017.01521 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee HN et al. Dendritic cells expressing immunoreceptor CD300f are critical for controlling chronic gut inflammation. J Clin Invest 127, 1905–1917, doi: 10.1172/JCI89531 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Parks BW et al. CD36, but not G2A, modulates efferocytosis, inflammation, and fibrosis following bleomycin-induced lung injury. J Lipid Res 54, 1114–1123, doi: 10.1194/jlr.M035352 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Tait JF & Smith C Phosphatidylserine receptors: role of CD36 in binding of anionic phospholipid vesicles to monocytic cells. J Biol Chem 274, 3048–3054, doi: 10.1074/jbc.274.5.3048 (1999). [DOI] [PubMed] [Google Scholar]
- 76.Szondy Z, Sarang Z, Kiss B, Garabuczi E & Koroskenyi K Anti-inflammatory Mechanisms Triggered by Apoptotic Cells during Their Clearance. Front Immunol 8, 909, doi: 10.3389/fimmu.2017.00909 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xu W et al. IL-10-producing macrophages preferentially clear early apoptotic cells. Blood 107, 4930–4937, doi: 10.1182/blood-2005-10-4144 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Fadok VA et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 101, 890–898, doi: 10.1172/JCI1112 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Freire-de-Lima CG et al. Apoptotic cells, through transforming growth factor-beta, coordinately induce anti-inflammatory and suppress pro-inflammatory eicosanoid and NO synthesis in murine macrophages. J Biol Chem 281, 38376–38384, doi: 10.1074/jbc.M605146200 (2006). [DOI] [PubMed] [Google Scholar]
- 80.Voll RE et al. Immunosuppressive effects of apoptotic cells. Nature 390, 350–351, doi: 10.1038/37022 (1997). [DOI] [PubMed] [Google Scholar]
- 81.Kim SJ, Gershov D, Ma X, Brot N & Elkon KB I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med 196, 655–665, doi: 10.1084/jem.20020542 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gardai SJ et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 123, 321–334, doi: 10.1016/j.cell.2005.08.032 (2005). [DOI] [PubMed] [Google Scholar]
- 83.Ogden CA et al. C1q and mannose binding lectin engagement of cell surface calreticulin and CD91 initiates macropinocytosis and uptake of apoptotic cells. J Exp Med 194, 781–795, doi: 10.1084/jem.194.6.781 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Oldenborg PA et al. Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054, doi: 10.1126/science.288.5473.2051 (2000). [DOI] [PubMed] [Google Scholar]
- 85.Barkal AA et al. CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396, doi: 10.1038/s41586-019-1456-0 (2019).Identification of the CD24-Siglec-10 interaction as a "don't-eat-me" signal and its possible value in cancer therapy.
- 86.Brown S et al. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature 418, 200–203, doi: 10.1038/nature00811 (2002). [DOI] [PubMed] [Google Scholar]
- 87.Barkal AA et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol 19, 76–84, doi: 10.1038/s41590-017-0004-z (2018).Identification of the MHC class I-LILRB1 interaction as a "don't-eat-me" signal and its possible value in cancer therapy.
- 88.Abram CL & Lowell CA Shp1 function in myeloid cells. J Leukoc Biol 102, 657–675, doi: 10.1189/jlb.2MR0317-105R (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Willingham SB et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A 109, 6662–6667, doi: 10.1073/pnas.1121623109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Werfel TA & Cook RS Efferocytosis in the tumor microenvironment. Semin Immunopathol 40, 545–554, doi: 10.1007/s00281-018-0698-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Weiskopf K et al. Engineered SIRPalpha variants as immunotherapeutic adjuvants to anticancer antibodies. Science 341, 88–91, doi: 10.1126/science.1238856 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chao MP et al. Calreticulin is the dominant pro-phagocytic signal on multiple human cancers and is counterbalanced by CD47. Sci Transl Med 2, 63ra94, doi: 10.1126/scitranslmed.3001375 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Azuma Y, Nakagawa H, Dote K, Higai K & Matsumoto K Decreases in CD31 and CD47 levels on the cell surface during etoposide-induced Jurkat cell apoptosis. Biol Pharm Bull 34, 1828–1834, doi: 10.1248/bpb.34.1828 (2011). [DOI] [PubMed] [Google Scholar]
- 94.Richards DM & Endres RG The mechanism of phagocytosis: two stages of engulfment. Biophys J 107, 1542–1553, doi: 10.1016/j.bpj.2014.07.070 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rosales C & Uribe-Querol E Phagocytosis: A Fundamental Process in Immunity. Biomed Res Int 2017, 9042851, doi: 10.1155/2017/9042851 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ma Z et al. Regulation of Rac1 activation by the low density lipoprotein receptor-related protein. J Cell Biol 159, 1061–1070, doi: 10.1083/jcb.200207070 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park D et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature 450, 430–434, doi: 10.1038/nature06329 (2007). [DOI] [PubMed] [Google Scholar]
- 98.Green DR, Oguin TH & Martinez J The clearance of dying cells: table for two. Cell Death Differ 23, 915–926, doi: 10.1038/cdd.2015.172 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Elliott MR & Ravichandran KS The Dynamics of Apoptotic Cell Clearance. Dev Cell 38, 147–160, doi: 10.1016/j.devcel.2016.06.029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sevajol M et al. The C-terminal polyproline-containing region of ELMO contributes to an increase in the life-time of the ELMO-DOCK complex. Biochimie 94, 823–828, doi: 10.1016/j.biochi.2011.11.014 (2012). [DOI] [PubMed] [Google Scholar]
- 101.Waterborg CEJ et al. Protective Role of the MER Tyrosine Kinase via Efferocytosis in Rheumatoid Arthritis Models. Front Immunol 9, 742, doi: 10.3389/fimmu.2018.00742 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Nienhuis HL et al. AGE and their receptor RAGE in systemic autoimmune diseases: an inflammation propagating factor contributing to accelerated atherosclerosis. Autoimmunity 42, 302–304, doi: 10.1080/08916930902831746 (2009). [DOI] [PubMed] [Google Scholar]
- 103.Marie-Anais F, Mazzolini J, Herit F & Niedergang F Dynamin-Actin Cross Talk Contributes to Phagosome Formation and Closure. Traffic 17, 487–499, doi: 10.1111/tra.12386 (2016). [DOI] [PubMed] [Google Scholar]
- 104.Stenmark H Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol 10, 513–525, doi: 10.1038/nrm2728 (2009). [DOI] [PubMed] [Google Scholar]
- 105.Rink J, Ghigo E, Kalaidzidis Y & Zerial M Rab conversion as a mechanism of progression from early to late endosomes. Cell 122, 735–749, doi: 10.1016/j.cell.2005.06.043 (2005). [DOI] [PubMed] [Google Scholar]
- 106.Chavrier P, Parton RG, Hauri HP, Simons K & Zerial M Localization of low molecular weight GTP binding proteins to exocytic and endocytic compartments. Cell 62, 317–329, doi: 10.1016/0092-8674(90)90369-p (1990). [DOI] [PubMed] [Google Scholar]
- 107.Vieira OV et al. Modulation of Rab5 and Rab7 recruitment to phagosomes by phosphatidylinositol 3-kinase. Mol Cell Biol 23, 2501–2514, doi: 10.1128/mcb.23.7.2501-2514.2003 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Flannagan RS, Jaumouille V & Grinstein S The cell biology of phagocytosis. Annu Rev Pathol 7, 61–98, doi: 10.1146/annurev-pathol-011811-132445 (2012). [DOI] [PubMed] [Google Scholar]
- 109.Rubino M, Miaczynska M, Lippe R & Zerial M Selective membrane recruitment of EEA1 suggests a role in directional transport of clathrin-coated vesicles to early endosomes. J Biol Chem 275, 3745–3748, doi: 10.1074/jbc.275.6.3745 (2000). [DOI] [PubMed] [Google Scholar]
- 110.Vieira OV et al. Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol 155, 19–25, doi: 10.1083/jcb.200107069 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Murray JT, Panaretou C, Stenmark H, Miaczynska M & Backer JM Role of Rab5 in the recruitment of hVps34/p150 to the early endosome. Traffic 3, 416–427 (2002). [DOI] [PubMed] [Google Scholar]
- 112.Kissing S et al. Vacuolar ATPase in phagosome-lysosome fusion. J Biol Chem 290, 14166–14180, doi: 10.1074/jbc.M114.628891 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lukacs GL, Rotstein OD & Grinstein S Determinants of the phagosomal pH in macrophages. In situ assessment of vacuolar H(+)-ATPase activity, counterion conductance, and H+ "leak". J Biol Chem 266, 24540–24548 (1991). [PubMed] [Google Scholar]
- 114.Harrison RE, Bucci C, Vieira OV, Schroer TA & Grinstein S Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol 23, 6494–6506, doi: 10.1128/mcb.23.18.6494-6506.2003 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Huynh KK et al. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J 26, 313–324, doi: 10.1038/sj.emboj.7601511 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Braun V et al. TI-VAMP/VAMP7 is required for optimal phagocytosis of opsonised particles in macrophages. EMBO J 23, 4166–4176, doi: 10.1038/sj.emboj.7600427 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Fairn GD & Grinstein S How nascent phagosomes mature to become phagolysosomes. Trends Immunol 33, 397–405, doi: 10.1016/j.it.2012.03.003 (2012). [DOI] [PubMed] [Google Scholar]
- 118.Collins RF, Schreiber AD, Grinstein S & Trimble WS Syntaxins 13 and 7 function at distinct steps during phagocytosis. J Immunol 169, 3250–3256, doi: 10.4049/jimmunol.169.6.3250 (2002). [DOI] [PubMed] [Google Scholar]
- 119.Xu H & Ren D Lysosomal physiology. Annu Rev Physiol 77, 57–80, doi: 10.1146/annurev-physiol-021014-071649 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Martinez J et al. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A 108, 17396–17401, doi: 10.1073/pnas.1113421108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 121.Martinez J et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol 17, 893–906, doi: 10.1038/ncb3192 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 122.Wong SW, Sil P & Martinez J Rubicon: LC3-associated phagocytosis and beyond. FEBS J 285, 1379–1388, doi: 10.1111/febs.14354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Heckmann BL, Boada-Romero E, Cunha LD, Magne J & Green DR LC3-Associated Phagocytosis and Inflammation. J Mol Biol 429, 3561–3576, doi: 10.1016/j.jmb.2017.08.012 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Heckmann BL & Green DR LC3-associated phagocytosis at a glance. J Cell Sci 132, doi: 10.1242/jcs.222984 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Martinez J et al. Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 533, 115–119, doi: 10.1038/nature17950 (2016).A demonstration that genetic ablation of LAP in myeloid cells promotes spontaneous lupus-like disease.
- 126.Heckmann BL et al. LC3-Associated Endocytosis Facilitates beta-Amyloid Clearance and Mitigates Neurodegeneration in Murine Alzheimer's Disease. Cell, doi: 10.1016/j.cell.2019.05.056 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Nakamura S & Yoshimori T New insights into autophagosome-lysosome fusion. J Cell Sci 130, 1209–1216, doi: 10.1242/jcs.196352 (2017). [DOI] [PubMed] [Google Scholar]
- 128.Pankiv S et al. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol 188, 253–269, doi: 10.1083/jcb.200907015 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.McEwan DG et al. PLEKHM1 regulates autophagosome-lysosome fusion through HOPS complex and LC3/GABARAP proteins. Mol Cell 57, 39–54, doi: 10.1016/j.molcel.2014.11.006 (2015). [DOI] [PubMed] [Google Scholar]
- 130.Tabata K et al. Rubicon and PLEKHM1 negatively regulate the endocytic/autophagic pathway via a novel Rab7-binding domain. Mol Biol Cell 21, 4162–4172, doi: 10.1091/mbc.E10-06-0495 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Heckmann BL et al. Liver X receptor alpha mediates hepatic triglyceride accumulation through upregulation of G0/G1 Switch Gene 2 expression. JCI Insight 2, e88735, doi: 10.1172/jci.insight.88735 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kidani Y & Bensinger SJ Liver X receptor and peroxisome proliferator-activated receptor as integrators of lipid homeostasis and immunity. Immunol Rev 249, 72–83, doi: 10.1111/j.1600-065X.2012.01153.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schulman IG Liver X receptors link lipid metabolism and inflammation. FEBS Lett 591, 2978–2991, doi: 10.1002/1873-3468.12702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Zhang X, Heckmann BL, Campbell LE & Liu J G0S2: A small giant controller of lipolysis and adipose-liver fatty acid flux. Biochim Biophys Acta Mol Cell Biol Lipids 1862, 1146–1154, doi: 10.1016/j.bbalip.2017.06.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.N AG et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity 31, 245–258, doi: 10.1016/j.immuni.2009.06.018 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rebe C et al. Induction of transglutaminase 2 by a liver X receptor/retinoic acid receptor alpha pathway increases the clearance of apoptotic cells by human macrophages. Circ Res 105, 393–401, doi: 10.1161/CIRCRESAHA.109.201855 (2009). [DOI] [PubMed] [Google Scholar]
- 137.Fond AM, Lee CS, Schulman IG, Kiss RS & Ravichandran KS Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J Clin Invest 125, 2748–2758, doi: 10.1172/JCI80300 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kiss RS, Elliott MR, Ma Z, Marcel YL & Ravichandran KS Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol 16, 2252–2258, doi: 10.1016/j.cub.2006.09.043 (2006). [DOI] [PubMed] [Google Scholar]
- 139.Yoon YS et al. PPARgamma activation following apoptotic cell instillation promotes resolution of lung inflammation and fibrosis via regulation of efferocytosis and proresolving cytokines. Mucosal Immunol 8, 1031–1046, doi: 10.1038/mi.2014.130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cunha LD et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 175, 429–441 e416, doi: 10.1016/j.cell.2018.08.061 (2018).Genetic ablation of LAP in myeloid cells of the tumor microenvironment promotes anti-cancer immunity in several model systems by the engagement of a type I interferon response.
- 141.Yokoyama K et al. Rab27a negatively regulates phagocytosis by prolongation of the actin-coating stage around phagosomes. J Biol Chem 286, 5375–5382, doi: 10.1074/jbc.M110.171702 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kuiper JW et al. Creatine kinase-mediated ATP supply fuels actin-based events in phagocytosis. PLoS Biol 6, e51, doi: 10.1371/journal.pbio.0060051 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lucin KM et al. Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron 79, 873–886, doi: 10.1016/j.neuron.2013.06.046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Baumann I et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum 46, 191–201, doi: (2002). [DOI] [PubMed] [Google Scholar]
- 145.Liu Z & Davidson A Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med 18, 871–882, doi: 10.1038/nm.2752 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Berden JH Lupus nephritis. Kidney Int 52, 538–558, doi: 10.1038/ki.1997.365 (1997). [DOI] [PubMed] [Google Scholar]
- 147.van Bruggen MC et al. Antigen specificity of anti-nuclear antibodies complexed to nucleosomes determines glomerular basement membrane binding in vivo. Eur J Immunol 27, 1564–1569, doi: 10.1002/eji.1830270636 (1997). [DOI] [PubMed] [Google Scholar]
- 148.Witting A, Muller P, Herrmann A, Kettenmann H & Nolte C Phagocytic clearance of apoptotic neurons by Microglia/Brain macrophages in vitro: involvement of lectin-, integrin-, and phosphatidylserine-mediated recognition. J Neurochem 75, 1060–1070, doi: 10.1046/j.1471-4159.2000.0751060.x (2000). [DOI] [PubMed] [Google Scholar]
- 149.Galloway DA, Phillips AEM, Owen DRJ & Moore CS Phagocytosis in the Brain: Homeostasis and Disease. Front Immunol 10, 790, doi: 10.3389/fimmu.2019.00790 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Mattson MP Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol 1, 120–129, doi: 10.1038/35040009 (2000). [DOI] [PubMed] [Google Scholar]
- 151.Heckmann BL, Tummers B & Green DR Crashing the computer: apoptosis vs. necroptosis in neuroinflammation. Cell Death Differ 26, 41–52, doi: 10.1038/s41418-018-0195-3 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Fricker M et al. MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J Neurosci 32, 2657–2666, doi: 10.1523/JNEUROSCI.4837-11.2012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Fuller AD & Van Eldik LJ MFG-E8 regulates microglial phagocytosis of apoptotic neurons. J Neuroimmune Pharmacol 3, 246–256, doi: 10.1007/s11481-008-9118-2 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ferguson TA & Green DR Autophagy and phagocytosis converge for better vision. Autophagy 10, 165–167, doi: 10.4161/auto.26735 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Kim JY et al. Noncanonical autophagy promotes the visual cycle. Cell 154, 365–376, doi: 10.1016/j.cell.2013.06.012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Nandrot EF & Dufour EM Mertk in daily retinal phagocytosis: a history in the making. Adv Exp Med Biol 664, 133–140, doi: 10.1007/978-1-4419-1399-9_16 (2010). [DOI] [PubMed] [Google Scholar]
- 157.Yang H et al. Activation of liver X receptor alleviates ocular inflammation in experimental autoimmune uveitis. Invest Ophthalmol Vis Sci 55, 2795–2804, doi: 10.1167/iovs.13-13323 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fox S, Leitch AE, Duffin R, Haslett C & Rossi AG Neutrophil apoptosis: relevance to the innate immune response and inflammatory disease. J Innate Immun 2, 216–227, doi: 10.1159/000284367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Juncadella IJ et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 493, 547–551, doi: 10.1038/nature11714 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Hodge S, Hodge G, Scicchitano R, Reynolds PN & Holmes M Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol 81, 289–296, doi: 10.1046/j.1440-1711.2003.t01-1-01170.x (2003). [DOI] [PubMed] [Google Scholar]
- 161.Vandivier RW et al. Impaired clearance of apoptotic cells from cystic fibrosis airways. Chest 121, 89S, doi: 10.1378/chest.121.3_suppl.89s (2002). [DOI] [PubMed] [Google Scholar]
- 162.Ley K, Miller YI & Hedrick CC Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol 31, 1506–1516, doi: 10.1161/ATVBAHA.110.221127 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Tabas I Consequences and therapeutic implications of macrophage apoptosis in atherosclerosis: the importance of lesion stage and phagocytic efficiency. Arterioscler Thromb Vasc Biol 25, 2255–2264, doi: 10.1161/01.ATV.0000184783.04864.9f (2005). [DOI] [PubMed] [Google Scholar]
- 164.Poon IK, Lucas CD, Rossi AG & Ravichandran KS Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 14, 166–180, doi: 10.1038/nri3607 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Schrijvers DM, De Meyer GR, Kockx MM, Herman AG & Martinet W Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arterioscler Thromb Vasc Biol 25, 1256–1261, doi: 10.1161/01.ATV.0000166517.18801.a7 (2005). [DOI] [PubMed] [Google Scholar]
- 166.Vucic E et al. Regression of inflammation in atherosclerosis by the LXR agonist R211945: a noninvasive assessment and comparison with atorvastatin. JACC Cardiovasc Imaging 5, 819–828, doi: 10.1016/j.jcmg.2011.11.025 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Li BZ, Zhang HY, Pan HF & Ye DQ Identification of MFG-E8 as a novel therapeutic target for diseases. Expert Opin Ther Targets 17, 1275–1285, doi: 10.1517/14728222.2013.829455 (2013). [DOI] [PubMed] [Google Scholar]
- 168.Cuchel M & Rader DJ Macrophage reverse cholesterol transport: key to the regression of atherosclerosis? Circulation 113, 2548–2555, doi: 10.1161/CIRCULATIONAHA.104.475715 (2006). [DOI] [PubMed] [Google Scholar]
- 169.Oram JF & Heinecke JW ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev 85, 1343–1372, doi: 10.1152/physrev.00005.2005 (2005). [DOI] [PubMed] [Google Scholar]
- 170.Tang C & Oram JF The cell cholesterol exporter ABCA1 as a protector from cardiovascular disease and diabetes. Biochim Biophys Acta 1791, 563–572, doi: 10.1016/j.bbalip.2009.03.011 (2009). [DOI] [PubMed] [Google Scholar]
- 171.Zhu X et al. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res 51, 3196–3206, doi: 10.1194/jlr.M006486 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Loirand G, Guerin P & Pacaud P Rho kinases in cardiovascular physiology and pathophysiology. Circ Res 98, 322–334, doi: 10.1161/01.RES.0000201960.04223.3c (2006). [DOI] [PubMed] [Google Scholar]
- 173.Liao X et al. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab 15, 545–553, doi: 10.1016/j.cmet.2012.01.022 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Razani B et al. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab 15, 534–544, doi: 10.1016/j.cmet.2012.02.011 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Davies SP, Reynolds GM & Stamataki Z Clearance of Apoptotic Cells by Tissue Epithelia: A Putative Role for Hepatocytes in Liver Efferocytosis. Front Immunol 9, 44, doi: 10.3389/fimmu.2018.00044 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Llacuna L et al. Growth arrest-specific protein 6 is hepatoprotective against murine ischemia/reperfusion injury. Hepatology 52, 1371–1379, doi: 10.1002/hep.23833 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Vives-Pi M, Rodriguez-Fernandez S & Pujol-Autonell I How apoptotic beta-cells direct immune response to tolerance or to autoimmune diabetes: a review. Apoptosis 20, 263–272, doi: 10.1007/s10495-015-1090-8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.O'Brien BA et al. A deficiency in the in vivo clearance of apoptotic cells is a feature of the NOD mouse. J Autoimmun 26, 104–115, doi: 10.1016/j.jaut.2005.11.006 (2006). [DOI] [PubMed] [Google Scholar]
- 179.Khalifeh-Soltani A et al. Mfge8 promotes obesity by mediating the uptake of dietary fats and serum fatty acids. Nat Med 20, 175–183, doi: 10.1038/nm.3450 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Khanna S et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One 5, e9539, doi: 10.1371/journal.pone.0009539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Maruyama K et al. Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am J Pathol 170, 1178–1191, doi: 10.2353/ajpath.2007.060018 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bossi F et al. C1q as a unique player in angiogenesis with therapeutic implication in wound healing. Proc Natl Acad Sci U S A 111, 4209–4214, doi: 10.1073/pnas.1311968111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bosurgi L et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science 356, 1072–1076, doi: 10.1126/science.aai8132 (2017).While the cytokines IL-4 and IL-13 are involved in the repair of tissue damage during helminth infections, this paper shows that efferocytosis of apoptotic cells by macrophages is also required for the repair response.
- 184.Weavers H, Evans IR, Martin P & Wood W Corpse Engulfment Generates a Molecular Memory that Primes the Macrophage Inflammatory Response. Cell 165, 1658–1671, doi: 10.1016/j.cell.2016.04.049 (2016).In a Drosophila model of tissue damage, efferocytosis of apoptotic cells is required to prime the participation of macrophages in tissue repair.
- 185.Beccafico S et al. Human muscle satellite cells show age-related differential expression of S100B protein and RAGE. Age (Dordr) 33, 523–541, doi: 10.1007/s11357-010-9197-x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zhang S et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab 29, 443–456 e445, doi: 10.1016/j.cmet.2018.12.004 (2019).During wound injury clearence of dying cells fills the phagocytic cell with a metabolite load nearly equal to that at rest. Here, the authors investigate how metabolic phagocytic signaling regulates the signature anti-inflammatory macrophage response.
- 187.Kawane K et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443, 998–1002, doi: 10.1038/nature05245 (2006). [DOI] [PubMed] [Google Scholar]
- 188.Park MC, Kwon YJ, Chung SJ, Park YB & Lee SK Liver X receptor agonist prevents the evolution of collagen-induced arthritis in mice. Rheumatology (Oxford) 49, 882–890, doi: 10.1093/rheumatology/keq007 (2010). [DOI] [PubMed] [Google Scholar]
- 189.Elliott MR & Ravichandran KS ELMO1 signaling in apoptotic germ cell clearance and spermatogenesis. Ann N Y Acad Sci 1209, 30–36, doi: 10.1111/j.1749-6632.2010.05764.x (2010). [DOI] [PubMed] [Google Scholar]
- 190.Burnier L et al. Gas6 deficiency in recipient mice of allogeneic transplantation alleviates hepatic graft-versus-host disease. Blood 115, 3390–3397, doi: 10.1182/blood-2009-02-206920 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Yan X et al. Annexin-V promotes anti-tumor immunity and inhibits neuroblastoma growth in vivo. Cancer Immunol Immunother 61, 1917–1927, doi: 10.1007/s00262-012-1250-4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Stach CM et al. Treatment with annexin V increases immunogenicity of apoptotic human T-cells in Balb/c mice. Cell Death Differ 7, 911–915, doi: 10.1038/sj.cdd.4400715 (2000). [DOI] [PubMed] [Google Scholar]
- 193.Schlegel J et al. MERTK receptor tyrosine kinase is a therapeutic target in melanoma. J Clin Invest 123, 2257–2267, doi: 10.1172/JCI67816 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Suresh K et al. CD36 mediates H2O2-induced calcium influx in lung microvascular endothelial cells. Am J Physiol Lung Cell Mol Physiol 312, L143–L153, doi: 10.1152/ajplung.00361.2016 (2017).Genetic loss or pharmacologic inhibition of CD36 attenuates the damage induced by hydrogen peroxide in lung and protects from ischemia-reperfusion injury.
- 195.Rider P, Voronov E, Dinarello CA, Apte RN & Cohen I Alarmins: Feel the Stress. J Immunol 198, 1395–1402, doi: 10.4049/jimmunol.1601342 (2017). [DOI] [PubMed] [Google Scholar]
- 196.Roh JS & Sohn DH Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw 18, e27, doi: 10.4110/in.2018.18.e27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Gong T, Liu L, Jiang W & Zhou R DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol 20, 95–112, doi: 10.1038/s41577-019-0215-7 (2020). [DOI] [PubMed] [Google Scholar]
- 198.Yu X, Feng B, He P & Shan L From Chaos to Harmony: Responses and Signaling upon Microbial Pattern Recognition. Annu Rev Phytopathol 55, 109–137, doi: 10.1146/annurev-phyto-080516-035649 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Brubaker SW, Bonham KS, Zanoni I & Kagan JC Innate immune pattern recognition: a cell biological perspective. Annu Rev Immunol 33, 257–290, doi: 10.1146/annurev-immunol-032414-112240 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rubartelli A & Lotze MT Inside, outside, upside down: damage-associated molecular-pattern molecules (DAMPs) and redox. Trends Immunol 28, 429–436, doi: 10.1016/j.it.2007.08.004 (2007). [DOI] [PubMed] [Google Scholar]
- 201.Broz P & Monack DM Newly described pattern recognition receptors team up against intracellular pathogens. Nat Rev Immunol 13, 551–565, doi: 10.1038/nri3479 (2013). [DOI] [PubMed] [Google Scholar]
- 202.Tang D, Kang R, Coyne CB, Zeh HJ & Lotze MT PAMPs and DAMPs: signal 0s that spur autophagy and immunity. Immunol Rev 249, 158–175, doi: 10.1111/j.1600-065X.2012.01146.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Mogensen TH Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev 22, 240–273, Table of Contents, doi: 10.1128/CMR.00046-08 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Takeuchi O & Akira S Pattern recognition receptors and inflammation. Cell 140, 805–820, doi: 10.1016/j.cell.2010.01.022 (2010). [DOI] [PubMed] [Google Scholar]
- 205.Le LQ et al. Mice lacking the orphan G protein-coupled receptor G2A develop a late-onset autoimmune syndrome. Immunity 14, 561–571, doi: 10.1016/s1074-7613(01)00145-5 (2001). [DOI] [PubMed] [Google Scholar]
- 206.Mike EV et al. Neuropsychiatric Systemic Lupus Erythematosus Is Dependent on Sphingosine-1-Phosphate Signaling. Front Immunol 9, 2189, doi: 10.3389/fimmu.2018.02189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Miyanishi M, Segawa K & Nagata S Synergistic effect of Tim4 and MFG-E8 null mutations on the development of autoimmunity. Int Immunol 24, 551–559, doi: 10.1093/intimm/dxs064 (2012). [DOI] [PubMed] [Google Scholar]
- 208.Potter PK, Cortes-Hernandez J, Quartier P, Botto M & Walport MJ Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J Immunol 170, 3223–3232, doi: 10.4049/jimmunol.170.6.3223 (2003). [DOI] [PubMed] [Google Scholar]
- 209.van Zoelen MA & van der Poll T Targeting RAGE in sepsis. Crit Care 12, 103, doi: 10.1186/cc6187 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Tian L et al. p85alpha recruitment by the CD300f phosphatidylserine receptor mediates apoptotic cell clearance required for autoimmunity suppression. Nat Commun 5, 3146, doi: 10.1038/ncomms4146 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Schittenhelm L, Hilkens CM & Morrison VL beta2 Integrins As Regulators of Dendritic Cell, Monocyte, and Macrophage Function. Front Immunol 8, 1866, doi: 10.3389/fimmu.2017.01866 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hanayama R, Miyasaka K, Nakaya M & Nagata S MFG-E8-dependent clearance of apoptotic cells, and autoimmunity caused by its failure. Curr Dir Autoimmun 9, 162–172, doi: 10.1159/000090780 (2006). [DOI] [PubMed] [Google Scholar]
- 213.Kruse K et al. Inefficient clearance of dying cells in patients with SLE: anti-dsDNA autoantibodies, MFG-E8, HMGB-1 and other players. Apoptosis 15, 1098–1113, doi: 10.1007/s10495-010-0478-8 (2010). [DOI] [PubMed] [Google Scholar]
- 214.Cohen PL & Shao WH Gas6/TAM Receptors in Systemic Lupus Erythematosus. Dis Markers 2019, 7838195, doi: 10.1155/2019/7838195 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.ten Kate MK & van der Meer J Protein S deficiency: a clinical perspective. Haemophilia 14, 1222–1228, doi: 10.1111/j.1365-2516.2008.01775.x (2008). [DOI] [PubMed] [Google Scholar]
- 216.Ramirez-Ortiz ZG et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol 14, 917–926, doi: 10.1038/ni.2670 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Walport MJ, Davies KA & Botto M C1q and systemic lupus erythematosus. Immunobiology 199, 265–285, doi: 10.1016/S0171-2985(98)80032-6 (1998). [DOI] [PubMed] [Google Scholar]
- 218.Zeng T et al. The detection of autoantibodies to ATP-binding cassette transporter A1 and its role in the pathogenesis of atherosclerosis in patients with systemic lupus erythematosus. Clin Biochem 45, 1342–1346, doi: 10.1016/j.clinbiochem.2012.06.009 (2012). [DOI] [PubMed] [Google Scholar]
- 219.Lutz SE et al. Contribution of pannexin1 to experimental autoimmune encephalomyelitis. PLoS One 8, e66657, doi: 10.1371/journal.pone.0066657 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Duman JG, Tu YK & Tolias KF Emerging Roles of BAI Adhesion-GPCRs in Synapse Development and Plasticity. Neural Plast 2016, 8301737, doi: 10.1155/2016/8301737 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Wium M, Paccez JD & Zerbini LF The Dual Role of TAM Receptors in Autoimmune Diseases and Cancer: An Overview. Cells 7, doi: 10.3390/cells7100166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Binder MD et al. Gas6 deficiency increases oligodendrocyte loss and microglial activation in response to cuprizone-induced demyelination. J Neurosci 28, 5195–5206, doi: 10.1523/JNEUROSCI.1180-08.2008 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Chu Y et al. Enhanced synaptic connectivity and epilepsy in C1q knockout mice. Proc Natl Acad Sci U S A 107, 7975–7980, doi: 10.1073/pnas.0913449107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Chen X et al. Apoptotic engulfment pathway and schizophrenia. PLoS One 4, e6875, doi: 10.1371/journal.pone.0006875 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Li Z & Weinman SA Regulation of Hepatic Inflammation via Macrophage Cell Death. Semin Liver Dis 38, 340–350, doi: 10.1055/s-0038-1670674 (2018). [DOI] [PubMed] [Google Scholar]
- 226.Bellan M et al. Gas6/TAM System: A Key Modulator of the Interplay between Inflammation and Fibrosis. Int J Mol Sci 20, doi: 10.3390/ijms20205070 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Elliott MR et al. Unexpected requirement for ELMO1 in clearance of apoptotic germ cells in vivo. Nature 467, 333–337, doi: 10.1038/nature09356 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Cattaneo M The platelet P2 receptors in inflammation. Hamostaseologie 35, 262–266, doi: 10.5482/HAMO-14-09-0044 (2015). [DOI] [PubMed] [Google Scholar]
- 229.Henson PM & Bratton DL Allergy: airway epithelial Rac1 suppresses allergic inflammation. Curr Biol 23, R104–106, doi: 10.1016/j.cub.2012.12.008 (2013). [DOI] [PubMed] [Google Scholar]
- 230.Good ME et al. Pannexin 1 Channels as an Unexpected New Target of the Anti-Hypertensive Drug Spironolactone. Circ Res 122, 606–615, doi: 10.1161/CIRCRESAHA.117.312380 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Liu H & Jiang D Fractalkine/CX3CR1 and atherosclerosis. Clin Chim Acta 412, 1180–1186, doi: 10.1016/j.cca.2011.03.036 (2011). [DOI] [PubMed] [Google Scholar]
- 232.Liu M, Tso P & Woods SC Receptor CD36 links a risk-associated allele to obesity and metabolic disorders. J Biol Chem 293, 13349–13350, doi: 10.1074/jbc.H118.004818 (2018).Pharmacological inhibition of integral membrane protein CD36 significantly reduces body weight gain and improves glucose tolerance in animals on high fat diet.
- 233.Boucher P & Herz J Signaling through LRP1: Protection from atherosclerosis and beyond. Biochem Pharmacol 81, 1–5, doi: 10.1016/j.bcp.2010.09.018 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Bhatia VK et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol 170, 416–426, doi: 10.2353/ajpath.2007.060406 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Freemerman AJ et al. Myeloid Slc2a1-Deficient Murine Model Revealed Macrophage Activation and Metabolic Phenotype Are Fueled by GLUT1. J Immunol 202, 1265–1286, doi: 10.4049/jimmunol.1800002 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Thorp EB Mechanisms of failed apoptotic cell clearance by phagocyte subsets in cardiovascular disease. Apoptosis 15, 1124–1136, doi: 10.1007/s10495-010-0516-6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Qi YY, Zhou XJ & Zhang H Autophagy and immunological aberrations in systemic lupus erythematosus. Eur J Immunol 49, 523–533, doi: 10.1002/eji.201847679 (2019). [DOI] [PubMed] [Google Scholar]
- 238.Duval C, Chinetti G, Trottein F, Fruchart JC & Staels B The role of PPARs in atherosclerosis. Trends Mol Med 8, 422–430, doi: 10.1016/s1471-4914(02)02385-7 (2002). [DOI] [PubMed] [Google Scholar]
- 239.Rogers MA et al. Dynamin-Related Protein 1 Inhibition Attenuates Cardiovascular Calcification in the Presence of Oxidative Stress. Circ Res 121, 220–233, doi: 10.1161/CIRCRESAHA.116.310293 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Schledzewski K et al. Deficiency of liver sinusoidal scavenger receptors stabilin-1 and - 2 in mice causes glomerulofibrotic nephropathy via impaired hepatic clearance of noxious blood factors. J Clin Invest 121, 703–714, doi: 10.1172/JCI44740 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Frasch SC et al. G2A Signaling Dampens Colitic Inflammation via Production of IFN-gamma. J Immunol 197, 1425–1434, doi: 10.4049/jimmunol.1600264 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Huveneers S, Truong H & Danen HJ Integrins: signaling, disease, and therapy. Int J Radiat Biol 83, 743–751, doi: 10.1080/09553000701481808 (2007). [DOI] [PubMed] [Google Scholar]
- 243.Marei H & Malliri A Rac1 in human diseases: The therapeutic potential of targeting Rac1 signaling regulatory mechanisms. Small GTPases 8, 139–163, doi: 10.1080/21541248.2016.1211398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Riuzzi F et al. RAGE in the pathophysiology of skeletal muscle. J Cachexia Sarcopenia Muscle 9, 1213–1234, doi: 10.1002/jcsm.12350 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Wu G et al. Molecular insights of Gas6/TAM in cancer development and therapy. Cell Death Dis 8, e2700, doi: 10.1038/cddis.2017.113 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Segawa K et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168, doi: 10.1126/science.1252809 (2014). [DOI] [PubMed] [Google Scholar]
- 247.Li H, Zhu H, Xu CJ & Yuan J Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501, doi: 10.1016/s0092-8674(00)81590-1 (1998). [DOI] [PubMed] [Google Scholar]
- 248.Weinlich R, Oberst A, Beere HM & Green DR Necroptosis in development, inflammation and disease. Nat Rev Mol Cell Biol 18, 127–136, doi: 10.1038/nrm.2016.149 (2017). [DOI] [PubMed] [Google Scholar]
- 249.Tummers B & Green DR Caspase-8: regulating life and death. Immunol Rev 277, 76–89, doi: 10.1111/imr.12541 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ingram JP et al. ZBP1/DAI Drives RIPK3-Mediated Cell Death Induced by IFNs in the Absence of RIPK1. J Immunol 203, 1348–1355, doi: 10.4049/jimmunol.1900216 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Kaiser WJ et al. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J Biol Chem 288, 31268–31279, doi: 10.1074/jbc.M113.462341 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Man SM, Karki R & Kanneganti TD Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 277, 61–75, doi: 10.1111/imr.12534 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Kayagaki N et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526, 666–671, doi: 10.1038/nature15541 (2015).Caspase-11 cleavage of gasdermin D is responsible for pyroptotic activation, IL1 processing, and spetic shock following Gram-negative infection.
- 254.Shi J et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665, doi: 10.1038/nature15514 (2015).Identification of Gasdermin D as a regulator of pyroptosis downstream of inflammatory caspase activation.
- 255.Ding J et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535, 111–116, doi: 10.1038/nature18590 (2016). [DOI] [PubMed] [Google Scholar]
- 256.Doll S et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98, doi: 10.1038/nchembio.2239 (2017).ACSL4 enriches cellular membranes with long polyunsaturated fatty acids required for ferroptosis.
- 257.Kagan VE et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90, doi: 10.1038/nchembio.2238 (2017).Discovery of a highly organized oxygenation center leading to oxidation of specific phospholipid species that direct cells towards ferroptosis.