

Graphical abstract

Abbreviations: ACC, drug accumulation; CAD, cationic amphiphilic drug; ER, endoplasmatic reticulum; PLD, phospholipidosis; Vd, volume of distribution; logP, distribution coefficient of non-charged species of compound between octanol and water; pKa, acid/base ionisation constant; hERG, human Ether-à-go-go-Related Gene, a potassium ion channel in heart; QSAR, quantitative structure activity relationship; CCR5, C-C chemokine receptor type 5
Keywords: COVID-19, Antiviral drugs, Drug repositioning, Lysosomotropism, Endosomal pathway, Broad spectrum antivirals
Highlights
-
•
Lysosomotropic drugs show moderate antiviral effects even on coronaviruses.
-
•
The antiviral activity is likely due to interference with endosomal pathway.
-
•
530 existing drugs were analysed for lysosomotropism, pharmacokinetics and toxicity.
-
•
36 drugs were identified that may possibly be suitable for repurposing for COVID-19.
-
•
Further research is needed to confirm their antiviral effects and safety limits.
Abstract
Given the speed of viral infection spread, repurposing of existing drugs has been given the highest priority in combating the ongoing COVID-19 pandemic. Only drugs that are already registered or close to registration, and therefore have passed lengthy safety assessments, have a chance to be tested in clinical trials and reach patients quickly enough to help in the current disease outbreak.
Here, we have reviewed available evidence and possible ways forward to identify already existing pharmaceuticals displaying modest broad-spectrum antiviral activity which is likely linked to their high accumulation in cells. Several well studied examples indicate that these drugs accumulate in lysosomes, endosomes and biological membranes in general, and thereby interfere with endosomal pathway and intracellular membrane trafficking crucial for viral infection. With the aim to identify other lysosomotropic drugs with possible inherent antiviral activity, we have applied a set of clear physicochemical, pharmacokinetic and molecular criteria on 530 existing drugs. In addition to publicly available data, we have also used our in silico model for the prediction of accumulation in lysosomes and endosomes. By this approach we have identified 36 compounds with possible antiviral effects, also against coronaviruses. For 14 of them evidence of broad-spectrum antiviral activity has already been reported, adding support to the value of this approach.
Presented pros and cons, knowledge gaps and methods to identify lysosomotropic antivirals, can help in the evaluation of many drugs currently in clinical trials considered for repurposing to target COVID-19, as well as open doors to finding more potent and safer alternatives.
1. Antiviral effects of highly accumulating lysosomotropic drugs
Currently available antiviral drugs were mainly designed to target a specific viral or human protein crucial for infection with a specific type of virus [1]. Considering the diversity and constant mutations of viruses, it will not come as a surprise if these antiviral drugs are shown to be inefficient against the new SARS-CoV-2 virus and thus not adequate for repurposing.
Certain marketed drugs seem to exhibit modest antiviral activity via the mechanism that is not specific for a single protein, but rather for a general mechanism of viral infection. For instance, chloroquine and hydroxychloroquine have been reported to affect endosomes, the critical point of viral entry into the cell, resulting in antiviral effect [2]. Various steps in the viral infection have been examined to understand the mechanism of (hydroxy)chloroquine’s activity, but it has remained not entirely clear. It has been reported that the drug’s effect of increasing the endosomal pH results in reduced capacity of the virus to infect the cell [3]. It has also been proposed that the antimalarial drug reduces the level of glycosylation of ACE2 in endosome critical for recognition of the SARS virus and the endosomal membrane [4]. Recent studies indicate possible interaction with sigma receptors as important for their antiviral activity [5]. Regardless, in a recent small clinical trial hydroxychloroquine demonstrated moderate effectiveness in treatment of COVID-19 patients, and the effect was even more pronounced in co-treatment with antibacterial azithromycin [6]. This led both drugs to numerous currently ongoing larger clinical trials as well as to heated debates in science and public on their potential benefits, mechanisms and safety.
The mentioned drugs, chloroquine, hydroxychloroquine and azithromycin, highly accumulate in lysosomes/endosomes and their membranes, reaching about 100-fold higher intracellular than their extracellular concentration [7,8], and thus belong to a group of lysosomotropic compounds (compounds that accumulate in lysosomes). We propose that the antiviral activity of (hydroxy)chloroquine and azithromycin is shared among all strong lysosomotropic drugs and is a consequence of their extremely high accumulation in cells and membranes, and subsequently of all the processes affected by this pharmacokinetic property.
Lysosomotropic compounds or cationic amphiphilic drugs (CADs) belong to various pharmacological classes but share the same physicochemical properties that enable them to accumulate in acidic compartments of cells (Fig. 1 ). Such compounds can pass the membrane in neutral form but as they come to a more acidic environment, they become protonated and as such cannot diffuse back through the membrane. Consequently, as the drug moves into more acidic compartment, the equilibrium between protonated and neutral forms is shifted towards the protonated form and a higher fraction of drug molecules become trapped inside the cell [8]. The highest concentration of lysosomotropic drugs is reached in lysosomes with pH of 4–5, and in late endosomes with pH of 5–6 [9]. For strong lysosomotropic drugs it was estimated that 50–70 % of intracellularly accumulated compound is stored in lysosomes and endosomes [10], leading to extreme concentrations in these compartments. While accumulating, such drugs are heavily loading in lysosomal and other biological membranes inside the cell due to their amphiphilic nature [11], meaning that the overall molecule is relatively lipophilic (often with logP ranging from 3 to 5) but also bears positive charge, which enables the molecule to bind close to the surface of the phospholipid bilayer [12,13].
Fig. 1.

Interaction of lysosomotropic cationic amphiphilic drugs (CADs) and coronavirus with membrane trafficking in the cell.
The impact of lysosomotropic drug accumulation on cells is obvious: lysosomes increase in volume [14], lysosomal function is impaired leading to downregulation of autophagy [15,16], endocytosis and the entire membrane trafficking in the cell is reduced [17]. Lysosomal and endosomal pH increases as a consequence of the overload of basic compounds [9,18]. Due to the cationic drug binding to phospholipid bilayer and change of the surface bilayer charge, the degradation of phospholipids is slowed [12] resulting in the accumulation of excess phospholipid membrane and vesicles inside the cell, which may lead to an adverse effect known as phospholipidosis [19]. If not too extreme, all these effects on cells are reversible upon the cessation of drug treatment [20].
In our previous studies on a set of 47 compounds we have shown that the extent of lysosomotropic accumulation in cells correlates with the compound’s extent of induction of phospholipidosis and lysosomal swelling [7]. The correlation is so apparent that it is even possible to determine the level of accumulation from the intensity of either of these processes caused by a drug. Strong correlation with accumulation was also shown for the inhibition of autophagy on a smaller set of compounds. Finally, accumulation in cells was also shown to correlate with their extent of binding to layers of phosphatidyl choline, the most abundant phospholipid in biological membranes [11].
The importance of intracellular membranes in coronavirus infection is immense. Firstly, viruses enter the cell by formation of membrane vesicles – endosomes, and subsequently enter cytoplasm through the endosomal membrane (Fig. 1). During the assembly of new virion particles, the virus is taking a portion of the membrane of endoplasmic reticulum (ER) to pack the RNA in a lipid envelope enriched with transmembrane proteins. New virions are then packed in an exocytic membrane vesicle, which ultimately fuses with the plasma membrane and releases virions outside of cells [21]. Therefore, it is conceivable that a drug that is heavily bound to cellular membranes, negatively affects the viral life cycle, if not even the structure of its lipid envelope itself. Since the assembly of virions relies on electrostatic forces between the lipid envelope, their proteins, and RNA inside it, it is likely that the overload of cationic drug in the ER membrane, that viral lipid envelope is made of, leads to serious disturbances of virion structure.
The evidence supporting antiviral activity of lysosomotropic cationic amphiphilic drugs (CADs) are described in various reports on individual drugs and have been extensively summarized by Salata et al. [22]. Chloroquine and amiodarone have so far been amongst the most widely studied CADs for their antiviral effects. A recent review on broad spectrum antiviral agents, summarized the data on antiviral activity of various drugs in the Drugvirus database [23] (https://drugvirus.info/). Even though the database is logically enriched in antiviral drugs, there are more than 20 CADs registered for various non-viral indications listed in the database with collected evidence of their broad antiviral activity in vitro, in vivo and in clinical studies, including activities on coronaviruses, influenza, zika and ebola virus. Among listed drugs with antiviral effects, the CADs include: psychoactive drugs (chlorpromazine, fluoxetine, clomipramine); antiarrhythmics (amiodarone); antimalarials (chloroquine, hydroxychloroquine, amodiaquine, mefloquine, quinacrine); channel blockers (amiodarone, verapamil, manidipine); antibacterial (azithromycin); estrogen receptor modulators (tamoxifen, raloxifene, toremifene); two antivirals, the only ones known to utilize CADs mechanism of antiviral action via the inhibition of endosomal pathway (arbidol (umifenovir) and tilorone (with additional activity of interferon induction)) [24,25].
It is noteworthy that there are more than 150 marketed and investigational drugs known to induce phospholipidosis [26], and a majority, but not all of them classify as lysosomotropic cationic amphiphilic drugs. Phospholipidosis is also the most common side effect of lysosomotropic compounds and a direct consequence of their binding to membranes [12] and reduction of vesicle trafficking in the cell [15,17]. Therefore, we anticipate that, among phospholipidosis inducers there are many registered drugs that may have intrinsic antiviral activity via the same mechanism as described for a handful of lysosomotropic compounds so far; by inhibiting endocytosis, increasing endosomal pH, and slowing down total intracellular membrane trafficking [22]. Their potency may be moderate but in combination with other treatments they can likely show synergistic effects. These drugs thus represent a pool of potential existing modest antiviral drugs that may prove useful as the first line of defence in current and possible new viral epidemic outbreaks.
Another possible positive contribution of CADs in COVID-19 could be their potential anti-inflammatory effects, which was for azithromycin and chloroquine as well as for a number of tool drugs found linked to their accumulation properties [17,20].
Apart from these potential beneficial antiviral and anti-inflammatory effects, it is important to be aware of potential safety issues linked to lysosomotropic drugs. CADs are often, but not always, linked to liver- and cardiotoxic effects [27,28]. SAR studies on hERG channel indicate that increasing basicity of many compounds increases the chance to block the channel resulting in cardiotoxicity [29]. Despite these effects, these drugs are still widely used, and several of them were true blockbusters not so long ago, for example antibacterial azithromycin, selective serotonin reuptake inhibitors (SSRI) antidepressants fluoxetine, citalopram and sertraline, as well as well-known antimalarial drug chloroquine. In repurposing for viral infections, additional types of toxicity may arise from their primary pharmacological targets, as they were developed to treat completely different diseases. We believe, however, that by careful analysis of pharmacological and toxicological properties of lysosomotropic CADs, new drugs among existing ones could be found that may be reasonably safe during short term treatment, at least for a group of COVID-19 patients that do not have pre-existing heart and liver conditions.
2. Analysis of marketed existing drugs for potential lysosomotropic accumulation and antiviral effects
We have now applied our in silico model for the prediction of lysosomotropic accumulation [30] on all marketed and investigational drugs that are known inducers of phospholipidosis [26,31], and that according to their physicochemical properties have the ability to get trapped by protonation in lysosomes (the strongest basic ionisation constant, pKa>7.5) [32]. The aim of this analysis was to find other strong lysosomotropic CADs which have a high likelihood of interacting with cell compartments and processes critical for viral infection cycle, such as endosomes, lysosomes and membranes in general. As additional proofs of their lysosomotropic nature and possibility of high accumulation in cells in vivo, we have considered the presence of tertiary nitrogen atoms in their structure, lipophilicity (logP), volume of distribution and half-life in plasma or blood in humans. All filtering criteria are listed in Table 1 and the results of this analysis are presented in Table 2 .
Table 1.
Criteria for the selection of drugs with high likelihood of lysosomotropic effects.
| Property | Parametera | Desirable range | Indication | Criterion evaluation |
|---|---|---|---|---|
| Induction of phospholipidosis | PLD induction (exp) | Positive |
|
Primary inclusion criterion |
| Accumulation in cells | ACC class (calc) | 2−5(3−5b) |
|
Primary inclusion criterion |
| Protonizability | pKa (basic) (exp or calc) | ≥ 7.5 |
|
Primary inclusion criterion |
| Physiological charge (calc) | ≥ 1 |
|
Additional | |
| Number of tertiary nitrogen atoms | ≥ 1 |
|
Additional | |
| Lipophilicity | logP (exp or calc) | 2.0−6.0 |
|
Additional |
| Volume of distribution | Vd (exp) | ≥ 10 L kg−1 |
|
Additional |
| Half-life in plasma/blood | t1/2 (exp) | ≥ 10 h |
|
Additional |
Footnote: a exp – experimental parameter, calc – calculated parameter; b – for compounds out of domain of the model; PLD - phospholipidosis.
Table 2.
Selection of drugs with highly likely lysosomotropic behaviour and probable antiviral effects.
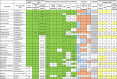 |
Footnote: Data sources: phospholipidosis [26,31], accumulation in cells calculated according to [30], molecular and phys-chem properties from PubChem [44] and DrugBank [63] (https://www.drugbank.ca/), hepatotoxicity from LiverTox [36], broad spectrum antiviral activity [23] (https://drugvirus.info/), pharmacokinetics and cardiotoxicity ([46,[48], [49], [50], [51], [52],54,55,[57], [58], [59], [60], [61], [62],53,47,56,45]). Green – data indicating lysosomotropism; orange – data indicating cardiotoxicity and hepatotoxicity; blue – data indicating safety; grey – approved drugs; yellow – data indicating broad spectrum antiviral effects; +* - drugs approved only in some countries; n.d. – no data.
From the compounds with experimental or clinical proofs of induction of phospholipidosis, we have selected those that according to our in silico prediction model for cellular accumulation are predicted to accumulate moderately to extremely high (accumulation (ACC) levels 2–5) for compounds within and at the borderline of the applicability domain for the model; and high to extremely high (ACC levels 3−5) for compounds outside the domain i.e. with less reliable prediction of accumulation intensity [30]. For several molecules in Table 2 the data on experimentally determined cellular accumulation is also provided (measured ACC) [7]. Third inclusion criterion was the strongest basic dissociation constant, pKa, higher than 7.5 which is an indication that the molecule can become protonated, and thus trapped, inside the acidic compartment in addition to eventually increasing lysosomal/endosomal pH [18,32].
In Table 2, all data marked green are in favour of the compound having lysosomotropic behaviour which may result in antiviral effects. In addition to the initial three inclusion criteria, we have considered beneficial and marked compounds which have physiological charge higher or equal 1, and one or more tertiary nitrogen atoms, which are the most common structural features of CADs [33]. Also marked were data on logP between 2 and 5, which indicates the capability of the compound to enter cells via diffusion through the membrane in neutral form, in addition to the ability of binding to the membrane [32,34]. Pharmacokinetic parameters, volume of distribution higher than 10 L kg−1 and half-life in blood/plasma equal or higher than 10 h, are also marked green as they indicate that the compound in its original form accumulates in tissues in vivo, and remains in the organism for a longer period [35].
Due to their strong primary pharmacology we have excluded psychoactive compounds and antiarrhythmics from our further analyses, as we think that these compounds, with strong effects on brain and hearth, are not suitable for repurposing for fighting viral infections. There is a strong possibility that they may cause rather specific and possibly dangerous side effects if needed to be given at higher doses than what is needed for their primary indication. It would, however, be important to know whether chronic use of antidepressants, or any other CADs, can in a positive or negative manner affect the activity of antiviral CADs. It is possible they may function as protection against viral infection, but conversely, in chronic use the body may adapt to constant exposure to lysosomotropic drugs, rendering CAD antivirals less effective. More research is needed on the effects of chronic usage of lysosomotropic drugs on antiviral activity of drugs exploiting the CAD mode of action.
In addition, although still in use, many CADs are associated with liver toxicity and cardiotoxicity in terms of arrythmia caused by QT interval prolongation [27,28], and thus we have added to our analysis known data on these adverse effects. Liver toxicity evaluation is taken from the LiverTox database (where A levels indicates the highest likelihood of liver toxicity, and E the lowest) [36]. For cardiotoxicity, we have marked positive (orange) if there is a reported effect of the compound on either QT interval prolongation/arrhythmia in humans or a positive result in in vitro hERG assay.
Finally, the existing information about broad spectrum antiviral activity of selected drugs has been taken from the DrugVirus database [23], including number of viruses that the drug was shown effective against, as well as whether there are also supporting in vivo and clinical data on their antiviral activity, and specifically data on activity against SARS-CoV, SARS-CoV-2 and MERS viruses.
In addition to looking at compounds passing both phospholipidosis and cell accumulation criteria we have also analysed compounds which failed one of these criteria, but still had pKa values higher than 7.5. These drugs, shown in Table 3 , are considered possible to exert CAD’s mode of antiviral action, but with less certainty than those in Table 2.
Table 3.
Selection of drugs with possible lysosomotropic behaviour and antiviral effects.
Footnote: Data sources: phospholipidosis [26,31], accumulation in cells calculated according to [30], molecular and phys-chem properties from PubChem [44] and DrugBank [63] (https://www.drugbank.ca/), hepatotoxicity from LiverTox [36], broad spectrum antiviral activity [23] (https://drugvirus.info/), pharmacokinetics and cardiotoxicity ([46,[48], [49], [50], [51], [52],54,55,[57], [58], [59], [60], [61], [62],53,47,56,45]). For colour legend see Table 2.
3. Existing drugs with probable antiviral activity via lysosomotropic mechanisms
530 drugs and compounds have been included in the analysis: 447 compounds with known potential to induce phospholipidosis (either positive or negative), 20 drugs with unknown or unclear phospholipidosis results and 63 additional antiviral drugs without phospholipidosis data (Fig. 2 ). All compounds were analysed by our in silico QSAR model for prediction of cellular accumulation, which than resulted in 103 compounds passing both phospholipidosis and accumulation criteria, 9 compounds passing ACC criterion with unknown phospholipidosis data, 24 compounds passing ACC criterion but not phospholipidosis, and 112 passing phosholipidosis criterion but not accumulation prediction. Compounds were then additionally filtered according to their pKa values and primary pharmacology, to find compounds less likely to induce serious side effects when used for a different indication than what they were developed for. This resulted in total of 26 drugs with potential for repurposing for viral infections due to the high likelihood of exhibiting antiviral effects as lysosomotropic drugs. Among drugs that failed in either phospholipidosis or accumulation criterion there are 10 compounds which passed other criteria and can be considered as additional drugs that may possibly induce the same effects.
Fig. 2.

Process of finding existing drugs with probable lysosomotropic mechanism of antiviral activity.
Drugs identified with high likelihood for antiviral effects due to CAD properties mainly belong to macrocyclic antibacterials, antimalarials, antihistaminics, antivirals and antiparasitic drugs. Since most, except antihistaminics, have non-human targets, they are among the drugs most acceptable for repurposing since their primary pharmacological activity will likely not pose a safety issue. Eight out of 26 identified drugs have already shown effectiveness against either SARS-CoV, SARS-CoV-2 or MERS viruses or two or all of them, adding support to the potential value of this antiviral strategy. These eight drugs are: promethazine, amodiaquine, chloroquine, mefloquine, tilorone, hydroxychloroquine, arbidol (umifenovir) and nelfinavir [23] (https://drugvirus.info/).
The drugs that passed all applied lysosomotropism criteria are: azithromycin, promethazine, cyclizine, chloroquine, clemastine, hydroxyzine, rifabutin and vicriviroc, and drugs that do not have data for one of the criteria but passed all the others are: chlorcyclizine, homochlorcyclizine and quinacrine.
It is important to consider that 15 out of 26 identified drugs were found either to induce QT prolongation/arrhythmia or interact with hERG channel in vitro, or both. This indicates that, even if proven effective against the virus, these drugs will likely need to be avoided in patients with heart conditions. Cautious approach is needed for six drugs that were found negative on QT prolongation and arrhythmia, as the lack of effect may still be linked to applied doses for their primary indication. In addition, higher likelihood of liver toxicity was found in six out of 26 drugs. Care should also be taken with identified antihistaminics in the group, as they share structural features with some lysosomotropic antidepressants, and some still have a mild sedative effect.
Out of the four antiviral drugs that may show CAD’s mode of action, two drugs have been claimed to utilize this mechanism: tilorone and arbidol (umifenovir). Even though tilorone was invented in the US as an interferon inducing broad spectrum antiviral, additional studies showed that its lysosomotropic potential is comparable to chloroquine, which likely contributes to its antiviral properties [25]. Tilorone is currently registered and used only in Russia and some neighbouring countries, where it is approved for indications such as influenza, acute respiratory viral infection, viral hepatitis, viral encephalitis and myelitis. Arbidol is registered in Russia and China for the treatment of influenza [24]. It is claimed to inhibit the membrane fusion of virus with the endosomal membrane, which possibly occurs via increasing the endosomal pH, and thereby preventing infection of the cell. The third antiviral, nelfinavir is a protease inhibitor drug approved for treatment of HIV infection, but also reported effective against a wide variety of viruses: Herpes simplex, SARS-CoV, hepatitis C, dengue and Chikungunya [23] (DrugVirus database). It is likely that its lysosomotropic properties may be responsible for this broad-spectrum activity. The last identified antiviral drug is vicriviroc, which was developed as nanomolar CCR5 antagonist for HIV infections and came to phase III clinical trials where it did not meet primary efficacy endpoints. When analysing the potential of repurposing such highly specific and potent antiviral drugs for fighting a different virus, it should be kept in mind that they would likely need a much higher concentration since their original target that they are specifically acting upon is not present in other viruses. Impact of strong lysosomotropic compounds on membranes and membrane trafficking often needs low micromolar concentrations [7]. It remains to be seen whether efficacious levels of any of CADs can be achieved in COVID-19 patients without trespassing into toxicity levels. In the case of CAD’s antiviral activity, it is worth investigating whether stereoselectivity could help to further separate activity and toxicity dose ranges. It has been shown previously that enantiomers (mirror image compounds) have different affinities for a range protein targets, including hERG [37,38]. If the antiviral effect of CADs is linked only to their physico-chemical properties, which are the same for both enantiomers, they would have the same activity, but may differ in the affinity for hERG. This would result in one enantiomer having a better safety profile than the other, with unchanged antiviral activity. 20/26 compounds from Table 2 possess at least one chiral centre, some even more than 10, meaning that there are many potential enantiomer pairs that could be examined from the perspective of obtaining increased safety margins.
Compounds listed in Table 3 are less likely to show antiviral effects caused by lysosmotropic behaviour (compared to those in Table 2) as they failed in one of the primary inclusion criteria. Three of them target respiratory system (bromhexine, ambroxol and formoterol), but should be cautiously assessed at which stages of the disease they may be most effective. Interestingly, in this group of compounds, we have also identified imatinib, which was proven effective against SARS-CoV and MERS-CoV viruses by inhibiting viral fusion and entry into the cells [39,40] and is currently being tested in a clinical trial on COVID-19 patients [41]. Its efficacy is speculated to be linked to its primary target Abl2 kinase, but it is conceivable that the observed effects are the consequence of its already proven lysosomotropic behaviour [42].
At the moment, there are more than 10 CADs currently being tested in numerous ongoing clinical trials on COVID-19, with chloroquine, hydroxychloroquine and azithromycin being most widely studied. FDA has recently revoked the emergency use authorization (EUA) to use hydroxychloroquine and chloroquine to treat COVID-19 outside clinical trials due to the lack of efficacy, and due to marked toxicity findings in patient populations treated in clinical trials so far [43]. Nevertheless, other clinical trials with these drugs are still ongoing [41]. In addition, other well known CADs are also being clinically tested, such as amiodarone, fluoxetine, chlorpromazine and fluvoxamine, but also other drugs with possible CAD properties such as imatinib, bromhexine, formoterol [41]. The number of clinical trials and diversity of test protocols will hopefully make it feasible to get a better picture in near future on potential clinical value of these compounds as broad spectrum antivirals and open door to the development of new safer CADs for antiviral purpose.
4. Conclusion
In this study, we have analysed 530 compounds with the aim to find existing registered drugs which may be useful in combating current COVID-19 pandemics. Specifically, we were looking for drugs capable of accumulating in endosomes and membranes and thereby possessing inherent antiviral activity. The drugs were analysed based on their known physicochemical and pharmacokinetic data, in addition to our in silico prediction of accumulation in lysosomes/endosomes. We have identified 26 drugs with high and 10 drugs with lower probability of showing antiviral effects due to their lysosomotropic behaviour. Broad spectrum antiviral activity has already been reported for some of these drugs and therefore, it is crucial to investigate their antiviral properties on SARS-CoV-2. Although they were not designed to be specific and may thus lack potency, these drugs may, either in combination or alone, be capable of reducing the extent of infection and helping patients avoid serious or long-term illness. Keeping in mind the low probability of finding a highly potent and specific antiviral drug for the ongoing pandemic, these drugs may represent at least a partial, but possibly one of the best antiviral pharmacotherapeutic solutions currently at hand.
It will be critical in further investigations of lysosomotropic drugs to find out which administration regimens would be useful. Although evidence of their antiviral activities is mounting, it is still unclear at which stage of disease development endosomal pathway disruption could play the most important role and whether the efficacious level of any of these drugs can be achieved in the body without significant toxic effects. Moreover, it should be determined whether prior exposure to lysosomotropic drugs could help prevent the disease or, on the contrary, induce adaptations in the body and reduce the efficacy of antivirals with lysosomotropic mechanism. Defining target patient populations, based on disease status, drug safety profiles and other factors need to be carefully investigated. Answers to these crucial questions are required for assessing the potential of these drugs as broad-spectrum antivirals, suitable for repurposing to treat COVID-19 or any future viral infection epidemics.
Declaration of Competing Interest
Authors declare no conflicts of interest.
Acknowledgements
This study has been funded by Karolinska Institutet, Stockholm, Sweden.
References
- 1.De Clercq E., Li G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016;29:695–747. doi: 10.1128/CMR.00102-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Bari M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017;5 doi: 10.1002/prp2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Savarino A., Boelaert J.R., Cassone A., Majori G., Cauda R. Effects of chloroquine on viral infections: an old drug against today’s diseases? Lancet Infect. Dis. 2003;3:722–727. doi: 10.1016/S1473-3099(03)00806-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vincent M.J., Bergeron E., Benjannet S., Erickson B.R., Rollin P.E., Ksiazek T.G., Seidah N.G., Nichol S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005;2:69. doi: 10.1186/1743-422X-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L., Tummino T.A., Hüttenhain R., Kaake R.M., Richards A.L., Tutuncuoglu B., Foussard H., Batra J., Haas K., Modak M., Kim M., Haas P., Polacco B.J., Braberg H., Fabius J.M., Eckhardt M., Soucheray M., Bennett M.J., Cakir M., Mcgregor M.J., Li Q., Meyer B., Roesch F., Vallet T., Mac Kain A., Miorin L., Moreno E., Naing Z.Z.C., Zhou Y., Peng S., Shi Y., Zhang Z., Shen W., Kirby I.T., Melnyk J.E., Chorba J.S., Lou K., Dai S.A., Barrio-Hernandez I., Memon D., Hernandez-Armenta C., Lyu J., Mathy C.J.P., Perica T., Pilla K.B., Ganesan S.J., Saltzberg D.J., Rakesh R., Liu X., Rosenthal S.B., Calviello L., Venkataramanan S., Liboy-Lugo J., Lin Y., Huang X.P., Liu Y., Wankowicz S.A., Bohn M., Safari M., Ugur F.S., Koh C., Savar N.S., Tran Q.D., Shengjuler D., Fletcher S.J., O’neal M.C., Cai Y., Chang J.C.J., Broadhurst D.J., Klippsten S., Sharp P.P., Wenzell N.A., Kuzuoglu-Ozturk D., Wang H.Y., Trenker R., Young J.M., Cavero D.A., Hiatt J., Roth T.L., Rathore U., Subramanian A., Noack J., Hubert M., Stroud R.M., Frankel A.D., Rosenberg O.S., Verba K.A., Agard D.A., Ott M., Emerman M., Jura N. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020 doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gautret P., Lagier J.C., Parola P., Hoang V.T., Meddeb L., Mailhe M., Doudier B., Courjon J., Giordanengo V., Vieira V.E., Dupont H.T., Honoré S., Colson P., Chabrière E., La Scola B., Rolain J.M., Brouqui P., Raoult D. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents. 2020 doi: 10.1016/j.ijantimicag.2020.105949. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 7.Easwaranathan A., Inci B., Ulrich S., Brunken L., Nikiforova V., Norinder U., Swanson S., Munic Kos V. Quantification of intracellular accumulation and retention of lysosomotropic macrocyclic compounds by high-throughput imaging of lysosomal changes. J. Pharm. Sci. 2019;108:652–660. doi: 10.1016/j.xphs.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 8.De Duve C., De Barsy T., Poole B., Trouet A., Tulkens P., Van Hoof F. Commentary. Lysosomotropic agents. Biochem. Pharmacol. 1974;23:2495–2531. doi: 10.1016/0006-2952(74)90174-9. [DOI] [PubMed] [Google Scholar]
- 9.Schmitt M.V., Lienau P., Fricker G., Reichel A. Quantitation of lysosomal trapping of basic lipophilic compounds using in vitro assays and in silico predictions based on the determination of the full pH profile of the endo-/lysosomal system in rat hepatocytes. Drug Metab. Dispos. 2019;47:49–57. doi: 10.1124/dmd.118.084541. [DOI] [PubMed] [Google Scholar]
- 10.Togami K., Chono S., Morimoto K. Subcellular distribution of azithromycin and clarithromycin in rat alveolar macrophages (NR8383) in vitro. Biol. Pharm. Bull. 2013;36:1494–1499. doi: 10.1248/bpb.b13-00423. [DOI] [PubMed] [Google Scholar]
- 11.Sanchez Garcia D., Sjödin M., Hellstrandh M., Norinder U., Nikiforova V., Lindberg J., Wincent E., Bergman Å., Cotgreave I., Munic Kos V. Cellular accumulation and lipid binding of perfluorinated alkylated substances (PFASs) - a comparison with lysosomotropic drugs. Chem. Biol. Interact. 2018;281:1–10. doi: 10.1016/j.cbi.2017.12.021. [DOI] [PubMed] [Google Scholar]
- 12.Kosol S., Schrank E., Krajačić M.B., Wagner G.E., Meyer N.H., Göbl C., Rechberger G.N., Zangger K., Novak P. Probing the interactions of macrolide antibiotics with membrane-mimetics by NMR spectroscopy. J. Med. Chem. 2012;55:5632–5636. doi: 10.1021/jm300647f. [DOI] [PubMed] [Google Scholar]
- 13.Gh M.S., Wilhelm M.J., Dai H.L. Azithromycin-induced changes to bacterial membrane properties monitored. ACS Med. Chem. Lett. 2018;9:569–574. doi: 10.1021/acsmedchemlett.7b00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Funk R.S., Krise J.P. Cationic amphiphilic drugs cause a marked expansion of apparent lysosomal volume: implications for an intracellular distribution-based drug interaction. Mol. Pharm. 2012;9:1384–1395. doi: 10.1021/mp200641e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashoor R., Yafawi R., Jessen B., Lu S. The contribution of lysosomotropism to autophagy perturbation. PLoS One. 2013;8 doi: 10.1371/journal.pone.0082481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mauthe M., Orhon I., Rocchi C., Zhou X., Luhr M., Hijlkema K.J., Coppes R.P., Engedal N., Mari M., Reggiori F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 2018;14:1435–1455. doi: 10.1080/15548627.2018.1474314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nujić K., Banjanac M., Munić V., Polančec D., Eraković Haber V. Impairment of lysosomal functions by azithromycin and chloroquine contributes to anti-inflammatory phenotype. Cell. Immunol. 2012;279:78–86. doi: 10.1016/j.cellimm.2012.09.007. [DOI] [PubMed] [Google Scholar]
- 18.Logan R., Funk R.S., Axcell E., Krise J.P. Drug-drug interactions involving lysosomes: mechanisms and potential clinical implications. Expert Opin. Drug Metab. Toxicol. 2012;8:943–958. doi: 10.1517/17425255.2012.691165. [DOI] [PubMed] [Google Scholar]
- 19.Shayman J.A., Abe A. Drug induced phospholipidosis: an acquired lysosomal storage disorder. Biochim. Biophys. Acta. 2013;1831:602–611. doi: 10.1016/j.bbalip.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Munić V., Banjanac M., Koštrun S., Nujić K., Bosnar M., Marjanović N., Ralić J., Matijašić M., Hlevnjak M., Eraković Haber V. Intensity of macrolide anti-inflammatory activity in J774A.1 cells positively correlates with cellular accumulation and phospholipidosis. Pharmacol. Res. 2011;64:298–307. doi: 10.1016/j.phrs.2011.03.011. [DOI] [PubMed] [Google Scholar]
- 21.J Alsaadi E.A., Jones I.M. Membrane binding proteins of coronaviruses. Future Virol. 2019;14:275–286. doi: 10.2217/fvl-2018-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salata C., Calistri A., Parolin C., Baritussio A., Palù G. Antiviral activity of cationic amphiphilic drugs. Expert Rev. Anti. Ther. 2017;15:483–492. doi: 10.1080/14787210.2017.1305888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Andersen P.I., Ianevski A., Lysvand H., Vitkauskiene A., Oksenych V., Bjørås M., Telling K., Lutsar I., Dumpis U., Irie Y., Tenson T., Kantele A., Kainov D.E. Discovery and development of safe-in-man broad-spectrum antiviral agents. Int. J. Infect. Dis. 2020;93:268–276. doi: 10.1016/j.ijid.2020.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boriskin Y.S., Leneva I.A., Pécheur E.I., Polyak S.J. Arbidol: a broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 2008;15:997–1005. doi: 10.2174/092986708784049658. [DOI] [PubMed] [Google Scholar]
- 25.Ekins S., Lingerfelt M.A., Comer J.E., Freiberg A.N., Mirsalis J.C., O’loughlin K., Harutyunyan A., Mcfarlane C., Green C.E., Madrid P.B. Efficacy of tilorone dihydrochloride against ebola virus infection. Antimicrob. Agents Chemother. 2018:62. doi: 10.1128/AAC.01711-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orogo A.M., Choi S.S., Minnier B.L., Kruhlak N.L. Construction and consensus performance of (Q)SAR models for predicting phospholipidosis using a dataset of 743 compounds. Mol. Inform. 2012;31:725–739. doi: 10.1002/minf.201200048. [DOI] [PubMed] [Google Scholar]
- 27.Dragovic S., Vermeulen N.P., Gerets H.H., Hewitt P.G., Ingelman-Sundberg M., Park B.K., Juhila S., Snoeys J., Weaver R.J. Evidence-based selection of training compounds for use in the mechanism-based integrated prediction of drug-induced liver injury in man. Arch. Toxicol. 2016;90:2979–3003. doi: 10.1007/s00204-016-1845-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun H., Xia M., Shahane S.A., Jadhav A., Austin C.P., Huang R. Are hERG channel blockers also phospholipidosis inducers? Bioorg. Med. Chem. Lett. 2013;23:4587–4590. doi: 10.1016/j.bmcl.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cavalluzzi M.M., Imbrici P., Gualdani R., Stefanachi A., Mangiatordi G.F., Lentini G., Nicolotti O. Human ether-à-go-go-related potassium channel: exploring SAR to improve drug design. Drug Discov. Today. 2020;25:344–366. doi: 10.1016/j.drudis.2019.11.005. [DOI] [PubMed] [Google Scholar]
- 30.Norinder U., Munic Kos V. QSAR models for predicting five levels of cellular accumulation of lysosomotropic macrocycles. Int. J. Mol. Sci. 2019:20. doi: 10.3390/ijms20235938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Goracci L., Ceccarelli M., Bonelli D., Cruciani G. Modeling phospholipidosis induction: reliability and warnings. J. Chem. Inf. Model. 2013;53:1436–1446. doi: 10.1021/ci400113t. [DOI] [PubMed] [Google Scholar]
- 32.Nadanaciva S., Lu S., Gebhard D.F., Jessen B.A., Pennie W.D., Will Y. A high content screening assay for identifying lysosomotropic compounds. Toxicol. In Vitro. 2011;25:715–723. doi: 10.1016/j.tiv.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 33.Rorig K.J., Ruben Z., Anderson S.N. Structural determinants of cationic amphiphilic amines which induce clear cytoplasmic vacuoles in cultured cells. Proc. Soc. Exp. Biol. Med. 1987;184:165–171. doi: 10.3181/00379727-184-42462. [DOI] [PubMed] [Google Scholar]
- 34.Lipinski C.A., Lombardo F., Dominy B.W., Feeney P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 35.Hanumegowda U.M., Wenke G., Regueiro-Ren A., Yordanova R., Corradi J.P., Adams S.P. Phospholipidosis as a function of basicity, lipophilicity, and volume of distribution of compounds. Chem. Res. Toxicol. 2010;23:749–755. doi: 10.1021/tx9003825. [DOI] [PubMed] [Google Scholar]
- 36.National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) 2012. LiverTox Clinical and Research Information on Drug-Induced Liver Injury [Online]. Available: https://www.ncbi.nlm.nih.gov/books/NBK547852/ [Accessed] [PubMed] [Google Scholar]
- 37.Lentini G., Cavalluzzi M.M., Habtemariam S. COVID-19, chloroquine repurposing, and cardiac safety concern: chirality might help. Molecules. 2020:25. doi: 10.3390/molecules25081834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grilo L.S., Carrupt P.A., Abriel H. Stereoselective inhibition of the hERG1 potassium channel. Front. Pharmacol. 2010;1:137. doi: 10.3389/fphar.2010.00137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dyall J., Coleman C.M., Hart B.J., Venkataraman T., Holbrook M.R., Kindrachuk J., Johnson R.F., Olinger G.G., Jr., Jahrling P.B., Laidlaw M., Johansen L.M., Lear-Rooney C.M., Glass P.J., Hensley L.E., Frieman M.B. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob. Agents Chemother. 2014;58:4885–4893. doi: 10.1128/AAC.03036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sisk J.M., Frieman M.B., Machamer C.E. Coronavirus S protein-induced fusion is blocked prior to hemifusion by Abl kinase inhibitors. J. Gen. Virol. 2018;99:619–630. doi: 10.1099/jgv.0.001047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Medicine, N. U. S. N. L. O. ClinicalTrials.gov [Online]. Available: https://clinicaltrials.gov/ [Accessed].
- 42.Fu D., Zhou J., Zhu W.S., Manley P.W., Wang Y.K., Hood T., Wylie A., Xie X.S. Imaging the intracellular distribution of tyrosine kinase inhibitors in living cells with quantitative hyperspectral stimulated Raman scattering. Nat. Chem. 2014;6:614–622. doi: 10.1038/nchem.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fda. Fda cautions against use of hydroxychloroquine or chloroquine for COVID-19 outside of the hospital setting or a clinical trial due to risk of heart rhythm problems [Online]. Available: https://www.fda.gov/drugs/drug-safety-and-availability/fda-cautions-against-use-hydroxychloroquine-or-chloroquine-covid-19-outside-hospital-setting-or [Accessed].
- 44.PubChem [Online]. Available: https://pubchem.ncbi.nlm.nih.gov/ [Accessed].
- 45.Administration, U. S. F. A. D. Drugs@FDA: FDA-Approved Drugs [Online] Available: https://www.accessdata.fda.gov/scripts/cder/daf/ [Accessed].
- 46.Bjorneboe M., Iversen O., Olsen S. Infective hepatitis and toxic jaundice in a municipal hospital during a five-year period. Incidence and prognosis. Acta Med. Scand. 1967;182:491–501. doi: 10.1111/j.0954-6820.1967.tb10873.x. [DOI] [PubMed] [Google Scholar]
- 47.Chen C.Y., Wang F.L., Lin C.C. Chronic hydroxychloroquine use associated with QT prolongation and refractory ventricular arrhythmia. Clin. Toxicol. (Phila) 2006;44:173–175. doi: 10.1080/15563650500514558. [DOI] [PubMed] [Google Scholar]
- 48.Crawford K.W., Li C., Keung A., Su Z., Hughes M.D., Greaves W., Kuritzkes D., Gulick R., Flexner C., Team A.A.S. Pharmacokinetic/pharmacodynamic modeling of the antiretroviral activity of the CCR5 antagonist Vicriviroc in treatment experienced HIV-infected subjects (ACTG protocol 5211) J. Acquir. Immune Defic. Syndr. 2010;53:598–605. doi: 10.1097/QAI.0b013e3181c9caac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ducharme J., Farinotti R. Clinical pharmacokinetics and metabolism of chloroquine. Focus on recent advancements. Clin. Pharmacokinet. 1996;31:257–274. doi: 10.2165/00003088-199631040-00003. [DOI] [PubMed] [Google Scholar]
- 50.Hancox J.C., Hasnain M., Vieweg W.V., Crouse E.L., Baranchuk A. Azithromycin, cardiovascular risks, QTc interval prolongation, torsade de pointes, and regulatory issues: a narrative review based on the study of case reports. Ther. Adv. Infect. Dis. 2013;1:155–165. doi: 10.1177/2049936113501816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kaneko T., Dougherty T.J., Magee T.V. Therapeutic areas II: cancer, Infectious diseases, inflammation & immunology and dermatology. Comprehensiv Med. Chem. II. 2007 [Google Scholar]
- 52.Krishna S., White N.J. Pharmacokinetics of quinine, chloroquine and amodiaquine. Clinical implications. Clin. Pharmacokinet. 1996;30:263–299. doi: 10.2165/00003088-199630040-00002. [DOI] [PubMed] [Google Scholar]
- 53.O’mara E., Kasserra C., Huddlestone J.R., Wan Y., Soni P., Caceres M., Medlock M., Morrison R., Devinsky O. Effect of vicriviroc on the QT/corrected QT interval and central nervous system in healthy subjects. Antimicrob. Agents Chemother. 2010;54:2448–2454. doi: 10.1128/AAC.01447-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ridley J.M., Milnes J.T., Hancox J.C., Witchel H.J. Clemastine, a conventional antihistamine, is a high potency inhibitor of the HERG K+ channel. J. Mol. Cell. Cardiol. 2006;40:107–118. doi: 10.1016/j.yjmcc.2005.09.017. [DOI] [PubMed] [Google Scholar]
- 55.Schran H.F., Petryk L., Chang C.T., O’connor R., Gelbert M.B. The pharmacokinetics and bioavailability of clemastine and phenylpropanolamine in single-component and combination formulations. J. Clin. Pharmacol. 1996;36:911–922. doi: 10.1002/j.1552-4604.1996.tb04758.x. [DOI] [PubMed] [Google Scholar]
- 56.Schrezenmeier E., Dörner T. Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat. Rev. Rheumatol. 2020;16:155–166. doi: 10.1038/s41584-020-0372-x. [DOI] [PubMed] [Google Scholar]
- 57.Shi J., Montay G., Bhargava V.O. Clinical pharmacokinetics of telithromycin, the first ketolide antibacterial. Clin. Pharmacokinet. 2005;44:915–934. doi: 10.2165/00003088-200544090-00003. [DOI] [PubMed] [Google Scholar]
- 58.Stanat S.J., Carlton C.G., Crumb W.J., Jr., Agrawal K.C., Clarkson C.W. Characterization of the inhibitory effects of erythromycin and clarithromycin on the HERG potassium channel. Mol. Cell. Biochem. 2003;254:1–7. doi: 10.1023/a:1027309703313. [DOI] [PubMed] [Google Scholar]
- 59.Taylor G., Houston J.B., Shaffer J., Mawer G. Pharmacokinetics of promethazine and its sulphoxide metabolite after intravenous and oral administration to man. Br. J. Clin. Pharmacol. 1983;15:287–293. doi: 10.1111/j.1365-2125.1983.tb01501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Traebert M., Dumotier B. Antimalarial drugs: QT prolongation and cardiac arrhythmias. Expert Opin. Drug Saf. 2005;4:421–431. doi: 10.1517/14740338.4.3.421. [DOI] [PubMed] [Google Scholar]
- 61.Van Haarst A.D., Van’ T Klooster G.A., Van Gerven J.M., Schoemaker R.C., Van Oene J.C., Burggraaf J., Coene M.C., Cohen A.F. The influence of cisapride and clarithromycin on QT intervals in healthy volunteers. Clin. Pharmacol. Ther. 1998;64:542–546. doi: 10.1016/S0009-9236(98)90137-0. [DOI] [PubMed] [Google Scholar]
- 62.Walker R.B., Kanfer I. Pharmacokinetics of cyclizine following intravenous administration to human volunteers. Eur. J. Pharm. Sci. 1996 [Google Scholar]
- 63.Wishart D.S., Feunang Y.D., Guo A.C., Lo E.J., Marcu A., Grant J.R., Sajed T., Johnson D., Li C., Sayeeda Z., Assempour N., Iynkkaran I., Liu Y., Maciejewski A., Gale N., Wilson A., Chin L., Cummings R., Le D., Pon A., Knox C., Wilson M. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. 2018;46:D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]